

Abstract
Protective efficacy against bovine herpesvirus 1 (BoHV-1) has been demonstrated to be induced by a plasmid encoding bovine neutrophil beta-defensin 3 (BNBD3) as a fusion construct with truncated glycoprotein D (tgD). However, in spite of the increased cell-mediated immune responses induced by this DNA vaccine, the clinical responses of BoHV-1-challenged cattle were not reduced over those observed in animals vaccinated with the plasmid encoding tgD alone; this might have been because the vaccine failed to improve humoral responses. We hypothesized that an alternative vaccine design strategy that utilized the DNA vaccine pMASIA-tgD as a complex with BNBD3 might improve humoral responses while maintaining robust Th1-type cell-mediated responses. C57BL/6 mice were vaccinated with pMASIA-tgD complexed with 0, 0.01875, 0.1875, or 1.875 nmol of a stable synthesized analog of BNBD3 (aBNBD3). The best results were seen in mice immunized with the vaccine composed of pMASIA-tgD complexed to 0.1875 nmol aBNBD3. In this group, humoral responses were improved, as evidenced by increased virus neutralization, tgD-specific early IgG1, and later IgG2a titers, while the strong cell-mediated immune responses, measured based on specific gamma interferon (IFN-γ)-secreting cells, were maintained relative to pMASIA-tgD. Modulation of the immune response might have been due in part to the effect of BNBD3 on dendritic cells (DCs). In vitro studies showed that murine bone marrow-derived DCs (BMDCs) pretreated with aBNBD3 were activated, as evidenced by CD11c downregulation, and were functionally mature, as shown by increased allostimulatory ability. Native, synthetic, and analog forms of BNBD3 were equally capable of inducing functional maturation of BMDCs.
INTRODUCTION
Bovine herpesvirus 1 (BoHV-1) is an economically important veterinary pathogen. Like other alphaherpesviruses, such as herpes simplex virus 1 (HSV-1) (or human herpesvirus 1 [HHV-1]) and HSV-2 in humans (1), pseudorabies virus (PrV) (or Suid herpesvirus 1 [SuHV-1]) in pigs, and equine herpesvirus 1 (EHV-1) and EHV-4 in horses, initial infection with BoHV-1 is typically followed by the establishment of viral latency (2–4). Along with hygienic measures, it is quite sensible to seek methods that prevent the initial infection; since vaccination is a primary method of prevention, it follows that development of an effective noninfectious preventative vaccine would be desirable.
DNA vaccines are noninfectious. Additionally, they are simple to design and economical to produce, thus making them attractive as veterinary vaccines (5). Immunization with a DNA vaccine results in endogenous host cell expression of the antigen, with subsequent antigen-specific immune responses (6, 7). In mice, this results in a predominantly Th1-type response, with induction of IgG2a antibodies and cytotoxic T lymphocytes (CTLs) (8). Additionally, antigen expressed by the host cells can be picked up by infiltrating antigen-presenting cells (APCs), such as dendritic cells (DCs) (9), or circulated as free antigen to stimulate humoral responses (10). DNA from immunization can also be found in directly transfected APCs (11), as free DNA in draining lymph, which can then transfect DCs in regional lymph nodes (11, 12), and in transfected macrophages in the peripheral blood (13). Major hurdles to the development of DNA vaccines for herpesviruses in general and for BoHV-1 in particular are that both cell-mediated and humoral responses are required (14) and humoral responses need to be increased over those that can be obtained with naked DNA (15). Achieving improvements in both arms of the immune response has proven to be a challenge and has led to many studies with attempts at various immune-enhancing strategies, including genetic adjuvanting (16–18), delivery by liposomes (19, 20), and addition or complexing of adjuvants to the plasmid DNA (8, 21).
Recently, we evaluated the potential of a genetic adjuvanting strategy for the BoHV-1 DNA vaccine pMASIA-tgD. In cattle, a DNA vaccine encoding the DC-chemotactic bovine neutrophil beta-defensin 3 (BNBD3) as a fusion construct with truncated glycoprotein D (tgD) showed protective efficacy against BoHV-1 through increased Th1-type cell-mediated responses (22). The vaccine was unable to improve the clinical responses of BoHV-1-challenged cattle over those observed in animals vaccinated with a DNA vaccine encoding the tgD antigen alone, however, which might have been because the vaccine failed to improve humoral responses. β-Defensins (BDs) are cationic, membrane-active, antimicrobial proteins of the innate immune system that participate in defense against microbiological pathogens (23). They are small peptides, 38 to 42 amino acids in length, characterized by an N-terminal α-helix and six conserved cysteine residues that form three disulfide bonds, defined as Cys1-Cys5, Cys2-Cys4, and Cys3-Cys6 (23–25). In cattle, 16 β-defensins have been discovered; 13 are produced by neutrophils and are known as bovine neutrophil β-defensin 1 (BNBD1) to BNBD13, of which BNBD3 is the most abundant (26).
We hypothesized that an alternative vaccine design strategy that utilized the DNA vaccine pMASIA-tgD as a complex with the cationic peptide BNBD3 might be able to improve the humoral response while maintaining a robust cell-mediated immune (CMI) response. Our hypothesis was based on the finding by Riedl et al. that, when a small cationic peptide fused to a short antigenic epitope was complexed with a DNA vaccine encoding a full-length antigen, the humoral immune response to the DNA-encoded antigen could be improved without loss of CMI responses, but only when the two components were complexed at a peptide/DNA ratio of 125:1 (27, 28). This phenomenon was not related to improved uptake of the DNA or subsequent expression of the antigen encoded by the DNA, and the authors were unable to explain the mechanism or whether and how it might be related to the cationic nature of the peptide. Enhanced serum antibody responses without loss of CTLs were also observed when the DNA-complexing adjuvant was in the form of cationic microparticles (29) or a cationic emulsion (8) or contained a cationic lipid such as dioleoyloxytrimethylammonium propane (DOTAP) (19). Additionally, it was found that, although macrophages could act as APCs after exposure to naked DNA, DCs could be transfected with plasmid DNA and subsequently act as APCs only when DNA was delivered in the presence of a cationic lipid (30). Thus, there does appear to be a benefit when the DNA-complexing adjuvant is of a cationic nature. The response to vaccination with a plasmid encoding BoHV-1 glycoprotein D (gD) has typically been of a Th1 cellular type (15), and achieving an improved humoral response could be difficult. In a murine model of HHV-2, the gD antigen delivered as a protein was a strong inducer of Th2 responses with high antibody titers, whereas the same antigen delivered by DNA vaccine induced responses with a Th1 bias (31, 32), a bias that was strengthened when the antigen was genetically adjuvanted with plasmid encoding the Th1 cytokine interleukin 12 (IL-12) (32, 33) and that could not be redirected by genetic adjuvanting with plasmids encoding Th2 cytokines IL-4 or IL-10 (33). Since the use of a BNBD as the cationic component in a peptide/DNA complexed BoHV-1 vaccine as a way to enhance humoral responses has not been studied, we evaluated the potential of this strategy.
The DNA vaccine pMASIA-tgD was complexed with increasing amounts of a stable synthetic analog of BNBD3 (aBNBD3) and then studied for its capacity to stimulate immune responses in mice in vivo. Furthermore, BNBD3 was examined for its effects on murine bone marrow-derived DCs (BMDCs) in vitro. The best results were seen in mice immunized with the vaccine composed of pMASIA-tgD complexed with aBNBD3 at a medium nanomolar peptide/DNA ratio of 125:1. Immunization with this complexed vaccine maintained robust Th1-type CMI responses, induced tgD-specific IgG1, and significantly improved IgG2a antibody responses. In vitro studies revealed that aBNBD3 activates and functionally matures murine BMDCs.
MATERIALS AND METHODS
BNBDs and plasmids.
BNBD3 peptides were chemically synthesized and then folded to form the defined β-defensin disulfide bonds (Cys1-Cys5, Cys2-Cys4, and Cys3-Cys6), as described previously (34). Native BNBD3 (nBNBD3) isolated from bovine neutrophils (26) was kindly provided by Micheal Selsted (University of California, Irvine, CA, USA). The amino acid sequences of the synthetic peptides synthetic BNBD3 (sBNBD3) (based on the published sequence with glutamine in amino acid positions 1 and 27; UniProtKB/Swiss-Prot accession number P46161) and aBNBD3 (an analog with glycine replacing glutamine in amino acid positions 1 and 27) are shown in relation to native BNBD3 and to human and murine β-defensin 2 (hBD2 and mBD2, respectively) (Fig. 1). The plasmids pMASIA and pMASIA-tgD (which encodes truncated glycoprotein D [tgD] of BoHV-1) were detailed previously (22). These plasmids were amplified in Escherichia coli JM109 cells and purified with Endofree Plasmid Giga kits (Qiagen, Montreal, PQ, Canada).
FIG 1.

