

Abstract
The need for novel therapeutics against Plasmodium falciparum is urgent due to recent emergence of multi-drug resistant malaria parasites. Since fatty acids are essential for both the liver and blood stages of the malarial parasite, targeting fatty acid biosynthesis is a promising strategy for combatting P. falciparum. We present a combined computational and experimental study to identify novel inhibitors of enoyl-acyl carrier protein reductase (PfENR) in the fatty acid biosynthesis pathway. A small-molecule database from ChemBridge was docked into three distinct PfENR crystal structures that provide multiple receptor conformations. Two different docking algorithms were used to generate a consensus score in order to rank possible small molecule hits. Our studies led to the identification of five low-micromolar pyrimidine dione inhibitors of PfENR.
Introduction
Malaria, caused by eurkaryotic protists of the genus Plasmodium, is currently one of the most deadly infectious diseases (1). Although there are four different species of Plasmodium known to infect humans (Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, and Plasmodium malariae), the most life threatening cases are caused by Plasmodium falciparum (2). Recently, malaria has become resistant to current treatments (3) including chloroquine, sulfadoxine/pyrimethamine, and artemisinin (4–6). As a result, the need for new antimalarial treatments is urgent.
Fatty acid synthase (FAS) is responsible for the biosynthesis of fatty acids (FAs) and is essential for the survival of P. falciparum in both the liver and blood stages (7). It was initially believed that the malaria parasite relied exclusively on exogenous fatty acids obtained from the infected organism (8, 9). This hypothesis was later refuted after the discovery of FAS machinery in the apicoplast, a non-photosynthetic organelle in the malarial parasite (10, 11). Recently, the strategy of targeting P. falciparum FAS in the asexual blood stage has been heavily debated. Yu and co-workers demonstrated that knockouts of various FAS components in P. falciparum and rodent parasite Plasmodium berghei do not inhibit blood-stage growth (12). While exogenous FAs are sufficient for membrane biogenesis in the blood stage (13), recent studies revealed that de novo biosynthesis is exclusively required in the liver stage of the malarial life cycle (12, 14). Few antimalarial drugs, including atovaquone, primaquine, and anti-folates, are effective against both the blood- and liver-stage parasites. Therefore targeting the FAS in the liver stage could serve as a valuable target for future prophylactic drugs (15, 16).
Since several antibiotics, including diazaborine (17), triclosan (18), thiolactomycin (19–21), and isoniazid (22, 23), have been used to target the FA pathway in other pathogens, other groups have similarly pursued FAS in P. falciparum (12, 24–26). P. falciparum segmented type II fatty acid synthase enzymes are structurally different from the functionally equivalent human type I FAS megasynthase, making FAS an promising target to combat malaria (27). The human type I FAS is a single, multi-domain protein, whereas P. falciparum contains a type II FAS comprised of discrete enzymes encoded by separate genes. Thus, antimalarial drugs targeting enzymes in the P. falciparum FAS would potentially be less toxic for humans due to the structural difference between the type II FAS in P. falciparum and type I FAS.
One possible strategy for disrupting FAS is to target the enoyl-acyl carrier protein (ACP) reductase (ENR, also frequently referred to as FabI), the rate-limiting enzyme in FA biosynthesis and the target of all known bacterial FAS inhibitors (18). ENR is responsible for the reduction of trans-2-acyl-ACP to acyl-ACP (Figure 1) (28, 29) using NADH as a cofactor. Isoniazid, an FDA-approved drug for the treatment for Mycobacterium tuberculosis targeting ENR (22), has been applied towards antimalarial ENR drug-discovery projects (12, 24–26), supporting this approach. Other studies have also focused on these targets (30).
Figure 1.
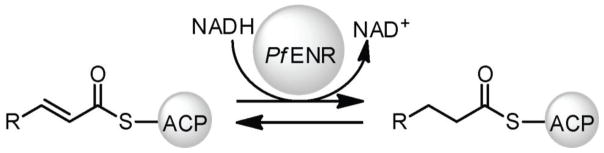
Reaction scheme for the reduction of trans-2-acyl-ACP to acyl-ACP by PfENR
Triclosan (TCL), a trichlorinated biphenyl ether (Figure 2, compound 1), was discovered as an inhibitor of Escherichia coli ENR (18), and was later found to similarly target PfENR (12, 24–26). Structural studies suggest that TCL binds PfENR at its acyl substrate-binding pocket (31), where it forms a stable ternary complex with the protein and the oxidized cofactor (NAD+). Additionally, several TCL-analogues have demonstrated inhibition of PfENR, and co-crystal structures of PfENR with these analogues (Figure 2, compounds 2-6) display similar binding conformations to the TCL-PfENR complex (32). The various pocket poses with TCL and its analogues bound to the acyl substrate-binding pocket offer an excellent starting point for structure-based drug design studies in PfENR.
Figure 2.
