

Abstract
As both a commensal and a major cause of healthcare-associated infections in humans, Enterococcus faecalis is a remarkably adaptable organism. We investigated how E. faecalis adapts in a mammalian host as a pathogen by characterizing changes in the transcriptome during infection in a rabbit model of subdermal abscess formation using transcriptional microarrays. The microarray experiments detected 222 and 291 differentially regulated genes in E. faecalis OG1RF at two and eight hours after subdermal chamber inoculation, respectively. The profile of significantly regulated genes at two hours post-inoculation included genes involved in stress response, metabolism, nutrient acquisition, and cell surface components, suggesting genome-wide adaptation to growth in an altered environment. At eight hours post-inoculation, 88% of the differentially expressed genes were down-regulated and matched a transcriptional profile consistent with a (p)ppGpp-mediated stringent response. Subsequent subdermal abscess infections with E. faecalis mutants lacking the (p)ppGpp synthetase/hydrolase RSH, the small synthetase RelQ, or both enzymes, suggest that intracellular (p)ppGpp levels, but not stringent response activation, influence persistence in the model. The ability of cells to synthesize (p)ppGpp was also found to be important for growth in human serum and whole blood. The data presented in this report provide the first genome-wide insights on E. faecalis in vivo gene expression and regulation measured by transcriptional profiling during infection in a mammalian host and show that (p)ppGpp levels affect viability of E. faecalis in multiple conditions relevant to mammalian infection. The subdermal abscess model can serve as a novel experimental system for studying the E. faecalis stringent response in the context of the mammalian immune system.
Introduction
Many Gram-positive cocci, including staphylococci, streptococci, and enterococci, maintain commensal relationships with mammals by occupying specific biological niches, yet maintain the potential to become pathogenic when colonizing alternate sites within the same hosts. The enterococci, which naturally reside in the human gastrointestinal tract, are distinct from staphylococci and streptococci in the diverse range of non-mammalian environments that they inhabit, from soil and water to plants and the guts of insects [1]. Enterococci tolerate extreme temperatures and growth conditions that are generally unfavorable for other bacteria [1]. The inherent adaptability of enterococci is apparent in the breadth of physiologies these organisms achieve in order to thrive in such disparate environments.
Enterococcus spp. are the second most common pathogens associated with healthcare-associated infections in the United States, having caused more than 5500 central line-associated bloodstream infections, 3100 catheter-associated urinary tract infections, and 2400 surgical site infections in 2009–2010, as reported to the National Healthcare Safety Network [2]. Enterococcus faecalis is the species responsible for the highest percentage of enterococcal infections [2]. A number of virulence factors encoded on both the E. faecalis chromosome and on mobile genetic elements have been identified and characterized [3]. However, many studies report a lack of correlation between the presence of virulence factors in the genomes of isolates and the types of infectious or non-infectious sources from which the isolates originated [4]–[8]. These data suggest that a complex combination of factors contribute to the virulence of E. faecalis.
To date, transposon mutagenesis [9] and recombinase-based in vivo expression technology (RIVET) [10]–[12] screens are the only techniques that have been used for in vivo studies to identify E. faecalis genetic determinants involved in infection on a genome-wide scale. Transcriptomics approaches, such as microarray and RNA sequencing (RNA-seq), offer an alternative to genetic screens for evaluating the contributions of bacterial genes to in vivo growth at specific time points. Transcriptional profiling using microarrays has been performed for over a decade on numerous bacterial pathogens harvested directly from mammalian hosts [13]–[20], demonstrating that this methodology is widely applicable across bacterial species and animal models. RNA-seq has also been used to determine high resolution in vivo transcriptomes of pathogenic bacteria collected from animal infection models [21]–[23]. RNA-seq was recently used to compare the transcriptome of E. faecalis from the intestinal tracts of gnotobiotic mice to that of cells grown in BHI broth [24]. However, no in vivo transcriptomics studies of E. faecalis in a pathogenic setting have been conducted.
To gain a comprehensive view of the E. faecalis transcriptional landscape during the early stages of infection, we used microarrays to analyze changes in gene expression over time in a rabbit model of subdermal abscess formation. Substantial portions of the genome were differentially regulated at the sampled time points, with the resulting transcriptional profile at eight hours post-inoculation unexpectedly revealing hallmarks of a (p)ppGpp-mediated stringent response. The in vivo transcriptomics data resulting from this work were compared to the collection of in vivo-activated promoters identified in our previous subdermal abscess model RIVET screen [11] to gain further insight on E. faecalis in vivo gene expression and regulation. Together, these data provide a global description of the transcriptional profiles in E. faecalis that support physiological adaptation to growth in a mammalian host as a pathogen.
Results and Discussion
Survival of E. faecalis OG1RF in subdermal chambers during the course of infection
Our goal in this study was to gain insight on the process of E. faecalis adaptation to a host environment as a pathogen during the early stages of infection. We previously used the rabbit subdermal abscess model for a RIVET genetic screen to identify promoters in E. faecalis that are specifically up-regulated in infection [11]. Since the model enables sampling of the same infection site over time and has been used for Staphylococcus aureus in vivo gene expression profiling [25], we used the model in this work to continue our investigations of E. faecalis OG1RF gene expression during infection. Chambers were inoculated to contain an average of 8.7 log10 CFU/ml OG1RF and were followed for 96 hours (Fig. 1). The number of OG1RF cells recovered from chambers showed an initial decrease of one log10 CFU/ml over the first two to four hours, followed by an additional drop of 0.4 log10 CFU/ml between four and eight hours. Cell recovery further declined to 6.3 log10 CFU/ml at 24 hours and remained at that level through the final sample collection at 96 hours post-inoculation. The 2.4 log10 CFU/ml decrease over the first 24 hours of infection suggests that bacteria were eliminated from the chamber by the local immune cell population, which was historically reported to be comprised of ∼10% polymorphonuclear cells and 90% mononuclear cells [26].
Figure 1. Recovery of E. faecalis OG1RF from subdermal abscesses during the course of infection.
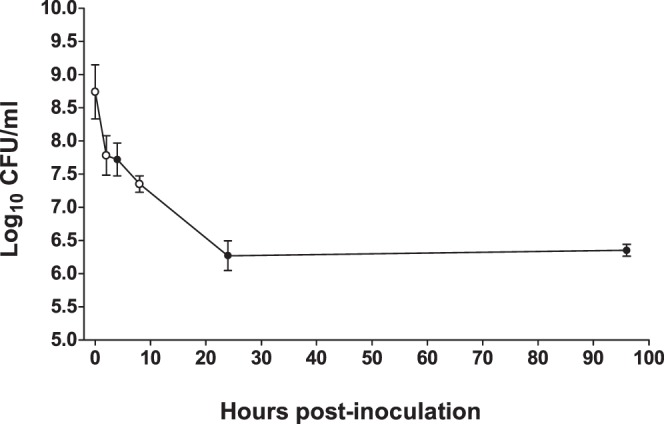
Two ml of E. faecalis OG1RF were inoculated into subdermal chambers from which two ml of serous fluid had been withdrawn. Dilutions of fluid aspirated from implanted chambers at the indicated time points following inoculation were plated to determine the number of viable E. faecalis CFU per ml recovered. The 0 h time points were calculated by dividing the total CFU in the two ml inocula (mean 1.65×1010 CFU, n = 4) by 30 ml, the approximate total volume of serous fluid in the implanted chamber. Results are reported as the log10 CFU/ml transformed values. Open circles indicate the time points analyzed by microarray (0, 2, and 8 hours). Symbols and error bars represent the mean ± SEM of n = 4 rabbits.
Use of microarray to detect differential gene expression in E. faecalis OG1RF during subdermal abscess infection
The RIVET screens that we and others previously carried out in rabbit subdermal chamber abscesses [11], murine bacteremia and peritonitis [10], the Manduca sexta gut [12], and G. mellonella larvae hemocoel [10] proved useful for identifying specific E. faecalis in vivo-activated promoters. However, these screens are limited by the low number of overlapping genes identified among different hosts [11], minimal data on the specific temporal expression patterns of in vivo-activated genes, and the inability to identify genes that are negatively regulated in vivo. As a result, we anticipated that genome-wide gene expression analyses would provide a novel perspective on the transcriptional changes that take place in E. faecalis in the transition from in vitro to in vivo pathogenic growth. Since OG1RF cells were challenged by the host immune response during the early hours of infection in the subdermal chambers, we decided to compare gene expression in the input inoculum with that of cells recovered at two and eight hours post-inoculation by microarray (Fig. 1, open circles).
Analysis of transcripts collected two hours post-inoculation revealed that 222 genes, representing 9.5% of the total OG1RF open reading frames (ORFs) assayed on the array, were differentially regulated by two-fold or greater (p-value <0.05) when compared to the initial inoculum (S1 Table). Of the 222 genes, 117 genes (53%) were up-regulated and 105 genes (47%) were down-regulated. At eight hours post-inoculation, 291 genes (12.4% of all analyzed OG1RF ORFs) were differentially regulated (S2 Table). Eighty-eight percent (n = 257) of these genes were down-regulated, while only 12% (n = 34) of genes were up-regulated. Only three up-regulated genes and 31 down-regulated genes were in common between the two and eight hour data sets (column H in S1 and S2 Tables), suggesting that the transcriptional profile in E. faecalis cells underwent a major shift between the two time points.
