

Abstract
Scedosporium boydii is an opportunistic filamentous fungus which may be responsible for a wide variety of infections in immunocompetent and immunocompromised individuals. This fungus belongs to the Scedosporium apiospermum species complex, which usually ranks second among the filamentous fungi colonizing the airways of patients with cystic fibrosis (CF) and may lead to allergic bronchopulmonary mycoses, sensitization, or respiratory infections. Upon microbial infection, host phagocytic cells release reactive oxygen species (ROS), such as hydrogen peroxide, as part of the antimicrobial response. Catalases are known to protect pathogens against ROS by detoxification of the hydrogen peroxide. Here, we investigated the catalase equipment of Scedosporium boydii, one of the major pathogenic species in the S. apiospermum species complex. Three catalases were identified, and the mycelial catalase A1 was purified to homogeneity by a three-step chromatographic process. This enzyme is a monofunctional tetrameric protein of 460 kDa, consisting of four 82-kDa glycosylated subunits. The potential usefulness of this enzyme in serodiagnosis of S. apiospermum infections was then investigated by an enzyme-linked immunosorbent assay (ELISA), using 64 serum samples from CF patients. Whatever the species involved in the S. apiospermum complex, sera from infected patients were clearly differentiated from sera from patients with an Aspergillus fumigatus infection or those from CF patients without clinical and biological signs of a fungal infection and without any fungus recovered from sputum samples. These results suggest that catalase A1 is a good candidate for the development of an immunoassay for serodiagnosis of infections caused by the S. apiospermum complex in patients with CF.
INTRODUCTION
Scedosporium boydii (formerly known as Pseudallescheria boydii) is one of the major pathogenic species within the Scedosporium apiospermum complex, which comprises four other species, namely, Scedosporium apiospermum sensu stricto, Scedosporium aurantiacum, Scedosporium minutisporum, and Scedosporium dehoogii, Scedosporium prolificans having been reassigned recently to the genus Lomentospora (Lomentospora prolificans) (1–4). These filamentous fungi are soilborne fungi that may cause a wide range of infections in humans, including subcutaneous mycetomas and ocular, bone, or joint infections resulting from traumatic inoculation of some fungal elements and infections of the respiratory tract (i.e., sinusitis and lung fungus ball), which are thought to be due to the inhalation of some airborne conidia (5–7). However, these fungi have gained attention during the past 2 decades mainly because of their recognition as common agents of colonization of the airways in patients with cystic fibrosis (CF). When appropriate culture media are used, the S. apiospermum species complex ranks second among the filamentous fungi recovered from respiratory specimens, with a prevalence ranging from 4.5 to 11.6% in patients (8–12). Although usually asymptomatic, this fungal colonization of the airways may sometimes lead to allergic bronchopulmonary mycoses, sensitization, or respiratory infections (8, 13). In addition, due to the propensity of these fungi to hematogenously disseminate in cases of immunodeficiency and to their low susceptibility to current antifungals, a prior colonization of the airways by these fungi may compromise the success of lung transplantation, and several cases of fatal infections have been reported in CF patients who had undergone lung transplantation and were colonized by species of the S. apiospermum complex (14–17).
Diagnosis of these infections mainly relies on cultivation of microorganisms from clinical samples on agar-based culture medium and, for deep-seated infections, on histopathological examination of fixed biopsy specimens. However, in tissue sections, species of the S. apiospermum complex cannot be differentiated from Aspergillus species and other hyaline hyphomycetes due to similar histomorphological patterns (6). Highly specific monoclonal antibodies which might allow the immunodetection of the fungus have been described by Thornton (18), but they are not commercially available. As for mycological examination, it requires skill and expertise and may lead to false-negative results for polymicrobial specimens like sputum samples because of the more rapid and more extensive growth of other molds frequently associated with this fungus, like Aspergillus fumigatus (19). Several molecular methods have been proposed for detection of the fungus from sputum samples (20–24), but as culture methods, they do not allow the differentiation between airway colonization and sensitization of the patient or respiratory infection in the CF context, which has important implications for patient management.
Detection of serum-specific antibodies may be a valuable alternative for diagnosis of a deep-seated S. boydii infection, and in the CF context, it remains the unique option for discriminating between airway colonization and a respiratory infection caused by species of the S. apiospermum complex. Nevertheless, there are no standardized methods to date, and this serodiagnosis is performed only in a few specialized laboratories by counterimmunoelectrophoresis (CIE) using homemade crude antigenic extracts (8). In these extracts, the relative amount of the different antigens is highly dependent on the strain used, the culture conditions, and the procedure used for preparation of the extracts. In addition, several proteins and cell wall polysaccharides are common to various pathogenic fungi. Therefore, cross-reactivity with other filamentous fungi such as A. fumigatus may occur, leading sometimes to false-positive results (6, 8). Because of this, identification of an antigen shared by species of the S. apiospermum complex and allowing specific antibody detection may be helpful.
Studies performed by Sarfati et al. (25) using recombinant antigens confirmed serum antibodies directed toward the mycelial catalase Cat1 of A. fumigatus as biological markers of Aspergillus infections. Catalases are ubiquitous antioxidant enzymes which catalyze the degradation of hydrogen peroxide. Therefore, these enzymes, which protect microorganisms against the reactive oxygen species (ROS) produced by the host phagocytic cells, have been largely studied as virulence factors, but also for their potential in serodiagnosis of the resulting infections.
Here, we report the purification and biochemical characterization of a mycelial catalase from S. boydii and its use for serodiagnosis.
MATERIALS AND METHODS
Culture conditions and preparation of fungal extracts.
Scedosporium boydii IHEM 15155 (Institute of Hygiene and Epidemiology-Mycology Section, Institute of Public Health, Brussels, Belgium) was used throughout this study. This strain was routinely maintained by cultivation on yeast extract-peptone-dextrose agar (YPDA) (containing in g/liter: yeast extract, 5; peptone, 5; glucose, 20; chloramphenicol, 0.5; and agar, 20) plates. After 9 days of incubation at 37°C, the mycelium was harvested by scraping the agar plates with sterile distilled water. Conidia were then separated from hyphae by filtration through 20-μm-pore-size nylon membranes, washed in sterile distilled water, and finally counted using a hemocytometer. They were then inoculated in yeast extract-peptone-dextrose (YPD) broth (500-ml flasks containing 200 ml YPD broth each) at a final density of 5 × 106 conidia per ml. After 7 days of incubation at 37°C without shaking, cultures were centrifuged at 2,000 × g for 20 min. The culture supernatant was sterilized by filtration through 0.2-μm-pore-size membranes, dialyzed against distilled water (in dialysis tubing with a 14,000-molecular-weight cutoff), and finally freeze-dried. The fungal mycelium was also collected and used to prepare somatic extracts after several washes in distilled water. In order to investigate the cellular distribution of catalases, different procedures were used for protein extraction.
A crude somatic extract was obtained by grinding the mycelium in liquid nitrogen followed by a mechanical disruption with glass beads (0.1 to 0.2 mm and 1 mm) with CO2 cooling (MSK disintegrator; Braun Melsungen, Melsungen, Germany). The suspension was then clarified by centrifugation at 50,000 × g for 30 min at 4°C, and the supernatant was stored at −20°C until used.