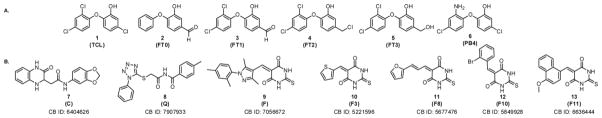
Structures of (A) known PfENR inhibitors docked into PfENR for verifying docking programs and (B) lead compounds inhibiting PfENR. The ChemBridge (CB) identification number is listed under the lead compounds.
Structure-based drug design is a powerful approach in drug discovery when the atomic structure of the target protein is known (33). Comprehensive computational and experimental studies elucidating both the structural (27, 32) and dynamic (34–38) nature of PfENR have been performed, with applications to drug discovery (39–42). With the advancement in computational and experimental technology, we can rationally and efficiently screen for novel drugs with a higher hit rate than the traditional and more expensive high-throughput library screens. In this study, we present a joint computational and experimental study that employs multiple PfENR crystal structures to account for receptor flexibility in the in silico screening process. Additionally a consensus of two independent docking programs, AutoDock Vina and Glide, was used to refine the experimental screens and reduce false positives. This protocol allowed us to identify new low-micromolar PfENR inhibitors that are reported for the first time from the ChemBridge small molecule repository.
Material and Methods
Preparation of ENR crystal structures
Systems based on three different crystal structures were prepared for docking simulations: PDB entries 1UH5 (27), 3LSY (32), and 3LT0 (32). The resolution of the structures is 2.2 Å, 2.9 Å and 2.0 Å, respectively. These crystal structures were chosen based on scoring of known inhibitors after docking into 17 PfENR crystal structures (unpublished data). 1UH5 contains triclosan (TCL; Figure 2, compound 1) and 3LSY and 3LT0 contain triclosan variants FT0 and FT1, respectively (Figure 2, compounds 2 and 3). All three structures contained an NAD+ cofactor. The missing substrate-binding loop residues (325–366) were built into all three crystal structures using Swiss model server (43) in order to have a complete protein model for MD simulations. For the AutoDock Vina screens, pdb2pqr (44, 45) was used to add hydrogen atoms to the first subunits of the crystal structure receptors. Ligands and cofactors were removed from the AutoDock Vina receptor files. Removal of the cofactor allowed us to probe binding to the cofactor binding site in addition to the substrate binding site. The AutoDock script (46) prepare_receptor4.py was used to prepare the final receptor pdbqt files. For the Glide screens, the receptors were prepared using the tools provided in the Maestro Protein Preparation Wizard (47) and the Glide Receptor Grid Generation (48, 49). Ligands 1 (TCL), 2, (FT0), and 3 (FT1) were removed from the Glide receptor grid files, while the cofactors were left in the binding site.
Docking of known ENR inhibitors
We evaluated the ability of both docking programs to dock triclosan (1) and five of its analogues (2-6) into the three crystal structures (27, 32) (Figure 3). The known inhibitors used in this analysis (compounds 1-6) are shown in Figure 2. Inhibitor sdf files from the following PDB entries were extracted: 1UH5 (TCL.sdf), 3LSY (FT0.sdf), 3LT0 (FT1.sdf), 3LT1 (FT2.sdf), 3LT2 (FT3.sdf), and 3LT3 (FB4.sdf). The LigPrep computer program (Schrödinger: LigPrep, Schrödinger, LLC, New York, NY, 2010) was used to add missing hydrogen atoms, generate all possible ionization states, as well as tautomers for compounds 1-6. The AutoDock script (46) prepare_ligand4.py was used to prepare the inhibitor pdbqt files for the AutoDock Vina screens. Due to the lack of the cofactor, the AutoDock Vina results were only analyzed in terms of deviation from known bound pose.
Figure 3.

Virtual screen workflow for identifying in silico small molecule hits for in vitro assay. (A) Three established PfENR structures were chosen for docking (PDB: 1UH5, 3LSY, 3LT0). (B) The docking protocols were evaluated for their ability to dock known inhibitors of PfENR. Inhibitors and cofactors were removed from the AutoDock Vina receptor files, while only inhibitors were removed from the Glide receptor grid files. The inhibitors were re-docked into these prepared structures and evaluated for their ability to dock known inhibitors by calculating the RMSD. (C) After establishing a validated docking program, 171,791 compounds from the ChemBridge EXPRESS-Pick compound collection were docked into the prepared ENR structures and a consensus in silico small molecule hit set was generated for in vitro testing.
To evaluate the ability of the docking programs to rank binders better than non-binders, a ROC-AUC (Receiver Operating Characteristics-Area Under the Curve) analysis (50) was performed on the three crystal structures. For this, we used Glide to dock the six known inhibitors (compounds 1-6) and the Schrödinger decoy library (1000 compounds with average molecular mass of ~400 Daltons (48, 49)). All compounds in the decoy set were assumed to be inactive. All 1006 compounds were docked into all three receptor structures. The compounds were ranked by their Glide XP docking scores, and AUC values were calculated from the ROC analysis.
