

SUMMARY
N6-methyl-adenosine (m6A) is the most abundant modification on messenger RNAs and is linked to human diseases, but its functions in mammalian development are poorly understood. Here we reveal the evolutionary conservation and function of m6A by mapping the m6A methylome in mouse and human embryonic stem cells. Thousands of messenger and long noncoding RNAs show conserved m6A modification, including transcripts encoding core pluripotency transcription factors. m6A is enriched over 3′ untranslated regions at defined sequence motifs, and marks unstable transcripts, including transcripts turned over upon differentiation. Genetic inactivation or depletion of mouse and human Mettl3, one of the m6A methylases, led to m6A erasure on select target genes, prolonged Nanog expression upon differentiation, and impaired ESC’s exit from self-renewal towards differentiation into several lineages in vitro and in vivo. Thus, m6A is a mark of transcriptome flexibility required for stem cells to differentiate to specific lineages.
INTRODUCTION
Reversible chemical modifications on messenger RNAs have emerged as prevalent phenomena that may open a new field of “RNA epigenetics”, akin to the diverse roles that DNA modifications play in epigenetics (reviewed by (Fu and He, 2012; Sibbritt et al., 2013)). N6-methyl-adenosine (m6A) is the most prevalent modification of mRNAs in somatic cells, and dysregulation of this modification has already been linked to obesity, cancer, and other human diseases (Sibbritt et al., 2013). m6A has been observed in a wide range of organisms, and the methylation complex is conserved across eukaryotes. In budding yeast, the m6A methylation program is activated by starvation and required for sporulation. In Arabidopsis thalania, the methylase responsible for m6A modification, MTA, is essential for embryonic development, plant growth and patterning, and the Drosophila homolog IME4 is expressed in ovaries and testes and is essential for viability (reviewed in (Niu et al., 2013)).
While m6A has been suggested to affect almost all aspects of RNA metabolism, the molecular function of this modification remains incompletely understood (Niu et al., 2013). Importantly, m6A modification(s) are reversible in mammalian cells. Two members of the alpha ketoglutarate-dependent dioxygenases protein family, fat-mass and obesity associated protein (FTO) and ALKBH5 have been shown to act as m6A demethylases (Jia et al., 2011; Zheng et al., 2013). Manipulating global m6A levels has implicated m6A modifications in a variety of cellular processes including nuclear RNA export, control of protein translation and splicing (reviewed in (Meyer and Jaffrey, 2014)). Recently, m6A modification has been suggested to play a role in controlling transcript stability as YTH domain family of “reader” proteins specifically bind m6A sites and recruit the transcripts to RNA decay bodies (Kang et al., 2014; Wang et al., 2014a).
Whereas the DNA methylome undergoes dramatic reprogramming during early embryonic life, the developmental origins and functions of m6A in mammals are incompletely understood. Furthermore, the degree of evolutionary conservation of m6A sites is not known in ESCs. To date, the functions of m6A in mammalian cells have only been examined by RNAi knockdown. Depletion of METTL3 and METTL14 in human cancer cell lines led to decreased cell viability and apoptosis, leading to the interpretation that m6A is important for cell viability (Dominissini et al., 2012; Liu et al., 2014). A recent study reported that depletion of Mettl3 inhibited mouse ESC proliferation and led to ectopic differentiation (Wang et al., 2014b). Here we assess the conservation of the m6A methylome at the level of gene targets and function in mouse and human ESCs. We report the consequences of genetic ablation of Mettl3 in mouse ESCs (mESCs) as well as depletion of METTL3 in human ESCs (hESCs). These experiments led to the unexpected finding that m6A and Mettl3 in particular are not required for ESC growth but are required for stem cells to adopt new cell fates.
RESULTS
Thousands of mESC transcripts bear m6A
To understand the role of the m6A RNA modification in early development, we mapped the locations of m6A modification across the transcriptome of mouse (mESC) and human (hESC) embryonic stem cells by m6A RIP-seq as described. (Dominissini et al., 2012; Meyer et al., 2012) (Methods). For each experiment, libraries were built for multiple biological replicates and concordant peaks for each experiment were used for subsequent bioinformatic analyses.
In mESCs, m6A-seq revealed a total of 9754 peaks in 5578 transcripts (average 2 peaks per transcript), including 5461 mRNAs (of 9923 mRNAs) and 117 lncRNAs. Due to the lower expression levels of lncRNA as a class, our approach likely underestimates the fraction of modified noncoding transcripts (Table S1). Thus, thousands of mESC transcripts, including mRNAs and lncRNAs, are m6A modified.
m6A in mRNAs of mESC core pluripotency factors
We found that mRNAs encoding the core pluripotency regulators in mESCs were modified with m6A (Dunn et al., 2014; Young, 2011), including Nanog, Klf4, Myc, Lin28, Med1, Jarid2 and Eed, whereas Pou5f1 (also known as Oct4) lacked m6A modification (Figure 1A, B). We confirmed m6A-seq results with independent m6A IP-qRT-PCR. (Figure S1A) and m6A-IP followed by Nanostring nCounter analysis (m6A-string) (Table S2). These validation results suggest that the m6A-seq data are accurate and robust. The top group of modified genes, based on degree of modification, was enriched for several functional groups, including: chordate embryonic development, embryonic development, gastrulation and cell cycle (Figure 1C). Thus, in mESCs, m6A targets include the ESC core pluripotency network and transcripts with dynamically controlled abundance during differentiation.
Figure 1. Topology and characterization of m6A target genes.
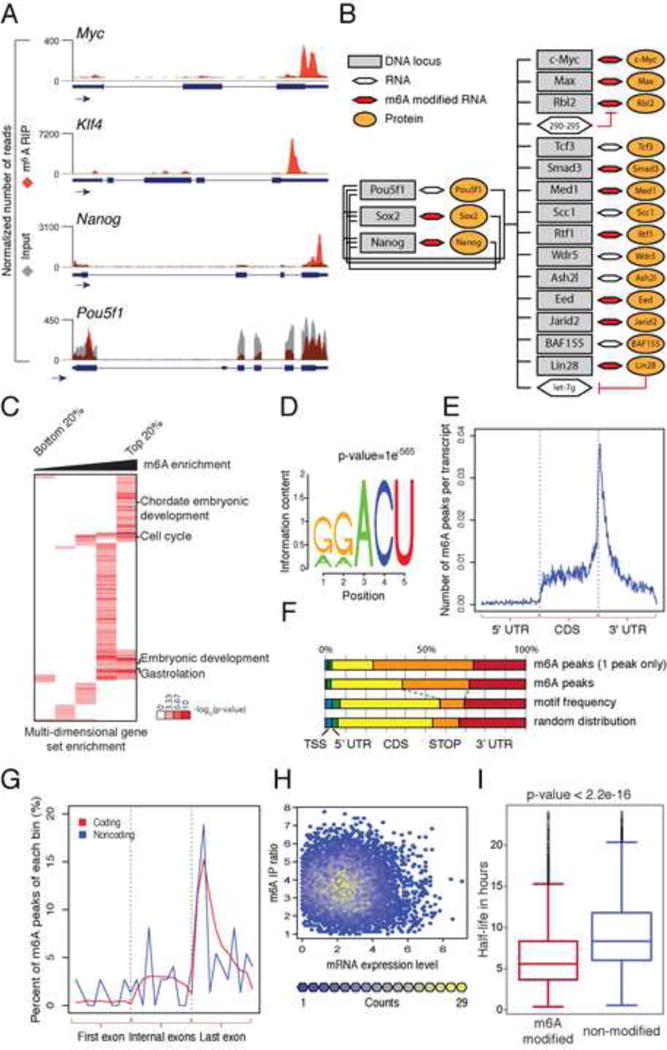
(A) UCSC Genome browser plots of m6A-seq reads along indicated mRNAs. Grey reads are from non-immunoprecipitated input libraries and red reads from anti-m6A immunoprecipitation libraries. The y-axis represents normalized number of reads. Blue thick boxes represent the open reading frame while the blue line represents the untranslated regions. See also Figure S1A and Table S1 and S2. (B) Model of genes involved in maintenance of stem cell state (adapted from Young et al., 2011). Red hexagons represent modified mRNAs. (C) Heatmap with log10 (p-vlaue) of gene set enrichment analysis for m6A modified genes. (D) Sequence motif identified after analysis of m6A enrichment regions. See also Figure S1B, S1C. (E) Normalized distribution of m6A peaks across 5′ UTR, CDS and 3′UTR of mRNAs for peaks common to all samples. (F) Graphical representation of frequency of m6A peaks and methylation motifs in genes, divided into 5 distinct regions. (G) Distribution of m6A peaks across the length of mRNAs (n=5070) and non-coding RNAs (n=51). See also Figure S1D, S1E, S1F, S1G and S1H. (H) Scatter plot representation of m6A enrichment score (on the X axis) and gene expression level (on the Y axis) for each m6A peak. See also Figure S1I. (I) Box plot representing the half-life for transcripts with at least one modification site and transcripts with no modification site identified. See also figure S1J and S1K.
