

Abstract
The global epidemic caused by the bacterial pathogen Mycobacterium tuberculosis continues unabated. Moreover, the only available vaccine against tuberculosis, Mycobacterium bovis bacillus Calmette-Guérin (BCG), demonstrates variable efficacy. To respond to this global threat, new animal models that mimic the pathological disease process in humans are required for vaccine testing. One new model, susceptible C3Heb/FeJ mice, is similar to human tuberculosis in that these animals are capable of forming necrotic tubercle granulomas, in contrast to resistant C3H/HeOuJ mice. In this study, we evaluated the impact of prior BCG vaccination of C3Heb/FeJ and C3H/HeOuJ mice on exposure to a low-dose aerosol of Mycobacterium tuberculosis W-Beijing strain SA161. Both BCG-vaccinated murine strains demonstrated reduced bacterial loads 25 days after infection compared to controls, indicating vaccine efficacy. However, during chronic infection, vaccine efficacy waned in C3H/HeOuJ but not in C3Heb/FeJ mice. Protection in vaccinated C3Heb/FeJ mice was associated with reduced numbers of CD11b+ Gr1+ cells, increased numbers of effector and memory T cells, and an absence of necrotic granulomas. BCG vaccine efficacy waned in C3H/HeOuJ mice, as indicated by reduced expression of gamma interferon (IFN-γ) and increased expressions of interleukin-17 (IL-17), IL-10, and Foxp3 by T cells compared to C3Heb/FeJ mice. This is the first murine vaccine model system described to date that can be utilized to dissect differential vaccine-derived immune efficacy.
INTRODUCTION
The global epidemic of tuberculosis results in 8 million new tuberculosis cases per year, with an annual projected increase of 3% (1). It is estimated that between 5 and 10% of healthy individuals are susceptible to tuberculosis, and of these individuals, 85% develop pulmonary disease (2). At present, the only available vaccine against tuberculosis, Mycobacterium bovis bacillus Calmette-Guérin (BCG), has proven unreliable to fully protect against pulmonary tuberculosis in adults (3). Furthermore, a thorough immunological explanation for the variability in the efficacy of BCG is absent (4). Therefore, understanding the specific protective properties of BCG is vital for developing a more efficacious tuberculosis vaccine.
The two small-animal models used most often for preclinical tuberculosis vaccine screening are the low-dose aerosol mouse and guinea pig models (5). Low-dose aerosol infection of guinea pigs with Mycobacterium tuberculosis produces a well-characterized disease that shares important morphological features with disease in humans, such as the development of necrotic granulomatous lesions (6). The majority of vaccine screening has been carried out with various mouse models due to their low cost and the wealth of immunological reagents, with the major drawback being the lack of tubercle necrosis formation.
C3Heb/FeJ mice are capable of forming necrotic, hypoxic tubercle granulomas, while control C3H/HeOuJ mice form nonnecrotic granulomas (7–9). The hallmark of human tuberculosis is the development of a primary necrotic granuloma (7–9). The ability to be able to precisely characterize the protective immune response induced by vaccination during Mycobacterium tuberculosis infection in C3Heb/FeJ mice would greatly improve the usefulness of this animal model for the testing and evaluation of urgently needed new vaccines.
In this study, we evaluated the impact of prior BCG vaccination on exposure of C3Heb/FeJ and C3H/HeOuJ mice to a low-dose aerosol of a W-Beijing strain of Mycobacterium tuberculosis. We evaluated the cellular influx and cytokine environment in mice made immune by prior BCG vaccination, in order to characterize how this might alter the composition of a protective immune response and granulomatous lesions in the lungs and spleens. Our results show that this is the first model system described to date that can be utilized to dissect differential vaccine-derived immune efficacy.
MATERIALS AND METHODS
Mice.
Specific-pathogen-free female C3Heb/FeJ and C3H/HeOuJ mice, 6 to 8 weeks old, were purchased from the Jackson Laboratories (Bar Harbor, ME). Mice were maintained in the biosafety level 3 facilities at Colorado State University and were given sterile water, chow, bedding, and enrichment for the duration of the experiments. The specific-pathogen-free nature of the mouse colonies was demonstrated by testing sentinel animals. All experimental protocols were approved by the Animal Care and Use Committee of Colorado State University.
Vaccination and infection.
Mice were vaccinated by subcutaneous injection with 106 CFU of BCG Pasteur (Glaxo 1077 strain) or phosphate-buffered saline (PBS) as a control. Using a Glas-Col aerosol generator (Glas-Col, Terre Haute, IN), calibrated to deliver 20 bacteria into the lungs, BCG-vaccinated and nonvaccinated mice were challenged 4 weeks later by a low-dose aerosol of the clinical isolate Mycobacterium tuberculosis W-Beijing strain SA161. Mycobacterium tuberculosis W-Beijing strain SA161 was kindly provided by K. Eisenach (University of Arkansas). Bacterial counts in the whole lungs (n = 5) and spleens (n = 5) at each time point of the study were processed. Briefly, bacterial loads were determined by plating serial dilutions of organ homogenates onto nutrient 7H11 agar. CFU were counted after incubation for 3 weeks at 37°C. Additional groups of mice were utilized for pathological analysis. Lungs from mice (n = 5) in each group were harvested for immunohistochemistry, histological, and flow cytometry analyses at 25 and 50 days postchallenge. The results shown in this study are representative of three experiments using 5 animals per group.
