

Abstract
Patient: Male, 9
Final Diagnosis: Hyperphosphatemic familial tumoral calcinosis
Symptoms: —
Medication: —
Clinical Procedure: Ortopantomography
Specialty: Dentistry
Objective:
Rare disease
Background:
Hyperphosphatemic familial tumoral calcinosis (HFTC) is to a rare autosomal recessive disorder characterized by cutaneous and sub-cutaneous calcified masses, usually adjacent to large joints. The aim of the current study was to report on the clinico-pathological features of a patient with HFCT, with emphasis on alterations in the jawbones and teeth and the subsequent therapeutic interventions.
Case Report:
A 13-year-old male patient with HFTC diagnosis came to our attention for dental anomalies and maxillary and mandibular hypoplasia. OPT highlighted multiple impacted teeth, short and bulbous teeth, and pulp chamber and canal obliterations. Lateral cephalometric radiograms pointed out retrusion of both jaws, skeletal class II malocclusion, and deep-bite. He underwent orthopedic, orthodontic, conservative, and surgical treatments, allowing the correction of maxillo-facial and dental abnormalities and dysmorphisms without adverse effects. The surgical samples were sent for conventional and confocal laser scanning microscope (CLSM) histopathological examination, which highlighted several metaplastic micro- and macro-calcifications in the soft tissues, and typical islands of homogenous, non-tubular, dentino-osteoid calcified structures in dentinal tissues.
Conclusions:
The management of maxillo-facial abnormalities in patients affected by HFTC is very difficult and, requires a combined therapeutic approach. To date, very few indications have been published in the literature.
MeSH Keywords: Body Dysmorphic Disorders, Rare Diseases, Hyperphosphatemia
Background
Familial tumoral calcinosis (FTC) is a clinically and genetically heterogeneous group of inherited disorders characterized by cutaneous and sub-cutaneous calcified masses, usually adjacent to large joints, such as hips, shoulders, and elbows [1]. FTC is an extremely rare condition, occurring without sex predilection, which has been mainly reported in patients from Africa and the Middle East and, to a lesser extent, in white individuals [2]. Although it was initially reported as an inherited, autosomal, dominant disease [3], more recent studies established that all forms of FTC show autosomal recessive inheritance [4–6]. Smack et al. first described normophosphatemic (NFTC) and hyperphosphatemic (HFTC) variants in 1996. NFTC seems to be caused by missense mutations in SAMD9 and initially manifests with non-specific erythematous rash and mucosal inflammation during the first year of life [7]. Inflammatory manifestations in the oral cavity (e.g., gingivitis) can be particularly debilitating [8]. The erythematous rash is usually followed by rapid development of calcified nodules with ulceration of the overlying skin and chalky exudate. NFTC occurs in the range of normal circulating levels of phosphate (2.5–4.5 mg/dl), as well as normal renal and intestinal resorption of phosphate [7]. Unremitting pain and infections are the major causes of morbidity. NFTC may resemble dystrophic calcinosis, an acquired form of skin calcinosis possibly complicating vascular diseases, cancer, or autoimmune disorders, in which calcifications occur as a consequence of prior tissue inflammation [9–12]. HFTC was initially associated with mutations in GALNT3 (2q24-q31), a gene encoding for a glycosyltransferase that plays a major role in regulating phosphate levels (phosphate homeostasis). The GALNT3 protein regulates the activity of the protein fibroblast growth factor 23 (FGF23), produced by osteocytes and inducing decreased renal phosphate resorption. More than 10 mutations have so far been reported in GALNT3, all resulting in a loss of function. Both familial and sporadic forms of HFTC have