Subcellular fractions were also prepared by grinding the mycelium in liquid nitrogen. The homogenate was then suspended in 10 ml of 150 mM phosphate-buffered saline (PBS) (pH 7.2). After vigorous shaking and successive centrifugations (10 min at 1,500 × g and then 30 min at 45,000 × g), the supernatant, which corresponds essentially to the cytosolic fraction, was concentrated by dialysis against polyethylene glycol (PEG) 35000. Meanwhile, the first centrifugation pellet (1,500 × g for 10 min) was suspended in 10 ml of PBS, ground with glass beads with CO2 cooling, and then clarified by centrifugation (45,000 × g for 30 min). The resulting supernatant was concentrated as described above, and the pellet, which corresponds to cell wall debris and intracellular organelles like peroxisomes, was resuspended in PBS, sonicated with three 30-s bursts at a setting of 8 and 70% duty cycle (Branson Sonifier 450; Fisher Scientific, Illkirch, France), and finally clarified by centrifugation (45,000 × g for 30 min). The pellet was discarded, and the supernatant (“peroxisomal” fraction) was concentrated.
Cultures were also performed at 37°C in YPD broth for various times ranging from 72 h to 10 days. At the end of each incubation period, the culture supernatant was collected, as was the mycelium, which was used to prepare somatic extracts. In addition, for some experiments, a cytosolic extract was also prepared from A. fumigatus strain CBS 113.26 as a comparison strain and control for the catalase activity assays.
The protein concentrations in these extracts were determined by the bicinchoninic acid assay.
Catalase activity assays.
Catalase activity was quantified by measuring the decrease in absorbance at 240 nm at 25°C after the addition of the fungal extracts or chromatographic fractions (100 μl) to 1.9 ml of 50 mM phosphate buffer (pH 7.2) containing 0.19 mM H2O2 (26). An enzyme unit was defined as the amount of enzyme that degrades 1 μM H2O2 per minute (ε = 43.6 M−1 cm−1), and specific activity was defined as the ratio between the enzyme activity and the total amount of protein in the extract. Catalase from bovine liver (Sigma-Aldrich, St. Louis, MO, USA) was used as a control.
Catalases were also visualized by negative staining after native polyacrylamide gel electrophoresis (PAGE) on 5 to 15% linear gradient gels as previously described for detection of A. fumigatus catalases (27). The ferricyanide-negative staining of Woodbury et al. (28) was used to locate bands corresponding to catalases. In some experiments, peroxidase activity was also investigated in the same gels as described by Wayne and Diaz (29).
Purification of catalase A1.
Catalase A1 was purified from the crude somatic extract by a three-step chromatographic procedure. For each step, chromatographic fractions were checked for catalase activity; then, positive fractions were analyzed by native PAGE and SDS-PAGE, and catalase A1-containing fractions were pooled.
(i) Anion exchange chromatography.
The crude somatic extract diluted in 20 mM Tris-HCl (pH 7.5) was applied on a DEAE-Trisacryl M (BioSepra, Villeneuve la Garenne, France) column. Elution was carried out using a linear NaCl gradient (0 to 250 mM) at a flow rate of 2 ml/min. The elution was monitored by UV absorbance at 280 nm.
(ii) Hydrophobic interaction chromatography.
Pooled fractions containing catalase A1 were diluted to a final concentration of 1.75 M by slow addition of phosphate buffer containing 4 M ammonium sulfate. After incubation for 30 min at 4°C and centrifugation at 4,000 × g for 15 min, the supernatant was applied to a phenyl-Sepharose 6 Fast Flow column (GE Healthcare Life Sciences, Uppsala, Sweden) previously equilibrated with 1.75 M ammonium sulfate in the same buffer. The sample was eluted with a stepwise gradient using decreasing ammonium sulfate concentrations (from 1.75 to 0.0 M with 0.25-M steps) in the same buffer at a flow rate of 2 ml/min, and the elution was monitored at 280 nm.
(iii) Gel filtration chromatography.
Proteins in pooled catalase A1-containing fractions were concentrated by ammonium sulfate (1.75 M) precipitation. After an overnight incubation at 4°C and centrifugation at 12,000 × g for 30 min, the pellet was resuspended in PBS and applied to a Sephacryl S300 column (GE Healthcare) equilibrated in the same buffer. Elution was carried out at a flow rate of 1.3 ml/min, and the elution was monitored at 280 nm. The molecular mass of catalase A1 was determined by calibration of the column with protein standards (high-molecular-weight gel filtration calibration kit from GE Healthcare).
Analytical methods and enzyme characterization. (i) Electrophoretic analysis.
SDS-PAGE was performed on 5 to 15% polyacrylamide gradient gels with Coomassie brilliant blue R250 or silver staining as described by Laemmli (30). The relative molecular mass of the purified catalase was estimated according to the molecular mass of protein markers (GE Healthcare).
(ii) Isoelectrophoresis.
The isoelectric point of catalase A1 was determined by isoelectric focusing (IEF) on precast gels LKB-IEF (3.5 to 9.5 and 4 to 6.5; GE Healthcare). After completion of electrophoresis, the gels were incubated for 20 min in a 1 mM solution of horseradish peroxidase in PBS, and hydrogen peroxide was added at a final concentration of 5 mM. After incubation for 10 min, washing in distilled water, and addition of 2 mM 3,3′-diaminobenzidine (DAB) in PBS, catalases appeared as unstained areas on a brown background. The pI was extrapolated from the migration of isoelectric point markers from GE Healthcare.
(iii) Effect of pH and temperature on catalase activity.
The pH stability of the catalase was determined by measuring the catalase activity in a range of pH (2.5 to 13) using 0.2 M sodium acetate buffer (pH 2.5 to 4.5), 66 mM sodium potassium phosphate buffer (pH 5 to 8), or 0.1 M glycine buffer (pH 9 to 13).
Heat stability was evaluated by measuring the residual enzyme activity after 1 to 15 min of incubation at different temperatures (37, 68, 80, and 100°C). The residual catalase activity was determined by densitometric determination after native PAGE and negative staining of the gels.
(iv) Catalytic properties of the catalase.
The effects of several catalase inhibitors were evaluated by UV spectrophotometry after incubation for 1 h with the purified enzyme (Table 1). Inhibitors of hemoproteins such as potassium cyanide (KCN) and sodium azide (NaN3) were tested at 10 mM final concentrations, whereas 3-amino-1,2,4-triazole (3-AT), a specific inhibitor of catalase, was tested at a 4 mM final concentration. Moreover, the effects of metallic ions Cu2+ and Hg2+ (10 mM), SDS (4%), and 2-mercaptoethanol (2-ME) (30 mM) were also evaluated. Stability of the enzyme in ethanol-chloroform was tested as described by Nadler et al. (31).
TABLE 1.
Effects of different reagents on catalase A1 activity
| Reagent (final concn) | Residual activity (%)a |
|---|---|
| Potassium cyanide (10 mM) | 0 |
| Sodium azide (10 mM) | 0 |
| 3-AT (4 mM) | 38 |
| Ethanol-chloroform (25%–15%) | 71 |
| Cu2+ (10 mM) | 52 |
| Hg2+ (10 mM) | 14 |
| SDS (4%) | 97 |
| 2-ME (30 mM) | 14 |
Residual activity was determined spectrophotometrically after incubation of the purified enzyme for 1 h in the presence of the different reagents tested.
(v) Glycosylation.