Amino acid sequence alignment of native and synthesized forms of BNBD3. sBNBD3 (published sequence [UniProtKB/Swiss-Prot accession number P46161] with glutamine in positions 1 and 27), aBNBD3 (analog in which glycine replaces glutamine in positions 1 and 27), and nBNBD3 (native BNBD3 sequence, with pyroglutamic acid [pE], the modified residue of glutamine, at the N terminus) were aligned with murine BD2 (mBD2) and human BD2 (hBD2). Aligned conserved cysteine residues are shown in the shaded vertical bars, and the β-defensin disulfide bonds (Cys1-Cys5, Cys2-Cys4, and Cys3-Cys6) are shown at the top.
Chemotaxis assay.
Biological activity of the synthesized BNBD3s was confirmed using a chemotaxis assay described previously (34). Chemotaxis of bovine immature DCs (iDCs) to synthetic BNBD3 was compared to that to native BNBD3 using a 96-well disposable chemotaxis system (ChemoTx system; Neuroprobe, Gaithersburg, MD, USA). Peptides diluted to 1,000, 100, 10, 1, 0.1, 0.01, or 0 ng/ml were placed in triplicate wells of the bottom chamber of the plate. Bovine monocytes were isolated from peripheral blood mononuclear cells (PBMCs) of four donor animals and were cultured with supernatants from bovine IL-4- and bovine granulocyte-macrophage colony-stimulating factor (GM-CSF)-transfected CHO cells (kindly provided by Merial Ltd., Lyon, France) for 3 days to generate iDCs, as verified by fluorescence-activated cell sorting (FACS) analysis. The monocyte-derived iDCs were labeled with calcein acetoxymethyl ester (Molecular Probes) and placed on top of the membrane above each BNBD3- or buffer-filled well, and the plates were incubated for 90 min at 37°C. Migrated cells were identified using a 96-well multilabel plate reader (Victor 3V multilabel counter; PerkinElmer Life and Analytical Sciences, Inc., Woodbridge, ON, Canada). The chemotactic index (CI) was calculated for each well by dividing the total fluorescence of each test well by the mean fluorescence of the buffer/control wells. A CI of ≥2 was considered statistically significant (P < 0.05) (35).
Preparation of BNBD3/DNA complexed vaccines and immunization of mice.
Cationic peptide/DNA complexed vaccines were prepared according to the method described by Riedl et al. (28), and the peptide concentrations were chosen based on the results of that work, in which a bell-shaped dose-response curve was shown, with optimal responses to the DNA-encoded antigen at a nanomolar peptide/DNA ratio of 125:1 (28). Accordingly, pMASIA-tgD (5 μg; 0.0015 nmol) was mixed with 0.01875 nmol (low), 0.1875 nmol (medium), or 1.875 nmol (high) of the cationic peptide aBNBD3 in Ca2+/Mg2+-free phosphate-buffered saline (PBS) (pH 7.4) (Gibco), in a final volume of 20 μl, for 60 min at room temperature (RT). The nanomolar peptide/DNA ratios were thus 12.5:1 for the low dose, 125:1 for the medium dose, and 1,250:1 for the high dose. DNA-peptide complex formation was visualized by agarose gel electrophoresis in an electrophoretic mobility shift assay (EMSA). Samples were loaded in 10% glycerol and were then analyzed on a 1% agarose gel containing 0.5 μg/ml ethidium bromide.
Groups of 6- to 8-week-old C57BL/6 mice (8 per group) were immunized twice, with a 4-week interval. Immunizations were delivered as two 10-μl intradermal injections into the base of the tail. Each 20-μl dose was formulated to contain 5 μg pMASIA (placebo), 5 μg pMASIA-tgD, 5 μg pMASIA-tgD (0.0015 nmol) plus 0.01875 nmol BNBD3 (low), 5 μg pMASIA-tgD plus 0.1875 nmol BNBD3 (medium), or 5 μg pMASIA-tgD plus 1.875 nmol BNBD3 (high).
Serology.
For enzyme-linked immunosorbent assays (ELISAs), 96-well polystyrene microtiter plates (Immulon 2; Thermo Electron Corp., Milford, MA, USA) were coated overnight with 0.05 μg of tgD per well, in sodium carbonate buffer, and then washed with PBS with 0.05% Tween-20. Fourfold serial dilutions of murine sera, starting at 1:40, were prepared in PBS containing 0.5% gelatin and were dispensed into plates. Following overnight incubation at 4°C, the plates were washed, and bound IgG1 and IgG2a were detected using biotinylated goat anti-mouse IgG1 and IgG2a antibodies (Caltag Laboratories, Burlingame, CA, USA) for 1 h at RT followed by streptavidin-alkaline phosphatase (AP) for 1 h at RT. Bound IgG was detected as described previously (22). All reactions were visualized with 0.01 M p-nitrophenyl phosphate (PNPP) (Sigma-Aldrich), and absorbance was read at 405 nm. ELISA titers were expressed as the inverse of the serum dilution that gave an absorbance (A) value 2 standard deviations above the value for serum from negative-control animals. BoHV-1-specific virus neutralization (VN) titers were determined by plaque reduction assays. Sera were serially diluted in 96-well plates (Corning), and the diluted samples were then mixed with 100 PFU of BoHV-1 per well for 1 h at 37°C. The serum-virus mixtures were transferred to duplicate MDBK cell monolayers and incubated for 48 h at 37°C. Viral plaques were visualized by staining each well with 0.5% crystal violet. Titers were expressed as the reciprocal of the highest dilution of serum that resulted in a 50% reduction in plaques relative to the serum-free positive virus control.
Enzyme-linked immunospot assay.
Nitrocellulose plates (96-well Multiscreen-HA; Millipore Corp., Bedford, MA, USA) were coated overnight at 4°C with 0.2 μg/well of anti-mouse gamma interferon (IFN-γ) or IL-5 IgG (BD Biosciences, San Jose, CA, USA). Plates were washed with PBS and then blocked with 1% (wt/vol) bovine serum albumin (BSA) (Sigma-Aldrich) for 2 h at 37°C. Splenocytes were isolated as described previously (36) and were resuspended at 1 × 107 cells per ml in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 50 μg/ml gentamicin, 1 mM l-glutamine, 10 mM HEPES, 1 mM nonessential amino acids, 1 mM sodium pyruvate (all from Life Technologies Inc., Burlington, ON, Canada), and 5 × 10−5 M 2-mercaptoethanol (Sigma-Aldrich). Splenocytes (106 cells/well) were added to triplicate wells containing medium or tgD (3 μg/ml) and were incubated at 37°C for 20 h. Plates were washed and then incubated with 2 μg/ml biotinylated rat anti-mouse IFN-γ or IL-5 IgG (BD Biosciences) for 1.5 h. Bound antibodies were detected using streptavidin-AP (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 1.5 h and were visualized using 5-bromo-4-chloro-3-indolylphosphate (BCIP)/nitroblue tetrazolium (NBT) (Sigma-Aldrich). Spots were counted, and the numbers of IFN-γ- or IL-5-secreting cells per 1 × 106 cells were expressed as the difference between the number of spots in the tgD-stimulated wells and the number of spots in the control wells, which contained splenocytes from the same animal with medium added in place of tgD.
Murine bone marrow-derived dendritic cell generation and stimulation.
BMDCs were prepared as described previously (37), with modifications. Bone marrow cells were flushed from femurs and tibiae of naive C57BL/6 mice and depleted of erythrocytes by lysis with Tris-ammonium chloride buffer (17 mM Tris, 144 mM NH4Cl [pH 7.2]). Subsequently, the cells were suspended at 1 × 106 cells/ml in RPMI 1640 medium supplemented with 10% FBS, 50 μg/ml gentamicin, 1 mM l-glutamine, 10 mM HEPES, 1 mM nonessential amino acids, 1 mM sodium pyruvate (all from Life Technologies Inc., Burlington, ON, Canada), 5 × 10−5 M 2-mercaptoethanol (Sigma-Aldrich), and 20 ng/ml murine GM-CSF (PeproTech Inc., Rocky Hill, NJ, USA) (DC medium). Cell suspensions (4 ml/well) were cultured in 6-well plates. On day 3, nonadherent cells (granulocytes, T cells, and B cells) were carefully removed and fresh DC medium was added. For the studies shown in Fig. 6, 50% of the medium was removed on days 5, 7, and 9 and replaced with fresh DC medium; also on day 9, cells were not treated or treated for 24 h with lipopolysaccharide (LPS) at a final concentration of 100 ng/ml or with aBNBD3 at a final concentration of 10, 100, or 1,000 ng/ml. On day 10, cells were harvested for FACS analysis or proliferation assays. For the studies shown in Fig. 7, BMDCs were harvested on day 5, resuspended at 1 × 105 cells/ml in DC medium, and dispensed (100 μl of cells/well) into U-bottom 96-well tissue culture plates. Cells were not treated or were treated for 18 h with LPS at a final concentration of 100 ng/ml, with nBNBD3, sBNBD3, or aBNBD3 at final concentrations of 10, 100, or 1,000 ng/ml, or with both LPS and nBNBD3, sBNBD3, or aBNBD3 at the aforementioned concentrations. Prior to their use in flow cytometric staining or mixed leukocyte reaction (MLR) assays, BMDCs were extensively washed at least three times with Ca2+/Mg2+-free PBS (pH 7.2) (Gibco, Invitrogen Canada Inc.).