been recognized and GALNT3 mutations having been identified in most instances and associated with loss of function of FGF23. Recent studies in patients without GALNT3 alterations demonstrated the occurrence of FGF23recessive mutations [5,13–15] or missense mutations of KL, a gene encoding for the klotho protein, also involved in the FGF23 signaling pathway. All such conditions result in clinical manifestations closely resembling HFTC [6]. HFTC generally is characterized by relatively late onset during the first to the third decade of life [16,17], and slowly growing calcified masses with predilection for the peri-articular tissues overlying large joints, the hip being more frequently involved. The calcified masses are asymptomatic, but progressively grow in a pseudo-neoplastic fashion, thus causing ulcers, pain, and secondary infections, which may occasionally become fatal. HFTC patients may also have pulmonary hypertension and lung restrictive diseases [2]. HFTC patients invariably have increased levels of circulating phosphate (>5–7 mg/dl) due to decreased fractional phosphate excretion through the renal proximal tubules and increased intestinal resorption [20]. Calcium serum levels usually are within the normal range, and 1,25-dihydroxyvitamin-D levels may be normal or inappropriately increased [21]. Dental manifestations, including pulp calcifications and obliteration of the pulp cavity, although occasionally reported, may be clinically prominent [18,19]. HFTC may simulate metastatic calcinosis, another acquired form of calcinosis, characterized by the deposition of calcified material and occasionally detected in patients with chronic renal failure, hyperparathyroidism, or sarcoidosis [5,13–15]. Morphologically, pseudo-tumoral calcification seem to develop in 2 major stages: an active phase, characterized by multinucleated giant cells and macrophages surrounding calcified deposits in the dermis, and a subsequent chronic or inactive phase, associated with prominent collagenous deposition [22]. Biochemical analyses of extruded calcified masses indicated that they are mainly composed of calcium hydroxyapatite, including amorphous calcium carbonate and phosphate [23]. An extensive list of therapeutic approaches can be found in the literature, the efficacy of which has never been definitively established. Steroids, colchicine, and aluminum hydroxide have been used, especially in the past [13,24,25], while successful treatment with diltiazem, a calcium channel blocker, has been reported in a few cases [26]. Bisphosphonates have also been occasionally and effectively administered [27]. Nevertheless, large or painful calcified masses may require surgical removal or laser therapy [28]. The aim of the current study was to report the clinico-pathological features of a patient with HFTC, with emphasis on alterations of the jawbones and teeth, and the subsequent therapeutic interventions.
Case Report
A 13-year-old male patient, who had been diagnosed with HFTC at the age of 9, came to our attention in the Oral Surgery Unit of the University of Bari Aldo Moro. His medical history showed the initial onset at the age of 7 and was characterized by long-standing night leg pain that was resistant to NSAID therapy. Subsequently, the patient developed mobile calcified masses in both hips and left elbow, which impaired joint mobility (Figure 1). Based on previous radiological and CT studies, which highlighted intra- and peri-articular radiolucent/radiopaque lesions (Figure 2), the provisional diagnoses of ossifying fibroma, osteogenesis imperfecta, and chronic osteomyelitis had been proposed.
Figure 1.
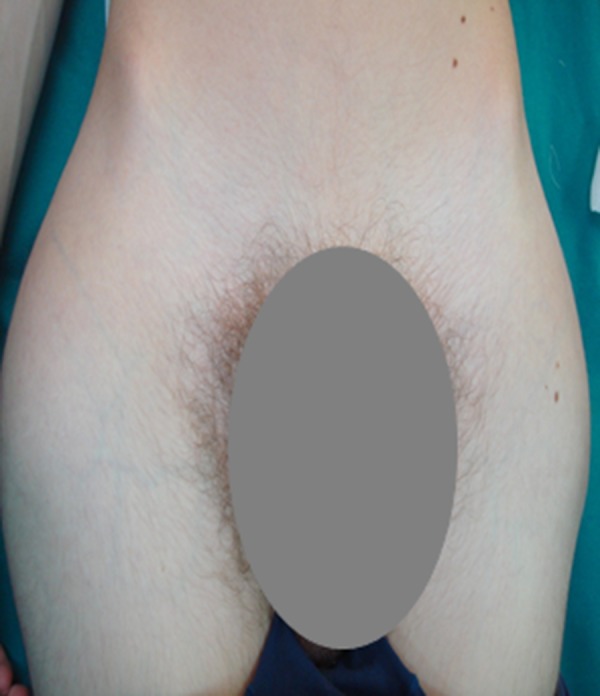
Clinical examination. Mobile calcified masses in both hips.
Figure 2.
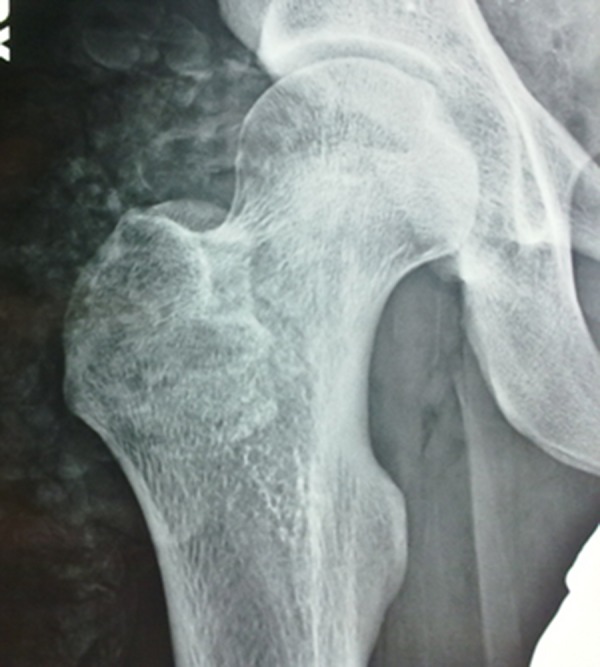
Right hip radiological examination. Intra- and periarticular radiolucent/radiopaque lesions.