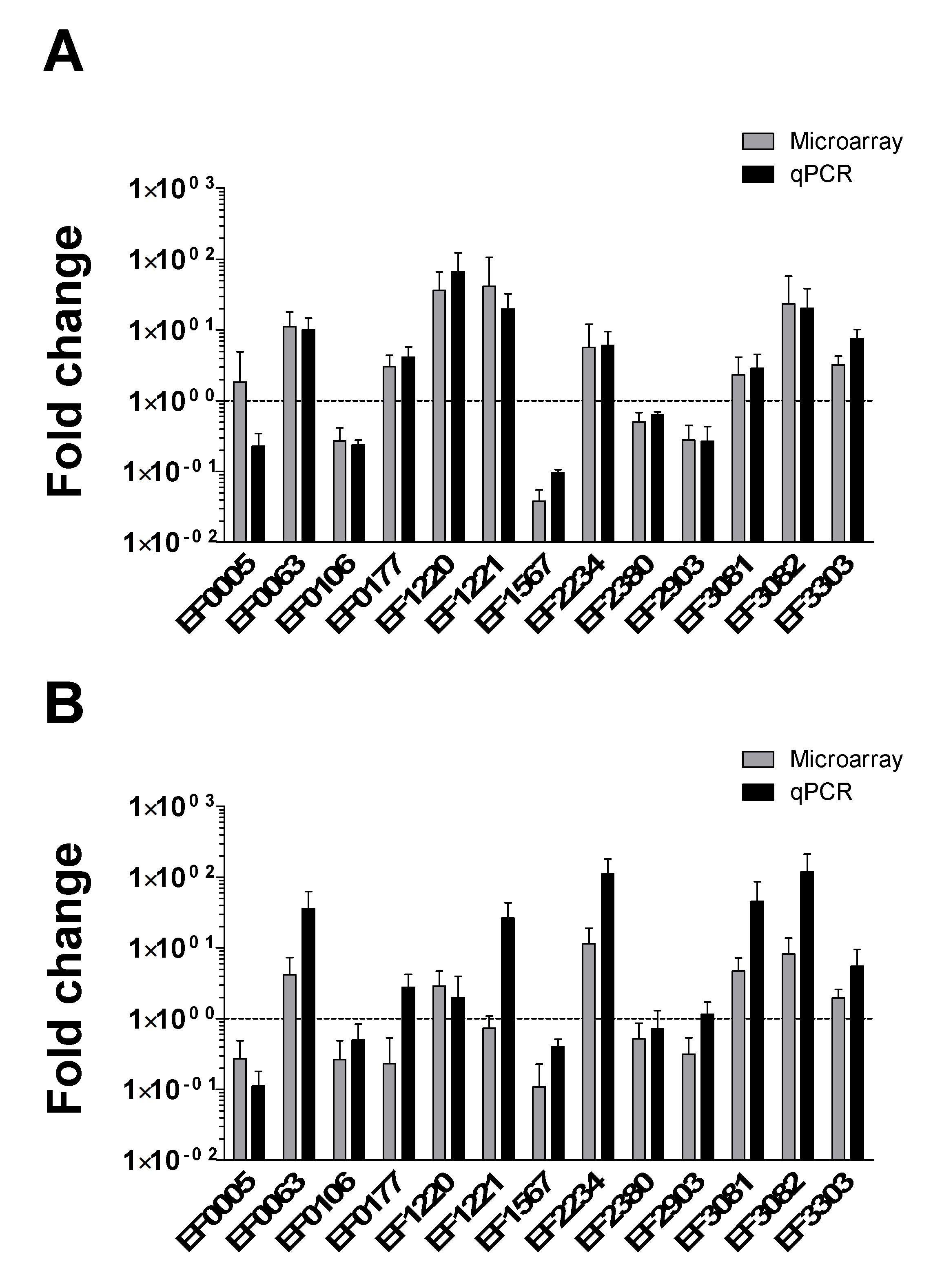
To validate the microarray results, quantitative PCR (qPCR) for 13 differentially regulated genes was performed on reverse-transcribed RNA collected from subdermal chambers at two and eight hours post-infection (S1 Fig.). Expression data for 12/13 genes at two hours and 11/13 genes at eight hours were concordant between the two techniques.
The significant differentially regulated genes at each time point are graphed in Fig. 2 according to their functional categories (S1 and S2 Tables), which were assigned following the scheme used by the J. Craig Venter Institute [27], [28] for the E. faecalis V583 genome [29]. Notably, 37% of the genes at two hours, and 24% of the genes at eight hours, encode proteins with annotations of hypothetical or unknown functions. The functional categories with the next-highest number of up- and down-regulated genes at two hours (Fig. 2A) were transport and binding proteins (n = 20) and DNA metabolism proteins (n = 11), respectively. The down-regulated genes at eight hours were distributed across all of the functional categories (Fig. 2B), with the highest number of genes occurring in the protein synthesis category (n = 40).
Figure 2. Functional categories of genes found by microarray to be differentially expressed in E. faecalis OG1RF in vivo during subdermal abscess infection.

The numbers of up- (right) and down-regulated (left) genes at two (top) or eight (bottom) hours post-inoculation are indicated on the horizontal axis.
E. faecalis OG1RF adaptation during the initial hours of growth in a mammalian host is the result of genome-wide transcriptional changes
The differentially regulated genes in the subdermal chambers at two hours revealed genome-wide transcriptional changes suggestive of cellular adaptation to growth in an altered environment. Groups of genes involved in stress response, metabolism, nutrient acquisition, and cell surface components were differentially expressed (S1 Table). Six E. faecalis stress response genes that were up-regulated at two hours in the subdermal chambers were similarly up-regulated upon in vitro exposure to defibrinated horse blood [30]. These genes included gls24 and glsB (EF0079–EF0080), the first of which contributes to virulence [31], [32] and both of which impact cellular stress response [33]; two universal stress proteins (EF1058 and EF1084); a hypothetical gene (EF1560); and clpB (EF2355), which confers thermotolerance and virulence in a Galleria mellonella infection model [34]. The genes encoding chaperone GroEL and chaperonin GroES (EF2633-2634) were also coordinately up-regulated in the rabbit, while the gene for the cold shock protein CspA (EF1367) was down-regulated.
Multiple operons contributing to metabolic processes were down-regulated at two hours. Five genes in the co-transcribed region spanning EF1561 to EF1568, which encode the enzymes of the shikimate pathway in E. faecalis, were among the most strongly down-regulated genes in the two hour microarray data set. Microbes and plants use the shikimate pathway to synthesize aromatic amino acids and aromatic secondary metabolites from carbohydrates through the precursor molecule chorismate [35]. Two genes in the EF0445-0450 operon dedicated to the synthesis of menaquinone from chorismate were correspondingly down-regulated at two hours. Vebø et al. also reported down-regulation of the EF1561-1568 and EF0445-0450 regions in E. faecalis V583 following incubation in horse blood [30]. Multiple arginine catabolism genes (EF0104, EF0106-0107) were also down-regulated at two hours (S1 Table).
Large numbers of genes in the transport and binding proteins functional category were up-regulated genes at two hours post-infection (Fig. 2A). This is consistent with a bacterial response to changing nutrient availabilities in the environment. The most highly up-regulated group of genes at two hours was EF1219 to EF1223 (S1 Table), which encodes components of a spermidine/putrescine transport system (EF1219-EF1221), adenine deaminase (EF1222), and a putative S-adenosylhomocysteine deaminase (EF1223). Adenine deaminase and polyamine transport systems have been shown to be active in other Gram positive species when extracellular adenine and polyamines, respectively, were abundant [36], [37]. The genes for four iron transport proteins were also up-regulated (EF0475-0476, EF3082, and EF3085). In particular, transcription of EF3082, a ferric ABC transporter binding protein, and EF3085, an ABC transporter membrane permease, was increased by 23.5- and 42.7-fold, respectively. Transcription of both genes was also significantly increased in cells grown in horse blood [30] and in broth under iron-limiting conditions [38].
E. faecalis produces a plethora of surface proteins [39], [40], including dozens of lipoproteins [41], that likely mediate interactions with the host. The surface proteins Ebp pili and Ace, a collagen binding protein, are important enterococcal virulence factors [42], but transcription was not significantly changed for either gene in the subdermal chambers at two hours. Nine lipoproteins were up-regulated at two hours. The differentially regulated lipoproteins belonged to the transport and binding proteins (EF0063, EF0176, EF0177, EF1221, EF2076, EF3082), cell envelope (EF0095), hypothetical proteins (EF1085), and protein fate (EF0685) functional categories (S1 Table). Several genes in the dlt operon [43], [44]–dltX (EF2750), dltA (EF2749), and dltC (EF2747)–which incorporates D-alanine into lipoteichoic acid and wall teichoic acid, were up-regulated at two hours post-inoculation. The gene for alanine racemase (EF0849), the enzyme that generates D-alanine from L-alanine and is integral for cell wall synthesis [45], was also up-regulated at two hours.
Our use of the subdermal abscess infection model in this work provided the benefit of studying E. faecalis transcription in response to the host innate immune system in the presence of a foreign body. However, a limitation of the model is that the results may not be predictive of E. faecalis gene expression patterns in other types of infection models, such as bacteremia or urinary tract infection, due to differences in nutrient availability in the respective environments (e.g., blood and urine). In vivo gene expression studies in such models would be of interest, as E. faecalis is a common cause of bloodstream and catheter-associated urinary tract infections [2]. Nevertheless, the early time-point in vivo transcriptional profiles presented here significantly expand on previous studies characterizing E. faecalis gene expression following in vitro incubation in biological fluids [30], [46], [47]. In the experiment that most closely resembled the conditions we tested, 549 genes in the vancomycin-resistant E. faecalis strain V583 were differentially expressed after incubation in 100% defibrinated horse blood for 30 minutes [30]. The adaptive response of V583 to blood includes transcriptional changes in genes coding for stress protection, cell envelope maintenance, metabolism, transport and binding, and amino acid biosynthesis proteins. Despite the lack of the host immune response in vitro, the difference in time points and strains studied, and bacterial exposure to blood in vitro and serous fluid in vivo, our two hour microarray data showed similar transcriptional responses (S1 Table and Fig. 2). The extensive involvement of genes that code for metabolism, biosynthesis, and transport and binding proteins in either experiment is indicative of bacterial physiological shifts in response to nutrient availability in the respective growth environments. Notably, proteins with functions in glycolysis/gluconeogenesis, virulence, stress, and iron acquisition were detected by proteomic analysis of S. aureus abscesses in neutropenic mice [48], suggesting that many of the same responses occur among Gram-positive pathogens during abscess infections.