FIG 6.
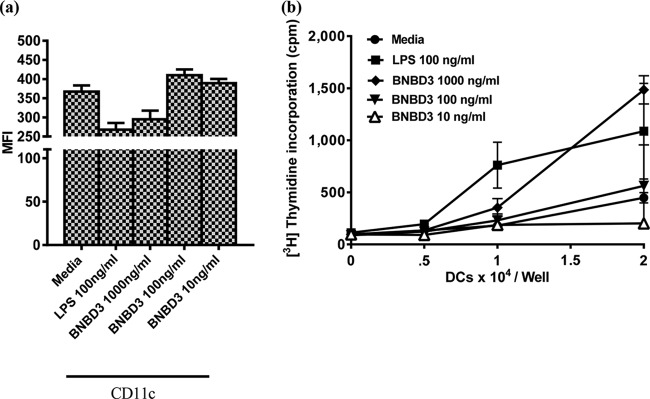
Phenotypic and proliferative changes effected by aBNBD3 treatment of mouse BMDCs. Day 9 BMDCs from C57BL/6 mice (n = 3) were cultured for 24 h in 6-well plates in the presence of aBNBD3 or LPS, and cells were extensively washed for use on day 10. (a) Fluorescence intensity of the expression of CD11c. Data are the geometric mean fluorescence intensity (MFI) ± standard error of the mean (SEM) of 1 well of cells from each of three mice. (b) Proliferation of mismatched splenocytes in an allogeneic MLR. Increasing numbers of BMDCs were incubated with 2 × 105 responder cells/well (BALB/c splenocytes) for 3 days. Proliferative responses were measured by the incorporation of 0.4 μCi/well [methyl-3H]thymidine for 18 h. Data are mean cpm ± SEM of results from triplicate wells. Similar results were obtained in a second independent experiment with nBNBD3.
FIG 7.

Activation/maturation measured by the stimulatory ability of BMDCs in the MLR assay after treatment with nBNBD3, aBNBD3, or sBNBD3, with or without LPS. Day 5 BMDCs (1 × 104 cells/well) from C56BL/6 mice (n = 4) were dispensed to U-bottom 96-well plates and treated for 18 h with LPS (100 ng/ml), BNBD3 (10, 100, or 1,000 ng/ml), or LPS (100 ng/ml) plus BNBD3 (1,000 ng/ml). (a to c) BMDCs treated with native BNBD3 (a), sBNBD3 (b), or aBNBD3 (c) were incubated with BALB/c splenocytes (1 × 105 cells/well) for 3 days. (d) The same splenocytes were cultured with medium alone or with aBNBD3 at 10, 100, or 1,000 ng/ml. Proliferative responses were measured by the incorporation of 1 μCi/well [methyl-3H]thymidine for the last 18 h of culture. Data are mean cpm + SEM of wells from each BMDC donor mouse (a to c) or mean cpm + SEM of wells from each treatment of splenocytes alone (d). Significant differences between groups are indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Flow cytometric analysis of stimulated BMDCs.
The BMDC phenotype was examined on day 10 after 24 h of incubation with the different treatments. Cells were washed and resuspended at 1 × 107 cells/ml in FACS buffer (PBS [pH 7.2], 0.1% BSA, 0.05% NaNH3) supplemented with 2% FBS, and 100-μl aliquots were cell surface stained for 30 min at 4°C with fluorescein isothiocyanate (FITC)-anti-mouse I-Ab (major histocompatibility complex class II [MHC-II], clone AF6-120.1; BD Biosciences), FITC-anti-mouse CD11c (clone HL3; BD Biosciences), FITC-anti-mouse CD40 (clone 3/23; BD Biosciences), or FITC-anti-mouse CD86 (clone GL1; BD Biosciences). Staining specificity was controlled with the appropriate isotype-matched antibody controls. Samples were acquired using a FACSCalibur flow cytometer, and the data were analyzed with CELLQuest software (BD Biosciences).
Mixed leukocyte reaction assay.
Allogeneic MLR assays were performed using mouse splenocytes as responder cells. Dendritic cells were cultured as described above. For Fig. 6, the BMDCs, untreated or treated with LPS or aBNBD3, were harvested on day 10, washed as described above, resuspended in proliferation medium (PM) made up of RPMI 1640 medium supplemented with 1 ng/ml dexamethasone, 10% FBS, 50 μg/ml gentamicin, 1 mM l-glutamine, 10 mM HEPES, 1 mM nonessential amino acids, 1 mM sodium pyruvate (all from Life Technologies Inc., Burlington, ON, Canada), and 5 × 10−5 M 2-mercaptoethanol (Sigma-Aldrich), and dispensed into U-bottom 96-well tissue culture plates to provide 0.5 × 104, 1 × 104, 1.5 × 104, or 2 × 104 cells/well. For Fig. 7, the BMDCs, untreated or treated with LPS or aBNBD3, were washed in the plates as described above and were resuspended in PM. Then, 2 × 106 responder cells/splenocytes (for Fig. 6) or 1 × 106 responder cells/splenocytes (for Fig. 7) from BALB/c mice in PM were dispensed, in 100-μl volumes, into wells containing treated or untreated BMDCs. Splenocytes alone were cultured in PM or with aBNBD3 at final concentrations of 10, 100, or 1,000 ng/ml. After 72 h in culture, the cells were pulsed with 0.4 μCi/well of [methyl-3H]thymidine (Amersham Biosciences, Baie d'Urfe, PQ, Canada). After an additional 18 h of culture, cells were collected with a Filtermate harvester and thymidine uptake was measured by scintillation counting with a TopCount NXT microplate scintillation counter (Packard Instrument Co., Meriden, CT, USA). Proliferative responses were reported in counts per minute.
Statistical analysis.
All data were analyzed with the aid of GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA). For ELISA and VN titers and enzyme-linked-immunospot (ELIspot) counts, the data were not normally distributed; therefore, the differences among groups were examined using the nonparametric Kruskal-Wallis test. If the results of analysis of variance (ANOVA) proved significant, then multiple post-test comparisons between medians were performed using Dunn's test or differences between the medians of two groups were examined using the Mann-Whitney U test. For in vitro MLR assays, the data were normally distributed; therefore, the differences among groups were analyzed by ANOVA followed by Tukey's multiple-comparison test in cases of significant ANOVA results, or differences between the means of two groups were examined using the unpaired Student t test. Differences between groups were considered significant if P values of <0.05 were obtained.
RESULTS
Functional and biological activities of synthesized peptides.
To verify the two synthesized forms of BNBD3 prior to their use in the peptide/DNA complexed vaccines, both synthesized peptides were assayed to ensure that they were biologically active and that the native β-defensin disulfide bonds were achieved during oxidation (folding) of the peptides (34). The attraction of bovine iDCs to the synthesized peptides sBNBD3 and aBNBD3 was compared with that of native BNBD3 by using a chemotaxis assay. Bovine iDCs migrated equally in response to synthesized peptides and native BNBD3, and the data gave characteristic bell-shaped dose-response curves with the same peak migration at 10 ng/ml (Fig. 2). Thus, both synthesized peptides were deemed suitable for use in the vaccine, as they were shown to be equivalent to native BNBD3 in chemotactic ability for iDCs, and the synthesized peptides most likely had the native conformation, since maximal cell migration occurred at the same concentration for all.
FIG 2.
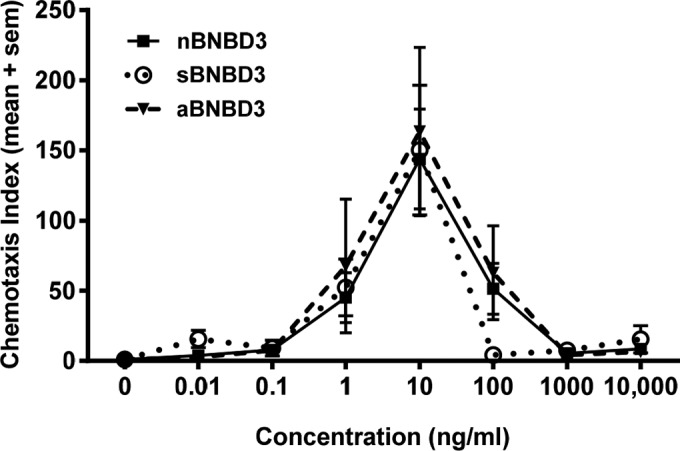
Chemotaxis of bovine iDCs in response to native and synthesized forms of BNBD3. Bovine iDCs were labeled with calcein acetoxymethyl ester and placed on a filter membrane (5-μm pores) above triplicate wells filled with either medium or 0.01, 0.1, 1, 10, 100, or 1,000 ng/ml of each BNBD3 peptide. The migration of cells toward medium or defensin was determined by reading the calcein fluorescence signal of migrated cells on the bottom of the filter after incubation at 37°C for 90 min. Data are expressed as the chemotactic index (CI) and are shown as the mean CI ± standard error of the mean (sem) for four donor animals (n = 4). sBNBD3 indicates the published sequence with glutamine in positions 1 and 27; in aBNBD3, glycine replaces glutamine in positions 1 and 27.