Following biochemical blood tests showing increased levels of phosphate (7,6 mg/dl) and parathyroid hormone (77.4 pg/ml), the diagnosis of HFTC was proposed and subsequently confirmed by the genetic test, which detected a homozygous transition mutation of the first GALNT3 intron in which the original A-T base pair was replaced by G-C. Molecular analyses extended to the parents showed that both the mother and father presented the same heterozygous mutation of the GALNT3 gene. The patient was subsequently treated by lanthanum carbonate hydrate (750 mg/die) and sodium ibandronate (150 mg once a month), which considerably reduced the size of the calcified nodules that were surgically removed. Following hospital admission, at oral examination we noticed several dental anomalies, such as enamel hypoplasia, maxillary and mandibular hypoplasia, and crossbite (Figure 3). Orthopantomography (OPT) highlighted multiple impacted teeth (teeth 13, 23, 33, and 43), short and bulbous teeth, and pulp chamber and canal obliterations (Figure 4).
Figure 3.
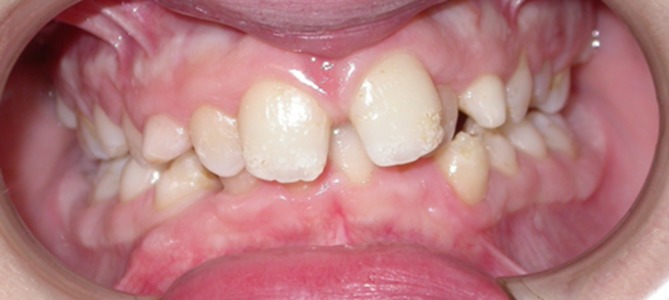
Oral examination. Dental anomalies such as enamel hypoplasia, malocclusion as lower dental overcrowding and crossbite of the 2.2.
Figure 4.
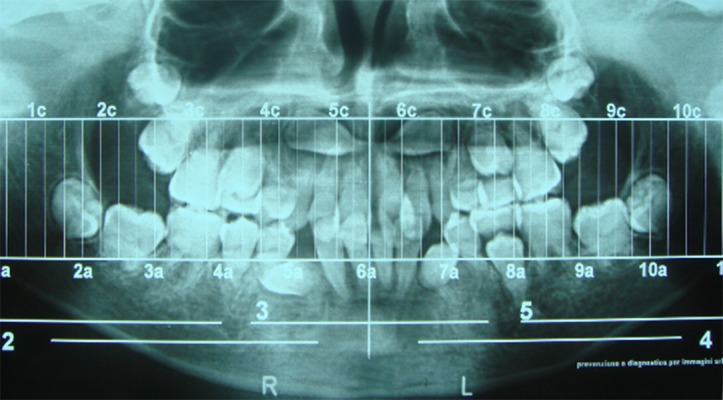
Pre-treatment OPT. Multiple impacted teeth (1.3, 2.3, 3.3, and 4.3), short and bulbous roots, and pulp chamber and canal obliterations.
Lateral cephalometric radiogram and Roth-Jarabak cephalometric analysis revealed retrusion of both jaws (SNA angle: 79°, SNB angle: 74.8°), skeletal class II malocclusion (ANB angle: 4.2°), and dental deep-bite. The patient, after providing informed consent for diagnostic and therapeutic procedures and possible use of biologic samples for research purposes, underwent conservative treatment consisting of dental composite sealing because to possibly decreased tooth decay risk due to enamel anomalies. Subsequently, we performed orthopedic treatments consisting of rapid palatal expansion to correct maxillary hypoplasia on the coronal plane and orthodontic treatments using a lingual arch to protrude the lower incisors and brackets on teeth buccal surfaces with the Straight Wire technique, using round nickel-titanium wires with small diameter.