Virtual screen of ChemBridge database
The virtual screen was performed using the ChemBridge EXPRESS-Pick compound collection (September 2012 update, http://www.chembridge.com/screening_libraries/). Compounds not available in sufficient quantities for reordering were removed from the dataset. The ChemBridge EXPRESS-Pick Collection contained 448,532 compounds which were selected by ChemBridge using novelty, diversity, drug-like properties, and chemical structure analyses as criteria. It covers a broad area of chemical space, offering diverse classes of compounds with analogues to support initial SAR work. The OpenEye filter program was used to remove undesirable compounds, with particular focus on removal of unwanted functional groups, reactive compounds and unwanted protecting group compounds. Filter default settings were used. Deviations from the default values included: compound molecular weight between 250 Da and 460 Da, removal of salts, duplicate structures and compounds with 3 or more (out of 4) Lipinski violations (51), removal of known aggregators (retention of “OE predicted aggregators”), predicted solubility cut-off “poorly” (or worse), and less than 10 rotatable bonds. As a result, 101,692 small molecules were obtained after applying these filters. Ligands were then prepared using LigPrep, adding missing hydrogen atoms, generating all possible ionization states, and tautomers. The final set used for virtual screening contained 171,791 compounds. Docking simulations were performed with both AutoDock Vina (52) as well as Glide (48, 49, 53). The AutoDock script (46) prepare_ligand4.py was used to prepare the ligand pdbqt files for the AutoDock Vina screens. Vina docking was performed into empty active sites, i.e. ligands and cofactors were removed from the AutoDock Vina receptor files. A docking grid of size 18.0 Å × 18.0 Å × 18.0 Å, centered on the position of the crystallographic ligand in the binding site, was used for docking. For Glide docking, the ligands were prepared using LigPrep. Glide docking was performed into cofactor-occupied active sites, i.e. only ligands had been removed from the Glide receptor files. The different docking protocols correspond to strategies of finding inhibitors that occupy either the crotonyl-CoA substrate binding site (in the case of Glide) or also the NAD+ cofactor binding site (in the case of AutoDock Vina). These different strategies were pursued to improve the likelihood of finding novel ENR inhibitors. The individual AutoDock Vina and Glide rankings were combined to form a consensus list of compounds that scored well with both methods, a method that has been reported previously to be successful in virtual screening (54).
Analogue Ligand-based Search from Lead Compound
Based on the experimental hit (Figure 2, compound 9), a ligand-based search of the ChemBridge database was performed with the goal of testing similar compounds for inhibition of PfENR. The sdf file connected to ChemBridge ID 7056672 (compound 9) was downloaded. We used Canvas (55, 56) for a substructure-based search on the key pyrimidine dione pharmacophore of 9. The resulting 2,545 compounds were ranked by drug-like properties. Fifteen compounds from that list were chosen for ordering and testing.
PfENR expression and purification
PfENR was subcloned into a pET28a plasmid and transformed into E. coli BL21 cells, which has been previously described (57). Cells were grown at 37 °C to a final OD of 1.0 without the addition of IPTG in terrific broth medium containing 100 mg/L kanamycin sulfate. The pellet was resuspended in lysis buffer (20 mM Tris/HCl buffer with 150 mM NaCl, pH 7.4), and supplemented with 0.1 mg/mL lysozyme (Worthington Biochemical Corp), 5 μg/mL DNAse I (Sigma), and 5 μg/mL RNAse (Worthington Biochemical Corp.). The supernatant was batch-bound with Ni-NTA resin (Qiagen) for an hour at 4 °C and washed with 20 mM Tris/HCl with 150 mM NaCl, pH 7.4. The protein was eluted from the resin using a step gradient of lysis buffer supplemented with 60 to 300 mM imidazole. PfENR was further purified via size exclusion chromatography using a HiPrep Sephacryl 16/60 S-200 (GE Healthcare) equilibrated with lysis buffer. PfENR was concentrated with a 10-kDa Amicon spin filter (Milipore) and stored at −20 C in 40% glycerol, 20 mM Tris/HCl buffer with 150 mM NaCl.
Validation of the in silico compounds by continuous assay
One-hundred and twenty small molecules identified as hits by our in silico analysis were purchased through ChemBridge and tested for PfENR inhibition. All ChemBridge compounds had greater than 90% purity as verified by spectra analysis. The final 100 μL reaction volume contained 50 μM NAD+, 20 mM Tris/HCl buffer, 150 mM NaCl (pH 7.4), 200 μM crotonyl-CoA, 100 μM NADH, and 0.05 μM ENR. Each inhibitor was evaluated for potency at a final concentration of 50 μM and took 5% of the reaction volume after dissolving in DMSO. PfENR was preincubated with NAD+ and inhibitor at 27 °C for 45 minutes and initiated with a final concentration of 200 μM crotonyl-CoA and 100 μM NADH cocktail following this preincubation period. PfENR inhibition was observed at 27 °C by monitoring the consumption of NADH via change in absorbance at 340 nm (εNADH = 6.22 mM−1 cm−1) (Perkin Elmer HTS 7000 Plus Bio Assay Reader) and repeated in triplicate.