Glycosylation of catalase A1 was first investigated by affinity chromatography on a concanavalin A (ConA)-conjugated Sepharose 4B column (GE Healthcare). Two hundred microliters of the crude extract was incubated for 30 min at 37°C with ConA-Sepharose. After centrifugation for 5 min at 4,000 × g and washing in PBS, glycosylated proteins were eluted with 0.2 M methyl α-d-mannopyranoside in PBS. After a further 30-min incubation at 37°C and centrifugation, the unbound fraction and eluted proteins were analyzed for catalase activity by native PAGE and negative staining.
Glycosylation was also investigated after electrophoretic transfer of proteins separated by SDS-PAGE on a Hybond-P membrane (GE Healthcare). After electrotransfer, the membrane was blocked overnight at 4°C with 5% bovine serum albumin (BSA) in PBS, washed three times with PBS, and then incubated for 1 h at 37°C with peroxidase-conjugated wheat germ agglutinin (WGA) (1 μg/ml) or ConA (3 μg/ml) from Sigma-Aldrich in 50 mM Tris buffer supplemented with 0.1 mM Ca2+ and 0.1 mM Mg2+. After washing, peroxidase was revealed with 0.5 mg/ml DAB in 0.1 M Tris buffer (pH 7.6) and 0.1% hydrogen peroxide.
Human sera.
A panel of 64 human serum samples was used to evaluate the usefulness of purified catalase A1 for serodiagnosis of infections caused by the S. apiospermum species complex. The samples were categorized into 3 groups based on the results of the mycological examination and antibody response against A. fumigatus and S. boydii crude extracts by routine serological procedures, i.e., CIE using crude somatic extracts and detection of anti-catalase antibodies by immunodiffusion assay (8): (i) sera from patients with CF without any filamentous fungus recovered from sputum samples during the 6 months preceding or following the blood sampling and without the serum antibodies directed toward A. fumigatus and S. boydii (group A; n = 20); (ii) sera from CF patients with A. fumigatus as the only filamentous fungus recovered from respiratory secretions and with a positive antibody response against A. fumigatus crude antigenic extract only (group B; n = 19); and (iii) sera from patients with the S. apiospermum species complex recovered from the clinical samples (S. boydii, S. apiospermum, or species not specified), but not A. fumigatus, and with a serological response against S. boydii antigenic extract (group C; n = 25). For group B, anti-A. fumigatus catalase antibodies were not detected by double immunodiffusion assays for 11 out of the 19 sera (B1 subgroup), whereas the remaining 8 sera exhibited such antibodies (B2 subgroup).
Enzyme-linked immunosorbent assay.
An enzyme-linked immunosorbent assay (ELISA) was performed by coating the wells of microtiter plates (Microlon 200, Greiner; Dutscher, Brumath, France) for 3 h at 37°C with purified catalase diluted in 50 mM carbonate-bicarbonate buffer (pH 9.6). After three washes with PBS, plates were blocked by overnight incubation at 4°C with a 10% BSA solution in PBS. Plates were then washed with PBS containing 0.05% Tween 20 (PBS-T), incubated with 100 μl of a 1:100 dilution of human sera diluted in PBS-T-BSA (0.3%) for 1 h at 37°C, and washed again with PBS-T. Horseradish peroxidase-conjugated goat anti-human IgG+A+M (H+L) (Invitrogen, Camarillo, CA) at a 1:10,000 dilution in PBS-T-BSA was added to each well (100 μl per well). After a further 1-h incubation at 37°C and washing, peroxidase was revealed using o-phenylenediamine tetrahydrochloride (Sigma-Aldrich) and 0.02% H2O2 in 0.15 M citrate-phosphate buffer (pH 5.0) (200 μl per well). After incubation at room temperature in the dark for 10 min, the reaction was stopped with 1 M H2SO4 (50 μl), and absorbance at 490 nm was quantified on a microplate reader (ELx800; Bio-Tek Instruments, Colmar, France). Controls consisted of omission of the antigenic extract or of human sera.
The ELISA cutoff value was calculated according to the following proven formula: control serum (group A) optical density (OD) values (mean plus 3 standard deviations).
The antibody titer for sera from group C patients was estimated using serial 2-fold dilutions of the sera starting from 1:400 up to 1:12,800.
Statistical analysis was performed using the Wilcoxon-Mann-Whitney test, and results were considered significantly different at a P value of <0.01.
RESULTS
Evidence for three catalases in S. boydii.
The presence of catalases in the crude somatic extract was first evidenced by spectrophotometric measurement of H2O2 degradation and further investigated by negative staining after native PAGE, which revealed catalases as unstained bands on a dark blue-green background. In this way, three bands with catalase activity were revealed for the S. boydii crude somatic extract, corresponding to proteins differing in their electrophoretic mobility, with a diffuse band into the upper part of the gel, designated A1 because of the similarity of its electrophoretic mobility with that of Cat1 of A. fumigatus, associated with two thin bands of greater electrophoretic mobility, designated A2 and A2′, which were very close, forming a doublet (Fig. 1A, lane 1). The three catalase bands were also detected by native PAGE and negative staining in the cytosolic extract and in the peroxisomal extract. However, catalase A1 was predominant in the cytosolic fraction, while catalases A2/A2′ were predominant in the peroxisomal fraction (data not shown).
FIG 1.
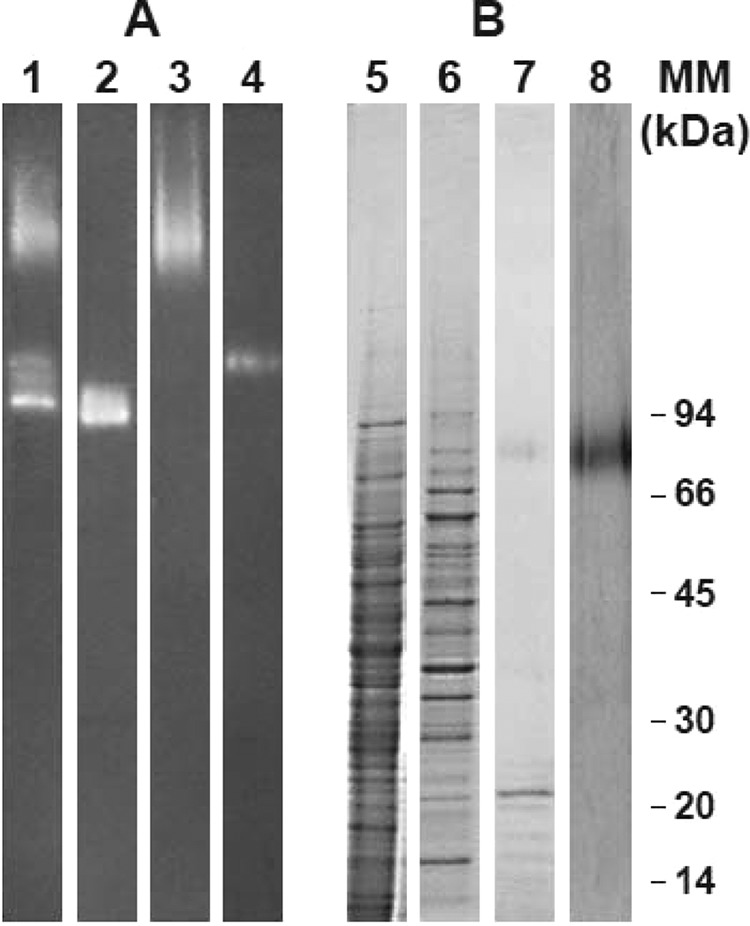
Native PAGE (A) and SDS-PAGE (B) analysis of S. boydii crude somatic extract and of the pooled catalase-containing fractions from the different chromatographic steps. Samples were loaded on native 5 to 15% polyacrylamide gels (A) or on SDS 5 to 15% polyacrylamide gels (B), which were developed using ferricyanide-negative (lanes 1 to 4), Coomassie blue (lanes 5 to 7), or silver (lane 8) staining. Lanes 1 and 5, crude somatic extract; lanes 2 to 4, pooled fractions from anion-exchange chromatography exhibiting H2O2-degrading activity; lanes 6 to 8, pooled catalase A1-containing fractions recovered from anion-exchange chromatography (lane 6), hydrophobic interaction chromatography (lane 7), or molecular size exclusion (lane 8). MM, molecular mass.