Identification of a stringent response transcriptional profile during subdermal abscess infection
By eight hours post-infection, at which point the bacterial cell numbers had dropped below the inoculum by 1.4 log10 CFU/ml (Fig. 1), we observed down-regulated transcription of many components of fundamental cellular pathways required for growth and replication (S2 Table). The down-regulated genes included: 23 ribosomal protein genes, eight aminoacyl tRNA ligase genes, five subunits of the ATP synthase F0 and F1 machinery, DNA polymerase III (δ’ subunit), DNA gyrase (B subunit), DNA topoisomerase (A subunit), replicative DNA helicase DnaB, DNA primase, the δ and Ω subunits of RNA polymerase, and sigma factor RpoD. Global shut-down of such processes is consistent with the stringent response elicited among bacteria during adverse conditions [49]. Indeed, over 21% (62/291) of the E. faecalis eight hour differentially regulated genes were identical to, or were the same types of genes as (e.g., ribosomal protein genes), the transcriptional profile of the E. coli stringent response induced after amino acid starvation (marked in blue font in S2 Table) [50]. The stringent response is governed by the accumulation of the metabolite (p)ppGpp during periods of stress that are unsuitable for growth, including nutrient deprivation [49]. One hundred twenty-three genes in E. faecalis OG1RF were found to be under (p)ppGpp-positive control during stringent response activation following 15 or 30 minute treatment with mupirocin [51]. Fifty-seven of the down-regulated genes and two of the up-regulated genes in the eight hour microarray (20.3%) overlapped this set of in vitro (p)ppGpp-controlled genes (marked in boldface text in S2 Table). From these data, we conclude that the population of OG1RF cells in the subdermal chambers at eight hours after inoculation is in a stringent response state. The low overlap of up-regulated genes between the mupirocin-induced and in vivo stringent response transcriptional profiles may be due, in part, to genes that were discarded during data analysis (see Materials and Methods). More so, the strong and rapid stringent response induction achieved under artificial conditions in vitro with mupirocin may have resulted in a greater number of differentially regulated genes overall. This has been observed in Escherichia coli, where gene expression patterns change directly in response to intracellular concentrations of ppGpp following stringent response induction [52].
It is important to note that the time at which the stringent response was activated cannot be discerned from these microarray data. An unknown source of stress in vivo, such as selective nutrient deprivation or one or more components of the host innate immune response (e.g., oxidative stress), may have contributed to in vivo activation of the stringent response in E. faecalis cells between two and eight hours post-inoculation. Alternatively, a minority of cells in the population may have entered a stringent response state much earlier. A situation like this has been reported to happen in E. coli, where persister cells form as a result of stochastic variation in (p)ppGpp levels [53]. In this scenario, a stringent response-induced subpopulation of E. faecalis in the subdermal abscesses would have contributed to the total gene expression measured in the two hour microarray, yet the stringent response signal would have been masked by the RNA patterns from the majority of the cells in the population, which revealed cellular adaptation to the altered environment. The transcriptome of stringent response-induced cells of such a subpopulation may have become measurable at eight hours due to the overall reduction of viable bacteria (Fig. 1) over the same period. However, the difference in viable bacteria between two and eight hours was only ∼0.5 log10 CFU/ml. Therefore, further experimentation will be required to test this hypothesis.
In E. faecalis, (p)ppGpp levels are maintained by two enzymes, the bifunctional synthetase/hydrolase RSH (also called RelA) and the small synthetase RelQ, which are encoded by genes EF1974 and EF2671, respectively [54], [55]. RSH produces (p)ppGpp during stress conditions, while RelQ maintains (p)ppGpp pools during homeostatic growth [55]. Neither gene was statistically differentially expressed in the microarray analyses (S1 and S2 Tables), which is consistent with the lack of detection of either gene in microarray experiments carried out previously on mupirocin-treated cells [51]. Using reverse transcription-qPCR, we found that transcript levels of rsh were unchanged (fold change 1.2±1.3 standard deviation, n = 5) and relQ were slightly increased (fold change 2.0±0.8 standard deviation, n = 5) at eight hours post-infection versus the inoculum. The relQ promoter was also found to be up-regulated in the subdermal abscess model in our previous RIVET screen [11], providing additional evidence that the locus is subject to transcriptional regulation under in vivo conditions. Geiger et al. recently showed that transcription of the S. aureus relQ locus, along with a second short (p)ppGpp synthase gene, relP, is strongly activated by the cell wall-targeting antibiotics vancomycin and ampicillin [56]. Similarly, the Bacillus subtilis (p)ppGpp synthase gene relQ (also known as ywaC) is transcriptionally up-regulated in response to alkaline shock [57]. These observations are consistent with the notion that the enzymes involved in (p)ppGpp metabolism are mostly under allosteric regulation.
Contribution of E. faecalis rsh and relQ to survival under in vivo and ex vivo conditions
During in vitro growth, deletion of rsh increases basal levels of ppGpp and simultaneously decreases GTP pools [58]. This is associated with a slow growth phenotype in unstressed conditions [55] and significant growth delays in 5% sodium chloride, acidic pH, and 2 mM hydrogen peroxide [55]. In contrast, deletion of relQ in OG1RF does not appear to affect basal pools of (p)ppGpp or GTP [58], but causes delayed onset of stringent response-related transcriptional changes [51] and increases susceptibility to vancomycin [55]. No virulence attenuation phenotypes were found when OG1RF Δrsh and ΔrelQ deletion strains were tested in Caenorhabditis elegans [55] or G. mellonella [51]. The OG1RF (p)ppGpp0 strain (ΔrshΔrelQ), however, is attenuated for virulence in both invertebrate models, survival in macrophages, and killing by vancomycin, norfloxacin, and ampicillin [51], [55], [58].
The contributions of the E. faecalis rsh and relQ genes to mammalian infection have not been studied previously. We used OG1RF Δrsh, ΔrelQ, ΔrshΔrelQ in-frame deletion mutants to evaluate the roles of RSH and RelQ, and thereby production of (p)ppGpp, in survival in the subdermal chambers. The percent survival of each strain, which was measured in the chambers during the initial hours of infection and then daily for four days post-infection, is shown in Fig. 3. The cell recovery in log10 CFU/ml is shown in S2 Fig. Since both the Δrsh and ΔrshΔrelQ strains are unable to mount a stringent response [55], these experimental infections allowed us to test the hypothesis that stringent response activation is necessary for the persistence phenotype observed for OG1RF in the subdermal chambers at 96 hours post-infection (Fig. 1).
Figure 3. Survival of E. faecalis OG1RF, Δrsh, ΔrelQ, and ΔrshΔrelQ in subdermal abscesses at (A) early and (B) late time points post-inoculation.
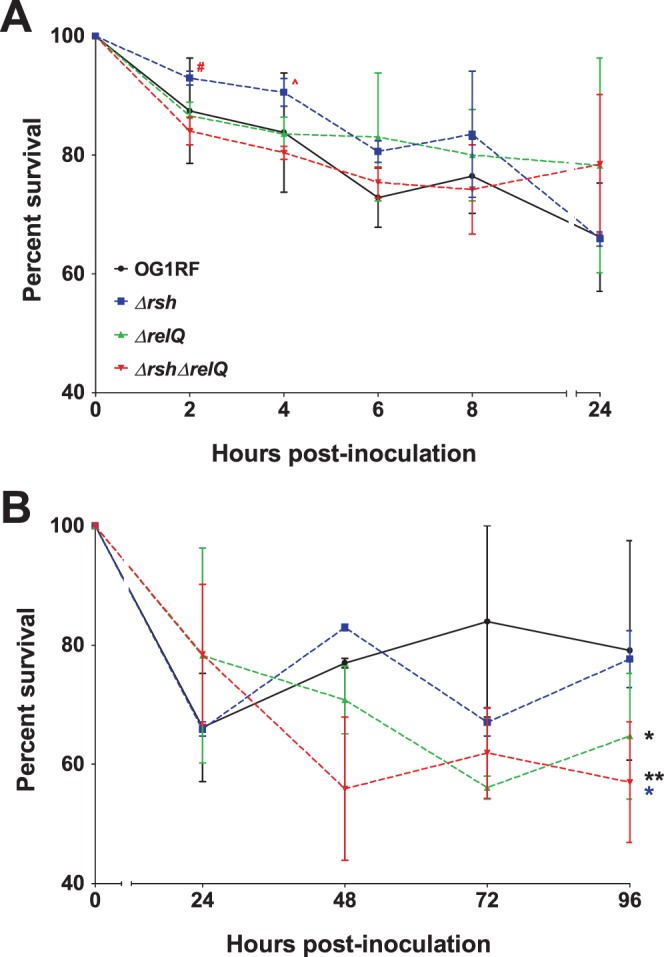
Subdermal abscess infections with the four strains were carried out as described in the text and in the legend of Fig. 1. Results are reported as percent survival with the 0 hour time point set to 100%. Values and error bars represent the mean ± SEM of n = 2 rabbits. Data from the same rabbits were separated into panels (A) and (B) for clarity. (A) Student’s t-test comparing Δrsh and ΔrshΔrelQ, α = 0.1: red #, p = 0.08; red ∧, p = 0.06. (B) One-way ANOVA followed by Tukey’s Multiple Comparison post-hoc test: black *, p<0.05 for OG1RF versus ΔrelQ; black **, p<0.01 for OG1RF versus ΔrshΔrelQ; blue *, p<0.05 for Δrsh versus ΔrshΔrelQ.