m6A location and motif in mESCs suggest a common mechanism shared with somatic cells
De novo motif analysis of mESC m6A sites specifically identified the previously described RRACU m6A sequence motif in somatic cells (Figure 1D, S1B) (reviewed in (Meyer and Jaffrey, 2014)). Furthermore, like somatic cells, m6A sites in mESC are significantly enriched near the stop codon and beginning of the 3′ UTR of protein coding genes (Figure 1E and 1F), as previously described for somatic mRNAs. Although the largest fraction of m6A sites was within the coding sequence (CDS, 35%), the stop codon neighborhood is most enriched, comprising 33% of m6A sites while representing 12% of the motif occurrence. In genes with only one modification site, this bias is even more pronounced (Figure 1F). Comparison of transcript read coverage between input and wild type revealed no bias for read accumulation around the stop codon in the input sample (Figure S1C).
In addition to the last exon, which often includes the stop codon and 3′-UTR, we found a strong bias for m6A modification occurring in long internal exons (median exon length of 737bp vs. 124 bp; P<2.2×10−16; two-sided Wilcoxon test), even when the number of peaks per exon was normalized for exon length or motif frequency (Figure S1D–F). These results suggest the possibility that processing of long exons is coupled mechanistically to m6A targeting through as yet unclear systems and/or that m6A modification itself may play a role in controlling long exon processing. The topological enrichment of m6A peaks surrounding stop codons in mRNAs is a poorly understood aspect of the m6A methylation system. We sought to understand if there was a topological enrichment or constraint on m6A modification in non-coding RNAs (ncRNAs), which lack stop codons. We parsed both classes of RNAs with three or more exons into three normalized bins including the 1st, all internal and last exon. We observed an enrichment of m6A near the last exon-exon splice junction for both coding and ncRNAs and toward 3′ end of single-exon genes (Figure 1G, S1G–H), suggesting that the 3′ enrichment of m6A peaks can occur independently of translation or splicing. Together, the location and sequence features we identified in mESCs suggest a mechanism for m6A deposition that is similar if not identical in somatic cells.
m6A is a mark for RNA turnover
We next tested if transcript levels are correlated with the presence of m6A modification. Comparison of m6A enrichment level versus the absolute abundance of RNAs revealed no correlation between level of enrichment and gene expression (Figure 1H). A separate, quartile based analysis found a higher percentage of m6A-modified transcripts in the middle quartiles of transcript abundance (Figure S1I). Thus, our analysis suggests that m6A modification is not simply a random modification that occurs on abundant cellular transcripts; rather, m6A preferentially marks transcripts expressed at a medium level.
To further define potential mechanisms of m6A function, we asked whether m6A-marked transcripts differ from unmodified transcripts at the level of transcription, RNA decay, or translation by leveraging published genome-wide datasets in mESCs. RNA polymerase II occupancy at the promoters encoding both unmodified and m6A-marked RNAs is similar (Figure S1J). In contrast, m6A-marked transcripts had significantly shorter RNA half-life — 2.5 hours shorter on average (p= < 2.2−16, Figure 1I), and increased rate of mRNA decay (average decay rate of 9 min vs. 5.4 min for m6A vs. unmodified, p= < 2.2−16). m6A modified transcripts have slightly lower translational efficiency than unmodified transcripts (1.32 vs. 1.51, respectively) (Ingolia et al., 2011) (Figure S1K). These results suggest that m6A is a chemical mark associated with transcript turnover.
Mettl3 knockout decreases m6A and promotes ESC self-renewal
To understand the role of m6A methylation in ESC biology, we chose to inactivate Mettl3, encoding one of the components of the m6A methylase complex. To date no genetic study of Mettl3 has been performed to rigorously define its requirement for m6A modification, as all studies have relied on knock down. We targeted Mettl3 by CRISPR-mediated gene editing, and generated several homozygous Mettl3 Knockout (KO) mESC lines. DNA sequencing confirmed homozygous stop codons that terminate translation within the first 75 amino acids, and immunoblot analysis confirmed the absence of Mettl3 protein (Figure 2A, Figure S2A). Two dimensional thin layer chromatography (2D-TLC) showed a significant (~60%) but incomplete reduction of m6A in Mettl3 KO mESC (Figure 2B and Figure S2B). Contrary to a recent publication (Wang et al., 2014b), Mettl3 KO slightly reduced but did not prevent the stable accumulation of Mettl14 (Figure S2C). These experiments provide formal genetic proof that Mettl3 is a major, but not the sole, m6A methylase in mESC.
Figure 2. Characterization of Mettl3 knock out cells.
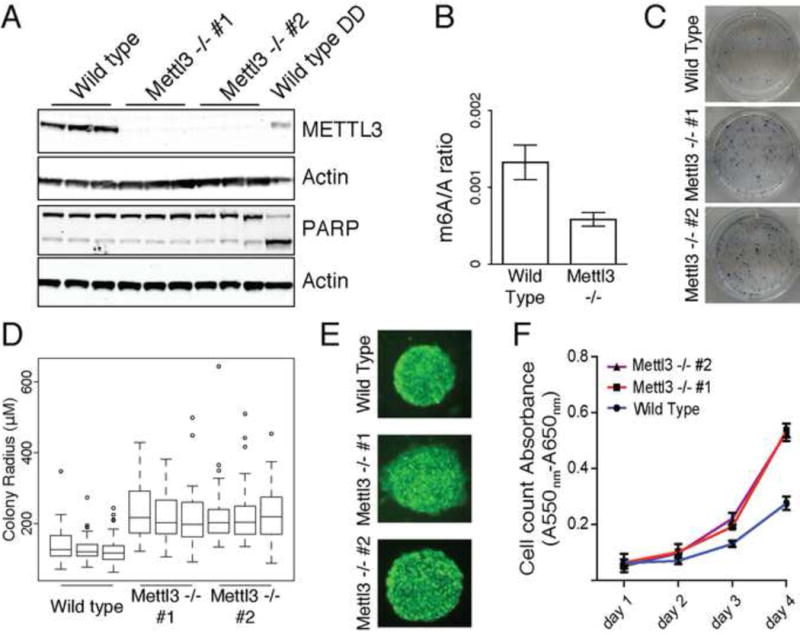
(A) Western blot for Mettl3 and PARP in wild type and two cell lines with CRISPR induced loss of protein (DD: DNA damaging agent). Actin is used as loading control. See also Figure S2A. (B) m6A ratio determined by 2D-TLC in wild type and Mettl3 KO. Error bars represent standard deviation of 3 biological replicates in all panels. See also Figure S2B and S2C. (C) Alkaline phosphatase staining of wild type and Mettl3 knock out cells. See also Figure S2D and S2E. (D) Box plot representation of colony radius for wild type and Mettl3 mutant cells. Experiments were performed in triplicate, with at least 50 colonies measured for each replicate. (E) Nanog staining of colonies of wild type and two cell lines with CRISPR induced loss of protein. (F) Cell proliferation assay of wild type and two cell lines with CRISPR induced loss of Mettl3 protein. See also Figure S2F, S2G and S2H.