PfENR inhibition assay
To evaluate the IC50 of lead compounds 7-13, NADH consumption was measured at varying concentrations in triplicate. The final 100 μL reaction volume contained 50 μM NAD+, 20 mM Tris/HCl buffer, 150 mM NaCl (pH 7.4), 200 μM crotonyl-CoA, 100 μM NADH, and 0.05 μM ENR. ENR was preincubated at 27 °C for 45 minutes with a final concentration of 50 μM NAD+, 20 mM Tris/HCl buffer, 150 mM NaCl (pH 7.4), and 0.9–75 μM inhibitor. Following this preincubation period, the reaction was initiated with a final concentration of 200 μM crotonyl-CoA and 100 μM NADH.
Results and Discussion
Docking of known ENR inhibitors: Glide yields high AUC values in ROC analysis
The protocol for the docking of known inhibitors mirrored the protocol of the virtual screen. Glide receptors contained cofactors while AutoDock Vina receptors had both the ligands and cofactors removed (Figure 3). Since the AutoDock Vina receptors have been prepared without the cofactor present, it is not expected to see good agreement between docked and known poses or even enrichment with respect to a set of decoys. Thus no ROC-AUC analysis was performed using AutoDock Vina. As expected, docked poses are found scattered all over the active site cavity because no cofactor is present to restrain the docked pose to the substrate part of the active site. However, the crystallographic TCL pose is recovered by Vina docking despite the absence of the cofactor. Figure 4A shows an overlay of one of the TCL docked poses with its crystallographic position with an RMSD of 0.87 Å and supports our AutoDock Vina methodology. A much more detailed analysis of the Glide docking results was possible since its receptors contained the crystallographic cofactor coordinates. Glide is thus expected to rank the known inhibitors high compared to the decoys. A ROC-AUC analysis yielded AUCs of 0.90, 0.92 and 0.92 for docking of 1006 compounds (six known inhibitors and 1000 decoys) into 1UH5, 3LSY and 3LT0 respectively. As a representative example, Figure 4B shows the ROC plot for 3LSY. These results suggest that Glide can differentiate actives from inactives in the presence of the cofactor.
Figure 4.
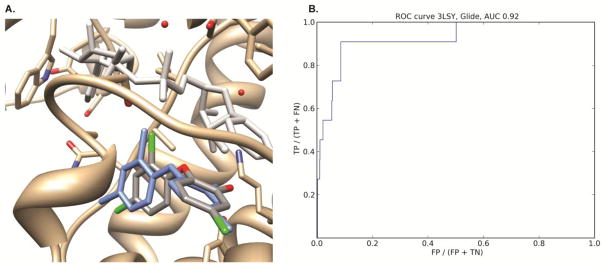
(A) AutoDock Vina docked pose of TCL into 1UH5. The ENR protein is displayed in gold, the crystallographic NAD cofactor in light grey and the crystallographic TCL inhibitor in blue. The Vina docked pose is colored according to element type. The cofactor was not present during the AutoDock Vina docking. The RMS deviation between the docked pose and the crystallographic TCL inhibitor is 0.87 Å. (B) ROC curve of docking six known actives and 1000 decoys into the 3LSY crystal structure. The area under the curve is 0.92.
In silico screen and compound selection
Three crystal structures of PfENR bound to triclosan (1) or triclosan variants (2-6) were used as receptors for the relaxed complex scheme docking protocol using a combination of AutoDock Vina and Glide. The ChemBridge Database was used as screening library. Both the AutoDock Vina and Glide XP docking results were ranked individually according to the predicted docking score. A consensus list of compounds containing high docking scores was generated from the Vina and Glide lists. The top 90 compounds from this list, as well as the top 15 scoring compounds from the individual AutoDock Vina and Glide lists, respectively, were selected for experimental investigation.
Confirmation of PfENR inhibition via enzymatic assays
We used a continuous assay to evaluate the inhibition of the 120 compounds recommended by our in silico studies. Without inhibitor, consumption of NADH was observed at 340 nm and a change in absorbance is observed and normalized to 1 (bar labeled DMSO in Figure 5). As a negative control, we tested cerulenin (CR), a commercially available inhibitor of β-ketoacyl-acyl-carrier protein synthase (KS-ACP-II) (Figure 5) (58). No inhibition of PfENR was observed, similar to the results obtained in the presence of DMSO alone. As a positive control, we tested TCL (1), a known PfENR inhibitor, and observed no change in NADH consumption indicative of ENR inhibition (Figure 5) (59). The inhibition of 120 compounds (ChemBridge) from the in silico screen at a final concentration of 50 μM (data not reported) were tested using this continuous assay. A change in absorbance at 340 nm below 30% inhibition was used as a criterion for the next set of inhibitor leads. The top 22 compounds from the 120 small molecule set were further evaluated for inhibition using a continuous assay. Most of the 22 compounds except compounds 7-9 exhibited a change in absorbance above 1 relative to the negative control (DMSO and CR) at a final concentration of 50 μM (Figure 5). We hypothesize that many of these compounds precipitate under the reaction conditions used resulting in a higher absorbance at 340 nm. Since TCL inhibits PfENR in the picomolar to nanomolar range (32, 60), we decided to test inhibitor concentrations that are in the mid-micromolar range for PfENR.