Histological analysis.
The right caudal lung lobe from each mouse was fixed with 10% formalin in PBS. Sections from these tissues were stained with hematoxylin-eosin or with Ziehl-Neelsen stain to detect acid-fast bacilli.
Immunohistochemistry.
Anti-Gr1 staining was performed as described previously (10). Briefly, paraffin-embedded slides were dewaxed with Histo-Clear (National Diagnostics, Atlanta, GA) and decreasing concentrations of ethanol. A pressure cooker was used to retrieve antigens in 1× DakoCytomation Target Retrieval solution (DakoCytomation, Carpinteria, CA), and endogenous peroxidases and alkaline phosphatase were inactivated in Bloxall (Vector Laboratories, Burlingame, CA) for 10 min. Thereafter, slides were blocked for 1 h with 2.5% normal goat serum (Vector Laboratories) and incubated overnight at 4°C with a 1/40 dilution of rat anti-Gr1 (clone RB6-8C5; eBioscience) or the rat IgG2b isotype control (clone eB149/1OH5; eBioscience), diluted in 2.5% goat serum. The secondary antibody, alkaline phosphatase-labeled goat anti-rat IgG (Santa Cruz Biotechnology, Dallas, TX), was diluted 1/1,200 in 2.5% goat serum and incubated for 1 h at room temperature. Finally, the reaction was developed for 20 min with Vector Red alkaline phosphatase substrate (Vector Laboratories), and slides were counterstained with Hematoxylin QS (Vector Laboratories) for 1 min.
Lung cell digestion.
Single-cell suspensions were prepared as described previously (11). Briefly, the lungs were perfused through the pulmonary artery with a solution containing PBS and heparin (50 U/ml; Sigma-Aldrich, St. Louis, MO), aseptically removed from the pulmonary cavity, placed into medium, and dissected. The dissected lung tissue was incubated with incomplete Dulbecco's modified Eagle's medium (DMEM) containing collagenase XI (0.7 mg/ml; Sigma-Aldrich) and type IV bovine pancreatic DNase (30 μg/ml; Sigma-Aldrich) for 30 min at 37°C. Digested lungs were further disrupted by gently pushing the tissue through a cell strainer (BD Biosciences, Lincoln Park, NJ). Thereafter, red blood cells were lysed with ammonium-chloride-potassium (ACK) buffer, and the remaining cells were washed and resuspended in complete DMEM.
Flow cytometric analysis.
For antibody staining of cell surface markers, cells were initially incubated with 10 μg/ml of FcBlock (anti-CD16/CD32, clone 93) for 20 min at 4°C. Thereafter, cells were stained with the following antibodies (all antibodies were from eBioscience, San Diego, CA) for 20 min at 4°C in the presence of FcBlock: anti-Gr1 (clone RB6-8C5), anti-CD11b (clone M1/70), anti-CD11c (clone N418), anti-CD4 (clone GK1.5), anti-CD44 (clone IM7), anti-CD25 (clone PC61.5), and anti-CD62L (MEL44). Cells were then washed in PBS, data acquisition of at least 100,000 events was performed by using an LSR-II instrument (BD), and data were analyzed by using FACSDiva version 6 (BD).
Intracellular cytokine staining.
Cells were initially stimulated for 4 h at 37°C with 1× cell stimulation cocktail (eBioscience) diluted in complete DMEM. Thereafter, cells were stained for cell surface markers as indicated above, fixed, and permeabilized according to the instructions provided by the manufacturer of the cell fixation/permeabilization kits for intracellular cytokine analysis (eBioscience). Thereafter, cells were incubated for 30 min at 4°C with FcBlock plus anti-interleukin-10 (IL-10) (clone JES5-16E3; eBioscience), anti-gamma interferon (IFN-γ) (clone XMG1.2; eBioscience), anti-Foxp3 (clones FJK-16S and NRRF-30; eBioscience), and anti-IL-17A and anti-IL-17F (clones TC11-18H10.1 and 9D3.1C8, respectively; Biolegend, San Diego, CA) or with the respective isotype controls. Data acquisition and analysis were performed as described above.
Statistical analysis.
Data were analyzed with GraphPad Prism version 4 (GraphPad software, San Diego, CA), using analysis of variance (ANOVA) and the Tukey posttest. Data are presented as the mean values (n = 5) ± standard errors of the means (SEM). A P value of <0.050 was considered significant.