Formation of peptide/DNA complexes.
To characterize the complexes of the positively charged aBNBD3 peptide with the negatively charged pMASIA-tgD DNA vaccine, a constant amount of pMASIA-tgD (5 μg; 0.0015 nmol) was mixed with increasing amounts of aBNBD3 and analyzed by EMSA (Fig. 3). Most of the pMASIA (Fig. 3, lane a) and pMASIA-tgD (Fig. 3, lane b) plasmids migrated as supercoiled DNA that moved farthest into the gel. Addition of 0.1875 nmol aBNBD3 (medium; nanomolar peptide/DNA ratio of 125:1) reduced the electrophoretic mobility of pMASIA-tgD (Fig. 3, lane c), and addition of 1.875 nmol aBNBD (high; ratio of 1,250:1) reduced the electrophoretic mobility of pMASIA-tgD to the point that the DNA did not migrate into the gel at all (Fig. 3, lane d).
FIG 3.
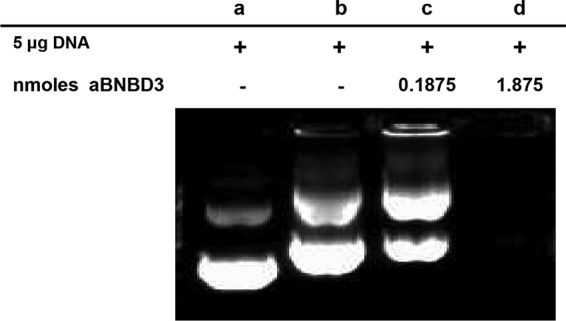
Electrophoretic mobility shift assay of pMASIA-tgD and aBNBD3 complexes. pMASIA-tgD DNA vaccine (5 μg) was complexed with aBNBD3 peptide at medium and high ratios, and the DNA-aBNBD3 complexes were visualized by agarose gel electrophoresis in the presence of ethidium bromide. Lane a, pMASIA (placebo); lane b, pMASIA-tgD; lane c, pMASIA-tgD complexed with 0.1875 nmol (medium) of aBNBD3; lane d, pMASIA-tgD DNA complexed with 1.8750 nmol (high) of aBNBD3. aBNBD3 is an analog in which glycine replaces glutamine in positions 1 and 27.
Optimization of peptide/DNA ratio based on immune responses of mice to complexed vaccines.
As an initial step, the optimal quantity of peptide in relation to the amount of DNA (peptide/DNA ratio) had to be established. To determine which peptide/DNA ratio would give the best results, in the first study mice were immunized with pMASIA-tgD (5 μg; 0.0015 nmol) alone or complexed with 0.01875 nmol (low), 0.1875 nmol (medium), or 1.875 nmol (high) of aBNBD3, to give nanomolar peptide/DNA ratios of 12.5:1, 125:1, and 1,250:1, respectively.
With the addition of aBNBD3 to pMASIA-tgD (Fig. 4), we observed optimal humoral and unchanged CMI responses to the DNA-encoded antigen at the medium peptide/DNA ratio of 125:1 (Fig. 4a to f). Compared to vaccination with pMASIA-tgD alone, the early tgD-specific IgG1 response (P < 0.05) (Fig. 4a) was increased and the later IgG2a (P < 0.05) (Fig. 4b) and VN (P < 0.01) (Fig. 4d) responses were increased by addition of the peptide at the medium peptide/DNA ratio. Addition of the peptide at the high peptide/DNA ratio resulted in significantly reduced IgG1 (P < 0.001) (Fig. 4a) and IgG2a (P < 0.001) (Fig. 4b) levels, compared to vaccination at the medium aBNBD3/pMASIA-tgD ratio, and reduced numbers of IFN-γ-secreting cells (P < 0.01) (Fig. 4e), compared to vaccination with pMASIA-tgD. There were no significant differences in the numbers of IFN-γ-secreting (Fig. 4e) or IL-5-secreting (Fig. 4f) cells from mice in the low or medium aBNBD3/pMASIA-tgD complexed vaccine groups, compared to mice vaccinated with pMASIA-tgD. These results suggested that aBNBD3 at the medium peptide/DNA ratio (0.1875 nmol peptide; 125:1 ratio), when complexed with pMASIA-tgD, improved the specific antibody response to tgD encoded by the DNA vaccine while maintaining the magnitude and balance of the cellular responses.
FIG 4.

First study of specific (tgD) immune responses in mice immunized with aBNBD3/pMASIA-tgD complexed vaccines. C57BL/6 mice (n = 8) were immunized twice intradermally with 5 μg pMASIA or pMASIA-tgD complexed with either 0 nmol, 0.01875 nmol (low), 0.1875 nmol (medium), or 1.8750 nmol (high) of aBNBD3 (analog in which glycine replaces glutamine in positions 1 and 27). (a to c) tgD-specific IgG1 titers 15 days after the second immunization (a) and IgG2a (b) and IgG (c) titers 1 month after the second immunization were determined by ELISA. ELISA titers are expressed as the reciprocal of the highest dilution resulting in a reading 2 standard deviations above the negative control. (d) One month after the second immunization, virus-neutralizing antibodies in serum were measured. VN titers are expressed as the reciprocal of the highest dilution of serum that resulted in <50% of cells displaying cytopathic effects. (e and f) The numbers of tgD-specific IFN-γ-secreting (e) and IL-5-secreting (f) splenic cells were measured by ELIspot assay. ELIspot results are expressed as the difference between the numbers of IFN-γ- or IL-5-secreting splenic cells in tgD-stimulated wells and medium-control wells per 106 cells. Bars represent the median values for each group, with interquartile ranges. Significant differences between groups are indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Immune responses in mice immunized with aBNBD3 complexed vaccines.
To confirm the merits of aBNBD3/pMASIA-tgD complexed vaccine, in the second study mice were immunized with pMASIA-tgD (5 μg; 0.0015 nmol) alone or complexed with 0.1875 nmol (medium) or 1.875 nmol (high) of aBNBD3, to give nanomolar peptide/DNA ratios of 125:1 and 1250:1, respectively. We again observed optimal humoral and unchanged CMI responses to the DNA-encoded antigen at the medium peptide/DNA ratio of 125:1 (Fig. 5a to f). The early tgD-specific IgG1 response (P < 0.05) (Fig. 5a) was increased and the later IgG2a (P < 0.001) (Fig. 5b) and VN (P < 0.05) (Fig. 5d) responses were increased by the addition of aBNBD3 at the medium peptide/DNA ratio, compared to pMASIA-tgD alone. There were no significant differences in the numbers of IFN-γ-secreting (Fig. 5e) or IL-5-secreting (Fig. 5f) cells. Only the group vaccinated with the medium aBNBD3/pMASIA-tgD ratio had significantly higher numbers of IFN-γ-secreting cells than the placebo group (pMASIA) (P < 0.05) (Fig. 5e). Again, we observed that use of the peptide at the high peptide/DNA ratio resulted in reduced humoral responses (Fig. 5a to d) and numbers of IFN-γ-secreting cells (Fig. 5e), compared to those after vaccination with pMASIA-tgD alone. These results are consistent with those of the first study and confirmed that pMASIA-tgD complexed with aBNBD3 at the medium peptide/DNA ratio (0.1875 nmol peptide; 125:1 ratio) improved the specific antibody response to tgD while maintaining the magnitude and balance of the cellular responses.
FIG 5.

Second study of specific (tgD) immune responses in mice immunized with aBNBD3/pMASIA-tgD complexed vaccines. C57BL/6 mice (n = 8) were immunized twice intradermally with 5 μg pMASIA or pMASIA-tgD complexed with either 0 nmol, 0.1875 nmol (medium), or 1.8750 nmol (high) of aBNBD3 (analog in which glycine replaces glutamine in positions 1 and 27). (a to c) tgD-specific IgG1 titers 15 days after the second immunization (a) and IgG2a (b) and IgG (c) titers 1 month after the second immunization were determined by ELISA. ELISA titers are expressed as the reciprocal of the highest dilution resulting in a reading 2 standard deviations above the negative control. (d) One month after the second immunization, virus-neutralizing antibodies in serum were measured. VN titers are expressed as the reciprocal of the highest dilution of serum that resulted in <50% of cells displaying cytopathic effects. (e and f) The numbers of tgD-specific IFN-γ-secreting (e) and IL-5-secreting (f) splenic cells were measured by ELIspot assay. ELIspot results are expressed as the difference between the numbers of IFN-γ- or IL-5-secreting splenic cells in tgD-stimulated wells and medium control wells per 106 cells. Bars represent the median values for each group, with interquartile ranges. Significant differences between groups are indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Effects of BNBD3 on maturation and activation of murine bone marrow-derived dendritic cells.