Figure 5.

Candidate small molecules screened in triplicate for inhibition of PfENR at a final concentration of 50 μM. (A) A set of small molecules taken from the 120 small molecule library tested for inhibition of PfENR against a known inhibitor for ENR, triclosan (TCL, 1) and commercially available inhibitor of β-ketoacyl-acyl-carrier protein synthase, cerulenin (CR). (B) Analogues of compound 9 (F) were tested for inhibition of PfENR at a final concentration of 50 μM.
Compounds 7-9 were further evaluated for their ability to inhibit PfENR by examining their IC50. Due to solubility issues, an IC50 curve was not attained for compounds 7-8. On the contrary, compound 9 was observed to have an average IC50 of 4.6 μM (Figure 6). IC50 measurements were not attempted for all other compounds due to lack of sufficient inhibition at 50 μM.
Figure 6.
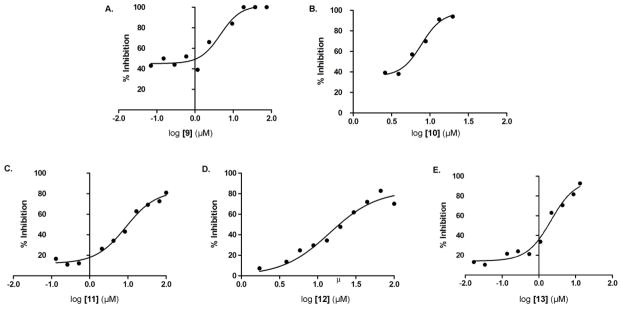
Average IC50 curves for PfENR inhibition repeated in triplicate for lead compound 9 and its analogues 10-13. The average IC50 calculated for compounds 9-13 are 4.6, 7.8, 8.8, 14.3 and 2.1 μM respectively.
To further evaluate the chemical moieties important for PfENR inhibition, analogues of 9 (F) were tested (Figure 5B). A continuous assay was used to evaluate the relative inhibition for each analogue of 9 (F). Compounds 10-13 exhibited minimal change in absorbance at 340 nm indicative of inhibition of PfENR. A change in absorbance at 340 nm below 0.7 was used as a criterion to further determine the IC50. The average IC50 was calculated for the 4 analogues (Figure 6). The average IC50 calculated for compounds 10-13 was 7.8, 8.8, 14.3 and 2.1 μM, respectively.
It is also interesting to note that ChemBridge compound 9 (F) only scored well within the AutoDock Vina protocol suggesting that it may at least in part occupy the cofactor binding site. Since different docking protocols were used for AutoDock Vina (no cofactor present during docking) and Glide (cofactor present during docking) this may highlight the value of targeting the cofactor part of the active site as opposed to solely focusing on competitive substrate binding site inhibitors.
Conclusion
A number of leads for PfENR inhibitors have been identified in a relaxed complex scheme virtual screen of the active site. The most potent leads, 9-13, were all pyrimidine diones with IC50 values in the ~1–15 μM range. All leads obey Lipinski’s rule of five, making them interesting drug-like chemical starting points. These results highlight the potential of combined computational and biochemical work to discover novel binding scaffolds. How these compounds bind to PfENR remains to be determined. Based on their high scoring performance in the AutoDock Vina docking protocol we speculate that 9-13 partly occupy the cofactor binding site, thus interacting with cofactor binding residues. Supplemental Figure 1 shows the best-predicted docking pose for compounds 9-13, obtained when the compound was docked into the 1UH5 structure. Further lead optimization will focus on elucidating the binding interactions by X-ray crystallography and development of nanomolar pyrimidine dione inhibitors. As a result of our current work, a drug discovery effort has been launched that may serve as a critical next step for structure-based drug design and optimization.
Supplementary Material
Acknowledgments
We would like to thank Dr. Jacob Durrant for helpful discussions and careful reading of the manuscript. Additionally we would like to thank Dr. Victoria Feher for her help with ordering the ChemBridge compounds. This work was supported by the National Institutes of Health NIH R21A090213, NIH R01GM094924, and NIH R01GM095970 (MDB), and the National Science Foundation, the National Institutes of Health, the Howard Hughes Medical Institute, the National Biomedical Computation Resource, and the NSF Supercomputer Centers (JAM). Computational resources were supported, in part, by the National Science Foundation grant PHY-0822283, the Center for Theoretical Biological Physics. S. L. was supported by the American Heart Association (12POST11570005) and the Center for Theoretical Biological Physics. L.T. was supported by the San Diego Match Fellowship and the University of California Interfaces Training Grant.