During orthodontic treatment and after bisphosphonates therapy discontinuation, the patient underwent surgical dental extraction (teeth 38, 48, and the impacted 43) and buccal gingivectomy in 43 region after extraction to restore gingival anatomy. Both dental and soft tissue surgical samples were sent for histological examination. The orthodontic treatment lasted about 3 years and, at the end, retainers were applied on both the upper and lower jaws to hold tooth position. At the end of the orthodontic therapy, extraction of both the impacted upper canines with associated cysts, enucleation, and extraction of decayed 47 were performed; the patient had 37 spontaneous exfoliations due to the presence of root anomalies; these specimens were also sent for histological examination. All samples were fixed in 10% buffered formalin for 48 h; subsequently, the teeth were divided into 2 parts, one of which was decalcified in Mielodec™, paraffin-embedded, and stained with hematoxylin-eosin and Picrosirius red, along with soft tissue samples. Undecalcified teeth were dehydrated in an ascending series of ethanol, infiltrated with propylene-oxide and Epon-Araldite resin, embedded in Epon-Araldite, and cut in 8–12-µm–thick sections with both an automatic rotative and a manual system of grinding, with water cooling. Histological examination was done using a Nikon Eclipse E600 microscope (Nikon Corporation, Tokyo, Japan) equipped with Argon-ion and Helium-Neon lasers emitting at 488 and 543 nm wavelengths, which allows both optical and confocal laser scanning analyses. The Nikon EZ C1 software (Nikon Corporation, ver. 2.10 Coord Automatisering) was used for bi-dimensional image processing.
Orthopedic-orthodontic treatments associated with extraction of impacted or abnormal teeth allowed the correction of skeletal class II by extension of the mandible and teeth alignment, thus achieving dental class I (Figures 5 and 6). Relevant adverse effects such as root resorption or osteonecrosis of the jaws were not noticed.
Figure 5.
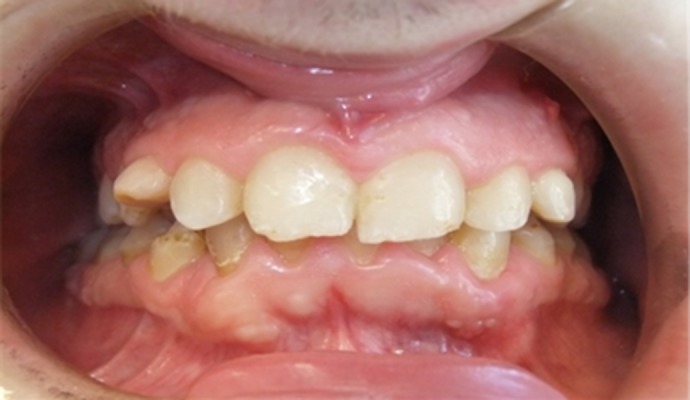
Clinical photograph at the end of the orthopedic, orthodontic, conservative, and surgical treatments. Teeth alignment and achievement of dental class I.
Figure 6.
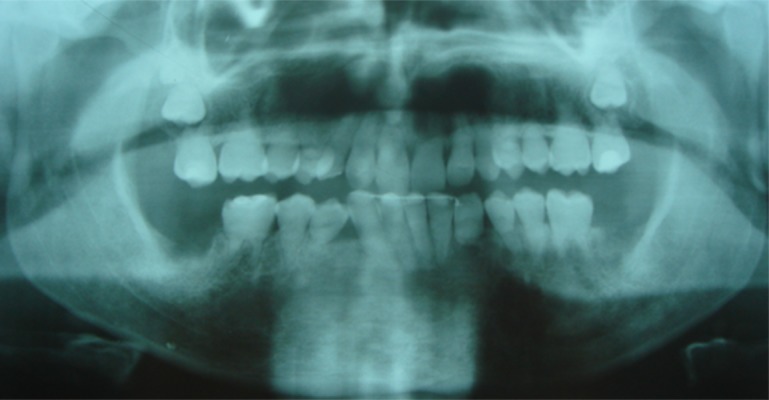
Post-treatment OPT. Teeth alignment and post-orthodontic canine-to-canine bonded retainer on the lower jaw.
Conventional histological examination of soft tissue specimens showed several metaplastic micro and macro-calcification with different grades of mineralization and aggregates in spherules in the surrounding connective tissue. At higher magnification we noticed the presence of calcified deposits in the context of minute nerve bundles of the buccal mucosa (Figure 7A, 7B).
Figure 7.