Catalase production was also investigated during the growth of S. boydii in YPD broth. Somatic extracts were prepared from cultures grown for 72 h to 10 days, and negative staining after native PAGE always revealed the three catalase bands whatever the age of the cultures. Catalase activity in these extracts was also quantified spectrophotometrically. Very low enzyme activity was detected during the first 72 h of cultures, and then catalase activity increased to reach a plateau (from <20 U/mg of proteins to 40 U/mg of proteins) at day 6 (data not shown).
Catalase activity was also seen in culture supernatant, but a single band corresponding to catalase A1 was seen on native PAGE, whatever the age of the cultures. However, enzyme activity in the culture supernatant remained low, the specific activity increasing gradually from 9 U/mg after 72 h of culture to 20 U/mg on day 10.
Purification of catalase A1.
Scedosporium boydii catalases were first separated by anion-exchange chromatography (Fig. 2A). Several chromatographic fractions exhibited catalase activity, and native PAGE analysis with ferricyanide staining confirmed the sequential elution of the three enzymes A2′, A1, and A2 (Fig. 1A, lanes 2 to 4). Catalase A1 was eluted with 120 to 145 mM NaCl (Fig. 1A, lane 2), but SDS-PAGE analysis of pooled catalase A1-containing fractions revealed numerous protein bands after Coomassie blue staining (Fig. 1B, lane 6). A second chromatographic step therefore was required, consisting of hydrophobic interaction chromatography. Catalase activity was detected in fractions eluted with 0.75 M and 1 M ammonium sulfate, and SDS-PAGE analysis of these fractions with silver staining revealed the disappearance of several protein bands together with enrichment in an 82-kDa species (Fig. 1B, lane 7). Purification of catalase A1 was achieved in a third chromatographic step consisting of molecular size exclusion (Fig. 2C), which suggested a 460-kDa molecular mass for the enzyme. SDS-PAGE of pooled catalase A1-containing fractions, which showed a single polypeptide band after silver staining, confirmed purification of the enzyme to homogeneity (Fig. 1B, lane 8).
FIG 2.

Purification of S. boydii catalase A1. (A) Fractionation of the crude somatic extract by anion-exchange chromatography on DEAE-Trisacryl. The locations of the different catalases as evidenced by the spectrophotometric detection of the enzyme activity and native PAGE analysis of the corresponding fractions with negative staining are indicated on the chromatogram. (B) Hydrophobic interaction chromatographic fractionation of pooled catalase A1-containing fractions from anion-exchange chromatography. Fractions containing catalase A1 are indicated on the chromatogram. (C) Molecular size exclusion chromatographic fractionation of pooled catalase A1-containing fractions recovered from hydrophobic interaction chromatography. Fractions containing catalase A1 are indicated on the chromatogram. AU, arbitrary units.
Biochemical properties of catalase A1.
As illustrated in Fig. 3A, native PAGE analysis with double staining according to Wayne and Diaz (29) did not reveal peroxidase activity for any of the catalases produced by S. boydii (lane 2), in contrast to that observed for one of the catalases produced by A. fumigatus CBS 113.26 (lane 1).
FIG 3.
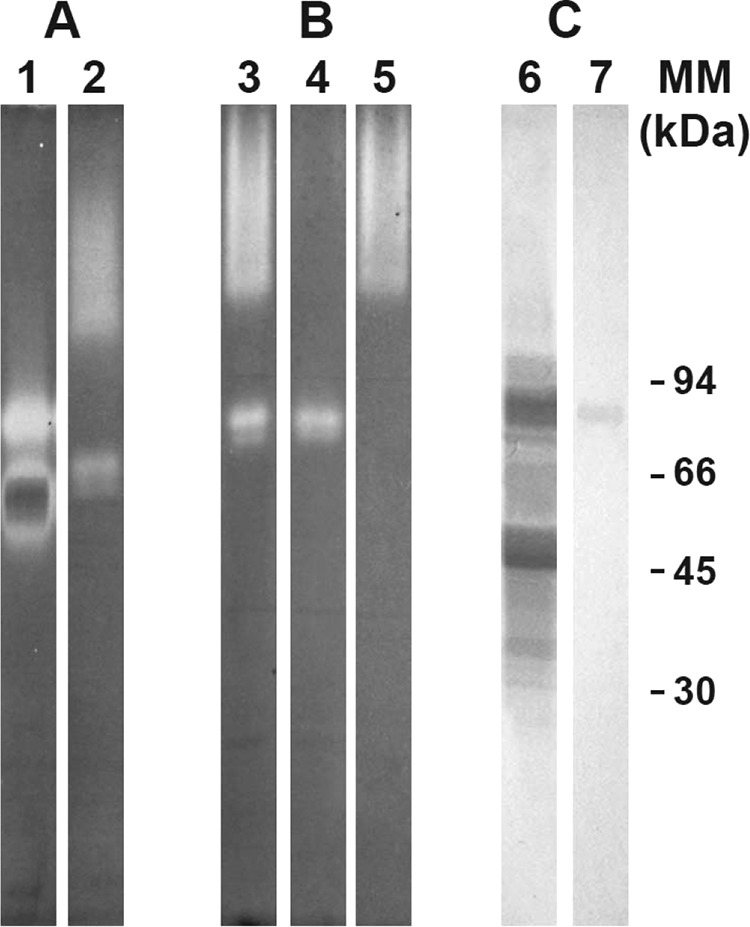
PAGE analysis of catalase A1. (A) Double staining according to Wayne and Diaz (29) after native PAGE analysis of crude somatic extracts from A. fumigatus CBS 113.26 (lane 1) and S. boydii IHEM 15155 (lane 2). (B) Ferricyanide-negative staining of native 5 to 15% polyacrylamide gels loaded with S. boydii crude somatic extract (lane 3), unbound fraction from affinity chromatography on concanavalin A-Sepharose (lane 4), and fraction eluted from the column with 0.2 M methyl α-d-mannopyranoside (lane 5). (C) S. boydii crude somatic extract (lane 6) and purified catalase A1 (lane 7) probed with peroxidase-concanavalin A after SDS-PAGE and Western blotting.