To study the potential mechanism by which aBNBD3 increased humoral responses, the effects of BNBD3 on maturation and functional activation of mouse BMDCs were examined. Phenotypic changes induced in BMDCs by different treatments were detected by analysis of DC markers, antigen presentation, and costimulation/activation molecule expression (CD11c, MHC-II, CD86, and CD40) using flow cytometry. Mean fluorescence intensity (MFI) of expression, used here to measure the quantities of molecules produced in response to stimulation, was not affected by treatment with aBNBD3 for any marker, with the exception of CD11c, for which increased MFI values, above results observed with LPS (100 ng/ml), were observed for the cells treated with aBNBD3 at 10 and 100 ng/ml (Fig. 6a). The MFI for cells treated with aBNBD3 at 1,000 ng/ml was lower and was comparable to that for cells matured with LPS at 100 ng/ml. With respect to the effects on numbers of cells, treatment with LPS (100 ng/ml) or any concentration (10, 100, or 1,000 ng/ml) of aBNBD3 or nBNBD3 resulted in equally high percentages (frequencies) of cells expressing CD11c (>80%) and equally moderate frequencies of cells expressing MHC-II (>30%) (data not shown). Treatment with LPS (100 ng/ml) but not aBNBD3 or nBNBD3 at any concentration resulted in strong upregulation of CD40 and CD86 costimulation/activation molecules, based on 5- to 10-fold increased percentages of cells expressing these molecules (data not shown).
Functional maturation and activation as a result of treatment with BNBD3 were determined by measuring changes in the allostimulatory capacity of treated BMDCs. Increases in proliferative ability, indicative of DC maturity, were observed with LPS (100 ng/ml) and with increasing numbers of DCs, which was expected (Fig. 6b). Treatment of BMDCs with aBNBD3 at 100 and 1,000 ng/ml also increased their proliferative ability over that of BMDCs cultured in medium alone, but only the higher concentration (1,000 ng/ml aBNBD3) tended to induce greater proliferation than LPS (100 ng/ml), and this was observed only when DCs were present at 2 × 104 cells/well (Fig. 6b). These results suggest that BMDC incubation with aBNBD3 at 1,000 ng/ml, and to a lesser extent at 100 ng/ml, results in functional maturation and activation, as shown by their increased allostimulatory ability. Additionally, we observed from the dose-response curve, as did Yu et al. (37), that a DC/responder cell ratio of 1:10 gave the best response in MLR assays (Fig. 6b). Therefore, a DC/responder cell ratio of 1:10 was used in subsequent studies. With respect to the effects on splenocytes alone, treatment with aBNBD3 at 10, 100, or 1,000 ng/ml did not induce proliferation, as evidenced by counts of less than 50 cpm (responder splenocytes in Fig. 6b) (data not shown).
To confirm that aBNBD3 stimulated functional maturation and activation of BMDCs and to elucidate differences (if any) in its activity versus that of nBNBD3 and that of sBNBD3, murine BMDCs were treated with nBNBD3 (Fig. 7a), sBNBD3 (Fig. 7b), or aBNBD3 (Fig. 7c) at 10, 100, and 1,000 ng/ml, with or without LPS at 100 ng/ml. Comparable stimulation and proliferation by the DCs treated with synthesized BNBD3 forms indicated that aBNBD3 and sBNBD3 acted equally to nBNBD3 and at concentrations of 100 ng/ml and 1,000 ng/ml acted equally to LPS to mature the DCs (Fig. 7a to c). Proliferation was greatest (equal to or better than that with LPS at 100 ng/ml) and significantly greater than the medium control with nBNBD3 at 100 ng/ml (P < 0.01) and 1,000 ng/ml (P < 0.01) (Fig. 7a), sBNBD3 at 100 ng/ml (P < 0.001) and 1,000 ng/ml (P < 0.001) (Fig. 7b), and aBNBD3 at 10 ng/ml (P < 0.01), 100 ng/ml (P < 0.01), and 1,000 ng/ml (P < 0.01) (Fig. 7c). Although the extensive washing of BMDCs prior to the addition of the mismatched responder splenocytes would have been expected to prevent any effect of residual BNBD3 on splenocytes in the MLR, we tested the effects of treatment with aBNBD3 at 10, 100, or 1,000 ng/ml and found that aBNBD3 did not induce proliferation of splenocytes (Fig. 7d). Combined stimulation with LPS and BNBD3 was tested to determine whether changes in maturation due to BNBD3 might be seen only with a second stimulation, as noted for other peptides (38). The combined treatment with BNBD3 at 1,000 ng/ml and LPS at 100 ng/ml tended to increase proliferation above that observed with LPS for all of the BNBD3 forms, and this difference was significant for sBNBD3 (P < 0.001) (Fig. 7b) and aBNBD3 (P < 0.05) (Fig. 7c). There were no differences in proliferation of BMDCs treated with nBNBD3, sBNBD3, or aBNBD3.
DISCUSSION
Control of BoHV-1 by infected animals is hindered by the virus' ability to achieve latency and to employ other mechanisms that promote viral evasion of the immune system (39; reviewed in reference 2). Thus, the ideal BoHV-1 vaccine, which would be effective and protective but also noninfectious, is sought. Responses to the ideal BoHV-1 vaccine should be broad-based (14) and should stimulate both cellular and humoral arms of the immune system (40). In our quest for this ideal vaccine, we previously studied the immune response-enhancing capacity of BNBD3 (34) in a DNA vaccine as a fusion construct with tgD of BoHV-1. This vaccination strategy increased cell-mediated immune responses, including the induction of CTLs, and was protective against BoHV-1, but these improvements did not result in improved clinical responses, compared to the DNA vaccine encoding tgD alone, which could have been because the vaccine did not concurrently increase humoral responses (22). We then hypothesized that the same cationic peptide, BNBD3, might improve the humoral responses without loss of robust CMI responses if it was formulated in a complex with pMASIA-tgD. This hypothesis was strongly influenced by the work of Riedl et al., who found that the humoral immune responses to a DNA-encoded antigen could be improved without loss of cellular responses when the DNA was complexed with a short cationic peptide at a nanomolar peptide/DNA ratio of 125:1 (27, 28). In the current study, aBNBD3, when used at a nanomolar peptide/DNA ratio of 125:1, enhanced the specific humoral responses of mice to tgD encoded by pMASIA-tgD, with robust Th-1-type CMI responses being maintained. In vitro, aBNBD3 activated BMDCs and functionally matured these cells, as evidenced by increased allostimulatory ability. There was no effect of amino acid substitutions in aBNBD3, as comparative studies with nBNBD3, sBNBD3, and aBNBD3 showed that all three peptides were equally capable of inducing functional maturation of BMDCs.
The slight changes in the amino acid sequence of sBNBD3 versus that of aBNBD3 theoretically afford aBNBD3 greater stability, and this might have accounted for its desirable ease of synthesis and greater yield than sBNBD3. sBNBD3 has an N-terminal glutamine residue, and these residues have shown a propensity to spontaneously cyclize over time to become pyroglutamate (41). This instability is reflected in subsequent reductions in the yield of the purified peptide with the correct amino acid sequence from the synthetic process. Since the conversion of glutamate to pyroglutamate has been found to occur in vivo (41), this might also cause the peptide to be unstable or unpredictable in an animal vaccine. Due to its potential in vivo instability and lower yield, sBNBD was deemed to be uneconomical and unsuitable as a vaccine component; therefore, only aBNBD3-complexed vaccine was selected for evaluation.
Chemotactic activity was unaffected by the amino acid substitutions in aBNBD3, and this peptide was used in the complexed vaccine. Although chemotactic activity for bovine iDCs does not guarantee that mouse iDCs would be attracted to aBNBD3, it is likely that they would be. Chemotactic activity of hBD2 for mouse and human CCR6-expressing cells (42) and of mBD4 (the mouse ortholog of human β-defensin 2) for human CCR2-expressing cells has been demonstrated (43). Thus, it is possible that pMASIA-tgD complexed with aBNBD3 at the medium DNA/peptide ratio of 125:1 effected an increase in the magnitude of the humoral responses by inducing chemotaxis of iDCs to the site of immunization. This in turn would increase the likelihood of uptake of the DNA vaccine by iDCs, followed by expression of the tgD antigen and subsequent presentation by DCs of tgD through both MHC-I and MHC-II pathways, leading to CMI and humoral responses, respectively (17, 18, 44, 45). We showed that chemotaxis of iDCs followed a bell-shaped curve in response to increasing concentrations of aBNBD3, in this study and in a previous study (34). It is possible that iDCs (and other immune cells) were optimally attracted to the concentration of aBNBD3 in the pMASIA-tgD vaccine complexed at the medium ratio of 125:1 and this translated to a favorable improvement in the humoral responses of the mice without changes in the cellular responses. In this scenario, fewer cells would be attracted to the vaccine complex with the low concentration of peptide (12.5:1), which might explain the lower humoral and cellular responses we observed. This would also explain the almost complete abrogation of in vivo immune responses in the group given the vaccine complexed to the highest peptide concentration (1250:1), as iDCs are so poorly chemoattracted to high concentrations of β-defensin. At high concentrations of β-defensin, iDCs are almost repulsed. Also, whereas the concentration of aBNBD3 in the pMASIA-tgD vaccine complexed at the medium nanomolar ratio of 125:1 may have made the vaccine attractive for uptake by infiltrating iDCs, the high concentration of peptide complexing pMASIA-tgD at the nanomolar ratio of 1,250:1 may have provided a sufficiently strong danger signal to target the vaccine for removal by extracellular processes (46), by endosomal or proteosomal degradation after cellular uptake (47), or by induction of cell death (48).