As observed with OG1RF (Figs. 1 and 3), recovery of the three in-frame deletion mutants decreased during early infection (Fig. 3A) and then persisted in the subdermal chambers for the duration of the infection (Fig. 3B). None of the strains, regardless of their ability to mount a stringent response, were cleared from the chambers. However, the (p)ppGpp mutant strains showed disparate phenotypes at the early (Fig. 3A) and prolonged (Fig. 3B) stages of infection. The rate of clearance of the Δrsh strain was diminished at two and four hours compared to the other strains. These differences were particularly pronounced with the ΔrshΔrelQ strain (Fig. 3A, α = 0.1, p = 0.08 and 0.06 at two and four hours, respectively). Between 48 to 96 hours, the four strains persisted at relatively stable levels that fluctuated daily (Fig. 3B). During that period, OG1RF persisted at a mean percent survival of 80.0±15.5% (standard deviation). The mean percent survival of Δrsh was nearly identical to OG1RF (75.9±8.0%), while the mean percent survival levels of ΔrelQ and ΔrshΔrelQ (63.9±10.1% and 58.3±11.4%, respectively) were significantly lower than OG1RF (p<0.05 for ΔrelQ and p<0.01 for ΔrshΔrelQ). The difference between the Δrsh and ΔrshΔrelQ strains was also significant (p<0.05).
We also compared growth and survival of OG1RF and the three (p)ppGpp-defective strains in human serum (Fig. 4A) and whole blood (Fig. 4B) ex vivo (Fig. 4). The growth kinetics and prolonged survival levels of OG1RF and the ΔrelQ strain were similar in serum and blood. In contrast, growth of the Δrsh strain at nine hours was significantly lower than growth of OG1RF in serum (Fig. 4A), but was significantly higher than growth of OG1RF in blood (Fig. 4B). By 24 hours, survival levels of the Δrsh strain were essentially indistinguishable from OG1RF and the ΔrelQ strain in either condition. Growth of the ΔrshΔrelQ strain was significantly lower than OG1RF at all nearly all time points studied in serum and blood.
Figure 4. Growth and survival of E. faecalis OG1RF, Δrsh, ΔrelQ, and ΔrshΔrelQ in human (A) serum and (B) whole blood ex vivo.
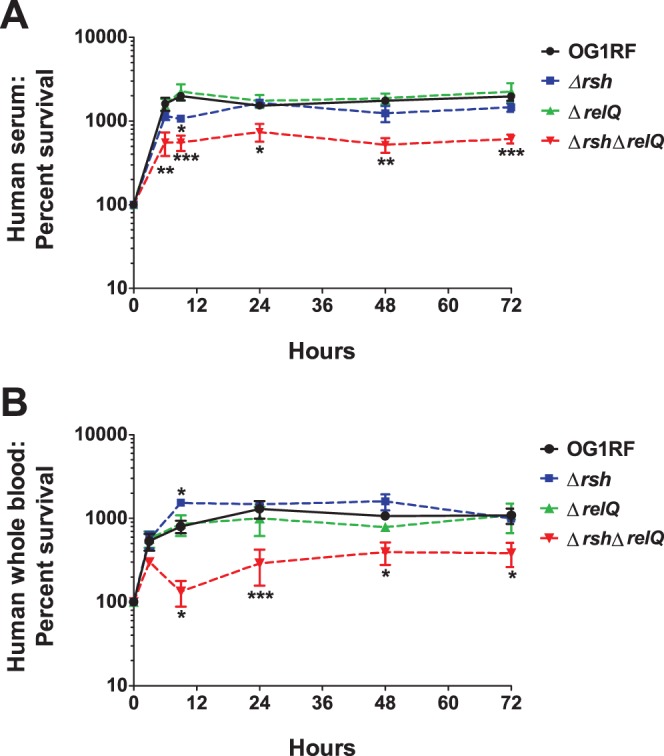
Overnight cultures of the four strains were diluted 1∶20 in serum or whole blood and incubated at 37°C. Dilutions of aliquots collected at 0, 3 (panel B), 6 (panel A), 9, 24, 48, and 72 hours were plated to assess bacterial survival. Results are reported as percent survival with the 0 hour time point set to 100%. Values and error bars represent the mean ± SEM of n = 3 independent replicates. Data were analyzed for statistical significance with two-way ANOVA followed by Dunnett’s multiple comparisons test. *, p<0.05; **, p<0.01; ***, p<0.001.
Taken together, these data show that intracellular (p)ppGpp levels affect viability of E. faecalis in multiple conditions relevant to mammalian infection. The ability of a strain to activate a stringent response does not influence persistence in this model, as evidenced by the fact that two strains that can mount a stringent response (OG1RF wild type and ΔrelQ) and two strains that cannot mount a stringent response (Δrsh and ΔrshΔrelQ) persisted in the subdermal chambers for the 96 hour infection. Gaca et al. previously reported impaired survival of the (p)ppGpp0 strain (ΔrshΔrelQ) in macrophages [51]. Consistent with this, our data confirm that the complete lack of (p)ppGpp impairs growth and survival in subdermal abscesses, serum, and blood. The different survival phenotypes for the ΔrelQ strain in the subdermal abscess model (Fig. 3B) and the ex vivo serum and blood experiments (Fig. 4) suggest the hypothesis that RelQ may contribute to host-pathogen interactions that were not intact in the ex vivo assays. Finally, the decreased clearance and increased growth rates of the Δrsh strain in subdermal abscesses (Fig. 3A) and in whole blood (Fig. 4B), respectively, support a model in which high basal levels of ppGpp, and correspondingly low levels of GTP, confer a protective advantage from the host innate immunity. Follow-up studies will be essential to decipher the complete role of (p)ppGpp in the pathogenesis of E. faecalis infections.
Insights on E. faecalis in vivo gene expression gained from complementary techniques
To gain additional insight on E. faecalis gene regulation in the subdermal abscess environment, we compared the microarray data from this study with our previous RIVET screen data set [11]. That screen identified 249 in vivo-activated promoters in E. faecalis OG1RF during the early hours of infection in the subdermal abscess model. One hundred fifty-six in vivo-activated promoters are predicted to generate transcripts from the sense strand, with the remainder of the promoters driving expression from the antisense strand. The subdermal abscess RIVET sense strand gene set was compared to the up-regulated genes from the microarray analysis at two and eight hours post-inoculation (Fig. 5A and Table 1). Ten genes were shared between the RIVET screen and either microarray condition, with a single gene, EF1672 (permease protein), activated in all three conditions tested. Interestingly, EF1672 was also one of the two up-regulated genes in the eight hour microarray that overlapped the set of genes regulated in a (p)ppGpp-dependent manner during in vitro stringent response activation (marked in boldface text in S2 Table) [51]. Overall, only a fraction of the RIVET clones overlapped with the differentially expressed genes (Fig. 5). Differences in the time points sampled (i.e., two and eight hours post-inoculation for microarray versus four, eight, and 24 hours post-inoculation for RIVET) and in the microarray and RIVET methodologies (e.g., RIVET clones may represent promoters that are expressed transiently or in only a minor subpopulation of cells) may account, in part, for the broad coverage of differentially regulated genes that comprise the two data sets. Regardless, the complementary and largely non-redundant outcome of pairing the data sets permitted the discrete identification of candidate genes that we hypothesize represent core genetic determinants necessary for E. faecalis adaptation to growth in a host (Fig. 5A and Table 1). Future functional studies of EF1672 and the other candidate core genes in Table 1 may lead to the identification of new targets for antimicrobial agents or vaccines for the treatment and prevention of enterococcal infections.
Figure 5. Overlap of differential gene expression between microarray and in vivo-activated transcripts identified by RIVET screen in the E. faecalis subdermal abscess model [11].
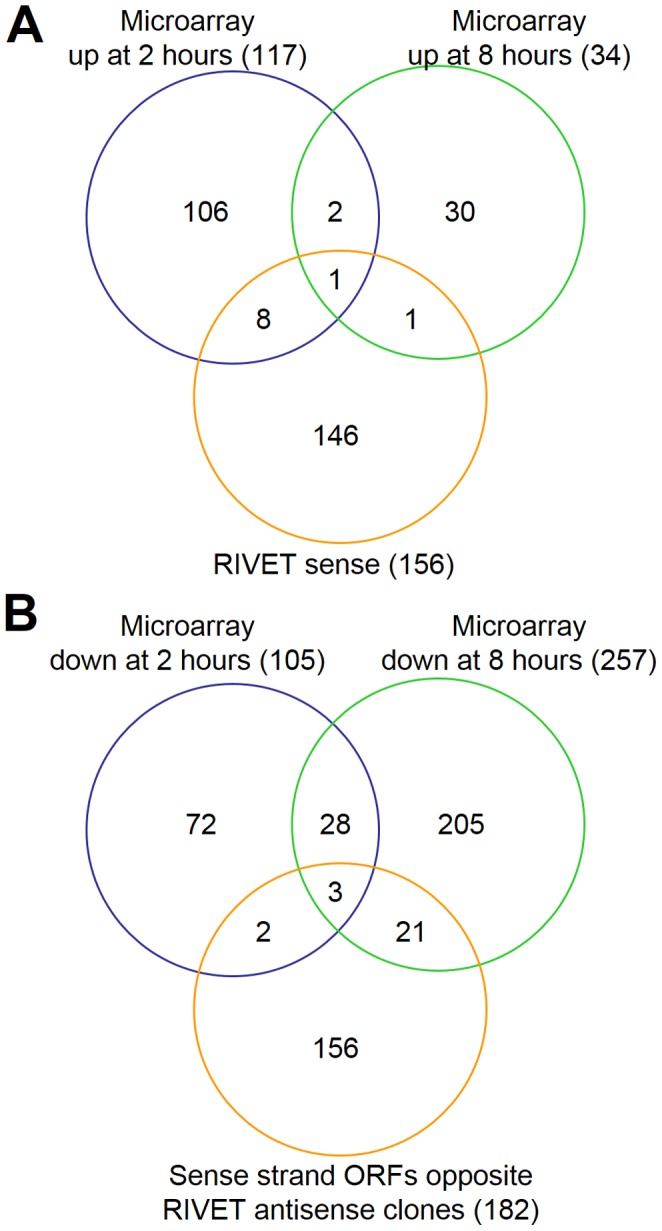
(A) Up-regulated genes identified by microarray compared with RIVET sense direction clones. (B) Down-regulated genes identified by microarray compared with sense strand ORFs that are complementary to RIVET antisense clones. See text and reference [11] for additional details on the subdermal abscess RIVET screen that was previously conducted. See Table 1 for the identities of the genes from microarray analyses that overlap with RIVET analysis genes. See column H of S1 and S2 Tables for the identities of genes found in both microarray analyses.