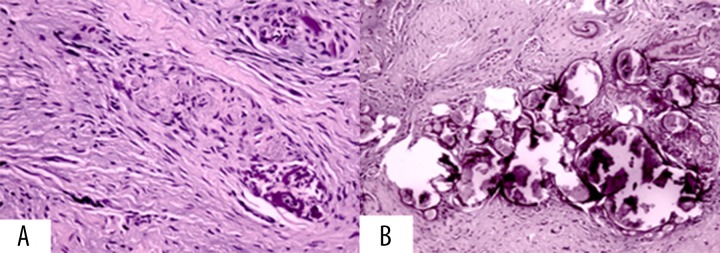
Soft tissues conventional histological examination. (A) Metaplastic micro- and macro-calcification with different grades of mineralization and aggregates in spherules in the surrounding connective tissue. (B) At higher magnification we noticed the presence of calcified deposits in minute nerve bundles of the buccal mucosa.
Microscopically, dental samples were characterized by typical islands of homogenous, non-tubular, dentino-osteoid calcified structures in dentinal tissues (Figure 8A). CSLM showed dentinal dysplasia with osteoid-like material intermingled with remnants of mature mucous connective tissue (Figure 8B).
Figure 8.
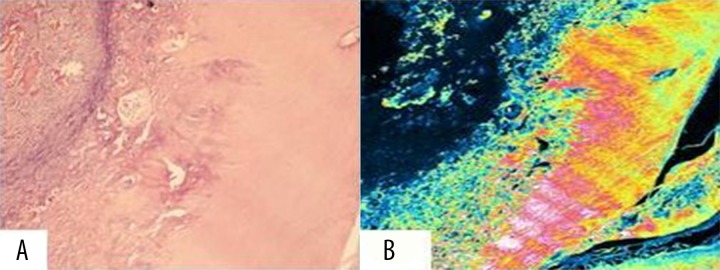
Hard tissues conventional histological and confocal laser scanning analyses. (A) Typical islands of homogenous, non-tubular, dentino-osteoid calcified structures in dentinal tissues. (B) Dentinal dysplasia with osteoid-like material, intermingled with remnants of mature mucous connective tissue.
Discussion
Familial tumoral calcinosis (FTC) is a rare autosomal recessive metabolic disorder characterized by progressive deposition of calcium phosphate crystals in periarticular spaces and soft tissues [29]. Teutschlaender [30] mentioned 2 French dermatologists, Giard and Duret, as the first to clinically describe FTC in 1898 and he himself described the same condition under the name of lipocalcinogranulomatosis in 1935. The current term, tumoral calcinosis, was proposed by Inclan et al. in 1943 and subsequently was widely adopted. FTC can occur in the setting of hyperphosphatemia (HFTC) or normophosphatemia (NFTC), the former resulting from distinct gene mutations involving FGF23, the FGF-23 glycosylating enzyme GALNT3, and the FGF23 co-receptor α-klotho (αKL) [29]. Clinically, HFTC is characterized by slowly growing calcified masses, mainly developing at periarticular sites, with predilection for the skin areas overlying large joints, particularly the hips. These lesions initially are asymptomatic but then become painful and may cause limitation of joint movements following increase in size (up to 1.5 kg). Ulceration may induce pain and is occasionally associated with secondary infections [1]. In the case reported here, the patient had long-standing night leg pain resistant to NSAID therapy, a still unreported sign of this disease. Furthermore, we also noticed maxillary and mandibular hypoplasia associated with dysmorphisms on coronal, axial, and sagittal planes, impacted teeth and other tooth anomalies such as short and bulbous shape, and pulp chamber and canal obliteration. While teeth anomalies have been occasionally reported [18,19], we were unable to find previous descriptions of the complex dysmorphic features detected in the current case. Patients affected by HFTC usually manifest hyperphosphatemia (7 mg/dl in the current case) due to increased intestinal and renal phosphate resorption [1], normo-calcemia, and normal or increased levels of 1,25-dihydroxyvitamin-D [20,21]. The differential diagnosis of HFTC includes: NFTC, in which the phosphate serum levels are lower and the calcium deposits are smaller; metastatic calcinosis, in which patients also show chronic renal failure, hyperparathyroidism and sarcoidosis; and hyperostosis-hyperphosphatemia syndrome (HHS). As GALNT3 mutations are present also in HHS [31], it may represent a different phenotypic expression of HFTC showing preferential bone involvement and lack of ectopic calcifications due to extra-articular calcium deposits. Nevertheless, the skeletal and extra-skeletal manifestations and the biochemical alterations of the previously mentioned diseases are rather unspecific, thus suggesting that genetic analyses may be necessary to distinguish HFTC from related disorders. Morphologically, calcified pseudo-tumoral masses in HFTC seem to develop