RESULTS
Course of M. tuberculosis infection in control and immune C3Heb/FeJ and C3H/HeOuJ mice.
The course of the experimental infection is shown in Fig. 1. In each case, control C3Heb/FeJ and C3H/HeOuJ and BCG-vaccinated mouse groups were exposed to aerosol infection with ∼20 CFU of the highly virulent M. tuberculosis W-Beijing strain SA161. Most vaccines to date have been tested by using other murine models, which do not result in primary granuloma necrosis after M. tuberculosis infection and which use laboratory strains (H37Rv or Erdman). Mice infected with M. tuberculosis SA161 were evaluated for bacterial loads in the lungs (Fig. 1A) and spleens (Fig. 1B) after 25 and 50 days of infection.
FIG 1.
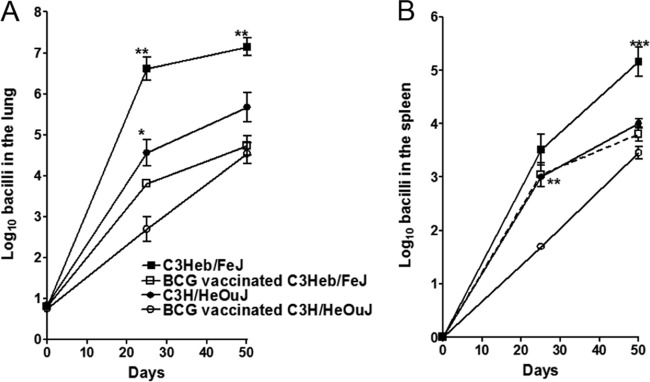
BCG-induced protection is lost in infected C3H/HeOuJ but not C3Heb/FeJ mice. Shown are bacterial counts in the lungs (A) and spleens (B) of control C3Heb/FeJ and C3H/HeOuJ mice as well as BCG-vaccinated immune C3Heb/FeJ and C3H/HeOuJ mice infected with a low-dose aerosol of M. tuberculosis W-Beijing strain SA161. CFU were determined on days 25 and 50 after infection by plating serial dilutions of organ homogenates onto nutrient 7H11 agar and counting CFU after 3 weeks of incubation at 37°C. For both murine strains, BCG vaccination resulted in a reduced bacterial burden at day 25 in comparison to the bacterial burden in nonvaccinated animals. However, protection in both the lungs and spleens of C3H/HeOuJ mice was lost at day 50 after infection. C3Heb/FeJ mice demonstrated strong BCG vaccine efficacy throughout infection. Results represent the average (n = 5) bacterial loads in each group and are expressed as log10 CFU (±SEM). *, P < 0.050; **, P < 0.010; ***, P < 0.001 (determined by ANOVA and the Tukey test).
Control C3Heb/FeJ mice showed an increase of ∼5.5 log10 CFU in the lungs over the first 25 days of infection (Fig. 1A), followed by a slight increase of 0.5 log10 CFU during the chronic phase of disease. Similarly, nonvaccinated C3H/HeOuJ mice showed an increase of 3.5 log10 CFU in the lungs over the first 25 days of infection (Fig. 1A), followed by a further increase of 1.0 log10 CFU during chronic disease. On day 25 after infection, BCG vaccination resulted in a statistically significant delay of bacterial growth in the lungs of both C3Heb/FeJ (P < 0.01) and C3H/HeOuJ (P < 0.05) mice. Remarkably, on day 50 after infection, the delay in bacterial growth in the lung was maintained only in BCG-vaccinated C3Heb/FeJ mice (P < 0.01), whereas no statistical significance was obtained when control and BCG-vaccinated C3H/HeOuJ mice were compared.
The increase in bacterial numbers in the spleens was not as high as that in the lungs (Fig. 1B). We show here that at day 50, BCG-vaccinated C3Heb/FeJ mice were more resistant than control animals (P < 0.001). BCG-vaccinated C3H/HeOuJ mice showed efficacy in the lungs and spleens at day 25 (P < 0.01); however, this delayed growth was lost 50 days later during chronic disease.
Altogether, these results demonstrate that BCG can delay the growth of M. tuberculosis in an immunocompetent yet highly susceptible murine strain such as C3Heb/FeJ. Furthermore, the BCG-induced delay in disease progression is maintained in these mice at least initially during the chronic phase of infection, whereas it is lost in C3H/HeOuJ mice. This result is consistent with our previous observations of resistant C57BL/6 mice (which do not develop necrosis) (12), in which BCG slowed down disease progression when mice were exposed to highly virulent Beijing strains.
Development of pathology in control and immune animals.