Table 1. Differentially regulated genes in subdermal abscesses identified by microarray that overlap with a previously conducted subdermal abscess RIVET screen [11].
| Category1 | Locus | Gene Description |
| 2 h microarray UP +8 h microarray UP+RIVET sense | ||
| EF1672 | permease protein | |
| 2 h microarray UP+RIVET sense | ||
| EF0031 | hypothetical protein | |
| EF0246 | ABC superfamily ATP binding cassette transporter, ABC protein | |
| EF0475 | ferrous iron transport protein A | |
| EF0697 | hypothetical protein | |
| EF0849 | alanine racemase | |
| EF1084 | universal stress protein | |
| EF2355 | chaperone protein ClpB | |
| EF2987 | integrating conjugative element protein | |
| 8 h microarray UP+RIVET sense | ||
| EF0383 | hypothetical protein | |
| 2 h microarray DOWN +8 hmicroarray DOWN+RIVET antisense | ||
| EF1360 | dihydroxyacetone kinase | |
| EF1614 | DNA topoisomerase subunit A | |
| EF2623 | P-ATPase superfamily P-type ATPase cadmium transporter | |
| 2 h microarray DOWN+RIVET antisense | ||
| EF0004 | recombination protein F | |
| EF1112 | exonuclease RexB | |
| 8 h microarray DOWN+RIVET antisense | ||
| EF0005 | DNA gyrase subunit B | |
| EF0013 | replicative DNA helicase DnaB | |
| EF0225 | 50S ribosomal protein L30 | |
| EF0226 | 50S ribosomal protein L15 | |
| EF0724 | glutamyl-tRNA(Gln) amidotransferase subunit C | |
| EF0801 | leucyl-tRNA synthetase | |
| EF0909 | oligopeptide ABC superfamily ATP binding cassette transporter, membrane protein | |
| EF0988 | cell division protein MraZ | |
| EF1003 | isoleucine–tRNA ligase | |
| EF1146 | DNA-directed RNA polymerase sigma subunit RpoE | |
| EF1361 | dihydroxyacetone kinase | |
| EF1522 | RNA polymerase sigma factor rpoD | |
| EF1550 | DNA-binding protein HU | |
| EF1580 | hypothetical protein | |
| EF1629 | ethanolamine ammonia-lyase large subunit | |
| EF2397 | elongation factor EF1B | |
| EF2398 | 30S ribosomal protein S2 | |
| EF2908 | group 2 glycosyl transferase | |
| EF3178 | succinyl-diaminopimelate desuccinylase | |
| EF3270 | glutathione-disulfide reductase | |
| EF3278 | hypothetical protein | |
Microarray UP and Microarray DOWN indicate that the locus was up- or down-regulated, respectively, in microarray analysis at the indicated time point.
The high number of in vivo-activated antisense promoter regions (n = 93) identified in the subdermal abscess RIVET screen [11] led us to hypothesize that E. faecalis may use global antisense gene regulation mechanisms during in vivo growth. We reasoned that if antisense transcripts predicted by the RIVET clones were acting to silence transcription, there might be a correlation between the down-regulated transcripts identified by microarray and the subdermal abscess RIVET antisense transcript list. Many of the 93 predicted antisense promoter regions from the RIVET screen overlap more than one sense strand ORF, resulting in a total of 182 sense strand ORFs potentially affected by an antisense transcript (see reference [11]). Therefore, we compared the down-regulated microarray gene lists to this set of 182 ORFs (Fig. 5B and Table 1). Twenty-six genes overlapped in the gene sets, with three genes (Table 1) shared in all three conditions. The majority of the remaining overlapping genes were from the eight hour microarray, ten of which were stringent response-related genes (Fig. 5B and Table 1). Two of these genes, EF0909 and EF2398, are complementary to confirmed antisense transcripts that are expressed in vivo during subdermal abscess infection [11]. In total, 4.8% of the down-regulated genes at two hours and 9.3% of down-regulated genes at eight hours corresponded with antisense clones identified in the subdermal abscess RIVET screen. Further studies are needed to determine whether these predicted sense-antisense interactions occur within E. faecalis and, if so, whether they are targets for RNase III cleavage, which has been proposed as a post-transcriptional regulatory mechanism in Gram-positive bacteria [59].
Conclusions
In conclusion, this report provides a comprehensive characterization of the gene expression changes in E. faecalis during the course of a subdermal abscess infection in a mammalian host. This is the first description of the use of microarrays to evaluate E. faecalis in vivo transcription in a vertebrate infection model (S1 and S2 Tables). Our analysis reveals that in vivo-grown E. faecalis cells existed in two distinct transcriptional states during the first several hours of infection (Figs. 2 and 4, and S1 and S2 Tables): the enterococci first adjusted to growth in a new environment followed by the surviving cells reaching a stringent response state. Our genetic data suggest that this physiologic state is important for survival in this environment and that it may have resulted from high levels of (p)ppGpp. In addition, the combined RIVET and microarray data analysis is in support of our hypothesis that E. faecalis may use antisense transcripts to globally silence expression of sense strand transcripts in vivo. Finally, our serendipitous observation of an E. faecalis in vivo stringent response transcriptome suggests that the subdermal chamber infection model can serve as a novel experimental system for studying the role of (p)ppGpp in the context of the mammalian immune system.
Materials and Methods
Bacterial strains, growth conditions, and enzymes
The E. faecalis OG1RF strain background [58], [60] was used for all experiments. OG1RF Δrsh (formerly called ΔrelA [58]), OG1RF ΔrelQ, and OG1RF ΔrshΔrelQ were described previously [55]. Bacteria were streaked onto Brain Heart Infusion (BHI, BD Bacto, Becton, Dickinson and Company, Sparks, MD) agar plates from glycerol stocks stored at -80°C and incubated at 37°C. Cultures for animal experiments were grown in trypsinized beef heart dialysate (BH) medium [61] under static conditions at 37°C. Lysozyme and mutanolysin were purchased from Sigma-Aldrich (St. Louis, MO).
Rabbit model of subdermal abscess formation
All animal procedures were carried out in accordance with the guidelines set forth by the Public Health Service Policy on Humane Care and Use of Laboratory Animals. The University of Minnesota Institutional Animal Care and Use Committee approved the protocol used in this work (protocol 0910A73332). Animals were euthanized with Beuthanasia-D and efforts were made to minimize suffering.
Subdermal chambers were implanted subcutaneously into the flanks of New Zealand white rabbits (2–3 kg, either sex) exactly as previously described [11]. The recovery period lasted a minimum of six weeks in order for the implanted chambers to be encapsulated with fibrous tissue and fill with approximately 30 ml of serous fluid. Microarray analysis samples were obtained from four rabbits in which chambers had been implanted 7 to 45 weeks prior to infection. The (p)ppGpp mutant strains were studied in two rabbits per strain in which chambers had been implanted 7 weeks prior to infection. To initiate infection, two ml of serous fluid were aspirated from the subdermal chamber and were replaced with 2 ml of inoculum prepared as described below for gene expression analyses. Bacterial recovery, measured as CFU/ml, was assessed as described below. Results are reported as the log10 CFU/ml transformed values or as percent survival with the bacterial load at the 0 hour time point set to 100%.
Subdermal chamber infections for survival counts and gene expression analyses
(A) Microarray experiments. A single colony of E. faecalis OG1RF was inoculated into 10–25 ml BH medium and incubated for ∼15–16 hours, then diluted 1∶5 into 25–50 ml of fresh BH medium and incubated for 2 more hours. Bacteria were harvested by centrifugation for 15–20 min at 6000 rpm in a Beckman JA-17 rotor at 4°C. Pelleted cells were resuspended to an optical density at a wavelength of 600 nm of ∼1.3–1.6 in KPBS, and 2 ml were used for each subdermal chamber infection. Two milliliter volumes were removed from the subdermal chambers at 2, 4, 8, 24, and 96 hours post-inoculation. Approximately 0.1–0.2 ml aliquots of the initial inoculum and each aspirate were used for serial dilution and plating to quantitate the bacterial load at each time point. Two milliliters of the initial inoculum, and the remainder of each aspirate (∼1.8 ml), were immediately added to 4 ml of RNAprotect Bacteria Reagent (Qiagen Inc., Valencia, CA), vortexed, processed according to the manufacturer’s instructions, flash-frozen in a dry ice/ethanol bath, and stored at −80°C until RNA extraction. (B) Infections with (p)ppGpp mutant strains. Infections with OG1RF and the OG1RF deletion strains Δrsh, ΔrelQ, and ΔrshΔrelQ were carried out as described above for microarray experiments, except that aspirates were collected from the subdermal chambers at 2, 4, 6, 8, 24, 48, 72, and 96 hours post-inoculation. Statistical comparisons were calculated with the Student’s t-test function in Microsoft Excel 2013 and one-way ANOVA followed by Tukey’s Multiple Comparison post-hoc test in Graphpad Prism (version 5.04). Significance was assessed at α = 0.05 unless otherwise stated.