Contrary to the expectation in the literature, the Mettl3 KO mESCs are viable and surprisingly demonstrated improved self-renewal. Mettl3 KO mESCs could be maintained indefinitely over months and exhibited low levels of apoptosis, similar to wild type mESCs, as judged by PARP cleavage and Annexin V flow cytometry (Figure 2A, Figure S2D). We next asked whether Mettl3 KO affected the ability of stem cells to remain pluripotent. Mettl3 KO mESC colonies were consistently larger than WT ESCs, and retained the round, compact ESC colony morphology with intense alkaline phosphatase staining comparable to wild type colonies as well as uniform expression of Nanog and Oct4 (Figure 2C, 2D, 2E, Figure S2E and data not shown). Quantitative cell proliferation assay confirmed the increased proliferation rate of KO over WT mESCs (Figure 2F). These observations suggest that Mettl3 KO enables enhanced mESC self-renewal. To rule out potential off-target effects from CRISPR-mediated gene targeting, we used an orthogonal approach to knockdown Mettl3 in mESCs. Two independent short hairpin RNAs (shRNAs) knocked down Mettl3 to ~20% (Figure S2F). 2D-TLC showed a ~40% loss of m6A in poly(A) RNAs (Figure S2G), and apoptosis assays confirmed lack of cell death induction. Importantly, Mettl3 depletion also increased mESC proliferation compared to control shRNA for one hairpin (Figure S2H). Thus, two independent approaches confirm that Mettl3 inactivation enhanced self-renewal of ESCs.
Mettl3 KO blocks directed differentiation in vitro and teratoma differentiation in vivo
These findings, coupled with the observation that modified genes tend to have a shorter half-life, suggest that Mettl3, and by extension m6A, is needed to fine-tune and limit the level of many ESC genes, including pluripotency regulators. Since Mettl3 KO cells are capable of self-renewal, we tested their capacity for directed differentiation in vitro toward two lineages: cardiomyocytes (CM) and the neural lineage. While the WT cells were able to generate beating CM (~50% of colonies), only ~3% of Mettl3 KO colonies of two independent clones produced beating CMs. Furthermore, differentiated colonies of Mettl3 KO cells retained high levels of Nanog expression but lacked expression of the CM structural protein Myh6, reflecting a larger number of cells that failed to exit the mESC program in the mutant cells. (Figure 3A and Supplemental Video 1 and 2). Similarly, upon directed differentiation to the neural lineage, we observed a marked difference between the ability of the two cells types to differentiate. To assay for neural differentiation we stained for Tuj1, a beta-3 tubulin expressed in mature and immature neurons. While ~53% of wild type colonies had Tuj1+ projections, less than 6% of Mettl3 KO colonies had Tuj1+ projections in both KO clones (Figure 3B). Additionally, differentiated Mettl3 KO cells showed an impaired ability to repress Nanog and activate Tuj1 mRNA (Figure 3B). To confirm the role of Mettl3 in mESC differentiation in vivo, we injected Mettl3 KO or wild type cells subcutaneously into the right or left flank respectively, of SCID/Beige mice (n=5). Both wild type and Mettl3 KO cells formed tumors consistent in morphology with teratomas. Mutant tumors tended to be larger, in accordance with mutant cell growth curves observed in vitro (Figure 3C). Histological analysis of H&E stained tumor sections revealed consistent differences between the two populations. While both groups of cells formed teratomas that contained some degree of differentiation into all three germ layers, the teratomas derived from KO cells were predominantly composed of poorly differentiated cells with very high mitotic indices and numerous apoptotic bodies, whereas wild type cells differentiated predominantly into neuroectoderm (Figure 3D). Analysis of adjacent sections revealed that the mutant teratomas have markedly higher staining of proliferation marker Ki67 and ESC protein Nanog, which highlight the poorly differentiated cells (Figure 3E, 3F and Figure S3A). Mettl3 KO tumors had higher levels of Nanog, Oct4 and Ki67 mRNAs and lower levels of Tuj1, Myh6 and Sox17 mRNAs (Figure S3B). These results suggest that insufficient m6A leads to a block in ESC differentiation and persistence of a stem-like, highly proliferative state.
Figure 3. Mettl3 loss of function impairs ESC ability to differentiate.
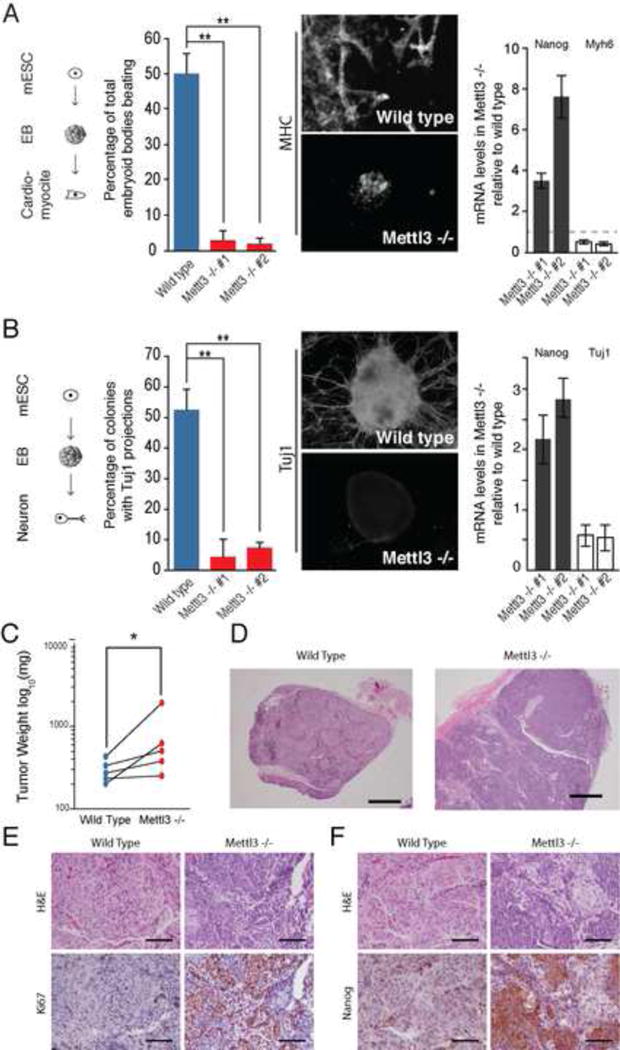
(A) Percentage of embryoid bodies with beating activity in Mettl3 KO and wild type control cells (right panel). Representative images of bodies stained for MHC and DAPI (center panel) and mRNA levels of Nanog and Myh6, measured by qRT-PCR, in Mettl3 KO cells in relation to wild type control cells. Error bars, standard deviation of 3 biological replicates in all panels. * represents p-value < 0.05, t-test (2 tailed). See also Movie S1 and S2. (B) Percentage of colonies with Tuj1 projections in Mettl3 KO and wild type control cells (right panel). Representative images of bodies stained for Tuj1 and DAPI (center panel) and mRNA levels of Nanog and Tuj1, measured by qRT-PCR, in Mettl3 KO cells in relation to wild type control cells. * represents p-value < 0.05, t-test (2 tailed). (C) Weight differences between teratomas generated from wild type and Mettl3 knock out cells. Tumors are paired by animal (n=5). (D) Representative sections of teratomas stained with hematoxylin and eosin at low magnification. Bar=1000 μm. See also Figure S3A. (E and F) Immunohistochemistry with antibody against Ki67 (E) and with antibody against Nanog (F). Bar represents 100 μm. See also Figure S3B.