We would like to acknowledge the UCSD Chemistry & Biochemistry Mass Spectrometry Facility; Professor Pieter Dorrestein and Dr. Jane Yang for providing the P. falciparum ENR plasmid; Dr. James La Clair, Dr. Joris Beld, and Christopher Vickery for manuscript design and input; and Dr. Nicolas Kosa for enzymatic assay and protein expression consulting.
References
- 1.Bourzac K. Infectious disease: Beating the big three. Nature. 2014;507:S4–7. doi: 10.1038/507s4a. [DOI] [PubMed] [Google Scholar]
- 2.Rich SM, Leendertz FH, Xu G, LeBreton M, Djoko CF, Aminake MN, Takang EE, Diffo JL, Pike BL, Rosenthal BM, Formenty P, Boesch C, Ayala FJ, Wolfe ND. The origin of malignant malaria. Proc Natl Acad Sci U S A. 2009;106:14902–7. doi: 10.1073/pnas.0907740106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tschan S, Kremsner PG, Mordmuller B. Emerging drugs for malaria. Expert opinion on emerging drugs. 2012;17:319–33. doi: 10.1517/14728214.2012.702754. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien C, Henrich PP, Passi N, Fidock DA. Recent clinical and molecular insights into emerging artemisinin resistance in Plasmodium falciparum. Curr Opin Infect Dis. 2011;24:570–7. doi: 10.1097/QCO.0b013e32834cd3ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nayyar GM, Breman JG, Newton PN, Herrington J. Poor-quality antimalarial drugs in southeast Asia and sub-Saharan Africa. The Lancet infectious diseases. 2012;12:488–96. doi: 10.1016/S1473-3099(12)70064-6. [DOI] [PubMed] [Google Scholar]
- 6.Klonis N, Crespo-Ortiz MP, Bottova I, Abu-Bakar N, Kenny S, Rosenthal PJ, Tilley L. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci U S A. 2011;108:11405–10. doi: 10.1073/pnas.1104063108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schrader FC, Glinca S, Sattler JM, Dahse HM, Afanador GA, Prigge ST, Lanzer M, Mueller AK, Klebe G, Schlitzer M. Novel type II fatty acid biosynthesis (FAS II) inhibitors as multistage antimalarial agents. ChemMedChem. 2013;8:442–61. doi: 10.1002/cmdc.201200407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elabbadi N, Ancelin ML, Vial HJ. Use of Radioactive Ethanolamine Incorporation into Phospholipids to Assess Invitro Antimalarial Activity by the Semiautomated Microdilution Technique. Antimicrob Agents Chemother. 1992;36:50–5. doi: 10.1128/aac.36.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vial HJ, Thuet MJ, Philippot JR. Phospholipid biosynthesis in synchronous Plasmodium falciparum cultures. The Journal of protozoology. 1982;29:258–63. doi: 10.1111/j.1550-7408.1982.tb04023.x. [DOI] [PubMed] [Google Scholar]
- 10.Ralph SA, van Dooren GG, Waller RF, Crawford MJ, Fraunholz MJ, Foth BJ, Tonkin CJ, Roos DS, McFadden GI. Tropical infectious diseases: metabolic maps and functions of the Plasmodium falciparum apicoplast. Nature reviews Microbiology. 2004;2:203–16. doi: 10.1038/nrmicro843. [DOI] [PubMed] [Google Scholar]
- 11.Waller RF, Keeling PJ, Donald RG, Striepen B, Handman E, Lang-Unnasch N, Cowman AF, Besra GS, Roos DS, McFadden GI. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc Natl Acad Sci U S A. 1998;95:12352–7. doi: 10.1073/pnas.95.21.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu M, Kumar TR, Nkrumah LJ, Coppi A, Retzlaff S, Li CD, et al. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell host & microbe. 2008;4:567–78. doi: 10.1016/j.chom.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qidwai T, Khan F. Antimalarial drugs and drug targets specific to fatty acid metabolic pathway of Plasmodium falciparum. Chem Biol Drug Des. 2012;80:155–72. doi: 10.1111/j.1747-0285.2012.01389.x. [DOI] [PubMed] [Google Scholar]
- 14.Vaughan AM, O’Neill MT, Tarun AS, Camargo N, Phuong TM, Aly AS, Cowman AF, Kappe SH. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cellular microbiology. 2009;11:506–20. doi: 10.1111/j.1462-5822.2008.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Derbyshire ER, Mota MM, Clardy J. The next opportunity in anti-malaria drug discovery: the liver stage. PLoS Pathog. 2011;7:e1002178. doi: 10.1371/journal.ppat.1002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazier D, Renia L, Snounou G. A pre-emptive strike against malaria’s stealthy hepatic forms. Nat Rev Drug Discov. 2009;8:854–64. doi: 10.1038/nrd2960. [DOI] [PubMed] [Google Scholar]