Changes in lung pathology and acid-fast staining are shown in Fig. 2 (left and right columns, respectively). As early as day 25 after infection, C3Heb/FeJ mice developed pulmonary necrotic lesions (Fig. 2A), and multiple extracellular clusters of bacilli were observed within these lesions (Fig. 2A, arrows). As previously reported (13), the lungs of control C3Heb/FeJ mice infected with M. tuberculosis SA161 exhibited progressive lesion development, and by day 50, much of the lung tissue was grossly consolidated, with inflammation and necrosis. In contrast, BCG-vaccinated C3Heb/FeJ mice had reduced numbers of lesions and clusters of extracellular bacilli, and remarkably, granuloma necrosis at both early and late time points was also abrogated (Fig. 2B). This is similar to our previous results obtained in guinea pigs, which also developed granuloma necrosis in response to M. tuberculosis infection unless they were previously BCG vaccinated (14). Overall, BCG vaccination in C3Heb/FeJ mice slowed disease progression.
FIG 2.

Changes in lung pathology in control and BCG-vaccinated C3H/HeOuJ and C3Heb/FeJ mice. Shown are representative photomicrographs of hematoxylin-eosin-stained slides (left) and acid-fast staining of slides (right) from the lungs of control or vaccinated mice. (A) As early as 25 days after infection, significant areas of necrosis were observed in C3Heb/FeJ mice. As disease progressed (day 50), areas of necrosis and bacterial burden (denoted by acid-fast staining) significantly increased in C3Heb/FeJ mice. Clusters of bacilli can be observed, which accumulated in areas of necrosis (arrows). (B) In contrast, BCG vaccination of C3Heb/FeJ mice significantly diminished necrosis and bacillary loads on both days 25 and 50 after infection. (D) BCG vaccination limited lesion size and the presence of acid-fast bacilli in C3H/HeOuJ mice temporarily 25 days after infection, and this protection was lost during chronic infection. (C) In control C3H/HeOuJ mice, bacillary numbers increased at day 50 albeit to a much lesser extent than in C3Heb/FeJ mice. Magnifications, ×4 (left) and ×100 (right).
As expected, the lungs of control C3H/HeOuJ mice had no significant necrosis (Fig. 2C) compared to the lungs of C3Heb/FeJ mice. Although BCG vaccination further reduced the numbers and size of granulomas 25 days after infection, this control was lost at day 50, as indicated by stable granuloma numbers and the presence of acid-fast staining in the lungs of C3H/HeOuJ mice (Fig. 2D).
Kinetics of the CD4+ T cell response.
Cells that migrated to the lungs of control, M. tuberculosis-infected, and BCG-vaccinated C3Heb/FeJ and C3H/HeOuJ mice were harvested and analyzed by flow cytometry. Specifically, cytokine-producing T cells, as well as the kinetics of activated Th1 (CD4+ IFN-γ-producing [IFN-γ+]), proinflammatory Th17 (CD4+ IL-17+), suppressive (CD4+ IL-10+), regulatory (CD4+ Foxp3+), and activated effector (CD4+ CD25+) T cell influx, were determined. As expected, at day 25, when bacterial burden (Fig. 1A) and pathology (Fig. 2) were already significantly different among C3Heb/FeJ and C3H/HeOuJ mice, higher percentages of CD4+ T cells were found in control and BCG-vaccinated C3Heb/FeJ mice (Fig. 3A). However, with respect to cytokine-producing T cells in control and BCG-vaccinated C3Heb/FeJ mice, overall lower percentages of IFN-γ, IL-17, IL-10, Foxp3, and CD25 cells were observed compared to control and BCG-vaccinated C3H/HeOuJ mice (Fig. 3B to F). The BCG-vaccinated C3Heb/FeJ mice demonstrated increased percentages of CD4+ T cells producing IFN-γ, IL-17, and regulatory T cell markers (Foxp3 and IL-10) (Fig. 3B to F), which was associated with reduced bacterial burden compared to that in control mice.
FIG 3.

Decreasing percentages of pulmonary CD4+ IFN-γ-producing cells with concomitant increasing percentages of Th17 and regulatory T cells during disease progression in C3H/HeOuJ mice. Shown are the percentages of pulmonary CD4+ T (A), Th1 (CD4+ IFN-γ+) (B), proinflammatory Th17 CD4+ IL-17+ (C), and regulatory CD4+ Foxp3+ LL-10+ (D to F) cells obtained from control C3Heb/FeJ and C3H/HeOuJ mice as well as BCG-vaccinated immune C3Heb/FeJ and C3H/HeOuJ mice infected with a low-dose aerosol of M. tuberculosis W-Beijing strain SA161, analyzed by flow cytometry. BCG-vaccinated C3Heb/FeJ mice demonstrated lower percentages of CD4+ T cells and cytokines (B to F), denoting bacterial control, than did control mice. As chronic disease progressed, the percentages of T cell cytokines in BCG-vaccinated C3Heb/FeJ mice significantly decreased as the bacterial burden declined (B to F). In contrast, percentages of T cell cytokines in control C3Heb/FeJ mice began to increase during chronic disease, indicative of attempted immune control of bacterial infection. Results represent the average (n = 5) percentages of cells (±SEM). *, P < 0.050 (determined by ANOVA and the Tukey posttest).