Mettl3 target genes in mESCs
The incomplete loss of bulk m6A in Mettl3 KO may result because Mettl3 is solely responsible for the methylation of a subset of genes or sites and/or Mettl3 functions in a redundant fashion with another methylase on all m6A-modified genes. To distinguish these possibilities, we mapped the m6A methylome in Mettl3 KO cells. Comparison of the methylomes of wild type vs. Mett3 KO mESCs revealed a global loss of methylation across m6A sites identified in wild type (Figure 4A). We detected changes in 3739 sites (in 3122 genes), including modification sites in Nanog mRNA. Thus, this unbiased analysis suggested a set of targets that rely more exclusively on Mettl3, including Nanog and other pluripotency mRNAs (Figure 4B and 4C) (Table S1). Gene Set Enrichment Analysis confirmed that Mettl3-target genes significantly overlap functional gene sets important for pluripotency, including targets of Ctnnb1 (4.43 ×10−6), targets of Smad2 or Smad3 (1.03 ×10−16), targets of Myc (9.20 ×10−10), targets of Sox2 (4.75 ×10−8), and targets of Nanog (7.18 × 10−8) (Figure 4C), and include five of eleven core ESC regulators such as Nanog, Rlf1, Jarid2, and Lin28 (Figure 4D). Independent validation by m6A RIP followed by Nanostring detection confirmed loss of m6A in Nanog and other mRNAs in KO vs. wild type mESCs (Figure 4E). Further, following transcription arrest by flavopiridol treatment, Nanog mRNA showed delayed turnover in Mettl3 KO cells compared to wild type, consistent with a requirement for m6A in Nanog mRNA turnover (Figure 4F). However, RNA-seq analysis of Mettl3 KO cells revealed modest perturbations in mRNA steady state levels with only ~300 genes demonstrating significant changes over 1.5 fold. Collectively, these results suggest ESC genes are under Mettl3 control, and m6A impacts ESC biology.
Figure 4. Impact of loss of Mettl3 on the mESC methylome.
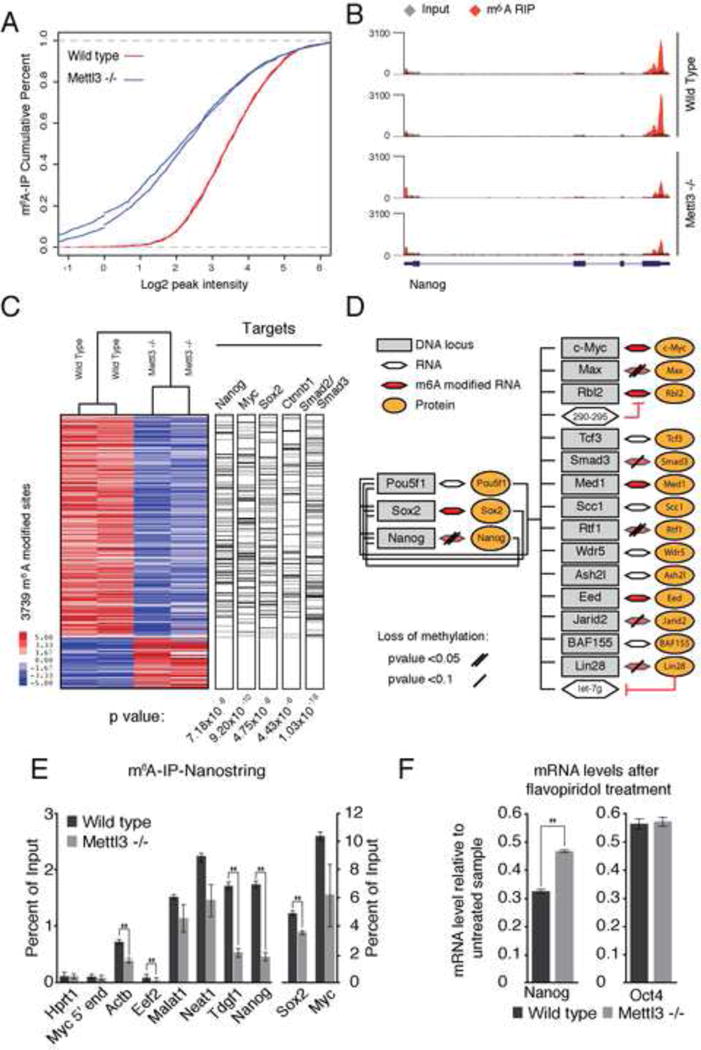
(A) Cumulative distribution function of log2 peak intensity of m6A modified sites. (B) Sequencing read density for input (grey) vs. in m6A IP (red) for Nanog. Y-axis represents normalized number of reads. Gene model as in Figure 1A. (C) Heatmap representing IP enrichment values for peaks with statistically significant difference between wild type and Mettl3 mutant. Bar to the right represent genes in each dataset with a >1.5 fold decrease in IP enrichment values. (D) Model of genes involved in maintenance of stem cell state (adapted from Young et al., 2011), representing transcripts with loss of m6A modification in Mettl3 −/− cells. (E) Percentage of input recovered after m6A IP measured by Nanostring for each mRNA. Error bars, standard deviation of 2 biological replicates. *, p-value < 0.05, t-test (2 tailed). (F) mRNA levels of Nanog and Oct4, measured by qRT-PCR, after PolII inhibition relative to untreated sample in wild type and Mettl3 KO cells. Error bars, standard deviation of 3 biological replicates. *, p-value < 0.05, t-test (2 tailed).
Wide spread m6A modification of human ESCs
The identification of thousands of m6A sites raises the challenge of defining the functional importance of each and every one of the sites. We reasoned that evolutionary conservation provides a powerful and comprehensive metric of function. To this end, we mapped m6A sites in hESCs and during endoderm differentiation to elucidate the patterns and potential conservation of m6A methylome (Figure 5A). In basal state hESCs (Time (T) = 0), m6A-seq identified 16943 peaks in 7871 genes representing 7530 coding and 341 non-coding RNAs. Upon differentiation towards endoderm (T = 48, “endoderm differentiation” thereafter), m6A-seq identified 15613 m6A peaks in 7195 genes representing 6909 coding and 286 non-coding RNAs (Table S3). As shown in Figure 5B, 11322 peaks (6004 genes) were common between the undifferentiated and differentiated hESCs, while 5348 (3979 genes) vs. 4087 peaks (3024 genes) were unique, respectively.
Figure 5. m6A-seq profiling of hESC during endoderm differentiation.
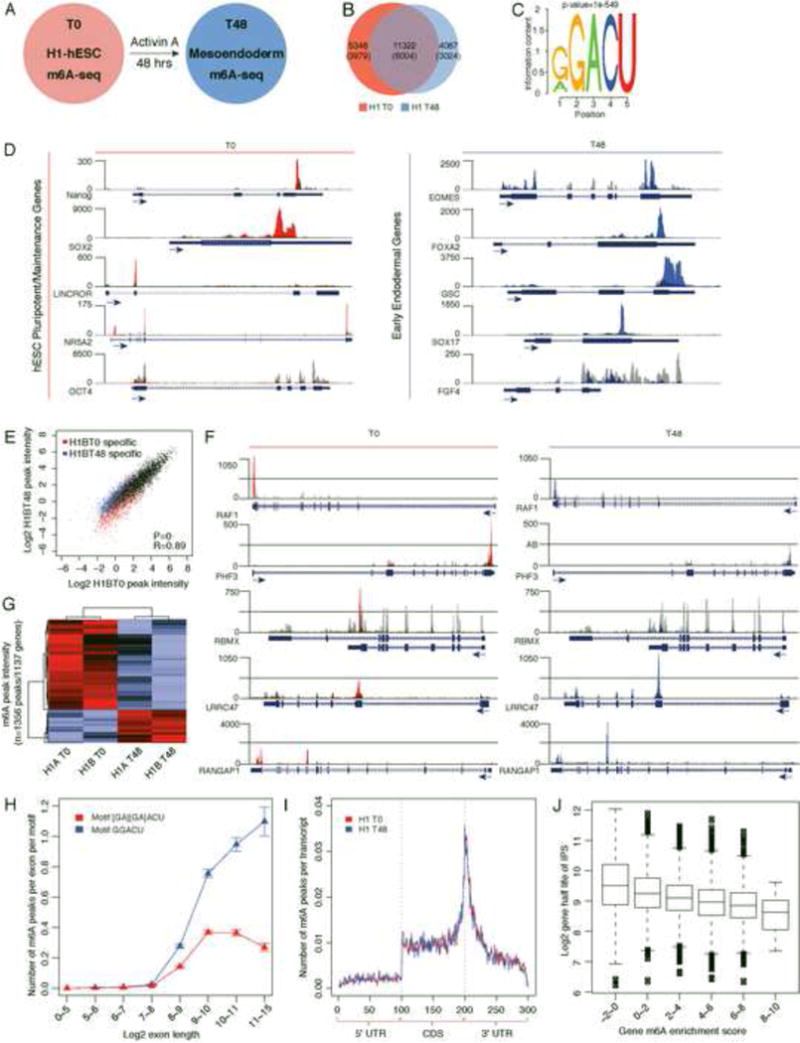
(A) m6A-seq was performed in resting (undifferentiated) human H1-ESCs (T0) and after 48hrs of Activin A induction towards endoderm (mesoendoderm) (T48). (B) Venn diagram of the overlap between high-confidence T0 and T48 m6A peaks. The number of genes in each category is shown in parenthesis. See also Table S3 and S4. (C) Sequence motif identified after analysis of m6A enrichment regions. (D) UCSC Genome browser plots of m6A-seq reads along indicated RNAs. Grey reads are from non-immunoprecipitated control input libraries and red (T0) or blue (T48) reads are from anti-m6A immunoprecipitation libraries. Y-axis represents normalized number of reads; X-axis is genomic coordinates. Key regulators of stem cell maintenance (left) and master regulators of endoderm differentiation (right) are represented. See also Figure S4A. (E) Scatterplot of m6A peak intensities between two different time points (T0 versus T48) of the same biological replicate with only “high-confidence” T0 or T48 specific peaks supported by both biological replicates highlighted. (F) UCSC Genome browser plots of m6A-seq reads along indicated mRNAs in undifferentiated (T0) versus differentiated cells (T48). The grey reads are from non-immunoprecipitated control input libraries. The red and blue reads are from the anti-m6A RIP of T=0 and T=48 samples respectively. (G) Differential intensities of m6A peaks (DMPIs) identify hESC cell states T0 vs T48hrs. Z score scaled Log2 peak intensities of DMPIs are color-coded according to the legend. The peaks and samples are both clustered by average linkage hierarchical clustering using 1-Pearson correlation coefficient of log2 peak intensity as the distance metric. (H) Number of peaks per exon normalized by the number of motifs (on sense strand) in the exon. The error bars represent standard deviations from 1000 times of bootstrapping. (I) The normalized distribution of m6A peaks across the 5′UTR, CDS, and 3′UTR of mRNAs for T0 and T48 m6A peaks. See also Figure S4B, S4C and S4D. (J) Box plot representing the half-life for transcripts, with transcripts separated according to enrichment score. See also Figure S4E, S4F and S4G.