RNA extraction and DNase treatment
RNA was prepared from subdermal aspirates as follows: frozen pellets were thawed, resuspended in 0.4 ml of RNase-free TE containing 50 mg/ml lysozyme and 1000 U/ml mutanolysin, homogenized using a hand-held motorized homogenizer with RNase-free pellet pestles (Fisher Scientific, Pittsburgh, PA), and incubated for 10 min at 37°C. Each sample was then split in half and extracted in duplicate with the RNeasy Mini Kit (Qiagen Inc.) according to the manufacturer’s instructions with an added homogenization step using the QIAshredder (Qiagen Inc.) immediately after the addition of Buffer RLT. RNA was eluted from each column with two 30 µl volumes of RNase-free water, and total RNA from duplicate extractions of each sample were pooled together. RNA from uninfected serous fluid was extracted with TRIzol Reagent (Invitrogen Corp., Carlsbad, CA) as suggested by the manufacturer immediately following aspiration from the subdermal chamber.
Contaminating DNA was removed using a TURBO DNA-free kit (Ambion, Austin, TX) following the rigorous protocol as directed by the manufacturer. RNA for microarray experiments was then ethanol precipitated in the presence of a glycogen carrier (Roche Applied Science, Indianapolis, IN) and resuspended in 10 µl RNase-free water; 1 µl was reserved for qPCR experiments.
Genomic DNA (gDNA) extraction and shearing
gDNA from a 50 ml culture of E. faecalis OG1RF grown overnight in Todd-Hewitt broth (BD Bacto, Becton, Dickinson and Company) was extracted using a Genomic-tip 500/G column (Qiagen, Inc.) as directed by the manufacturer. Precipitated DNA was resuspended in 1.7 ml of 10 mM Tris-Cl, pH 8.5. Approximately 425 µg of gDNA were sheared by nebulization with nitrogen gas, essentially as previously described [62], [63], at 25 psi to obtain fragments of 0.8–1.5 kb. Sheared DNA was ethanol precipitated, resuspended in 0.2 ml sterile water, and stored at -20°C until use in microarray experiments.
Microarray design and printing
The 3,229 70-mer oligonucleotides representing ORFs from E. faecalis, including 3093 chromosomal ORFs of strain V583, that were used as probes on the microarrays in this study have been described in detail elsewhere [64]. Approximately 2340 protein coding ORFs (87%) of the OG1RF genome, as annotated in our previously published RAST server annotation [11], were represented on the slides. Oligos were spotted in quadruplicate on GAPS II coated slides (Corning Incorporated, Corning, New York) with a Biorobotics Microgrid II spotter (Digilab, Inc., Holliston, MA) at the University of Minnesota Genomics Center. Spotted arrays were UV-crosslinked 24 hours after printing and were stored in a dessicator. Slides were incubated at 42°C in prehybridization buffer immediately before use, as described in the Corning GAPS II manual.
Microarray probe preparation and hybridization
Sheared gDNA, which was used as a reference in the microarray experiments [62], was labeled with Cy3-dUTP (GE Healthcare Life Sciences, Piscataway, NJ) using the BioPrime Array CGH Genomic Labeling Module kit (Invitrogen Corp.). Total RNA (9 µl) was reverse transcribed and labeled with Alexa Fluor 647-aha-dUTP using the SuperScript Direct cDNA Labeling System (Invitrogen Corp.). Labeled nucleic acids were purified with the QIAquick PCR purification kit (Qiagen Inc.) and eluted with two 30 µl volumes of 10 mM Tris-Cl, pH 8.5, that were pooled together. Labeled cDNA was mixed with 0.5 µg labeled gDNA, dried down to ∼10 µl in a speedvac, and mixed with ∼40 µl of hybridization probe solution that contained a final concentration of 50% formamide, 5X SSC, 0.1 mg/ml salmon sperm DNA, and 0.1% SDS. The probe was incubated at 95°C for 5 min and cooled at room temperature. Hybridizations were performed at 42°C for 17–19 hours in a MAUI hybridization system (BioMicro Systems, Salt Lake City, UT) using the appropriate MAUI mixers. Slides were then washed twice with 2X SSC, 0.5% SDS, once with 1X SSC, and once with 0.1X SSC before being dried by low-speed centrifugation for 10 min. Slides were scanned on a ScanArray 5000 scanner at a resolution of 10 µm (Perkin Elmer, Waltham, MA). A control experiment was performed with RNA extracted from uninfected chamber aspirate to assess background hybridization from the eukaryotic component of samples. Fluorescence was minimal (data not shown), so the contribution of eukaryotic RNA to the measured signal intensities in experiments containing bacterial RNA was considered negligible.
Microarray data analysis
The microarray experimental design employed a reference sample design, in which a constant amount of labeled gDNA served as the common reference sample for cDNA samples across all time points studied [62], [65]. This experimental design is advantageous over cDNA/cDNA pair-wise comparison experimental designs for time course studies because it reduces the number of hybridization arrays and amount of RNA per sample required [62]. Scanned images were imported into BlueFuse version 3.6 or earlier (BlueGnome Ltd., Cambridge, UK), where spots were picked using Bayesian statistical modeling to extract the fluorescence intensities. Data were then filtered based upon intensity, Bluefuse confidence, and spot size. Due to a malfunction during slide printing, block 1 spots were uninterpretable and were removed from the analysis at this step. Replicate spots were fused by averaging the intensities of high quality spots. The filtered raw values were exported for further analysis in Expressionist Analyst version 5.3.2 or earlier software (Genedata Inc., Basel, Switzerland). The ratio of the cDNA intensity to the gDNA intensity for each fused spot was calculated and the data from all chips were quantile normalized, as previously described for analyzing gene expression in bacterial systems [62]. Principal components analysis was used to visualize variability among biological replicates while ensuring they clustered together by time point (data not shown). The fold change for each gene was calculated by dividing the cDNA/gDNA ratio obtained at either of the sampled time points (i.e., two or eight hours post-inoculation) by the corresponding cDNA/gDNA ratio of the input inoculum (i.e., zero hours) for each biological replicate. Individual fold changes were then averaged to obtain the final average fold change. The values for down-regulated genes are reported as the negative inverse of the calculated fold-changes, unless otherwise specified. Paired t-tests comparing the cDNA/gDNA ratio for each gene in each time point analysis (i.e., 0 versus 2 hours, 0 versus 8 hours) were performed. Results were filtered for genes showing at least a 2-fold change in expression with a p-value of less than 0.05 and a false discovery rate p-value of less than 0.05. For ease of analysis, data from only those spots known to represent genes in the genome of E. faecalis OG1RF, as determined by cross-referencing the gene lists with our previously published RAST genome annotation [11], were included in the final analysis.
Bioinformatics analysis
Bioinformatics analysis of the gene lists were performed with the Database for Annotation, Visualization and Integrated Discovery (DAVID, v6.7, http://david.abcc.ncifcrf.gov/) [66], [67]. Uniprot Accession numbers were entered to create working gene lists that were analyzed with the KEGG and GOTERM_BP_FAT functions. Results were viewed using DAVID’s Functional Annotation Clustering method. Additional analysis of pathways and gene functions in E. faecalis was performed by searching the KEGG Pathway Map and Brite hierarchy functions available at www.genome.jp/kegg/ [68], [69].
Microarray data accession number
Microarray data from this study has been submitted to the Gene Expression Omnibus (GEO) database [70], accession number GSE22391, to comply with the MIAME standards.
Quantitative PCR primers, cDNA, and amplification
Oligonucleotide sequences for primers used in qPCR assays are listed in S3 Table. DNase-treated total RNA was reverse-transcribed with random primers using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen Corp.). qPCR was carried out on an iCycler equipped with an iQ5 real-time detection system (Bio-Rad Laboratories, Inc., Hercules, CA) with iQ SYBR Green Supermix (Bio-Rad Laboratories, Inc.). Each reaction was performed in triplicate and the average Ct value was used in calculations. DNase-treated RNA was used as a template in control reactions to confirm removal of DNA. EF0886, which was identified in the microarray analysis as being a gene with non-changing expression at the analyzed time points, was used as a reference gene. The fold change for each sample was calculated using the Pfaffl method [71], and the average fold change for each gene assayed was obtained by averaging the fold changes from all biological replicates.
Blood and serum survival experiments
Human whole blood was obtained from the University of Rochester Medical Center blood bank. Serum was collected from centrifuged whole blood that was coagulated with 20 mM CaCl2. Overnight cultures of OG1RF and the (p)ppGpp mutant strains grown in BHI broth were diluted 1∶20 in whole blood or serum and incubated at 37°C. Aliquots collected at 0, 3 (blood only), 6 (serum only), 9, 24, 48, and 72 hours were serially diluted and plated on tryptic soy agar to assess bacterial survival. The ΔrshΔrelQ strain caused blood to clot by 24 hours through an as-yet-uncharacterized mechanism (C. Colomer-Winter and J. A. Lemos, unpublished data). Sonication of the clotted blood yielded an increase of one log10 CFU/ml in viable counts relative to clotted blood; therefore, quantitative culture results for ΔrshΔrelQ strain were adjusted accordingly. Experiments were performed in triplicate and the results were averaged. Results are reported as percent survival with the 0 hour time point set to 100%. Statistical comparisons were calculated with two-way ANOVA followed by Dunnett’s multiple comparisons test in Graphpad Prism. Significance was assessed at α = 0.05.
Supporting Information
Reverse transcription-qPCR validation of selected differentially expressed genes identified by microarray analysis. E. faecalis OG1RF RNA extracted from two and eight hour post-infection subdermal chamber aspirates was reverse transcribed with random hexamers. The resulting cDNAs were used as templates in qPCR experiments. EF0886, which was shown to be stably expressed across time points in the microarray experiments, was used as a reference gene to calculate relative fold change for each gene shown. Data are the mean ± standard deviation of four biological replicates for microarray values and three biological replicates for qPCR values. Down-regulated genes have fold-changes less than one (dotted line). (A) Two hours post-inoculation, (B) Eight hours post-inoculation.