SDS-PAGE analysis of the purified enzyme revealed a molecular mass of 82 kDa (Fig. 1B, lane 8), and a 4.2 pI was determined by isoelectric focusing (data not shown). In addition, after chromatographic fractionation of the crude extract on ConA-Sepharose 4B, bands corresponding to catalases A2/A2′ were detected in the unbound fractions (Fig. 3B, lane 4), whereas catalase A1 was eluted from the column using 0.2 M methyl α-d-mannopyranoside (Fig. 3B, lane 5), thus suggesting that the enzyme was glycosylated. This was confirmed by SDS-PAGE analysis of the purified enzyme followed by Western blotting and incubation of the blot with peroxidase-conjugated ConA (Fig. 3C, lane 7).
Catalase A1 exhibited activity over a broad range of pH values (5 to 10). Moreover, pretreatment of purified catalase A1 at 80°C for 5 min resulted in 80% inhibition of the enzyme activity, whereas it was not affected by heating for 5 min at 68°C (data not shown). In addition, catalase A1 was completely inactivated by KCN and NaN3, but 62% and 29% reductions were also seen in enzyme activity after 1 h of incubation with 3-AT or after ethanol-chloroform treatment, respectively (Table 1). Moreover, SDS had no effect on enzyme activity, whereas 2-ME strongly inhibited the purified catalase A1. Finally, a 48 to 86% reduction in enzyme activity was observed in the presence of the heavy metal ions Cu2+ and Hg2+.
Sensitivity and specificity of anti-catalase A1 ELISA.
The potential usefulness of purified catalase A1 in serodiagnosis of infections caused by the S. apiospermum species complex was investigated by an ELISA.
As shown in Fig. 4, the highest OD values were obtained for sera from CF patients with an S. apiospermum complex infection (group C patients), i.e., patients with recovery of species of the S. apiospermum complex but not A. fumigatus from clinical samples and with a positive serological response against S. boydii but not A. fumigatus by CIE. The median and geometric mean OD values for group C patients were 2.264 and 2.253, respectively, with OD values ranging from 1.471 to 3.188.
FIG 4.
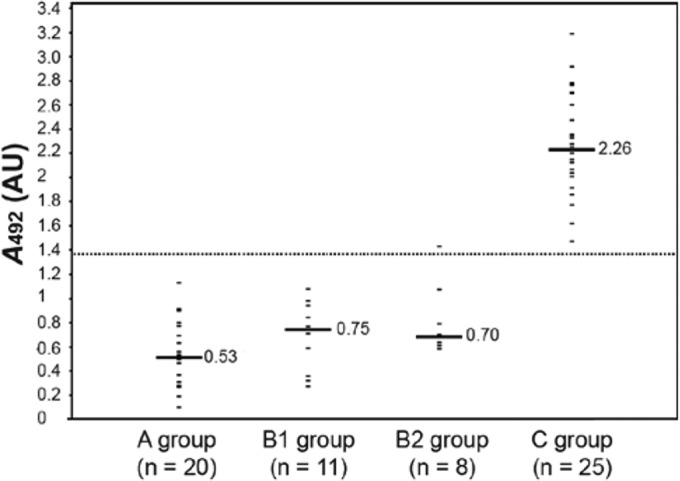
ELISA reactivity of sera from infected or noninfected CF patients with immobilized purified catalase A1 from S. boydii IHEM 15155. Sera were obtained from CF patients without clinical or biological signs of fungal infections and without any fungus recovered from sputum samples (group A) and with A. fumigatus the sole filamentous fungus recovered from sputum samples and with serum antibodies directed toward A. fumigatus but not S. boydii by routine procedures (group B: B1, patients without anti-A. fumigatus catalase antibodies; B2, patients with anti-A. fumigatus catalase antibodies) and CF patients colonized by species of the S. apiospermum complex and exhibiting serum antibodies directed toward S. boydii but not A. fumigatus (group C). The cutoff (dotted line) and median OD values (solid lines) are indicated.
The specificity of the ELISA was assessed using (i) sera from CF patients with no filamentous fungi recovered from respiratory secretions and no serum antibodies against A. fumigatus or the S. apiospermum species complex (group A) and (ii) sera from patients with recovery of A. fumigatus but not the S. apiospermum species complex from clinical samples and with a positive serological response against A. fumigatus and not S. boydii by CIE (group B). Results showed median and geometric mean OD values of 0.530 and 0.479, respectively, with OD values ranging from 0.369 to 1.129, for sera from group A patients, whereas values were 0.7 and 0.779, respectively, with OD values ranging from 0.701 to 1.429, for group B patients. In the latter group, reactivity with S. boydii purified catalase A1 was not higher for sera which showed the presence of anti-A. fumigatus catalase antibodies by immunodiffusion assay (median and geometric mean OD values of 0.750 and 0.631 for group B1, versus 0.7 and 0.78 for group B2).
Results were also analyzed statistically. Sera from patients with S. apiospermum infection (group C) were clearly differentiated from sera from group A patients (no airway colonization or infection by molds, P < 10−4) or group B patients (patients infected by A. fumigatus but without anti-A. fumigatus catalase antibodies, P < 10−4, or patients with A. fumigatus infection and the presence of serum anti-A. fumigatus catalase antibodies, P < 10−4). Interestingly, sera from group A patients might not be differentiated from sera from group B1 or B2 patients (P = 0.06 and P = 0.20, respectively).
According to the cutoff value of 1.38, a 100% sensitivity and 97.44% specificity of this ELISA were determined (Table 2). Positive and negative predictive values were 96.15% and 100%, respectively.
TABLE 2.
Performances of the ELISA detection of anti-S. boydii catalase A1 antibodies for serodiagnosis of infections caused by the S. apiospermum complex in CF patientsa
| Serum sample result | No. of patients with ELISA result from group: |
||||
|---|---|---|---|---|---|
| A (n = 20) | B (n = 19) | B1 (n = 11) | B2 (n = 8) | C (n = 25) | |
| Positive | 0 | 1 | 0 | 1 | 25 |
| Negative | 20 | 18 | 11 | 7 | 0 |
Sensitivity, 100%; specificity, 97.44%; positive predictive value, 96.15%; negative predictive value, 100%. Serum samples were obtained from CF patients without clinical or biological signs of fungal infections and without any fungus recovered from sputum samples (group A), with A. fumigatus the sole filamentous fungus recovered from sputum samples and with serum antibodies directed toward A. fumigatus but not S. boydii by routine procedures (group B: B1, patients without anti-A. fumigatus catalase antibodies; B2, patients with anti-A. fumigatus catalase antibodies), and CF patients colonized by species of the S. apiospermum complex and exhibiting serum antibodies directed toward S. boydii but not A. fumigatus (group C).
Finally, group C patients were classified according to the species identified within the S. apiospermum complex or the number of precipitin lines obtained by CIE using an S. boydii crude antigenic extract, and the geometric mean of anti-S. boydii catalase antibody titers was calculated for each subpopulation. The geometric mean titer was 4,810 for the total population, with geometric titers ranging from 1,600 to 12,800, without any significant difference between subpopulations.
DISCUSSION
During microbial infection, an inflammatory reaction occurs in the respiratory tract, resulting in an influx of host phagocytic cells with production of reactive oxygen species, particularly hydrogen peroxide. Catalases are enzymes involved in the detoxification of the hydrogen peroxide, and therefore they are considered virulence factors, allowing the pathogen to escape the host immune response.