Complex formation was monitored by EMSA, and we observed a reduction of electrophoretic mobility of the complexed DNA versus that of naked DNA. This reduction of electrophoretic mobility became more pronounced as the amount of peptide (added) was increased. This phenomenon has been described by others (49). How formation of a complex of DNA with a cationic component translates into a benefit to the in vivo immune response is currently unknown. Increased transfection of cells by complexed DNA is one mechanism that would be expected to enhance the magnitude of the immune responses. Others have reported, however, that, while complexes at higher peptide/DNA ratios showed enhanced transfection efficacy in vitro, the same complexes resulted in suppressed immunogenicity in vivo (28). Typically, the optimal amount of peptide for the complex delivered in vivo had to be much lower than the amount that gave the best transfection efficiency in vitro (28, 50). Interestingly, Riedl et al. also found the optimal cationic peptide/DNA ratio in vivo to be 125:1 (28); thus, our findings are in good agreement. Another reason for the low responses at the high ratio in vivo could be that, after uptake of the complex by the cells, the DNA was unable to be fully released from the complex. The higher concentration of the peptide also could have hampered the movement of DNA to the nucleus (50) or “trapped” the DNA in the endosomes, where ultimately the DNA would be degraded by lysosomal enzymes following merging of the lysosomes with the late endosomes (51). In vivo, naked DNA can be degraded by serum nuclease; at the optimal ratio, aBNBD3 might have provided protection to the DNA, as has been observed for cationic liposomes (52), cationic copolymers (53), and cationic peptides (54).
Due to its high molecular weight and negative charge, DNA does not easily cross cell membranes (46). Complexing with aBNBD3 may have condensed the DNA, making its size and shape more attractive for cellular uptake (55, 56). In addition, aBNBD3 may have increased the binding and entry of the DNA into cells across the negatively charged cell membranes, as has been reported for cationic liposomes (57) and cationic peptides (54, 58). It is also possible that aBNBD3 acted on iDCs as a cell-penetrating peptide (CPP). This idea is supported by the finding that many cationic CPPs are taken up with their cargo through macropinocytosis (59, 60); the resulting macropinosomes may acidify but do not intersect with lysosomes, and thus lysosomal degradation is avoided (61). It has been reported that human iDCs in the presence of hBD2 undergo actin-driven plasma membrane ruffling and uptake of peptide suggestive of macropinocytosis (62). aBNBD3 may use the same mechanism to gain entry into DCs, thus facilitating transfer of DNA into, and protection of DNA within, the cells. Following transfer, aBNBD3 might then carry the DNA as a passenger molecule into the nucleus (63), as has been described for the cationic antimicrobial peptide human LL-37 (64).
We then hypothesized that aBNBD3 might be acting through effects on DCs. As a first step in elucidating potential mechanisms through which delivery of β-defensin in a complexed vaccine might have influenced immune responses, we evaluated the in vitro effects of BNBD3 on BMDCs. When we treated BMDCs in vitro with aBNBD3, we observed only partial, maturation-type, phenotypic changes relative to LPS treatment, but the aBNBD3-treated cells that displayed the lowest CD11c levels (comparable to LPS results) were the most capable of inducing proliferation, a characteristic displayed by functionally mature DCs. Although there are conflicting reports of up- or downregulation of CD11c with maturation (65, 66), and upregulation has been associated with increased DC survival rates (66), our results are in agreement with those that found downregulation of CD11c expression to be a sign of activation and to be correlated with functional maturity (67). Increased proliferative ability of T cells exposed to DCs treated with aBNBD3 may provide the best clue regarding mechanisms for increased antibody levels, as this can indicate induction of CD4+ T helper cell responses (68), providing increased help to B cells that would result in increased antibody responses. Additionally, as we observed increased stimulation by BNBD3 when combined with LPS, secondary stimulation by BNBD3 may also be involved in the mechanism of activation/maturation of BMDCs (38). Another possibility for how the complexed vaccine might have affected the humoral responses, particularly with respect to the increase in levels of antibodies of the IgG2a isotype, could be from aBNBD3 directly acting on the natural killer (NK) cell subset (69) or indirectly acting on the iDCs with subsequent effects on the NK cell subset (70). Since activation of NK cells has been shown to increase antigen-specific IgG2a responses (71), the increased IgG2a levels that we observed might have occurred as a result of aBNBD3-induced activity of NK cells.
In summary, we showed enhanced humoral responses to a DNA vaccine, without loss of cell-mediated responses, with the addition of aBNBD3 as a complex with the DNA vaccine at the nanomolar peptide/DNA ratio of 125:1. This was evident based on the induction of IFN-γ-secreting cells, an increase in early IgG1 antibody production followed by an increase in IgG2a antibody production, and an increase in the virus-neutralizing ability of the antibodies, all of which are desirable. Since both robust antibody and CMI responses are desired for protection from BoHV-1 infection and this strategy does result in both, the results of this study support our hypothesis and indicate a direction for future studies that could be undertaken for a DNA vaccination strategy to protect cattle from challenge with BoHV-1.
ACKNOWLEDGMENTS
This research was supported by grants from the Natural Sciences and Engineering Research Council of Canada, the British Columbia Beef Cattle Industry Development Council, the Saskatchewan Agriculture Development Fund, and the Alberta Livestock Industry Development Fund.
We thank the staff of the Vaccine and Infectious Diseases Organization Animal Care Unit for handling of the mice and the samples and Marlene Snider, Natasa Arsic, and Yuri Popowich for technical help.
Footnotes
This is manuscript 717 of the Vaccine and Infectious Diseases Organization-InterVac journal series.
REFERENCES
- 1.Rouse BT, Kaistha SD. 2006. A tale of 2 α-herpesviruses: lessons for vaccinologists. Clin Infect Dis 42:810–817. doi: 10.1086/500141. [DOI] [PubMed] [Google Scholar]
- 2.Muylkens B, Thiry J, Kirten P, Schynts F, Thiry E. 2007. Bovine herpesvirus 1 infection and infectious bovine rhinotracheitis. Vet Res 38:181–209. doi: 10.1051/vetres:2006059. [DOI] [PubMed] [Google Scholar]
- 3.Ludwig H. 1983. Bovine herpesviruses, p 135–214. In Roizman B. (ed), The herpesviruses, vol 2 Plenum Press, New York, NY. [Google Scholar]
- 4.Pellett PE, Roizman B. 2013. Herpesviridae, p 1802–1822. In Knipe DM, Howley PM (ed), Fields virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 5.Davis HL, McCluskie MJ. 1999. DNA vaccines for viral diseases. Microbes Infect 1:7–21. doi: 10.1016/S1286-4579(99)80009-4. [DOI] [PubMed] [Google Scholar]
- 6.Gurunathan S, Klinman DM, Seder RA. 2000. DNA vaccines: immunology, application, and optimization. Annu Rev Immunol 18:927–974. doi: 10.1146/annurev.immunol.18.1.927. [DOI] [PubMed] [Google Scholar]
- 7.Weiner DB, Kennedy RC. 1999. Genetic vaccines. Sci Am 281:50–57. doi: 10.1038/scientificamerican0799-50. [DOI] [PubMed] [Google Scholar]
- 8.Ott G, Singh M, Kazzaz J, Briones M, Soenawan E, Ugozzoli M, O'Hagan DT. 2002. A cationic sub-micron emulsion (MF59/DOTAP) is an effective delivery system for DNA vaccines. J Control Release 79:1–5. doi: 10.1016/S0168-3659(01)00545-4. [DOI] [PubMed] [Google Scholar]