Many master regulators of hESC maintenance and differentiation are modified with m6A
As we observed for mESC, transcripts encoding many hESC master regulators, including human NANOG, SOX2, and NR5A2, were m6A modified. Like mESC, the transcripts for OCT4 (POUF51) in hESC did not harbor an m6A modification (Figure 5D). These results show that in both organisms the core-pluripotency/maintenance genes are under the regulatory influence of the m6A pathway. We also identified human specific lncRNAs with known roles in hESC maintenance such as LINC-ROR and MEGAMIND/TUNA to contain m6A modification(s) (Figure 5D; Figure S4A) (Lin et al., 2014; Loewer et al., 2010). Upon induction of differentiation, we observed transcripts encoded by several key regulators of endodermal differentiation also to have m6A modifications including EOMES and FOXA2 (Figure 5D). Gene ontology (GO) analyses of methylated genes in undifferentiated hESCs, and after endodermal differentiation, were significantly enriched in biological functions such as regulation of transcription (FDR=1.2×10−14), chordate embryonic development (FDR=1.1×10−4), and regulation of cell morphogenesis (FDR=0.01).
Upon differentiation toward endoderm, 1356 peaks in 1137 genes showed quantitative differences of at least 1.5 fold in m6A intensity, after normalization for input transcript abundance (Figure 5E and 5F, Table S4). The majority of these differential m6A sites represented quantitative differences at existing sites (i.e. 59.1% of the peaks were called in both time points), rather then state-specific de novo appearance or erasure of modification (Figure 5G). This is consistent with the observation that 74.9% of sites in the hESCs overlapped those observed in HEK293T data (Meyer et al., 2012) and the minimal changes in m6A sites observed in a recent survey of m6A pattern across cell types (Schwartz et al., 2014). We suggest that transcripts exhibit dynamic differential peak m6A methylation intensity largely at “hard wired sites” during differentiation under the conditions examined and when compared to other tissue types.
Conserved features of m6A modifications spanning different species
We found that three salient features of the m6A methylome are conserved in hESCs. First, m6A sites in hESCs are also dominated by the RRACU motif seen in mESC and somatic cells (Dominissini et al., 2012; Meyer et al., 2012) (Figure 5C). There was also a strong preference of targeting long-internal exons at the RRACU motif even after normalizing for exon length and number of m6A motifs (Figure 5H). Second, there was a significant enrichment in m6A peaks at 3′ end of transcripts, near the stop codon of coding genes or the last exon in non-coding RNAs (Figure 5I, Figure S4B, S4C and S4D). Furthermore, the topology of m6A modification is preserved upon endodermal differentiation (Figure 5I). As in mESCs, moderate to lowly expressed genes have higher probability of becoming methylated (Figure S4E). Lastly, hESC m6A is not correlated with transcription rate as judged by GRO-seq (Sigova et al., 2013), but is strongly anti-correlated with measured mRNA half-life in human pluripotent cells (Neff et al., 2012), strongly suggesting that m6A modification also marks RNA turnover in hESCs (Figure 5J, Figure S4F and S4G).
Evolutionary conservation and divergence of the m6A epi-transcriptomes of human and mouse ESCs
Previous studies suggested significant conservation of m6A modified genes between mouse and human in somatic cell types, but the comparisons are limited by non-matched tissue types (Dominissini et al., 2012; Meyer et al., 2012). We were thus interested in examining the evolutionary conservation of human and mouse ESC m6A methylomes. At the gene level, 69.4% (3609 of 5204) of hESC genes are also m6A modified in the orthologus mouse gene (p-value= 8.3×10−179; Fisher exact test) (Figure 6A; Table S5). Furthermore, we identified 632 conserved m6A peak sites (46.1%) between hESCs and mESCs (Table S6). Notably, conserved sites tended to have higher m6A peak intensities compared to m6A peak sites that are not conserved (Figure 6B and 6C, p-values=1.3×10−15 and 8.7×10−23 for hESC or mESC, respectively; Wilcoxon test). Commonly methylated genes can demonstrate m6A modification sites at identical site(s) such as GLI1, similar but not identical locations such as SOX2, or m6A site at different exons, such as CHD6 (Figure 6D, 6E and 6F, Table S4). Our data thus reveal a substantial overlap at the gene level, suggesting broad functional significance of m6A modification in ESCs in both species. At the same time, we also observed numerous species-specific m6A patterns that may contribute to specific aspects of ESC biology (Schnerch et al., 2010).
Figure 6. Evolutionary conservation and divergence of the m6A epi-transcriptomes of human and mouse ESCs.