Although control and BCG-vaccinated C3H/HeOuJ mice showed lower overall percentages of CD4+ T cells than did C3Heb/FeJ mice, cytokine-producing cells were present in higher percentages (Fig. 3B to F). It was evident that both control and BCG-vaccinated C3H/HeOuJ mice showed diminished percentages of activated CD4+ CD25+ IFN-γ-producing cells. Moreover, the sharp decline in percentages CD4+ IFN-γ-producing cells was associated with increased percentages of proinflammatory (CD4+ IL-17+) and regulatory (CD4+ Foxp3+ cells producing IL-10) T cells. Also of interest is the fact that during chronic infection, significant differences between the controls and BCG-vaccinated mice were not evident, which supports the waning of BCG vaccine efficacy and increased bacterial burden during chronic infection in the C3H/HeOuJ model.
As reported previously for C57BL/6 mice succumbing to infection with highly virulent M. tuberculosis Beijing strains, the reduced numbers of protective T cells could be attributed to increasing numbers of T regulatory cells (12). However, this was not the case for control C3HeB/FeJ mice, as numbers of T regulatory cells actually declined during disease progression (Fig. 3E), resembling the tendency observed for protective T cells. Declining numbers of NK1.1 and γδ T cells were also observed in nonvaccinated C3HeB/FeJ mice (data not shown).
Kinetics of the CD4+ effector and memory T cell responses.
Cells that migrated to the lungs of infected, control, and BCG-vaccinated C3Heb/FeJ and C3H/HeOuJ mice were harvested and analyzed by flow cytometry. Specifically, levels of cytokine-producing T cells, as well as the kinetics of effector (CD4+ CD44hi CD62lo) and memory (CD4+ CD44hi CD62Lhi) T cell influx, were determined. Figure 4 shows a comparison of C3Heb/FeJ and C3H/HeOuJ mouse expansions of effector and memory cells from control and BCG-vaccinated animals (Fig. 4A and D). During early infection, the percentages of total effector cells (Fig. 4A) and subsets of cytokine-producing cells (Fig. 4B and C) in C3H/HeOuJ mice were higher than those in C3Heb/FeJ mice. However, both mouse strains showed a decline in effector immunity during chronic disease. BCG-vaccinated C3Heb/FeJ mice expressed increased effector immunity capable of producing IFN-γ and IL-17, which was associated with bacterial clearance (Fig. 4B and C). On the contrary, the percentage of total effector cells producing IFN-γ (Fig. 4B) decreased during disease progression in both BCG-vaccinated and control C3H/HeOuJ mice.
FIG 4.

Increased percentages of effector and memory IFN-γ-and IL-17-producing cells confer BCG-induced protection in C3Heb/FeJ mice. Shown are the percentages of pulmonary effector (CD44hi CD62Llo) and memory (CD44hi CD62Lhi) cells obtained from control C3Heb/FeJ and C3H/HeOuJ mice as well as BCG-vaccinated immune C3Heb/FeJ and C3H/HeOuJ mice infected with a low-dose aerosol of M. tuberculosis W-Beijing strain SA161, analyzed by flow cytometry. During chronic infection, BCG-vaccinated C3Heb/FeJ mice expressed increased numbers of effector and memory cells capable of producing IFN-γ and IL-17, which was associated with bacterial clearance (D to F). In contrast, BCG-vaccinated C3H/HeOuJ mice showed reduced levels of IFN-γ- and IL-17-producing memory T cells during chronic infection. Results represent the average (n = 5) percentages of cells (±SEM). *, P < 0.050; **, P < 0.010; ***, P < 0.001 (determined by ANOVA and the Tukey posttest).
Memory immunity in all the groups assayed was nominal (Fig. 4D). This was not surprising, as the deficit of BCG itself is the lack of induction of memory immunity (15), and most is present in the spleen. Conversely, the percentage of memory T cells capable of producing IFN-γ and IL-17 in BCG-vaccinated C3Heb/FeJ mice increased during chronic infection (Fig. 4D to F). In contrast, as infection progressed, vaccinated C3H/HeOuJ mice showed reduced levels of IFN-γ and IL-17 production in memory cells (Fig. 4E and F).
Kinetics of the granulocyte immune response.
Neutrophil granulocytes have been implicated to have a role in tuberculosis disease. Neutrophils have an intimate relationship with the primary granuloma in that they continuously degranulate in the necrotic core in an attempt to kill extracellular bacilli. However, this degranulation results in tissue damage by expansion of the necrotic core in the granuloma. Different populations of neutrophils have different potential functions, such as CD11b GR-1hi cells capable of degranulation and CD11b GR-1int myeloid-derived cells capable of T cell suppression.