- 9.Casares S, Inaba K, Brumeanu TD, Steinman RM, Bona CA. 1997. Antigen presentation by dendritic cells after immunization with DNA encoding a major histocompatibility complex class II-restricted viral epitope. J Exp Med 186:1481–1486. doi: 10.1084/jem.186.9.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michel ML, Davis HL, Schleef M, Mancini M, Tiollais P, Whalen RG. 1995. DNA-mediated immunization to the hepatitis B surface antigen in mice: aspects of the humoral response mimic hepatitis B viral infection in humans. Proc Natl Acad Sci U S A 92:5307–5311. doi: 10.1073/pnas.92.12.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torres CA, Iwasaki A, Barber BH, Robinson HL. 1997. Differential dependence on target site tissue for gene gun and intramuscular DNA immunizations. J Immunol 158:4529–4532. [PubMed] [Google Scholar]
- 12.Davis HL. 1997. Plasmid DNA expression systems for the purpose of immunization. Curr Opin Biotechnol 8:635–646. doi: 10.1016/S0958-1669(97)80041-9. [DOI] [PubMed] [Google Scholar]
- 13.Chattergoon MA, Robinson TM, Boyer JD, Weiner DB. 1998. Specific immune induction following DNA-based immunization through in vivo transfection and activation of macrophages/antigen-presenting cells. J Immunol 160:5707–5718. [PubMed] [Google Scholar]
- 14.Babiuk LA, van Drunen Littel-van den Hurk S, Tikoo SK. 1996. Immunology of bovine herpesvirus 1 infection. Vet Microbiol 53:31–42. doi: 10.1016/S0378-1135(96)01232-1. [DOI] [PubMed] [Google Scholar]
- 15.Manoj S, Griebel PJ, Babiuk LA, van Drunen Littel-van den Hurk S. 2003. Targeting with bovine CD154 enhances humoral immune responses induced by a DNA vaccine in sheep. J Immunol 170:989–996. doi: 10.4049/jimmunol.170.2.989. [DOI] [PubMed] [Google Scholar]
- 16.Sin JI, Kim JJ, Ugen KE, Ciccarelli RB, Higgins TJ, Weiner DB. 1998. Enhancement of protective humoral (Th2) and cell-mediated (Th1) immune responses against herpes simplex virus-2 through co-delivery of granulocyte-macrophage colony-stimulating factor expression cassettes. Eur J Immunol 28:3530–3540. doi:. [DOI] [PubMed] [Google Scholar]
- 17.Biragyn A, Belyakov IM, Chow YH, Dimitrov DS, Berzofsky JA, Kwak LW. 2002. DNA vaccines encoding human immunodeficiency virus-1 glycoprotein 120 fusions with proinflammatory chemoattractants induce systemic and mucosal immune responses. Blood 100:1153–1159. doi: 10.1182/blood-2002-01-0086. [DOI] [PubMed] [Google Scholar]
- 18.Biragyn A, Surenhu M, Yang D, Ruffini PA, Haines BA, Klyushnenkova E, Oppenheim JJ, Kwak LW. 2001. Mediators of innate immunity that target immature, but not mature, dendritic cells induce antitumor immunity when genetically fused with nonimmunogenic tumor antigens. J Immunol 167:6644–6653. doi: 10.4049/jimmunol.167.11.6644. [DOI] [PubMed] [Google Scholar]
- 19.Perrie Y, Frederik PM, Gregoriadis G. 2001. Liposome-mediated DNA vaccination: the effect of vesicle composition. Vaccine 19:3301–3310. doi: 10.1016/S0264-410X(00)00432-1. [DOI] [PubMed] [Google Scholar]
- 20.Commander NJ, Brewer JM, Wren BW, Spencer SA, Macmillan AP, Stack JA. 2010. Liposomal delivery of p-ialB and p-omp25 DNA vaccines improves immunogenicity but fails to provide full protection against B. melitensis challenge. Genet Vaccines Ther 8:5. doi: 10.1186/1479-0556-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenland JR, Letvin NL. 2007. Chemical adjuvants for plasmid DNA vaccines. Vaccine 25:3731–3741. doi: 10.1016/j.vaccine.2007.01.120. [DOI] [PubMed] [Google Scholar]
- 22.Mackenzie-Dyck S, Kovacs-Nolan J, Snider M, Babiuk LA, van Drunen Littel-van den Hurk S. 2014. Inclusion of the bovine neutrophil beta-defensin 3 with glycoprotein D of bovine herpesvirus-1 in a DNA vaccine modulates immune responses of mice and cattle. Clin Vaccine Immunol 21:463–477. doi: 10.1128/CVI.00696-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diamond G, Zasloff M, Eck H, Brasseur M, Maloy WL, Bevins CL. 1991. Tracheal antimicrobial peptide, a cysteine-rich peptide from mammalian tracheal mucosa: peptide isolation and cloning of a cDNA. Proc Natl Acad Sci U S A 88:3952–3956. doi: 10.1073/pnas.88.9.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoover DM, Chertov O, Lubkowski J. 2001. The structure of human β-defensin-1: new insights into structural properties of β-defensins. J Biol Chem 276:39021–39026. doi: 10.1074/jbc.M103830200. [DOI] [PubMed] [Google Scholar]
- 25.Selsted ME, Ouellette AJ. 2005. Mammalian defensins in the antimicrobial immune response. Nat Immunol 6:551–557. doi: 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- 26.Selsted ME, Tang YQ, Morris WL, McGuire PA, Novotny MJ, Smith W, Henschen AH, Cullor JS. 1993. Purification, primary structures, and antibacterial activities of β-defensins, a new family of antimicrobial peptides from bovine neutrophils. J Biol Chem 268:6641–6648. [PubMed] [Google Scholar]
- 27.Riedl P, Reimann J, Schirmbeck R. 2006. Complexes of DNA vaccines with cationic, antigenic peptides are potent, polyvalent CD8+ T-cell-stimulating immunogens. Methods Mol Med 127:159–169. doi: 10.1385/1-59745-168-1:159. [DOI] [PubMed] [Google Scholar]
- 28.Riedl P, Reimann J, Schirmbeck R. 2004. Peptides containing antigenic and cationic domains have enhanced, multivalent immunogenicity when bound to DNA vaccines. J Mol Med 82:144–152. doi: 10.1007/s00109-003-0502-3. [DOI] [PubMed] [Google Scholar]
- 29.Singh M, Briones M, Ott G, O'Hagan D. 2000. Cationic microparticles: a potent delivery system for DNA vaccines. Proc Natl Acad Sci U S A 97:811–816. doi: 10.1073/pnas.97.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rouse RJ, Nair SK, Lydy SL, Bowen JC, Rouse BT. 1994. Induction in vitro of primary cytotoxic T-lymphocyte responses with DNA encoding herpes simplex virus proteins. J Virol 68:5685–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sin JI, Ayyavoo V, Boyer J, Kim J, Ciccarelli RB, Weiner DB. 1999. Protective immune correlates can segregate by vaccine type in a murine herpes model system. Int Immunol 11:1763–1773. doi: 10.1093/intimm/11.11.1763. [DOI] [PubMed] [Google Scholar]
- 32.Sin JI, Kim JJ, Arnold RL, Shroff KE, McCallus D, Pachuk C, McElhiney SP, Wolf MW, Pompa-de Bruin SJ, Higgins TJ, Ciccarelli RB, Weiner DB. 1999. IL-12 gene as a DNA vaccine adjuvant in a herpes mouse model: IL-12 enhances Th1-type CD4+ T cell-mediated protective immunity against herpes simplex virus-2 challenge. J Immunol 162:2912–2921. [PubMed] [Google Scholar]
- 33.Sin JI, Kim JJ, Boyer JD, Ciccarelli RB, Higgins TJ, Weiner DB. 1999. In vivo modulation of vaccine-induced immune responses toward a Th1 phenotype increases potency and vaccine effectiveness in a herpes simplex virus type 2 mouse model. J Virol 73:501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackenzie-Dyck S, Attah-Poku S, Juillard V, Babiuk LA, van Drunen Littel-van den Hurk S. 2011. The synthetic peptides bovine enteric β-defensin (EBD), bovine neutrophil β-defensin (BNBD) 9 and BNBD 3 are chemotactic for immature bovine dendritic cells. Vet Immunol Immunopathol 143:87–107. doi: 10.1016/j.vetimm.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 35.Yang D, Chen Q, Chertov O, Oppenheim JJ. 2000. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J Leukoc Biol 68:9–14. [PubMed] [Google Scholar]
- 36.Mapletoft JW, Oumouna M, Kovacs-Nolan J, Latimer L, Mutwiri G, Babiuk LA, van Drunen Littel-van den Hurk S. 2008. Intranasal immunization of mice with a formalin-inactivated bovine respiratory syncytial virus vaccine co-formulated with CpG oligodeoxynucleotides and polyphosphazenes results in enhanced protection. J Gen Virol 89:250–260. doi: 10.1099/vir.0.83300-0. [DOI] [PubMed] [Google Scholar]
- 37.Yu H, Huang H, Xiang J, Babiuk LA, van Drunen Littel-van den Hurk S. 2006. Dendritic cells pulsed with hepatitis C virus NS3 protein induce immune responses and protection from infection with recombinant vaccinia virus expressing NS3. J Gen Virol 87:1–10. doi: 10.1099/vir.0.81423-0. [DOI] [PubMed] [Google Scholar]