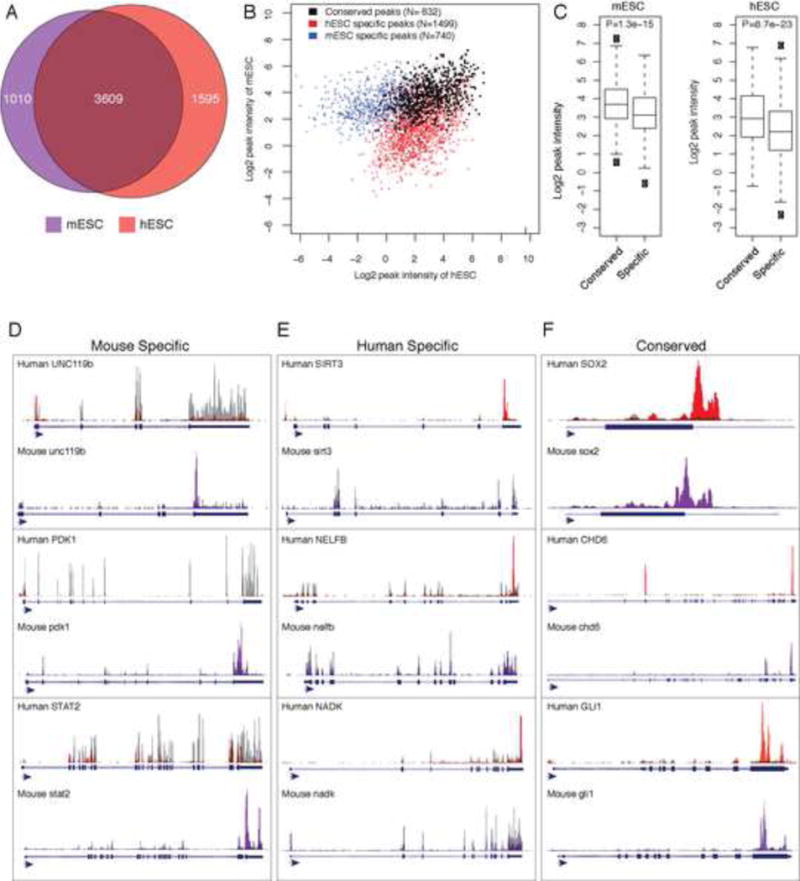
(A) Venn diagram showing a 62% overlap between methylated genes in M. musculus (purple) and H. sapiens (red) embryonic stem cells (p value= 3.5 × 10−92; Fisher exact test). See also Table S5 and S6. (B) The m6A peaks that could be mapped to orthologous genomic windows between mouse and human were identified. The intensities of m6A-seq signals in human and mouse ESCs were shown for m6A peaks found to be unique in mouse (blue), unique in human (red), and conserved between human and mouse (black). (C) Boxplot of peak intensities of m6A sites conserved (“common”) or not conserved (“specific”) in mouse and human ESCs. (p values=1.3×10−15 and 8.7×10−23 respectively). (D to F) UCSC Genome browser plots of m6A-seq reads along indicated mRNAs. The grey reads are from non-immunoprecipitated control input libraries and the purple and red reads are from the anti-m6A RIP of mESCs and hESCs (T0) respectively. (D) Mouse-specific m6A modifications are represented. (E) Human-specific m6A modifications ESCs are represented. (F) Conserved m6A modifications at gene and site level are represented. Genes such CHD6 have a conserved m6A peak location at its 3′UTR as well as mouse and human specific m6A peaks at conserved but distinct exons.
METTL3 is required for hESC differentiation
To address the function of m6A in hESCs, we generated hESC colonies with stable knockdown of METTL3 and shRNA control (Figure 7A). Knockdown of METTL3 in hESCs resulted in reduction in METTL3 mRNA levels and reduction in m6A level (Figure 7B, 7C and Figure S5B, S5C). METTL3-depleted hESCs could be stably maintained, suggesting the dispensability of METTL3 for hESC self-renewal or viability. Strikingly, differentiation of METTL3-depleted hESCs into neural stem cells (NSCs) by dual inhibition of SMAD signaling, using Dorsomorphin and SB-431542 revealed a block in neuronal differentiation (Methods). While 44% (+/−3.5% s.d.) of the control cells were Sox1 positive, only 10% (+/−3.1% s.d) of the METTL3-depleted were Sox1 positive (Figure S5A).
Figure 7. METTL3 is required for normal human ESC endoderm differentiation. Model of METTL3 function(s).
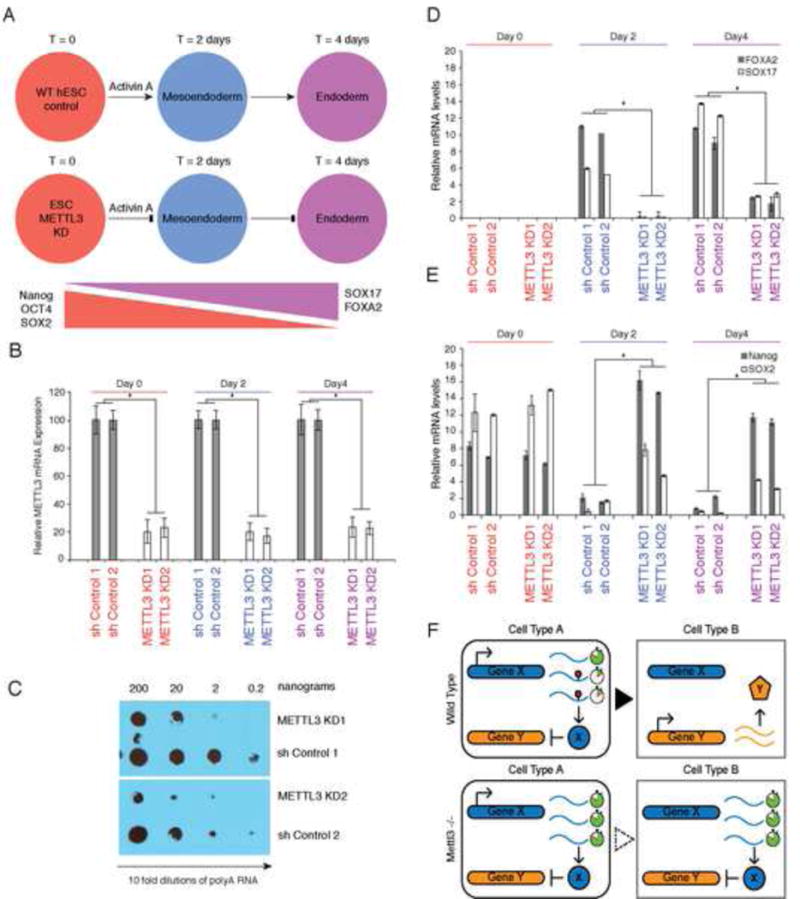
(A) hESC cells were transfected with anti-METTL3 shRNA (KD) as well control shRNA and stable hESC colonies were obtained after drug selection. Two independent clones were subjected to endodermal differentiation with Activin A and examined at various indicated time points. A schematic of the trends of gene expression for indicated markers of stem maintenance and endoderm differentiation is also shown. See also figure S5A. (B) Levels of METTL3 mRNA in hESC cells with control shRNA versus anti-METTL3 shRNA (KD) across the three indicated time points during endodermal differentiation (n=2 independent generated ES cell knockdown and control clones shown). In all panels, Error bars represent standard deviation across 3 replicates per time point; *, p value<0.05 t-test (2 tailed) between different clones. See also Figure S5B. (C) Anti-m6A dot blot was performed on 10x fold dilutions of polyA selected RNA from hESC cells derived from control shRNA versus anti-METTL3 shRNA clones. See also Figure S5C. (D and E) mRNA levels of endodermal and stem maintenance/marker genes. qRT-PCR was performed on indicated genes and time points (n=2 independently generated ES cell knockdown and control clones shown). See also Figure S5D. (F) Model: m6A marks transcripts for faster turn-over. Upon transition to new cell fate, m6A marked transcripts are readily removed to allow the expression of new gene expression networks. In the absence of m6A, the unwanted presence of transcripts will disturb the proper balanced required for cell fate transitions.
Similarly, knockdown of METTL3, in three independently generated hES colony clones selected for METTL3 knockdown, led to a profound block in endodermal differentiation at day 2 and day 4 based on failure to express the endoderm markers EOMES and FOXA2 compared to either two shRNA control colony clones (Figure 7D) or wildtype hESCs (Figure S5D). Consistently, METTL3-depleted ESCs retain high levels of expression of the master regulators NANOG and SOX2 throughout the differentiation time course in contrast to their diminishing expression in wild type cells (Figure 7E and S5E). These results indicate that METTL3 and m6A control differentiation of human embryonic stem cells.
DISCUSSION
m6A methylome in ES cells
Our analysis of the ESC m6A methylome in mouse and human cells reveals extensive m6A modification of ESC genes, including most key regulators of ESC pluripotency and lineage control. However, this observation does not mean that m6A is uniquely tied to the pluripotency network. Because m6A marks moderately expressed transcripts that need to be turned over in a timely fashion, such genes in ESCs likely include many regulators of pluripotency and lineage determination. The pattern and sequence motif associated with ESC m6A are similar if not identical to those previously reported in somatic cells, suggesting a single mechanism that deposits m6A modification in early embryonic life. This invariant mechanism for m6A contrasts with the complexity of 5-methyl-cytosine in DNA and histone lysine methylations that undergo extensive reprogramming with distinct rules in pluripotent vs. somatic cells.