A major difference between the M. tuberculosis-infected C3Heb/FeJ and C3H/HeOuJ mice is that the C3Heb/FeJ mice showed increased percentages of CD11b+ GR-1int (Fig. 5A) and CD11b+ GR-1hi (Fig. 5B) cells. Moreover, BCG vaccination of C3Heb/FeJ mice resulted in a significant reduction of both granulocytic populations (Fig. 5A and B), which can be observed in the contour plots (Fig. 5C). The effect of BCG vaccination on the influx of Gr1+ cells in C3Heb/FeJ mice was confirmed by immunohistochemistry (Fig. 5D). The paucity of Gr1+ cells in vaccinated C3Heb/FeJ mice contrasted with the extensive and coalescing Gr1+ staining throughout the consolidated, necrotic lung of nonvaccinated C3Heb/FeJ mice (Fig. 4C). Altogether, these results suggest that BCG-induced protection in C3Heb/FeJ mice is associated with reduced granulocytic influx and tissue damage.
FIG 5.

BCG vaccination curtails the influx of Gr1hi and Gr1int cells present in areas of primary granuloma necrosis in C3Heb/FeJ mice. Shown are percentages of pulmonary CD11b+ Gr1int (A) and CD11b+ Gr1hi (B) cells obtained from control C3Heb/FeJ and C3H/HeOuJ mice as well as BCG-vaccinated immune C3Heb/FeJ and C3H/HeOuJ mice infected with a low-dose aerosol of M. tuberculosis W-Beijing strain SA161, analyzed by flow cytometry. Control C3Heb/FeJ and C3H/HeOuJ mice had higher percentages of both populations of GR-1+ cells. (A) As the disease progressed, the numbers of Gr1int cells increased in control C3Heb/FeJ mice but not in C3H/HeOuJ mice (A). Interestingly, BCG-vaccinated C3Heb/FeJ mice showed a reduced influx of Gr1+ cells (A and B), which was also observed in the flow cytometric contour plots (C) and confirmed by immunohistochemistry (D). Results represent the average (n = 5) percentages of cells (±SEM). *, P < 0.050; **, P < 0.010; ***, P < 0.001 (determined by ANOVA and the Tukey posttest).
DISCUSSION
The results of this study show that BCG vaccination delayed early disease progression in both murine strains during the peak of acquired immunity. However, during chronic infection, vaccine efficacy waned as bacterial loads increased, and the lung pathology of vaccinated C3H/HeOuJ mice became similar to that of control animals. On the contrary, C3Heb/FeJ mice demonstrated strong BCG vaccine efficacy during chronic infection. Thus, while BCG vaccination of C3H/HeOuJ mice clearly had a positive protective effect against early growth of the SA161 strain, this effect was transient and not true protection during later disease. We have previously shown a similar phenomenon in C57BL/6 mice infected with W-Beijing strain SA161 or HN878 (12), in which BCG vaccination resulted in initial protection; however, this protection was lost by 60 days after infection. This raises the very troubling possibility that BCG-based vaccines may likely be ineffective in areas of the world where W-Beijing and potentially other M. tuberculosis families of high-virulence isolates are increasingly prevalent. Our serious concerns on this matter are discussed in more detail elsewhere (16, 17).
The fact that susceptible C3Heb/FeJ but not resistant C3H/HeOuJ mice were protected by BCG vaccination at later times of infection might seem paradoxical. An interesting possibility could be that after vaccination, BCG survives longer in susceptible C3Heb/FeJ mice than in resistant C3H/HeOuJ ones. The efficacy of BCG has been shown to be dependent on its ability to replicate in vivo (18). Furthermore, in humans, BCG's control in the regional lymph node is related to the host's immune status, and susceptible individuals actually become infected during BCG vaccination (19). Thus, it is envisioned that susceptible C3Heb/FeJ mice would slowly clear this live vaccine and that a better memory T cell response than the response in resistant mice would ensue. BCG clearance could have been slower in C3Heb/FeJ than in C3H/HeOuJ mice, which translated into clear differences in adaptive immunity between the two murine strains.
However, other studies (20–22) suggest that the main differences in BCG vaccination in different animal models are due to the substrain of BCG utilized, which leads to different qualitative and quantitative immune responses while resulting in similar reductions in the bacterial burden after challenge with an M. tuberculosis strain. Our study contradicts previous reports by Kramnik's laboratory which demonstrated similar adaptive T cell responses between congenic murine strains differing in the sst locus (15). This is not surprising, since the initial BCG vaccine was administered by the intravenous route (15), while the BCG vaccine in our study was administered by the subcutaneous route.