- 17.Roujeinikova A, Sedelnikova S, de Boer GJ, Stuitje AR, Slabas AR, Rafferty JB, Rice DW. Inhibitor binding studies on enoyl reductase reveal conformational changes related to substrate recognition. J Biol Chem. 1999;274:30811–7. doi: 10.1074/jbc.274.43.30811. [DOI] [PubMed] [Google Scholar]
- 18.Heath RJ, Yu YT, Shapiro MA, Olson E, Rock CO. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J Biol Chem. 1998;273:30316–20. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi T, Yamamoto O, Sasaki H, Kawaguchi A, Okazaki H. Mechanism of action of the antibiotic thiolactomycin inhibition of fatty acid synthesis of Escherichia coli. Biochem Biophys Res Commun. 1983;115:1108–13. doi: 10.1016/s0006-291x(83)80050-3. [DOI] [PubMed] [Google Scholar]
- 20.Jackowski S, Murphy CM, Cronan JE, Jr, Rock CO. Acetoacetyl-acyl carrier protein synthase. A target for the antibiotic thiolactomycin. J Biol Chem. 1989;264:7624–9. [PubMed] [Google Scholar]
- 21.Nishida I, Kawaguchi A, Yamada M. Effect of thiolactomycin on the individual enzymes of the fatty acid synthase system in Escherichia coli. J Biochem. 1986;99:1447–54. doi: 10.1093/oxfordjournals.jbchem.a135614. [DOI] [PubMed] [Google Scholar]
- 22.Baldock C, Rafferty JB, Sedelnikova SE, Baker PJ, Stuitje AR, Slabas AR, Hawkes TR, Rice DW. A mechanism of drug action revealed by structural studies of enoyl reductase. Science. 1996;274:2107–10. doi: 10.1126/science.274.5295.2107. [DOI] [PubMed] [Google Scholar]
- 23.Parikh SL, Xiao G, Tonge PJ. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry. 2000;39:7645–50. doi: 10.1021/bi0008940. [DOI] [PubMed] [Google Scholar]
- 24.Freundlich JS, Wang F, Tsai HC, Kuo M, Shieh HM, Anderson JW, et al. X-ray structural analysis of Plasmodium falciparum enoyl acyl carrier protein reductase as a pathway toward the optimization of triclosan antimalarial efficacy. J Biol Chem. 2007;282:25436–44. doi: 10.1074/jbc.M701813200. [DOI] [PubMed] [Google Scholar]
- 25.Kumar G, Parasuraman P, Sharma SK, Banerjee T, Karmodiya K, Surolia N, Surolia A. Discovery of a rhodanine class of compounds as inhibitors of Plasmodium falciparum enoyl-acyl carrier protein reductase. J Med Chem. 2007;50:2665–75. doi: 10.1021/jm061257w. [DOI] [PubMed] [Google Scholar]
- 26.McLeod R, Muench SP, Rafferty JB, Kyle DE, Mui EJ, Kirisits MJ, Mack DG, Roberts CW, Samuel BU, Lyons RE, Dorris M, Milhous WK, Rice DW. Triclosan inhibits the growth of Plasmodium falciparum and Toxoplasma gondii by inhibition of apicomplexan Fab I. International journal for parasitology. 2001;31:109–13. doi: 10.1016/s0020-7519(01)00111-4. [DOI] [PubMed] [Google Scholar]
- 27.Pidugu LS, Kapoor M, Surolia N, Surolia A, Suguna K. Structural basis for the variation in triclosan affinity to enoyl reductases. Journal of Molecular Biology. 2004;343:147–55. doi: 10.1016/j.jmb.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 28.Ward WH, Holdgate GA, Rowsell S, McLean EG, Pauptit RA, Clayton E, et al. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry. 1999;38:12514–25. doi: 10.1021/bi9907779. [DOI] [PubMed] [Google Scholar]
- 29.Oliveira JS, Vasconcelos IB, Moreira IS, Santos DS, Basso LA. Enoyl reductases as targets for the development of anti-tubercular and anti-malarial agents. Curr Drug Targets. 2007;8:399–411. doi: 10.2174/138945007780058942. [DOI] [PubMed] [Google Scholar]
- 30.Belluti F, Perozzo R, Lauciello L, Colizzi F, Kostrewa D, Bisi A, Gobbi S, Rampa A, Bolognesi ML, Recanatini M, Brun R, Scapozza L, Cavalli A. Design, synthesis, and biological and crystallographic evaluation of novel inhibitors of Plasmodium falciparum enoyl-ACP-reductase (PfFabI) J Med Chem. 2013;56:7516–26. doi: 10.1021/jm400637m. [DOI] [PubMed] [Google Scholar]
- 31.Lu X, Huang K, You Q. Enoyl acyl carrier protein reductase inhibitors: a patent review (2006 – 2010) Expert Opin Ther Pat. 2011;21:1007–22. doi: 10.1517/13543776.2011.581227. [DOI] [PubMed] [Google Scholar]
- 32.Maity K, Bhargav SP, Sankaran B, Surolia N, Surolia A, Suguna K. X-ray crystallographic analysis of the complexes of enoyl acyl carrier protein reductase of Plasmodium falciparum with triclosan variants to elucidate the importance of different functional groups in enzyme inhibition. IUBMB Life. 2010;62:467–76. doi: 10.1002/iub.327. [DOI] [PubMed] [Google Scholar]