(JPG)
{kind=link}
Recovery of OG1RF, Δrsh, ΔrelQ, and ΔrshΔrelQ from subdermal abscesses at (A) early and (B) late time points post-inoculation. Subdermal abscess infections with the four strains were carried out as described in the text and in the legend of Fig. 1. Values and error bars represent the mean ± SEM of n = 2 rabbits. Data from the same rabbits were separated into panels (A) and (B) for clarity.
(TIF)
E. faecalis OG1RF in vivo differentially regulated genes identified via microarray analysis at two hours post-infection of subdermal chambers. The fold change values for up- and down-regulated genes are shown in green and red fonts, respectively. Column H lists the corresponding positive and negative fold changes, highlighted in green and red, respectively, from the eight hour microarray.
(XLS)
E. faecalis OG1RF in vivo differentially regulated genes identified via microarray analysis at eight hours post-infection of subdermal chambers. The fold change values for up- and down-regulated genes are shown in green and red fonts, respectively. Column H lists the corresponding positive and negative fold changes, highlighted in green and red, respectively, from the two hour microarray. Genes with text in blue font are identical or highly similar to E. coli stringent response genes [50]. Genes in boldface text were also differentially expressed in E. faecalis in a (p)ppGpp-dependent manner during stringent response activation following 15 or 30 minute treatment with mupirocin [51].
(XLS)
List of oligonucleotides.
(DOCX)
Acknowledgments
The authors gratefully acknowledge Christopher Kristich for advice on the microarray experiments, Archana Deshpande for her assistance and advice with the microarray experiments, Tracy Bergemann for her valuable input on the microarray data analysis, Jacqueline Abranches for sharing sequences for qPCR primers, and Aaron Barnes for critical reading of the manuscript.
This work was carried out in part using software provided by the University of Minnesota Supercomputing Institute.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. Microarray data from this study has been submitted to the Gene Expression Omnibus (GEO) database, accession number GSE22391, to comply with the MIAME standards. The GEO series accession number for the RNA-seq data is GSE58467.
Funding Statement
This research was supported by Award Number R01AI58134 from the National Institute of Allergy and Infectious Diseases to GMD. KLF was supported by Award Number F32AI082881 from the National Institute of Allergy and Infectious Diseases, Award Number T32DE007288 from the National Institute of Dental and Craniofacial Research, and Award Number 10POST3290026 from the American Heart Association. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases, the National Institute of Dental and Craniofacial Research, the National Institutes of Health, or the American Heart Association. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Lebreton F, Willems RJL, Gilmore MS (2014) Enterococcus Diversity, Origins in Nature, and Gut Colonization. In: Gilmore MS, Clewell DB, Ike Y, Shankar N, editors. Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. Boston. [PubMed]
- 2. Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, et al. (2013) Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol 34:1–14. [DOI] [PubMed] [Google Scholar]
- 3. Sava IG, Heikens E, Huebner J (2010) Pathogenesis and immunity in enterococcal infections. Clin Microbiol Infect 16:533–540. [DOI] [PubMed] [Google Scholar]
- 4. Creti R, Imperi M, Bertuccini L, Fabretti F, Orefici G, et al. (2004) Survey for virulence determinants among Enterococcus faecalis isolated from different sources. J Med Microbiol 53:13–20. [DOI] [PubMed] [Google Scholar]
- 5. Coque TM, Patterson JE, Steckelberg JM, Murray BE (1995) Incidence of hemolysin, gelatinase, and aggregation substance among enterococci isolated from patients with endocarditis and other infections and from feces of hospitalized and community-based persons. J Infect Dis 171:1223–1229. [DOI] [PubMed] [Google Scholar]
- 6. Dupre I, Zanetti S, Schito AM, Fadda G, Sechi LA (2003) Incidence of virulence determinants in clinical Enterococcus faecium and Enterococcus faecalis isolates collected in Sardinia (Italy). J Med Microbiol 52:491–498. [DOI] [PubMed] [Google Scholar]
- 7. Bittencourt de Marques E, Suzart S (2004) Occurrence of virulence-associated genes in clinical Enterococcus faecalis strains isolated in Londrina, Brazil. J Med Microbiol 53:1069–1073. [DOI] [PubMed] [Google Scholar]
- 8. Johansson D, Rasmussen M (2013) Virulence factors in isolates of Enterococcus faecalis from infective endocarditis and from the normal flora. Microb Pathog 55:28–31. [DOI] [PubMed] [Google Scholar]
- 9. Maadani A, Fox KA, Mylonakis E, Garsin DA (2007) Enterococcus faecalis mutations affecting virulence in the Caenorhabditis elegans model host. Infect Immun 75:2634–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hanin A, Sava I, Bao Y, Huebner J, Hartke A, et al. (2010) Screening of in vivo activated genes in Enterococcus faecalis during insect and mouse infections and growth in urine. PLoS One 5:e11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frank KL, Barnes AM, Grindle SM, Manias DA, Schlievert PM, et al. (2012) Use of recombinase-based in vivo expression technology to characterize Enterococcus faecalis gene expression during infection identifies in vivo-expressed antisense RNAs and implicates the protease Eep in pathogenesis. Infect Immun 80:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holt JF, Kiedrowski MR, Frank KL, Du J, Guan C, et al. (2014) Enterococcus faecalis 6-phosphogluconolactonase is required for both commensal and pathogenic interactions with Manduca sexta. Infect Immun Epub ahead of print. [DOI] [PMC free article] [PubMed]
- 13. Orihuela CJ, Radin JN, Sublett JE, Gao G, Kaushal D, et al. (2004) Microarray analysis of pneumococcal gene expression during invasive disease. Infect Immun 72:5582–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sebbane F, Lemaitre N, Sturdevant DE, Rebeil R, Virtaneva K, et al. (2006) Adaptive response of Yersinia pestis to extracellular effectors of innate immunity during bubonic plague. Proc Natl Acad Sci U S A 103:11766–11771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, et al. (2009) The Listeria transcriptional landscape from saprophytism to virulence. Nature 459:950–956. [DOI] [PubMed] [Google Scholar]
- 16. Xu Q, Dziejman M, Mekalanos JJ (2003) Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro . Proc Natl Acad Sci U S A 100:1286–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stintzi A, Marlow D, Palyada K, Naikare H, Panciera R, et al. (2005) Use of genome-wide expression profiling and mutagenesis to study the intestinal lifestyle of Campylobacter jejuni . Infect Immun 73:1797–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Talaat AM, Lyons R, Howard ST, Johnston SA (2004) The temporal expression profile of Mycobacterium tuberculosis infection in mice. Proc Natl Acad Sci U S A 101:4602–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Graham MR, Virtaneva K, Porcella SF, Gardner DJ, Long RD, et al. (2006) Analysis of the transcriptome of group A Streptococcus in mouse soft tissue infection. Am J Pathol 169:927–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chaffin DO, Taylor D, Skerrett SJ, Rubens CE (2012) Changes in the Staphylococcus aureus transcriptome during early adaptation to the lung. PLoS One 7:e41329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Skvortsov TA, Ignatov DV, Majorov KB, Apt AS, Azhikina TL (2013) Mycobacterium tuberculosis transcriptome profiling in mice with genetically different susceptibility to tuberculosis. Acta naturae 5:62–69. [PMC free article] [PubMed] [Google Scholar]
- 22. Mandlik A, Livny J, Robins WP, Ritchie JM, Mekalanos JJ, et al. (2011) RNA-Seq-based monitoring of infection-linked changes in Vibrio cholerae gene expression. Cell Host Microbe 10:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jorth P, Trivedi U, Rumbaugh K, Whiteley M (2013) Probing bacterial metabolism during infection using high-resolution transcriptomics. J Bacteriol 195:4991–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lindenstrauss AG, Ehrmann MA, Behr J, Landstorfer R, Haller D, et al. (2014) Transcriptome analysis of Enterococcus faecalis toward its adaption to surviving in the mouse intestinal tract. Arch Microbiol 196:423–433. [DOI] [PubMed] [Google Scholar]
- 25. Yarwood JM, McCormick JK, Paustian ML, Kapur V, Schlievert PM (2002) Repression of the Staphylococcus aureus accessory gene regulator in serum and in vivo . J Bacteriol 184:1095–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tight RR, Prior RB, Perkins RL, Rotilie CA (1975) Fluid and penicillin G dynamics in polyethylene chambers implanted subcutaneously in rabbits. Antimicrob Agents Chemother 8:495–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tanenbaum DM, Goll J, Murphy S, Kumar P, Zafar N, et al. (2010) The JCVI standard operating procedure for annotating prokaryotic metagenomic shotgun sequencing data. Stand Genomic Sci 2:229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Torrieri R, Oliveira FS, Oliveira G, Coimbra RS (2012) Automatic assignment of prokaryotic genes to functional categories using literature profiling. PLoS One 7:e47436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, et al. (2003) Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis . Science 299:2071–2074. [DOI] [PubMed] [Google Scholar]
- 30. Vebø HC, Snipen L, Nes IF, Brede DA (2009) The transcriptome of the nosocomial pathogen Enterococcus faecalis V583 reveals adaptive responses to growth in blood. PLoS One 4:e7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Teng F, Nannini EC, Murray BE (2005) Importance of gls24 in virulence and stress response of Enterococcus faecalis and use of the Gls24 protein as a possible immunotherapy target. J Infect Dis 191:472–480. [DOI] [PubMed] [Google Scholar]