Protection in vaccinated C3Heb/FeJ mice was associated with increased percentages of effector and memory T cells. On the contrary, C3H/HeOuJ mice expressed reduced percentages of protective T cells with concomitant increases in percentages of Th17 and Foxp3+ cells compared to those in C3Heb/FeJ mice. BCG vaccine efficacy waned in C3H/HeOuJ mice coincident with reduced expression of IFN-γ by T cells and was replaced with increased expressions of IL-17, IL-10, and Foxp3. In previous studies using C57BL/6 mice, we reported that BCG-induced protection waned with specific alterations of the adaptive immune response, such as increased numbers of regulatory T cells (12), concomitant with reduced numbers of central memory T cells. Furthermore, in a reinfection model (20), we previously observed that the protective immune response decreased with increasing numbers of PD1+ CD4 T cells, a possible indication of exhaustion in a chronic disease such as tuberculosis. Using congenic strains of mice, such as B6.C3H-sst1, in which the C3Heb/FeJ sst locus was introgressed into the C57BL/6 background, significant changes in the kinetics of necrosis induction were reported (15, 23). Therefore, other loci in C57BL/6 mice seem to participate in the regulation of the immune response against tuberculosis.
A major difference between controls and vaccinated C3Heb/FeJ mice was the absence of necrosis and a reduced influx of CD11b+ Gr1+ cells in the latter. We currently believe that these two events are related, as our previous results showed that the influx of these inflammatory cells is present only in murine strains undergoing necrosis in response to M. tuberculosis infection (10). In the guinea pig model of tuberculosis, BCG vaccination also results in a delayed development of necrosis (14). Furthermore, BCG-induced protection correlated with a reduced influx of MIL4+ heterophils and increased numbers of effector and memory IFN-γ-producing cells (14). It is currently unknown if MIL4+ heterophils are the guinea pig equivalent of murine CD11b+ Gr1+ cells, as these two cellular populations were the major differences attributed to BCG vaccination in the guinea pig and murine models, respectively.
Necrosis development is an important aspect of tuberculosis pathogenesis and transmission. Bacteria residing in necrotic granulomas have access to an abundant source of carbon in the form of cholesterol and triglycerides (24), and this could explain why bacilli grow to such high numbers in these lesions. Furthermore, antibiotic penetration is significantly reduced in necrotic granulomas (25), which could partially explain the protracted antibiotic therapy required to eradicate tuberculosis. Finally, as necrotic granulomas liquefy and rupture into the airways (26, 27), bacilli encounter an efficient transmission route. Thus, necrosis prevention could be considered an effective antitransmission vaccination strategy. Clearly, BCG vaccination has not been able to protect humans against pulmonary tuberculosis and transmission (3). Therefore, we expect that at some point, BCG-induced necrosis protection would also wane in C3Heb/FeJ mice. However, we propose that differential expression of vaccine-induced efficacy in C3Heb/FeJ and C3H/HeOuJ mice would be a good model to evaluate continuous protection against tuberculosis-induced necrosis and nonnecrosis.
ACKNOWLEDGMENTS
This work was supported by NIH Innovator Award grant numbers 1DP2OD006450 and AI081959. This study was also funded under the American Recovery and Reinvestment Act of 2009.
REFERENCES
- 1.World Health Organization. 2011. Global tuberculosis control: WHO report 2011, p viii, 122–246. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.North RJ, Jung YJ. 2004. Immunity to tuberculosis. Annu Rev Immunol 22:599–623. doi: 10.1146/annurev.immunol.22.012703.104635. [DOI] [PubMed] [Google Scholar]
- 3.Pitt JM, Blankley S, McShane H, O'Garra A. 2013. Vaccination against tuberculosis: how can we better BCG? Microb Pathog 58:2–16. doi: 10.1016/j.micpath.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Fine PE. 1995. Variation in protection by BCG: implications of and for heterologous immunity. Lancet 346:1339–1345. doi: 10.1016/S0140-6736(95)92348-9. [DOI] [PubMed] [Google Scholar]
- 5.Orme IM. 2006. Preclinical testing of new vaccines for tuberculosis: a comprehensive review. Vaccine 24:2–19. doi: 10.1016/j.vaccine.2005.07.078. [DOI] [PubMed] [Google Scholar]
- 6.Basaraba RJ. 2008. Experimental tuberculosis: the role of comparative pathology in the discovery of improved tuberculosis treatment strategies. Tuberculosis (Edinb) 88:S35–S47. doi: 10.1016/S1472-9792(08)70035-0. [DOI] [PubMed] [Google Scholar]