in a 2-step process: a phagic phase, involving multinucleated giant cells and macrophages surrounding the calcified deposits in the peri-articular soft tissues; and a chronic or inactive phase associated with dense fibrous tissue deposition [22]. Conventional histological examination of the soft tissue specimens showed several metaplastic micro-and macro-calcifications, with variable mineralization, and aggregates in spherules in the surrounding connective tissues. At higher magnification, we noticed calcified deposits in the small nerve bundles of the buccal mucosa. Microscopically, the dental samples were characterized by typical islands of homogenous, non-tubular, dentino-osteoid calcified structures in dentinal tissues. CSLM showed dentinal dysplasia with osteoid-like material, intermingled with remnants of mature mucous connective tissue. Surgical removal or laser therapy may be indicated for cosmetic purposes or pain relief [28]. Medical treatments are mostly based on limited clinical evidence and may be inconsistent or frustrating [29]. They are generally aimed at decreasing intestinal absorption of phosphates and mostly consist of dietary phosphate deprivation combined with aluminum and magnesium-based phosphate binders. Long-term treatments with acetazolamide, a carbonic anhydrase inhibitor with phosphaturic effects [32–34], resulted in inconstant resolution of larger HFTC lesions [35, 36], while successful treatments with diltiazem, a calcium channel blocker, or bisphosphonates have been reported in a few cases [26,27]. In addition, experimental medical treatments consisting of FGF23 or FGF23-agonists have been proposed in animal models to possibly increase phosphate renal excretion and lower serum phosphate; if proven effective, such treatments may be used in subjects with GALNT3 or FGF23 mutations [29]. Based on the case reported here, the administration of lanthanum carbonate hydrate (750 mg/die) and sodium ibandronate (150 mg once a month) induced decrease in size of the calcified nodules, which required subsequent surgical removal to achieve proper articular function. The management of maxillo-facial abnormalities in patients with HFTC was made difficult by the multiplicity and diversity of tooth alterations, the simultaneous disregulation of bone metabolism, and the concurrent drug therapies. The presence of enamel anomalies increased caries risk, bulbous roots, and pulp calcifications did not allow endodontic treatments of caries complications and the young age of the patient contraindicated the extraction of permanent teeth, thus suggesting the application of dental sealants for caries prevention as a better treatment option. In addition, orthopedic-orthodontic treatments were difficult as the shorter bulbous dental roots hindered the use of conventional forces and promoted the use of continuous forces of lower strength to possibly avoid adverse effects such as root resorption. Finally, surgical treatment, including the extraction of impacted teeth, required discontinuation of oral bisphosphonates therapy to avoid severe, well-known, and frequent complications such as osteonecrosis of the jaws. In our patient we did not observe surgical treatment failure or adverse effects by observing a “drug-holiday” of at least 3 months before and after surgery.
Conclusions
The management of maxillo-facial abnormalities in patients with hyperphosphatemic tumoral calcinosis is very difficult, requires a combined therapeutic approach and, to date, very few indications have been published and accepted in the literature. Nevertheless, treatment guidelines are a major goal to possibly improve quality of life for these patients.
Acknowledgments
We acknowledge the participation of the study patient and his family.
Footnotes
Competing interests
The authors declare that they have no conflicting financial interests.