Here, we showed that S. boydii produces three mycelial catalases, i.e., A1, A2, and A2′, the first exhibiting high similarity to A. fumigatus Cat1, which is known as a valuable diagnostic marker for aspergillosis (25, 27). Purified catalase A1 was seen on SDS-PAGE as a single polypeptide chain of 82 kDa under reducing or nonreducing conditions, whereas gel filtration suggested a molecular mass of 460 kDa for the native protein. Likewise, affinity chromatography on immobilized ConA, as well as Western blotting experiments, demonstrated the glycosylation of the enzyme. Together, these results suggest that catalase A1 is a tetrameric protein consisting of four 82-kDa glycosylated subunits, structural features that are similar to those of A. fumigatus Cat1, which differs from catalase A1 by the 90-kDa molecular size of its subunits (27). Likewise, Aspergillus niger produces a 385-kDa catalase called CatR, made of four identical 97-kDa subunits (32), and Aspergillus nidulans produces a 360-kDa catalase called CatB, consisting of four identical 90-kDa subunits (33).
The apparent molecular mass of 82 kDa estimated by SDS-PAGE and the lack of effect of β-mercaptoethanol suggest the absence of intercatenary or intracatenary disulfide bonds. Interestingly, no cysteine residues were found in the amino acid sequence of A. nidulans CatB (33). In addition, the pI of S. boydii catalase A1 was in the range of 4.1 to 4.3. Previously characterized fungal catalases have a predicted pI ranging from 4.8 (CatB from A. nidulans) to 7.0 (Cta1p from Saccharomyces cerevisiae) (34, 35). Thus, S. boydii catalase A1 is one of the most acidic fungal catalases known so far.
Some biochemical properties of the enzyme were also evaluated, including susceptibility to different catalase inhibitors and the presence of an associated peroxidase activity. Our results are consistent with those obtained for the atypical catalases CatR from A. niger and Cat1 from A. fumigatus, which retain about 70% of their activity after ethanol-chloroform treatment and are quite resistant to SDS treatment (27, 32). Moreover, contrary to the results obtained with A. fumigatus mycelial extract, we did not find any catalase peroxidase in S. boydii mycelial extract, and catalase A1 in particular did not exhibit peroxidase activity. Consequently, S. boydii catalase A1 can be classified in clade 2 of the catalase phylogenetic tree (36, 37), which corresponds to the so-called atypical monofunctional catalases characterized by large subunits, a broad pH range, susceptibility to 3-AT, and resistance to SDS and ethanol-chloroform (38), like Escherichia coli HP-II catalase (39), A. niger CatR (40), and Neurospora crassa Cat1 (41). Moreover, detection of catalase A1 in the culture supernatant demonstrates its secretion in the environment, therefore indicating that it belongs to clade B of fungal catalases, which comprise secreted monofunctional catalases (42, 43).
In CF, a major concern regarding the clinical relevance of the isolation of molds from respiratory secretions (44) remains. Recently, by combining the results of several biological tests, including a sputum real-time Aspergillus PCR, sputum galactomannan, total serum IgE level, and specific serum IgE and IgG levels, Baxter et al. (45) highlighted the importance of a specific IgG for diagnosis of an Aspergillus respiratory infection in A. fumigatus-colonized CF patients. Besides allergic bronchopulmonary aspergillosis (ABPA) and sensitization, which are characterized by an elevated total serum IgE titer and the presence of serum-specific anti-A. fumigatus IgE, the presence of serum-specific anti-A. fumigatus IgG allows the differentiation between noninfected patients and patients with Aspergillus bronchitis. Currently, CIE is the unique method for detection of serum antibodies against species of the S. apiospermum complex (8). However, there are currently no antigenic extracts commercially available for this serodiagnosis, which is performed only in a few specialized laboratories using nonstandardized homemade antigenic extracts. In addition, the numerous proteins and polysaccharides shared between molds may lead to immune cross-reactions, particularly between A. fumigatus and Scedosporium species, which are the most common molds colonizing/infecting CF patients, and therefore to inaccurate interpretation of positive serological results.
Serum anti-catalase antibodies have been known as valuable markers for serodiagnosis of Aspergillus infections since the work of Tran van Ky et al. (46), and this was confirmed during the past decade using recombinant proteins. Several recombinant antigens were compared in enzyme-linked immunosorbent assays by Sarfati et al. (25), and recombinant catalase showed a high potential in the serodiagnosis of all forms of aspergillosis in both immunocompetent and immunocompromised patients. In addition, regarding patients with CF, the detection of anti-A. fumigatus catalase antibodies has been shown to be associated with a clinical or functional deterioration (47). Because of this and considering the high similarity between the biochemical products of A. fumigatus Cat1 and S. boydii catalase A1, we investigated the potential application of catalase A1 for specific antibody detection in CF patients.
Sera from CF patients classified according to mycological and serological results were compared by ELISA. Our results showed 100% sensitivity and a very high specificity (97.44%). Patients infected by the S. apiospermum species complex were clearly differentiated from noninfected patients (without any filamentous fungus recovered from respiratory secretions and without serum antibodies directed toward A. fumigatus or the S. apiospermum complex). Likewise, they were easily differentiated from patients infected by A. fumigatus (recovery of A. fumigatus but no Scedosporium species from respiratory secretions, the presence of serum anti-A. fumigatus IgG, and a negative response by CIE using an S. boydii mycelial extract). Only one of these patients was positive by an ELISA with S. boydii purified catalase A1. These results suggest that catalase A1 is a good candidate for the development of an immunoassay for serodiagnosis of infections caused by the S. apiospermum complex in CF patients.
No differences were observed in the antibody titer with the causative species (i.e., S. boydii or S. apiospermum), indicating that S. boydii purified catalase A1 may be used to detect infections caused by, at least, the two major species within the S. apiospermum complex. Due to the very low frequency of the other species of the complex in our center, a multicenter study is needed to investigate the interest of this serological method for patients colonized by S. aurantiacum or S. minutisporum. In addition, no relationship was observed between the antibody titer and the number of precipitin lines by CIE, which is not surprising since a purified enzyme was used here as an antigen instead of a mixture of proteins and polysaccharides. Nevertheless, the positive reaction observed with all CIE-positive sera also suggests that catalase A1 is a major antigen.
Although serum anti-catalase antibodies have long been reported in A. fumigatus as diagnostic markers of Aspergillus infections, specificity toward other fungal respiratory infections in the CF context has not been investigated. Here, we show that even if catalases are shared by all oxygen-tolerating organisms, there are enough epitope differences to develop an efficient, sensitive, and specific serological test. Due to the limitations of our purification procedure, which is time-consuming, and the small amounts of catalases in the mycelial extracts, the cloning and sequencing of the catalase A1-encoding gene are currently being performed in order to produce a recombinant protein which will be used to develop a standardized serological test for diagnosis of infections caused by the S. apiospermum complex.
ACKNOWLEDGMENT
All authors are members of the ECMM (European Confederation of Medical Mycology)/ISHAM (International Society for Human and Animal Mycology) Fungal Respiratory Infections in Cystic Fibrosis (Fri-CF) working group.