We identified a general and conserved topological enrichment of m6A sites at the 3′ end of genes among single-exon and multi-exon mRNAs as well as ncRNAs. Thus, neither the stop codon nor the last exon-exon splice junction can alone explain the observed m6A topology in RNA. However, all species examined to date including Saccharomyces cerevisae and A. thalania exhibit a strong 3′ bias in m6A localization, suggesting an evolutionary constraint that may target the m6A modification to the 3′ ends of genes regardless of gene structure or coding potential. This bias may be achieved by preferential m6A methylases recruitment to 3′ sites or preferential action of demethylases in upstream regions of the transcript. Although the role of demethylases cannot be excluded, the observation of 3′ end m6A bias in S. cerevisiae, which lacks known m6A demethylases argues against the latter mechanism (Bodi et al., 2012; Jia et al., 2011; Schwartz et al., 2013; Zheng et al., 2013). The functional importance of m6A location vs. its specific molecular outcome need to be addressed in future studies.
Mettl3 selectively targets mRNAs including pluripotency regulators
While several studies had approached Mettl3 function by RNAi knock down (Dominissini et al., 2012; Fustin et al., 2013; Liu et al., 2014; Wang et al., 2014b), genetic ablation of Mettl3 KO allowed us to examine the true loss-of-function phenotypes. The importance of using definitive genetic models is highlighted by recent studies in the DNA methylation field where shRNA experiments led to mis-assigned functions of Ten-eleven translocation (TET) proteins that were later recognized in genetic knockouts (Dawlaty et al., 2013; Dawlaty et al., 2011). We found that both Mettl3 KO and depletion led to incomplete reduction of the global levels m6A in both mESCs and hESCs, demonstrating redundancy in m6A methylases. However, m6A profiling in Mettl3 KO cells revealed a subset of targets, approximately 33% of m6A peaks, that are preferentially dependent on Mettl3, and these included Nanog, Sox2, and additional pluripotency genes. A second m6A methylase, Mettl14, was described during the preparation of this manuscript.
RNAi knockdown of Mettl3 in somatic cancer cells led to apoptosis (Dominissini et al., 2012), and one study reported ectopic differentiation of mESC with Mettl3 depletion (Wang et al., 2014b). In contrast, we found that Mettl3 KO does not affect mESC cell viability or self-renewal, and in fact mESC renewed at an improved rate. The differences in phenotype observed could potentially be explained by different dependency on m6A modified RNAs in different cell types, acute versus chronic inactivation, or RNAi off target effects. m6A methylome analysis in different cell types with Mettl3 inactivation may shed light on these differences in the future.
Conservation of m6A methylome in mammalian ESCs
The conserved methylation patterns of many ESC master regulators and the shared phenotype observed upon inactivation of METTL3 suggest that METTL3 operates to control stem cell differentiation. It is known that human and mouse ESCs are not equivalent (Schnerch et al., 2010), and are cultured in different conditions. By focusing in on orthologous genes, we were able to catalog both shared and species-specific methylation sites. The observation that certain methylation sites are modified whenever a target transcript is expressed in both species, despite cell state or culture differences, argues that these modification events have been preserved under strong purifying selection during evolution. Our comparative genomic analyses also pave the way to further understand potential biological differences between mouse and human ESCs at the level of m6A epitranscriptome, given the unique patterns of some methylation sites between the species.
RNA “anti-epigenetics”: m6A as a mark of transcriptome flexibility
Stem cell gene expression programs need to balance fidelity and flexibility. On one hand, stem cell genes need sufficient stability to maintain self-renewal and pluripotency over multiple cell generations, but on the other hand, gene expression needs to change dynamically and rapidly in response to differentiation cues. It has been proposed that ESC gene expression programs are in constant flux between competing fates, and pluripotency is a statistical average (Loh and Lim, 2011; Montserrat et al., 2013; Shu et al., 2013). We found that mRNAs with m6A tend to have a shorter half-life, and Nanog and Sox2 mRNAs could not be properly down-regulated with differentiation in Mettl3-deficient mESC and hESC. However, Mettl3 deficiency has only modest effects on steady state gene expression, which could arise from the non-stoichiometric nature of the m6A modification. The application of methods that can determine level of modification of each RNA species will allow us to answer these questions (Harcourt et al., 2013; Liu et al., 2013). Mettl3 KO mESCs have enhanced self-renewal but hindered differentiation, concomitant with decreased ability to down regulate ESC mRNAs. WTAP, a conserved Mettl3 interacting partner from yeast to human cells (Horiuchi et al., 2013; Schwartz et al., 2014), is also required for endodermal and mesodermal differentiation (Fukusumi et al., 2008). The observed phenotypes in ESC and teratomas are all the more notable because we have significantly reduced but not eliminated m6A.
Our findings suggest a model where m6A serves as the necessary flexibility factor to counter balance epigenetic fidelity—a RNA “anti-epigenetics” (Figure 7F). m6A marks a wide range of transcripts, including ESC fate determinants to limit their level of expression, and ensure their continual degradation so that cells can rapidly transition between gene expression programs. In ESC, m6A is required for cells to rapidly exit the pluripotent state upon differentiation. The inability to exit the stem cell state and continued proliferation upon insufficient m6A offers a potential explanation for the association of FTO with human cancers (Loos and Yeo, 2013). METTL3 depletion also leads to elongation of the circadian clock (Fustin et al., 2013), suggesting a role for m6A in resetting the transcriptome. In yeast, m6A is active during meiosis (Clancy et al., 2002), where diploid gene expression programs are reset to generate haploid offspring. We propose that m6A makes the transition between cell states possible by facilitating a reset mechanism between stages, as occurs in ESCs and likely other cell types. In contrast to epigenetic mechanisms that provide cellular memory of gene expression states, m6A enforces the transience of genetic formation – helping cells to forget the past and thereby embrace the future.
EXPERIMENTAL PROCEDURES
For full details, see Extended Experimental Procedure.
Mouse cell culture and differentiation
J-1 murine embryonic stem cells were grown under typical feeder free ES cell culture conditions. For cardiomyocyte formation, mESCs were differentiated in cardiomyocyte differentiation media and scored on day 12. For neuron formation, mESCs were differentiated in MEF and ITSFn medium and scored after 10 days in ITSFn medium. For the cell proliferation assay 5000 cells where cultured in 24 well plates and the assay performed according to the manufacturer’s protocol (MTT assay). For the single colony assays and Nanog staining, 1000 cells where cultured per well, on a six well plate. For alkaline phosphatase staining, cells were stained according to the manufacturer’s protocol (Vector Blue Alkaline Phosphatase Substrate Kit).
hESCs cell culture, transfection and differentiation
H1 (WA01) cells were cultured in feeder-free conditions as described (Sigova et al., 2013). Stable hESC lines were created that expressed shMETTL3 RNA or scrambled shRNA by transfecting hESCs with plasmids encoding shMETTL3 or scrambled shRNA and a puromycin resistance gene. Cells were treated with puromycin for six days beginning two days after transfection. For each shRNA, two independent puromycin-resistant colonies were picked and expanded. Endodermal differentiation was then induced by Activin A, as described (Sigova et al., 2013). Day 2 and Day 4 of differentiation were measured from the time that Activin was added. Puromycin was removed from the media one day prior to endodermal differentiation.
RNA m6A IP and m6A methylation IP RNA-sequencing analysis
Libraries generated with iCLIP adaptors where separated by barcode, and perfectly matching reads were collapsed. Sequencing reads were mapped using TopHat (Trapnell et al., 2009). A non-redundant mm9 transcriptome was assembled from UCSC RefSeq genes, UCSC genes, and predictions from (Ulitsky et al., 2011) and (Guttman et al., 2011). For human datasets, the Ensembl genes (release 64) was used. Search for enriched peaks was performed by scanning each gene using 100-nucleotide sliding windows, and calculating an enrichment score for each sliding window (Dominissini et al., 2012). HOMER software package (Heinz et al., 2010) was used for de novo discovery of the methylation motif.
CRISPR-mediated Mettl3 knockout
gRNA sequences where chosen and designed a CRISPR design tool (Hsu et al., 2013). Plasmids for guide RNA were co-nucleofected, with a human codon optimized Cas9 expression plasmid and a plasmid with a puromycine resistance cassette. Cells were plated at low density for single colony isolation and selected single colonies tested by western blot for loss of protein.