References:
- 1.Sprecher E. Familial Tumoral Calcinosis: From Characterization of a Rare Phenotype to the Pathogenesis of Ectopic Calcification. J Invest Dermatol. 2010;130:652–60. doi: 10.1038/jid.2009.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carmichael KD, Bynum JA, Evans EB. Familial tumoral calcinosis: a forty-year follow-up on one family. J Bone Joint Surg Am. 2009;91:664–71. doi: 10.2106/JBJS.G.01512. [DOI] [PubMed] [Google Scholar]
- 3.Lyles KW, Burkes EJ, Ellis GJ, et al. Genetic transmission of tumoral calcinosis: autosomal dominant with variable clinical expressivity. J Clin Endocrinol Metab. 1985;60:1093–96. doi: 10.1210/jcem-60-6-1093. [DOI] [PubMed] [Google Scholar]
- 4.Topaz O, Indelman M, Chefetz I, et al. A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis. Am J Hum Genet. 2006;79:759–64. doi: 10.1086/508069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benet-Pages A, Lorenz-Depiereux B, Zischka H, et al. FGF23 is processed by proprotein convertases but not by PHEX. Bone. 2004;35:455–62. doi: 10.1016/j.bone.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Ichikawa S, Sorenson AH, Austin AM, et al. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology. 2009;150:2543–50. doi: 10.1210/en.2008-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metzker A, Eisenstein B, Oren J, Samuel R. Tumoral calcinosis revisited – common and uncommon features. Report of ten cases and review. Eur J Pediatr. 1988;147:128–32. doi: 10.1007/BF00442209. [DOI] [PubMed] [Google Scholar]
- 8.Gal G, Metzker A, Garlick J, Gold Y, Calderon S. Head and neck manifestations of tumoral calcinosis. Oral Surg Oral Med Oral Pathol. 1994;77:158–66. doi: 10.1016/0030-4220(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 9.Atzeni F, Sarzi-Puttini P, Bevilacqua M. Calcium deposition and associted chronic diseases (atherosclerosis, difuse idiopathic skeletal hyperostosis, and others) Rheum Dis Clin North Am. 2006;32:413–26. doi: 10.1016/j.rdc.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Touart DM, Sau P. Cutaneous deposition diseases. Part II. J Am Acad Dermatol. 1998;39:527–44. doi: 10.1016/s0190-9622(98)70001-5. [DOI] [PubMed] [Google Scholar]
- 11.Conlin PA, Jimenez-Quintero LP, Rapini RP. Osteomas of the skin revisited: a clinicopathologic review of 74 cases. Am J Dermatopathol. 2002;24:479–83. doi: 10.1097/00000372-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 12.Boulman N, Slobodin G, Rozenbaum M, Rsner I. Calcinosis in rheumatic diseases. Semin Arthritis Rheum. 2005;34:805–12. doi: 10.1016/j.semarthrit.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 13.Araya K, Fukumoto S, Backenroth R, et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:5523–27. doi: 10.1210/jc.2005-0301. [DOI] [PubMed] [Google Scholar]
- 14.Chefetz I, Kohno K, Izumi H, et al. GALNT3, a gene associated with hyperphosphatemic familial tumoral calcinosis, is transcriptionally regulated by extracellular phosphate and modulates matrix metalloproteinase activity. Biochim Biophys Acta. 2009;1792:61–67. doi: 10.1016/j.bbadis.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larsson T, Davis SI, Garringer HJ, et al. Fibroblast growth factor-23 mutants causing familial tumoral calcinosis are differentially processed. Endocrinology. 2005;146:3883–91. doi: 10.1210/en.2005-0431. [DOI] [PubMed] [Google Scholar]
- 16.Prince MJ, Schaeffer PC, Goldsmith RS, Chausmer AB. Hyperphosphatemic tumoral calcinosis: association with elevation of serum 1,25-dihydroxycholecalciferol concentrations. Ann Intern Med. 1982;96:586–91. doi: 10.7326/0003-4819-96-5-586. [DOI] [PubMed] [Google Scholar]
- 17.Slavin RE, Wen J, Kumar D, Evans EB. Familial tumoral calcinosis. A clinical, histopathologic, and ultrastructural study with an analysis of its calcifying process and pathogenesis. Am J Surg Pathol. 1993;17:788–802. [PubMed] [Google Scholar]
- 18.Burkes EJ, Jr, Lyles KW, Dolan EA, et al. Dental lesions in tumoral calcinosis. J Oral Pathol Med. 1991;20:222–27. doi: 10.1111/j.1600-0714.1991.tb00423.x. [DOI] [PubMed] [Google Scholar]