REFERENCES
- 1.Gilgado F, Cano J, Gené J, Guarro J. 2005. Molecular phylogeny of the Pseudallescheria boydii species complex: proposal of two new species. J Clin Microbiol 43:4930–4942. doi: 10.1128/JCM.43.10.4930-4942.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilgado F, Cano J, Gené J, Sutton DA, Guarro J. 2008. Molecular and phenotypic data supporting distinct species statuses for Scedosporium apiospermum and Pseudallescheria boydii and the proposed new species Scedosporium dehoogii. J Clin Microbiol 46:766–771. doi: 10.1128/JCM.01122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilgado F, Gené J, Cano J, Guarro J. 2010. Heterothallism in Scedosporium apiospermum and description of its teleomorph Pseudallescheria apiosperma sp. nov. Med Mycol 48:122–128. doi: 10.3109/13693780902939695. [DOI] [PubMed] [Google Scholar]
- 4.Lackner M, de Hoog SG, Yang L, Ferreira Moreno L, Ahmed SA, Andreas F, Kaltseis J, Nagl M, Lass-Flörl C, Risslegger B, Rambach G, Speth C, Robert V, Buzina W, Chen S, Bouchara JP, Cano-Lira JF, Guarro J, Gené J, Fernández Silva F, Rosa H, Haase G, Havlicek V, Garcia-Hermoso D, Meis JF, Hagen F, Kirchmair M, Rainer J, Schwabenbauer K, Zoderer M, Meyer W, Gilgado F, Schwabenbauer K, Vicente VA, Piecková E, Regenermel M, Rath PM, Steinmann J, Wellington de Alencar X, Symoens F, Tintelnot K, Ulfig K, Velegraki A, Tortorano AM, Giraud S, Sara M, Rigler-Hohenwarter K, Hernando F, Ramirez-Garcia A, Pellón A, et al. 25 July 2014. Proposed nomenclature for Pseudallescheria, Scedosporium and related genera. Fungal Divers doi: 10.1007/s13225-014-0295-4. [DOI] [Google Scholar]
- 5.Guarro J, Kantarcioglu AS, Horré R, Rodriguez-Tudela JL, Cuenca Estrella M, Berenguer J, de Hoog GS. 2006. Scedosporium apiospermum: changing clinical spectrum of a therapy-refractory opportunist. Med Mycol 44:295–327. doi: 10.1080/13693780600752507. [DOI] [PubMed] [Google Scholar]
- 6.Cortez KJ, Roilides E, Quiroz-Telles F, Meletiadis J, Antachopoulos C, Knudsen T, Buchanan W, Milanovich J, Sutton DA, Fothergill A, Rinaldi MG, Shea YR, Zaoutis T, Kottilil S, Walsh TJ. 2008. Infections caused by Scedosporium spp. Clin Microbiol Rev 21:157–197. doi: 10.1128/CMR.00039-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kantarcioglu AS, de Hoog GS, Guarro J. 2012. Clinical characteristics and epidemiology of pulmonary pseudallescheriasis. Rev Iberoam Micol 29:1–13. doi: 10.1016/j.riam.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Cimon B, Carrère J, Vinatier JF, Chazalette JP, Chabasse D, Bouchara JP. 2000. Clinical significance of Scedosporium apiospermum in patients with cystic fibrosis. Eur J Clin Microbiol Infect Dis 19:53–56. doi: 10.1007/s100960050011. [DOI] [PubMed] [Google Scholar]
- 9.Masoud-Landgraf L, Badura A, Eber E, Feierl G, Marth E, Buzina W. 2014. Modified culture method detects a high diversity of fungal species in cystic fibrosis patients. Med Mycol 52:179–186. doi: 10.3109/13693786.2013.792438. [DOI] [PubMed] [Google Scholar]
- 10.Horré R, Marklein G. 2009. Isolation and clinical significance of Pseudallescheria and Scedosporium species. Med Mycol 47:415–421. doi: 10.1080/13693780902801259. [DOI] [PubMed] [Google Scholar]
- 11.Horré R, Marklein G, Siekmeier R, Nidermajer S, Reiffert SM. 2009. Selective isolation of Pseudallescheria and Scedosporium species from respiratory tract specimens of cystic fibrosis patients. Respiration 77:320–324. doi: 10.1159/000167419. [DOI] [PubMed] [Google Scholar]
- 12.Harun A, Gilgado F, Chen SC, Meyer W. 2010. Abundance of Pseudallescheria/Scedosporium species in the Australian urban environment suggests a possible source for scedosporiosis including the colonization of airways in cystic fibrosis. Med Mycol 48(Suppl 1):S70–S76. doi: 10.3109/13693786.2010.515254. [DOI] [PubMed] [Google Scholar]
- 13.Vázquez-Tsuji O, Campos Rivera T, Rondán Zárate A, Mirabal García M. 2006. Endobronchitis by Scedosporium apiospermum in a child with cystic fibrosis. Rev Iberoam Micol 23:245–248. doi: 10.1016/S1130-1406(06)70054-7. [DOI] [PubMed] [Google Scholar]
- 14.Symoens F, Knoop C, Schrooyen M, Denis O, Estenne M, Nolard N, Jacobs F. 2006. Disseminated Scedosporium apiospermum infection in a cystic fibrosis patient after double-lung transplantation. J Heart Lung Transplant 25:603–607. doi: 10.1016/j.healun.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 15.Morio F, Horeau-Langlard D, Gay-Andrieu F, Talarmin JP, Haloun A, Treilhaud M, Despins P, Jossic F, Nourry L, Danner-Boucher I, Pattier S, Bouchara JP, Le Pape P, Miègeville M. 2010. Disseminated Scedosporium/Pseudallescheria infection after double-lung transplantation in patients with cystic fibrosis. J Clin Microbiol 48:1978–1982. doi: 10.1128/JCM.01840-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borghi E, Iatta R, Manca A, Montagna MT, Morace G. 2010. Chronic airway colonization by Scedosporium apiospermum with a fatal outcome in a patient with cystic fibrosis. Med Mycol 48(Suppl 1):S108–S113. doi: 10.3109/13693786.2010.504239. [DOI] [PubMed] [Google Scholar]
- 17.Hirschi S, Letscher-Bru V, Pottecher J, Lannes B, Jeung MY, Degot T, Santelmo N, Sabou AM, Herbrecht R, Kessler R. 2012. Disseminated Trichosporon mycotoxinivorans, Aspergillus fumigatus and Scedosporium apiospermum co-infection after lung and liver transplantation in a cystic fibrosis patient. J Clin Microbiol 50:4168–4170. doi: 10.1128/JCM.01928-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thornton CR. 2009. Tracking the emerging human pathogen Pseudallescheria boydii by using highly specific monoclonal antibodies. Clin Vaccine Immunol 16:756–764. doi: 10.1128/CVI.00061-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borman AM, Palmer MD, Delhaes L, Carrère J, Favennec L, Ranque S, Gangneux JP, Horré R, Bouchara JP. 2010. Lack of standardization in the procedures for mycological examination of sputum samples from CF patients: a possible cause for variations in the prevalence of filamentous fungi. Med Mycol 48(Suppl 1):S88–S97. doi: 10.3109/13693786.2010.511287. [DOI] [PubMed] [Google Scholar]