- 7.Kramnik I, Dietrich WF, Demant P, Bloom BR. 2000. Genetic control of resistance to experimental infection with virulent Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 97:8560–8565. doi: 10.1073/pnas.150227197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, Higgins DE, Daly MJ, Bloom BR, Kramnik I. 2005. Ipr1 gene mediates innate immunity to tuberculosis. Nature 434:767–772. doi: 10.1038/nature03419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harper J, Skerry C, Davis SL, Tasneen R, Weir M, Kramnik I, Bishai WR, Pomper MG, Nuermberger EL, Jain SK. 2012. Mouse model of necrotic tuberculosis granulomas develops hypoxic lesions. J Infect Dis 205:595–602. doi: 10.1093/infdis/jir786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obregón-Henao A, Henao-Tamayo M, Orme IM, Ordway DJ. 2013. Gr1(int)CD11b+ myeloid-derived suppressor cells in Mycobacterium tuberculosis infection. PLoS One 8:e80669. doi: 10.1371/journal.pone.0080669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Junqueira-Kipnis AP, Turner J, Gonzalez-Juarrero M, Turner OC, Orme IM. 2004. Stable T-cell population expressing an effector cell surface phenotype in the lungs of mice chronically infected with Mycobacterium tuberculosis. Infect Immun 72:570–575. doi: 10.1128/IAI.72.1.570-575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ordway DJ, Shang S, Henao-Tamayo M, Obregon-Henao A, Nold L, Caraway M, Shanley CA, Basaraba RJ, Duncan CG, Orme IM. 2011. Mycobacterium bovis BCG-mediated protection against W-Beijing strains of Mycobacterium tuberculosis is diminished concomitant with the emergence of regulatory T cells. Clin Vaccine Immunol 18:1527–1535. doi: 10.1128/CVI.05127-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C, Basaraba RJ, Orme IM. 2007. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol 179:522–531. doi: 10.4049/jimmunol.179.1.522. [DOI] [PubMed] [Google Scholar]
- 14.Ordway D, Henao-Tamayo M, Shanley C, Smith EE, Palanisamy G, Wang B, Basaraba RJ, Orme IM. 2008. Influence of Mycobacterium bovis BCG vaccination on cellular immune response of guinea pigs challenged with Mycobacterium tuberculosis. Clin Vaccine Immunol 15:1248–1258. doi: 10.1128/CVI.00019-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan BS, Pichugin AV, Jobe O, Helming L, Eruslanov EB, Gutiérrez-Pabello JA, Rojas M, Shebzukhov YV, Kobzik L, Kramnik I. 2007. Progression of pulmonary tuberculosis and efficiency of bacillus Calmette-Guerin vaccination are genetically controlled via a common sst1-mediated mechanism of innate immunity. J Immunol 179:6919–6932. doi: 10.4049/jimmunol.179.10.6919. [DOI] [PubMed] [Google Scholar]
- 16.Orme IM. 2011. Development of new vaccines and drugs for TB: limitations and potential strategic errors. Future Microbiol 6:161–177. doi: 10.2217/fmb.10.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orme IM. 2010. The Achilles heel of BCG. Tuberculosis (Edinb) 90:329–332. doi: 10.1016/j.tube.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 18.Andersen P, Doherty TM. 2005. The success and failure of BCG—implications for a novel tuberculosis vaccine. Nat Rev Microbiol 3:656–662. doi: 10.1038/nrmicro1211. [DOI] [PubMed] [Google Scholar]
- 19.Norouzi S, Aghamohammadi A, Mamishi S, Rosenzweig SD, Rezaei N. 2012. Bacillus Calmette-Guerin (BCG) complications associated with primary immunodeficiency diseases. J Infect 64:543–554. doi: 10.1016/j.jinf.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henao-Tamayo M, Obregón-Henao A, Ordway DJ, Shang S, Duncan CG, Orme IM. 2012. A mouse model of tuberculosis reinfection. Tuberculosis (Edinb) 92:211–217. doi: 10.1016/j.tube.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Irwin SM, Goodyear A, Keyser A, Christensen R, Troudt JM, Taylor JL, Bohsali A, Briken V, Izzo AA. 2008. Immune response induced by three Mycobacterium bovis BCG substrains with diverse regions of deletion in a C57BL/6 mouse model. Clin Vaccine Immunol 15:750–756. doi: 10.1128/CVI.00018-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minassian AM, Ronan EO, Poyntz H, Hill AV, McShane H. 2011. Preclinical development of an in vivo BCG challenge model for testing candidate TB vaccine efficacy. PLoS One 6:e19840. doi: 10.1371/journal.pone.0019840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pichugin AV, Yan BS, Sloutsky A, Kobzik L, Kramnik I. 2009. Dominant role of the sst1 locus in pathogenesis of necrotizing lung granulomas during chronic tuberculosis infection and reactivation in genetically resistant hosts. Am J Pathol 174:2190–2201. doi: 10.2353/ajpath.2009.081075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, Visser A, Wang W, Hsu FF, Wiehart U, Tsenova L, Kaplan G, Russell DG. 2010. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med 2:258–274. doi: 10.1002/emmm.201000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prideaux B, Dartois V, Staab D, Weiner DM, Goh A, Via LE, Barry CE III, Stoeckli M. 2011. High-sensitivity MALDI-MRM-MS imaging of moxifloxacin distribution in tuberculosis-infected rabbit lungs and granulomatous lesions. Anal Chem 83:2112–2118. doi: 10.1021/ac1029049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. 2009. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol 10:943–948. doi: 10.1038/ni.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O'Garra A. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]