Determination of m6A levels
2D-TLC was performed as described by (Jia et al., 2011). For dot-blots, the indicated amounts of RNA were applied to the membrane and cross-linked by UV. The m6A primary antibody was added at a concentration of 1:500. The membrane was incubated with the secondary antibody and exposed to an auto-radiographic film. m6A RNA mass-spectrometry was performed as described in the Extended Experimental Procedures.
Dataset comparison
Mouse Pol II occupancy data, mRNA half life and Protein translation efficiency were obtained from (Ingolia et al., 2011; Rahl et al., 2010; Sharova et al., 2009). Plotting and statistical tests were performed in R. Multi-dimensional gene set enrichment analysis over DAVID Gene Ontology terms and stem cell gene sets (Wong et al., 2008) were performed using Genomica (Segal et al., 2003).
Teratoma generation and histopathology
Mettl3 wild type and mutant cells were subcutaneously injected into 8-week-old female SCID/Beige mice (Charles River). Four weeks after injection, the mice were euthanized and the tumors harvested. All animal studies were approved by Stanford University IACUC guidelines. For histological analysis, slides were stained with hematoxylin and eosin (H&E) or stained by immunohistochemistry (IHC) with VECTASTAIN ABC Kit and DAB Peroxidase Substrate Kit following the manufacturer’s instructions. Analyses were performed by a boarded veterinary pathologist (DMB).
Supplementary Material
Acknowledgments
We thank C. He, C. Mason, S. Schwartz, A. Regev, J. M. Claycomb, N. Van Wittenberghe, B.D. Howard and members of the Chang and Giallourakis labs for discussions, and assistance. We thank H.E. Arda and S.K. Kim, for help with FACS analysis, and A. Memmelaar for his expertise in graphic arts. Supported by California Institute for Regenerative Medicine and NIH R01-CA118750 (H.Y.C.), the MGH Start-Up Funds and MGH ECOR grant 2013A051178 (C.C.G.), NIH grant DK090122 (A.C.M.), and the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA Research Award (Y.X.). P.J.B. is the Kenneth G. and Elaine A. Langone Fellow of the Damon Runyon Cancer Research Foundation. Y.X. is an Alfred Sloan Foundation Research Fellow. H.Y.C. is an Early Career Scientist of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
PJB and HYC conceived of the mESC studies; PJB, LL, DMB, EL, ACC, RAF, MW, YX and HYC designed and performed experiments in the mouse system. ACM, YX, CCG conceived of the hESC studies; BM, BH, KD, CZ, KL, PD, MW, ACM, YX, CCG designed and performed experiments in the human system. JW, KQ, JZ analyzed the data. PJB, BM, JK, YX, CCG and HYC wrote the paper with input from all authors.
References
- Bodi Z, Zhong S, Mehra S, Song J, Graham N, Li H, May S, Fray RG. Adenosine Methylation in Arabidopsis mRNA is Associated with the 3′ End and Reduced Levels Cause Developmental Defects. Front Plant Sci. 2012;3:48. doi: 10.3389/fpls.2012.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy MJ, Shambaugh ME, Timpte CS, Bokar JA. Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res. 2002;30:4509–4518. doi: 10.1093/nar/gkf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Breiling A, Le T, Raddatz G, Barrasa MI, Cheng AW, Gao Q, Powell BE, Li Z, Xu M, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24:310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi SW, Page DC, et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell. 2011;9:166–175. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Dunn SJ, Martello G, Yordanov B, Emmott S, Smith AG. Defining an essential transcription factor program for naive pluripotency. Science. 2014;344:1156–1160. doi: 10.1126/science.1248882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, He C. Nucleic acid modifications with epigenetic significance. Curr Opin Chem Biol. 2012;16:516–524. doi: 10.1016/j.cbpa.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukusumi Y, Naruse C, Asano M. Wtap is required for differentiation of endoderm and mesoderm in the mouse embryo. Dev Dyn. 2008;237:618–629. doi: 10.1002/dvdy.21444. [DOI] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, et al. RNA-Methylation-Dependent RNA Processing Controls the Speed of the Circadian Clock. Cell. 2013;155:793–806. doi: 10.1016/j.cell.2013.10.026. [DOI] [PubMed] [Google Scholar]
- Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt EM, Ehrenschwender T, Batista PJ, Chang HY, Kool ET. Identification of a selective polymerase enables detection of N(6)-methyladenosine in RNA. J Am Chem Soc. 2013;135:19079–19082. doi: 10.1021/ja4105792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T, Hamakubo T. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem. 2013 doi: 10.1074/jbc.M113.500397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Jeong SJ, Kim KN, Baek IJ, Chang M, Kang CM, Park YS, Yun CW. A novel protein, Pho92, has a conserved YTH domain and regulates phosphate metabolism by decreasing the mRNA stability of PHO4 in Saccharomyces cerevisiae. Biochem J. 2014;457:391–400. doi: 10.1042/BJ20130862. [DOI] [PubMed] [Google Scholar]
- Lin N, Chang KY, Li Z, Gates K, Rana ZA, Dang J, Zhang D, Han T, Yang CS, Cunningham TJ, et al. An Evolutionarily Conserved Long Noncoding RNA TUNA Controls Pluripotency and Neural Lineage Commitment. Mol Cell. 2014;53:1005–1019. doi: 10.1016/j.molcel.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. Rna. 2013 doi: 10.1261/rna.041178.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH, Garber M, Curran M, Onder T, Agarwal S, et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet. 2010;42:1113–1117. doi: 10.1038/ng.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KM, Lim B. A precarious balance: pluripotency factors as lineage specifiers. Cell Stem Cell. 2011;8:363–369. doi: 10.1016/j.stem.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Loos RJ, Yeo GS. The bigger picture of FTO-the first GWAS-identified obesity gene. Nat Rev Endocrinol. 2013 doi: 10.1038/nrendo.2013.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montserrat N, Nivet E, Sancho-Martinez I, Hishida T, Kumar S, Miquel L, Cortina C, Hishida Y, Xia Y, Esteban CR, et al. Reprogramming of human fibroblasts to pluripotency with lineage specifiers. Cell Stem Cell. 2013;13:341–350. doi: 10.1016/j.stem.2013.06.019. [DOI] [PubMed] [Google Scholar]
- Neff AT, Lee JY, Wilusz J, Tian B, Wilusz CJ. Global analysis reveals multiple pathways for unique regulation of mRNA decay in induced pluripotent stem cells. Genome Res. 2012;22:1457–1467. doi: 10.1101/gr.134312.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y, Zhao X, Wu YS, Li MM, Wang XJ, Yang YG. N6-methyl-adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics. 2013;11:8–17. doi: 10.1016/j.gpb.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnerch A, Cerdan C, Bhatia M. Distinguishing between mouse and human pluripotent stem cell regulation: the best laid plans of mice and men. Stem Cells. 2010;28:419–430. doi: 10.1002/stem.298. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, Tabach Y, Mikkelsen TS, Satija R, Ruvkun G, et al. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013;155:1409–1421. doi: 10.1016/j.cell.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5′ Sites. Cell Rep. 2014;8:284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Shapira M, Regev A, Pe’er D, Botstein D, Koller D, Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MS. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 2009;16:45–58. doi: 10.1093/dnares/dsn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu J, Wu C, Wu Y, Li Z, Shao S, Zhao W, Tang X, Yang H, Shen L, Zuo X, et al. Induction of pluripotency in mouse somatic cells with lineage specifiers. Cell. 2013;153:963–975. doi: 10.1016/j.cell.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibbritt T, Patel HR, Preiss T. Mapping and significance of the mRNA methylome. Wiley Interdiscip Rev RNA. 2013;4:397–422. doi: 10.1002/wrna.1166. [DOI] [PubMed] [Google Scholar]
- Sigova AA, Mullen AC, Molinie B, Gupta S, Orlando DA, Guenther MG, Almada AE, Lin C, Sharp PA, Giallourakis CC, et al. Divergent transcription of long noncoding RNA/mRNA gene pairs in embryonic stem cells. Proc Natl Acad Sci U S A. 2013;110:2876–2881. doi: 10.1073/pnas.1221904110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–1550. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014a;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014b;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.