- 38.Lillard JW Jr, Boyaka PN, Chertov O, Oppenheim JJ, McGhee JR. 1999. Mechanisms for induction of acquired host immunity by neutrophil peptide defensins. Proc Natl Acad Sci U S A 96:651–656. doi: 10.1073/pnas.96.2.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Favoreel HW, Van Minnebruggen G, Van de Walle GR, Ficinska J, Nauwynck HJ. 2006. Herpesvirus interference with virus-specific antibodies: bridging antibodies, internalizing antibodies, and hiding from antibodies. Vet Microbiol 113:257–263. doi: 10.1016/j.vetmic.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Manoj S, Babiuk LA, van Drunen Littel-van den Hurk S. 2003. Immunization with a dicistronic plasmid expressing a truncated form of bovine herpesvirus-1 glycoprotein D and the amino-terminal subunit of glycoprotein B results in reduced gB-specific immune responses. Virology 313:296–307. doi: 10.1016/S0042-6822(03)00325-8. [DOI] [PubMed] [Google Scholar]
- 41.Liu YD, Goetze AM, Bass RB, Flynn GC. 2011. N-terminal glutamate to pyroglutamate conversion in vivo for human IgG2 antibodies. J Biol Chem 286:11211–11217. doi: 10.1074/jbc.M110.185041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rohrl J, Yang D, Oppenheim JJ, Hehlgans T. 2010. Specific binding and chemotactic activity of mBD4 and its functional orthologue hBD2 to CCR6-expressing cells. J Biol Chem 285:7028–7034. doi: 10.1074/jbc.M109.091090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rohrl J, Yang D, Oppenheim JJ, Hehlgans T. 2010. Human β-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J Immunol 184:6688–6694. doi: 10.4049/jimmunol.0903984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. 2000. Immunobiology of dendritic cells. Annu Rev Immunol 18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 45.Gil-Torregrosa BC, Lennon-Dumenil AM, Kessler B, Guermonprez P, Ploegh HL, Fruci D, van Endert P, Amigorena S. 2004. Control of cross-presentation during dendritic cell maturation. Eur J Immunol 34:398–407. doi: 10.1002/eji.200324508. [DOI] [PubMed] [Google Scholar]
- 46.Kawabata K, Takakura Y, Hashida M. 1995. The fate of plasmid DNA after intravenous injection in mice: involvement of scavenger receptors in its hepatic uptake. Pharm Res 12:825–830. doi: 10.1023/A:1016248701505. [DOI] [PubMed] [Google Scholar]
- 47.Martin ME, Rice KG. 2007. Peptide-guided gene delivery. AAPS J 9:E18–E29. doi: 10.1208/aapsj0901003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerashchenko OL, Zhuravel EV, Skachkova OV, Khranovska NN, Filonenko VV, Pogrebnoy PV, Soldatkina MA. 2013. Biologic activities of recombinant human-beta-defensin-4 toward cultured human cancer cells. Exp Oncol 35:76–82. [PubMed] [Google Scholar]
- 49.Licciardi M, Campisi M, Cavallaro G, Cervello M, Azzolina A, Giammona G. 2006. Synthesis and characterization of polyaminoacidic polycations for gene delivery. Biomaterials 27:2066–2075. doi: 10.1016/j.biomaterials.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 50.Lehto T, Simonson OE, Mager I, Ezzat K, Sork H, Copolovici DM, Viola JR, Zaghloul EM, Lundin P, Moreno PM, Mae M, Oskolkov N, Suhorutsenko J, Smith CI, Andaloussi SE. 2011. A peptide-based vector for efficient gene transfer in vitro and in vivo. Mol Ther 19:1457–1467. doi: 10.1038/mt.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wattiaux R, Laurent N, Wattiaux-De Coninck S, Jadot M. 2000. Endosomes, lysosomes: their implication in gene transfer. Adv Drug Deliv Rev 41:201–208. doi: 10.1016/S0169-409X(99)00066-6. [DOI] [PubMed] [Google Scholar]
- 52.Zelphati O, Szoka FC Jr. 1996. Mechanism of oligonucleotide release from cationic liposomes. Proc Natl Acad Sci U S A 93:11493–11498. doi: 10.1073/pnas.93.21.11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pedone E, Cavallaro G, Richardson SC, Duncan R, Giammona G. 2001. α,β-Poly(asparthylhydrazide)-glycidyltrimethylammonium chloride copolymers (PAHy-GTA): novel polymers with potential for DNA delivery. J Control Release 77:139–153. doi: 10.1016/S0168-3659(01)00459-X. [DOI] [PubMed] [Google Scholar]
- 54.Lingnau K, Riedl K, von Gabain A. 2007. IC31 and IC30, novel types of vaccine adjuvant based on peptide delivery systems. Expert Rev Vaccines 6:741–746. doi: 10.1586/14760584.6.5.741. [DOI] [PubMed] [Google Scholar]
- 55.Niidome T, Takaji K, Urakawa M, Ohmori N, Wada A, Hirayama T, Aoyagi H. 1999. Chain length of cationic α-helical peptide sufficient for gene delivery into cells. Bioconjug Chem 10:773–780. doi: 10.1021/bc990012d. [DOI] [PubMed] [Google Scholar]
- 56.Haines AM, Irvine AS, Mountain A, Charlesworth J, Farrow NA, Husain RD, Hyde H, Ketteringham H, McDermott RH, Mulcahy AF, Mustoe TL, Reid SC, Rouquette M, Shaw JC, Thatcher DR, Welsh JH, Williams DE, Zauner W, Phillips RO. 2001. CL22: a novel cationic peptide for efficient transfection of mammalian cells. Gene Ther 8:99–110. doi: 10.1038/sj.gt.3301314. [DOI] [PubMed] [Google Scholar]
- 57.Foged C, Arigita C, Sundblad A, Jiskoot W, Storm G, Frokjaer S. 2004. Interaction of dendritic cells with antigen-containing liposomes: effect of bilayer composition. Vaccine 22:1903–1913. doi: 10.1016/j.vaccine.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 58.Wyman TB, Nicol F, Zelphati O, Scaria PV, Plank C, Szoka FC Jr. 1997. Design, synthesis, and characterization of a cationic peptide that binds to nucleic acids and permeabilizes bilayers. Biochemistry 36:3008–3017. doi: 10.1021/bi9618474. [DOI] [PubMed] [Google Scholar]
- 59.Wadia JS, Stan RV, Dowdy SF. 2004. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med 10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 60.Heitz F, Morris MC, Divita G. 2009. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol 157:195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Conner SD, Schmid SL. 2003. Regulated portals of entry into the cell. Nature 422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 62.Morgera F, Pacor S, Creatti L, Antcheva N, Vaccari L, Tossi A. 2011. Effects on antigen-presenting cells of short-term interaction with the human host defence peptide β-defensin 2. Biochem J 436:537–546. doi: 10.1042/BJ20101977. [DOI] [PubMed] [Google Scholar]
- 63.Zanta MA, Belguise-Valladier P, Behr JP. 1999. Gene delivery: a single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc Natl Acad Sci U S A 96:91–96. doi: 10.1073/pnas.96.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sandgren S, Wittrup A, Cheng F, Jonsson M, Eklund E, Busch S, Belting M. 2004. The human antimicrobial peptide LL-37 transfers extracellular DNA plasmid to the nuclear compartment of mammalian cells via lipid rafts and proteoglycan-dependent endocytosis. J Biol Chem 279:17951–17956. doi: 10.1074/jbc.M311440200. [DOI] [PubMed] [Google Scholar]
- 65.Labeur MS, Roters B, Pers B, Mehling A, Luger TA, Schwarz T, Grabbe S. 1999. Generation of tumor immunity by bone marrow-derived dendritic cells correlates with dendritic cell maturation stage. J Immunol 162:168–175. [PubMed] [Google Scholar]
- 66.Rivas-Caicedo A, Soldevila G, Fortoul TI, Castell-Rodriguez A, Flores-Romo L, Garcia-Zepeda EA. 2009. Jak3 is involved in dendritic cell maturation and CCR7-dependent migration. PLoS One 4:e7066. doi: 10.1371/journal.pone.0007066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Singh-Jasuja H, Thiolat A, Ribon M, Boissier MC, Bessis N, Rammensee HG, Decker P. 2013. The mouse dendritic cell marker CD11c is down-regulated upon cell activation through Toll-like receptor triggering. Immunobiology 218:28–39. doi: 10.1016/j.imbio.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 68.Asano Y, Komuro T, Tada T. 1990. Synergistic T-T cell interaction present in alloreactivity: determination of ‘MLR helper’ T cell subsets. Int Immunol 2:1203–1211. doi: 10.1093/intimm/2.12.1203. [DOI] [PubMed] [Google Scholar]
- 69.Chalifour A, Jeannin P, Gauchat JF, Blaecke A, Malissard M, N′Guyen T, Thieblemont N, Delneste Y. 2004. Direct bacterial protein PAMP recognition by human NK cells involves TLRs and triggers alpha-defensin production. Blood 104:1778–1783. doi: 10.1182/blood-2003-08-2820. [DOI] [PubMed] [Google Scholar]
- 70.Amakata Y, Fujiyama Y, Andoh A, Hodohara K, Bamba T. 2001. Mechanism of NK cell activation induced by coculture with dendritic cells derived from peripheral blood monocytes. Clin Exp Immunol 124:214–222. doi: 10.1046/j.1365-2249.2001.01550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koh CY, Yuan D. 2000. The functional relevance of NK-cell-mediated upregulation of antigen-specific IgG2a responses. Cell Immunol 204:135–142. doi: 10.1006/cimm.2000.1703. [DOI] [PubMed] [Google Scholar]