- 20.Castelli MV, Buitrago MJ, Bernal-Martinez L, Gomez-Lopez A, Rodriguez-Tudela JL, Cuenca-Estrella M. 2008. Development and validation of a quantitative PCR assay for diagnosis of scedosporiosis. J Clin Microbiol 46:3412–3416. doi: 10.1128/JCM.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouchara JP, Hsieh HY, Croquefer S, Barton R, Marchais V, Pihet M, Chang TC. 2009. Development of an oligonucleotide array for direct detection of fungi in sputum samples from patients with cystic fibrosis. J Clin Microbiol 47:142–152. doi: 10.1128/JCM.01668-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu Q, van den Ende AH, de Hoog GS, Li R, Accoceberry I, Durand-Joly I, Bouchara JP, Hernandez F, Delhaès L. 2011. Reverse line blot hybridisation screening of Pseudallescheria/Scedosporium species in patients with cystic fibrosis. Mycoses 54(Suppl 3):5–11. doi: 10.1111/j.1439-0507.2011.02108.x. [DOI] [PubMed] [Google Scholar]
- 23.Harun A, Blyth CC, Gilgado F, Middleton P, Chen SCA, Meyer W. 2011. Development and validation of a multiplex PCR for detection of Scedosporium spp. in respiratory tract specimens from patients with cystic fibrosis. J Clin Microbiol 49:1508–1512. doi: 10.1128/JCM.01810-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massire C, Buelow DR, Zhang SX, Lovari R, Matthews HE, Toleno DM, Ranken RR, Hall TA, Metzgar D, Sampath R, Blyn LB, Ecker DJ, Gu Z, Walsh TJ, Hayden RT. 2013. PCR followed by electrospray ionization mass spectrometry for broad-range identification of fungal pathogens. J Clin Microbiol 51:959–966. doi: 10.1128/JCM.02621-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarfati J, Monod M, Recco P, Sulahian A, Pinel C, Candolfi E, Fontaine T, Debeaupuis JP, Tabouret M, Latgé JP. 2006. Recombinant antigens as diagnostic markers for aspergillosis. Diagn Microbiol Infect Dis 55:279–291. doi: 10.1016/j.diagmicrobio.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 26.Beers RF Jr, Sizer IW. 1952. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem 195:133–140. [PubMed] [Google Scholar]
- 27.López-Medrano R, Ovejero MC, Calera JA, Puente P, Leal F. 1995. An immunodominant 90-kilodalton Aspergillus fumigatus antigen is the subunit of a catalase. Infect Immun 63:4774–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woodbury W, Spencer AK, Stahmann MA. 1971. An improved procedure using ferricyanide for detecting catalase isoenzymes. Anal Biochem 44:301–305. doi: 10.1016/0003-2697(71)90375-7. [DOI] [PubMed] [Google Scholar]
- 29.Wayne LG, Diaz GA. 1986. A double staining method for differentiating between two classes of mycobacterial catalase in polyacrylamide electrophoresis gels. Anal Biochem 157:89–92. doi: 10.1016/0003-2697(86)90200-9. [DOI] [PubMed] [Google Scholar]
- 30.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 31.Nadler V, Goldberg I, Hochman A. 1986. Comparative study of bacterial catalases. Biochim Biophys Acta 882:234–241. doi: 10.1016/0304-4165(86)90160-1. [DOI] [Google Scholar]
- 32.Mosavi-Movahedi AA, Wilkinson AE, Jones MN. 1987. Characterization of Aspergillus niger catalase. Int J Biol Macromol 9:327–332. doi: 10.1016/0141-8130(87)90003-1. [DOI] [Google Scholar]
- 33.Calera JA, Sánchez-Weatherby J, López-Medrano R, Leal F. 2000. Distinctive properties of the catalase B of Aspergillus nidulans. FEBS Lett 475:117–120. doi: 10.1016/S0014-5793(00)01637-9. [DOI] [PubMed] [Google Scholar]
- 34.Kawasaki L, Wysong D, Diamond R, Aguirre J. 1997. Two divergent catalase genes are differentially regulated during Aspergillus nidulans development and oxidative stress. J Bacteriol 179:3284–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen G, Rapatz W, Ruis H. 1988. Sequence of the Saccharomyces cerevisiae CTA1 gene and amino acid sequence of catalase A derived from it. Eur J Biochem 176:159–163. doi: 10.1111/j.1432-1033.1988.tb14263.x. [DOI] [PubMed] [Google Scholar]
- 36.Klotz MG, Loewen PC. 2003. The molecular evolution of catalatic hydroperoxidases: evidence for multiple lateral transfer of genes between prokaryota and from bacteria into eukaryota. Mol Biol Evol 20:1098–1112. doi: 10.1093/molbev/msg129. [DOI] [PubMed] [Google Scholar]
- 37.Zamocky M, Furtmüller PG, Obinger C. 2008. Evolution of catalases from bacteria to humans. Antioxid Redox Signal 10:1527–1548. doi: 10.1089/ars.2008.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loewen PC, Klotz MG, Hassett DJ. 2000. Catalase—an “old” enzyme that continues to surprise us. ASM News 66:76–82. [Google Scholar]
- 39.Switala J, O'Neil JO, Loewen PC. 1999. Catalase HP from Escherichia coli exhibits enhanced resistance to denaturation. Biochemistry 38:3895–3901. doi: 10.1021/bi982863z. [DOI] [PubMed] [Google Scholar]
- 40.Fowler T, Rey MW, Vähä-Vahe P, Power SD, Berka RM. 1993. The catR gene encoding a catalase from Aspergillus niger primary structure and elevated expression through increased gene copy number and use of a strong promoter. Mol Microbiol 9:989–998. doi: 10.1111/j.1365-2958.1993.tb01228.x. [DOI] [PubMed] [Google Scholar]
- 41.Chary P, Natvig DO. 1989. Evidence for three differentially regulated catalase genes in Neurospora crassa: effects of oxidative stress, heat shock, and development. J Bacteriol 171:2646–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson CH, Klotz MG, York JL, Kruft V, McEwen JE. 2002. Redundancy, phylogeny and differential expression of Histoplasma capsulatum catalases. Microbiology 148:1129–1142. [DOI] [PubMed] [Google Scholar]
- 43.Giles SS, Stajich JE, Nichols C, Gerrald QD, Alspaugh JA, Dietrich F, Perfect JR. 2006. The Cryptococcus neoformans catalase gene family and its role in antioxidant defense. Eukaryot Cell 5:1447–1459. doi: 10.1128/EC.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chotirmall SH, McElvaney NG. 2014. Fungi in the cystic fibrosis lung: Bystanders or pathogens? Int J Biochem Cell Biol 52:161–173. doi: 10.1016/j.biocel.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 45.Baxter CG, Dunn G, Jones AM, Webb K, Gore R, Richardson MD, Denning DW. 2013. Novel immunologic classification of aspergillosis in adult cystic fibrosis. J Allergy Clin Immunol 132:560–566. doi: 10.1016/j.jaci.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 46.Tran van Ky P, Biguet J, Vaucelle T. 1968. Study on an antigenic fraction of Aspergillus fumigatus supporting a catalase activity. Consequence on the immunologic diagnosis of aspergillosis. Rev Immunol Ther Antimicrob 32:37–52. (In French.) [PubMed] [Google Scholar]
- 47.Schønheyder H, Jensen T, Laessøe IH, Høiby N, Koch C. 1988. Serum antibodies to Aspergillus fumigatus catalase in patients with cystic fibrosis. Eur J Clin Microbiol Infect Dis 7:40–44. doi: 10.1007/BF01962169. [DOI] [PubMed] [Google Scholar]