- 19.Campagnoli MF, Pucci A, Garelli E, et al. Familial tumoral calcinosis and testicular microlithiasis associated with a new mutation of GALNT3 in a White family. J Clin Pathol. 2006;59:440–42. doi: 10.1136/jcp.2005.026369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White KE, Larsson TE, Econs MJ. The roles of specific genes implicated as circulating factors involved in normal and disordered phosphate homeostasis: frizzled related protein-4, matrix extracellular phosphoglycoprotein, and fibroblast growth factor 23. Endocr Rev. 2006;27:221–41. doi: 10.1210/er.2005-0019. [DOI] [PubMed] [Google Scholar]
- 21.Steinherz R, Chesney RW, Eisenstein B, et al. Elevated serum calcitriol concentrations do not fall in response to hyperphosphatemia in familial tumoral calcinosis. Am J Dis Child. 1985;139:816–19. doi: 10.1001/archpedi.1985.02140100078036. [DOI] [PubMed] [Google Scholar]
- 22.Veress B, Malik MO, El Hassan AM. Tumoral lipocalcinosis: a clinicopathological study of 20 cases. J Pathol. 1976;119:113–18. doi: 10.1002/path.1711190206. [DOI] [PubMed] [Google Scholar]
- 23.Boskey AL, Vigorita VJ, Sencer O, et al. Chemical, microscopic, and ultra-structural characterization of the mineral deposits in tumoral calcinosis. Clin Orthop Relat Res. 1983;178:258–69. [PubMed] [Google Scholar]
- 24.Nakagawa T, Takaiwa T. Calcinosis cutis in juvenile dermatomyositis responsive to aluminium hydroxide treatment. J Dermatol. 1993;20:558–60. doi: 10.1111/j.1346-8138.1993.tb01338.x. [DOI] [PubMed] [Google Scholar]
- 25.Fuchs D, Fruchter L, Fishel B, et al. Colchicine suppression of local inflammation due to calcinosis in dermatomyositis and progressive systemic sclerosis. Clin Rheumatol. 1986;5:527–30. [PubMed] [Google Scholar]
- 26.Ichiki Y, Akiyama T, Shimozawa N, et al. An extremely severe case of cutaneous calcinosis with juvenile dermatomyositis, and successful treatment with diltiazem. Br J Dermatol. 2001;144:894–97. doi: 10.1046/j.1365-2133.2001.04153.x. [DOI] [PubMed] [Google Scholar]
- 27.Mukamel M, Horev G, Mimouni M. New insight into calcinosis of juvenile dermatomyositis: a study of composition and treatment. J Pediatr. 2001;138:763–66. doi: 10.1067/mpd.2001.112473. [DOI] [PubMed] [Google Scholar]
- 28.Chamberlain AJ, Walker NP. Successful palliation and significant remission of cutaneous calcinosis in CREST syndrome with carbon dioxide laser. Dermatol Surg. 2003;29:968–70. doi: 10.1046/j.1524-4725.2003.29261.x. [DOI] [PubMed] [Google Scholar]
- 29.Farrow EG, Imel EA, White KE. Hyperphosphatemic familial tumoral calcinosis (FGF23, GALNT3 and αKlotho) Best Pract Res Clin Rheumatol. 2011;25:735–47. doi: 10.1016/j.berh.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tseutschlaender O. Uber progressive lipogranulomatose der muskulatur. Zugleich ein beitrag zur pathogenese der myopathia osteoplystica progressive. Klin Wochenschr. 1935;13:451–53. [Google Scholar]
- 31.Mikati MA, Melhem RE, Najjar SS. The syndrome of hyperostosis and hyperphosphatemia. J Pediatr. 1981;99:900–4. doi: 10.1016/s0022-3476(81)80013-3. [DOI] [PubMed] [Google Scholar]
- 32.Lufkin EG, Wilson DM, Smith LH, et al. Phosphorus excretion in tumoral calcinosis: response to parathyroid hormone and acetazolamide. J Clin Endocrinol Metab. 1980;50:648–53. doi: 10.1210/jcem-50-4-648. [DOI] [PubMed] [Google Scholar]
- 33.Knox FG, Haas JA, Lechene CP. Effect of parathyroid hormone on phosphate reabsorption in the presence of acetazolamide. Kidn Int. 1976;10:216–20. doi: 10.1038/ki.1976.100. [DOI] [PubMed] [Google Scholar]
- 34.Sinha TK, Allen DO, Queener SF, et al. Effects of acetazolamide on the renal excretion of phosphate in hypoparathyroidism and pseudohypoparathyroidism. J Lab Clin Med. 1977;89:1188–97. [PubMed] [Google Scholar]
- 35.Yamaguchi T, Sugimoto T, Imai Y, et al. Successful treatment of hyperphosphatemic tumoral calcinosis with long-term acetazolamide. Bone. 1995;16:247S–50S. doi: 10.1016/8756-3282(95)00019-a. [DOI] [PubMed] [Google Scholar]
- 36.Garringer HJ, Fisher C, Larsson TE, et al. The role of mutant UDP-N-acetyl-alpha-Dgalactosamine- polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab. 2006;91:4037–42. doi: 10.1210/jc.2006-0305. [DOI] [PubMed] [Google Scholar]