- 32. Nannini EC, Teng F, Singh KV, Murray BE (2005) Decreased virulence of a gls24 mutant of Enterococcus faecalis OG1RF in an experimental endocarditis model. Infect Immun 73:7772–7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Giard JC, Rince A, Capiaux H, Auffray Y, Hartke A (2000) Inactivation of the stress- and starvation-inducible gls24 operon has a pleiotrophic effect on cell morphology, stress sensitivity, and gene expression in Enterococcus faecalis . J Bacteriol 182:4512–4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de Oliveira NE, Abranches J, Gaca AO, Laport MS, Damaso CR, et al. (2011) clpB, a class III heat-shock gene regulated by CtsR, is involved in thermotolerance and virulence of Enterococcus faecalis . Microbiology 157:656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Herrmann KM, Weaver LM (1999) The Shikimate Pathway. Annu Rev Plant Physiol Plant Mol Biol 50:473–503. [DOI] [PubMed] [Google Scholar]
- 36. Nygaard P, Duckert P, Saxild HH (1996) Role of adenine deaminase in purine salvage and nitrogen metabolism and characterization of the ade gene in Bacillus subtilis . J Bacteriol 178:846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shah P, Romero DG, Swiatlo E (2008) Role of polyamine transport in Streptococcus pneumoniae response to physiological stress and murine septicemia. Microb Pathog 45:167–172. [DOI] [PubMed] [Google Scholar]
- 38. Lopez G, Latorre M, Reyes-Jara A, Cambiazo V, Gonzalez M (2012) Transcriptomic response of Enterococcus faecalis to iron excess. Biometals 25:737–747. [DOI] [PubMed] [Google Scholar]
- 39. Benachour A, Morin T, Hebert L, Budin-Verneuil A, Le Jeune A, et al. (2009) Identification of secreted and surface proteins from Enterococcus faecalis . Can J Microbiol 55:967–974. [DOI] [PubMed] [Google Scholar]
- 40. Bøhle LA, Riaz T, Egge-Jacobsen W, Skaugen M, Busk OL, et al. (2011) Identification of surface proteins in Enterococcus faecalis V583. BMC Genomics 12:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reffuveille F, Leneveu C, Chevalier S, Auffray Y, Rince A (2011) Lipoproteins of Enterococcus faecalis: bioinformatic identification, expression analysis and relation to virulence. Microbiology 157:3001–3013. [DOI] [PubMed] [Google Scholar]
- 42. Paganelli FL, Willems RJ, Leavis HL (2012) Optimizing future treatment of enterococcal infections: attacking the biofilm? Trends Microbiol 20:40–49. [DOI] [PubMed] [Google Scholar]
- 43. Fabretti F, Theilacker C, Baldassarri L, Kaczynski Z, Kropec A, et al. (2006) Alanine esters of enterococcal lipoteichoic acid play a role in biofilm formation and resistance to antimicrobial peptides. Infect Immun 74:4164–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Le Jeune A, Torelli R, Sanguinetti M, Giard JC, Hartke A, et al. (2010) The extracytoplasmic function sigma factor SigV plays a key role in the original model of lysozyme resistance and virulence of Enterococcus faecalis . PLoS One 5:e9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Priyadarshi A, Lee EH, Sung MW, Nam KH, Lee WH, et al. (2009) Structural insights into the alanine racemase from Enterococcus faecalis . Biochim Biophys Acta 1794:1030–1040. [DOI] [PubMed] [Google Scholar]
- 46. Shepard BD, Gilmore MS (2002) Differential expression of virulence-related genes in Enterococcus faecalis in response to biological cues in serum and urine. Infect Immun 70:4344–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vebø HC, Solheim M, Snipen L, Nes IF, Brede DA (2010) Comparative genomic analysis of pathogenic and probiotic Enterococcus faecalis isolates, and their transcriptional responses to growth in human urine. PLoS One 5:e12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Attia AS, Cassat JE, Aranmolate SO, Zimmerman LJ, Boyd KL, et al. (2013) Analysis of the Staphylococcus aureus abscess proteome identifies antimicrobial host proteins and bacterial stress responses at the host-pathogen interface. Pathog Dis. [DOI] [PMC free article] [PubMed]
- 49. Potrykus K, Cashel M (2008) (p)ppGpp: still magical? Annu Rev Microbiol 62:35–51. [DOI] [PubMed] [Google Scholar]
- 50. Traxler MF, Summers SM, Nguyen HT, Zacharia VM, Hightower GA, et al. (2008) The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli . Mol Microbiol 68:1128–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gaca AO, Abranches J, Kajfasz JK, Lemos JA (2012) Global transcriptional analysis of the stringent response in Enterococcus faecalis . Microbiology 158:1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Traxler MF, Zacharia VM, Marquardt S, Summers SM, Nguyen HT, et al. (2011) Discretely calibrated regulatory loops controlled by ppGpp partition gene induction across the ‘feast to famine’ gradient in Escherichia coli . Mol Microbiol 79:830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maisonneuve E, Castro-Camargo M, Gerdes K (2013) (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154:1140–1150. [DOI] [PubMed] [Google Scholar]
- 54. Yan X, Zhao C, Budin-Verneuil A, Hartke A, Rince A, et al. (2009) The (p)ppGpp synthetase RelA contributes to stress adaptation and virulence in Enterococcus faecalis V583. Microbiology 155:3226–3237. [DOI] [PubMed] [Google Scholar]
- 55. Abranches J, Martinez AR, Kajfasz JK, Chavez V, Garsin DA, et al. (2009) The molecular alarmone (p)ppGpp mediates stress responses, vancomycin tolerance, and virulence in Enterococcus faecalis . J Bacteriol 191:2248–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Geiger T, Kastle B, Gratani FL, Goerke C, Wolz C (2014) Two small (p)ppGpp synthases in Staphylococcus aureus mediate tolerance against cell envelope stress conditions. J Bacteriol 196:894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nanamiya H, Kasai K, Nozawa A, Yun CS, Narisawa T, et al. (2008) Identification and functional analysis of novel (p)ppGpp synthetase genes in Bacillus subtilis . Mol Microbiol 67:291–304. [DOI] [PubMed] [Google Scholar]
- 58. Gaca AO, Kajfasz JK, Miller JH, Liu K, Wang JD, et al. (2013) Basal levels of (p)ppGpp in Enterococcus faecalis: the magic beyond the stringent response. MBio 4:e00646–00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lasa I, Toledo-Arana A, Dobin A, Villanueva M, de los Mozos IR, et al. (2011) Genome-wide antisense transcription drives mRNA processing in bacteria. Proc Natl Acad Sci U S A 108:20172–20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dunny GM, Brown BL, Clewell DB (1978) Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc Natl Acad Sci U S A 75:3479–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roggiani M, Schlievert PM (2000) Purification of Streptococcal Pyrogenic Exotoxin A. In: Evans TJ, editor. Septic Shock Methods and Protocols. Totowa, N. J.: Humana Press. pp. 59–66. [DOI] [PubMed]
- 62. Mehra S, Lian W, Jayapal KP, Charaniya SP, Sherman DH, et al. (2006) A framework to analyze multiple time series data: a case study with Streptomyces coelicolor . J Ind Microbiol Biotechnol 33:159–172. [DOI] [PubMed] [Google Scholar]
- 63.Roe B, Crabtree J, Khan A (1996) DNA Isolation and Sequencing: Essential Techniques Series. New York: J. Wiley and Sons.
- 64. Solheim M, Aakra A, Snipen LG, Brede DA, Nes IF (2009) Comparative genomics of Enterococcus faecalis from healthy Norwegian infants. BMC Genomics 10:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Churchill GA (2002) Fundamentals of experimental design for cDNA microarrays. Nat Genet 32 Suppl: 490–495 [DOI] [PubMed] [Google Scholar]
- 66. Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. [DOI] [PubMed] [Google Scholar]
- 67. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, et al. (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4:P3. [PubMed] [Google Scholar]
- 68. Kanehisa M, Goto S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, et al. (2014) Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42:D199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reverse transcription-qPCR validation of selected differentially expressed genes identified by microarray analysis. E. faecalis OG1RF RNA extracted from two and eight hour post-infection subdermal chamber aspirates was reverse transcribed with random hexamers. The resulting cDNAs were used as templates in qPCR experiments. EF0886, which was shown to be stably expressed across time points in the microarray experiments, was used as a reference gene to calculate relative fold change for each gene shown. Data are the mean ± standard deviation of four biological replicates for microarray values and three biological replicates for qPCR values. Down-regulated genes have fold-changes less than one (dotted line). (A) Two hours post-inoculation, (B) Eight hours post-inoculation.
(JPG)
Recovery of OG1RF, Δrsh, ΔrelQ, and ΔrshΔrelQ from subdermal abscesses at (A) early and (B) late time points post-inoculation. Subdermal abscess infections with the four strains were carried out as described in the text and in the legend of Fig. 1. Values and error bars represent the mean ± SEM of n = 2 rabbits. Data from the same rabbits were separated into panels (A) and (B) for clarity.
(TIF)
E. faecalis OG1RF in vivo differentially regulated genes identified via microarray analysis at two hours post-infection of subdermal chambers. The fold change values for up- and down-regulated genes are shown in green and red fonts, respectively. Column H lists the corresponding positive and negative fold changes, highlighted in green and red, respectively, from the eight hour microarray.
(XLS)
E. faecalis OG1RF in vivo differentially regulated genes identified via microarray analysis at eight hours post-infection of subdermal chambers. The fold change values for up- and down-regulated genes are shown in green and red fonts, respectively. Column H lists the corresponding positive and negative fold changes, highlighted in green and red, respectively, from the two hour microarray. Genes with text in blue font are identical or highly similar to E. coli stringent response genes [50]. Genes in boldface text were also differentially expressed in E. faecalis in a (p)ppGpp-dependent manner during stringent response activation following 15 or 30 minute treatment with mupirocin [51].
(XLS)
List of oligonucleotides.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. Microarray data from this study has been submitted to the Gene Expression Omnibus (GEO) database, accession number GSE22391, to comply with the MIAME standards. The GEO series accession number for the RNA-seq data is GSE58467.