- 33.Andricopulo AD, Salum LB, Abraham DJ. Structure–based drug design strategies in medicinal chemistry. Current topics in medicinal chemistry. 2009;9:771–90. doi: 10.2174/156802609789207127. [DOI] [PubMed] [Google Scholar]
- 34.Lee HM, Singh NJ. Understanding Drug-Protein Interactions in Escherichia coli FabI and Various FabI Inhibitor Complexes. B Korean Chem Soc. 2011;32:162–8. [Google Scholar]
- 35.Pasqualoto KFM, Ferreira MMC. Application of a receptor pruning methodology to the enoyl-ACP reductase from Escherichia coli (Fabl) Qsar Comb Sci. 2006;25:629–36. [Google Scholar]
- 36.Singh NJ, Shin D, Lee HM, Kim HT, Chang HJ, Cho JM, Kim KS, Ro S. Structural basis of triclosan resistance. Journal of Structural Biology. 2011;174:173–9. doi: 10.1016/j.jsb.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 37.Yang L, Liu Y, Sternberg C, Molin S. Evaluation of enoyl-acyl carrier protein reductase inhibitors as Pseudomonas aeruginosa quorum-quenching reagents. Molecules. 2010;15:780–92. doi: 10.3390/molecules15020780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindert S, McCammon JA. Dynamics of Plasmodium falciparum enoyl-ACP reductase and implications on drug discovery. Protein Sci. 2012;21:1734–45. doi: 10.1002/pro.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicola G, Smith CA, Lucumi E, Kuo MR, Karagyozov L, Fidock DA, Sacchettini JC, Abagyan R. Discovery of novel inhibitors targeting enoyl-acyl carrier protein reductase in Plasmodium falciparum by structure-based virtual screening. Biochem Biophys Res Commun. 2007;358:686–91. doi: 10.1016/j.bbrc.2007.04.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morde VA, Shaikh MS, Pissurlenkar RR, Coutinho EC. Molecular modeling studies, synthesis, and biological evaluation of Plasmodium falciparum enoyl-acyl carrier protein reductase (PfENR) inhibitors. Molecular diversity. 2009;13:501–17. doi: 10.1007/s11030-009-9141-0. [DOI] [PubMed] [Google Scholar]
- 41.Frecer V, Megnassan E, Miertus S. Design and in silico screening of combinatorial library of antimalarial analogs of triclosan inhibiting Plasmodium falciparum enoyl-acyl carrier protein reductase. Eur J Med Chem. 2009;44:3009–19. doi: 10.1016/j.ejmech.2008.12.028. [DOI] [PubMed] [Google Scholar]
- 42.Shah P, Siddiqi MI. 3D-QSAR studies on triclosan derivatives as Plasmodium falciparum enoyl acyl carrier reductase inhibitors. Sar Qsar Environ Res. 2010;21:527–45. doi: 10.1080/1062936X.2010.502297. [DOI] [PubMed] [Google Scholar]
- 43.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 44.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W5. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W7. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry. 2009;30:2785–91. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–34. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 48.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 49.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–9. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 50.Lee HS, Choi J, Kufareva I, Abagyan R, Filikov A, Yang Y, Yoon S. Optimization of high throughput virtual screening by combining shape-matching and docking methods. J Chem Inf Model. 2008;48:489–97. doi: 10.1021/ci700376c. [DOI] [PubMed] [Google Scholar]
- 51.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 52.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–96. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 54.Lindert S, Zhu W, Liu YL, Pang R, Oldfield E, Andrew McCammon J. Farnesyl diphosphate synthase inhibitors from in silico screening. Chem Biol Drug Des. 2013 doi: 10.1111/cbdd.12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duan J, Dixon SL, Lowrie JF, Sherman W. Analysis and comparison of 2D fingerprints: insights into database screening performance using eight fingerprint methods. J Mol Graph Model. 2010;29:157–70. doi: 10.1016/j.jmgm.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 56.Sastry M, Lowrie JF, Dixon SL, Sherman W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J Chem Inf Model. 2010;50:771–84. doi: 10.1021/ci100062n. [DOI] [PubMed] [Google Scholar]
- 57.Perozzo R, Kuo M, Sidhu AS, Valiyaveettil JT, Bittman R, Jacobs WR, Jr, Fidock DA, Sacchettini JC. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J Biol Chem. 2002;277:13106–14. doi: 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- 58.Choi KH, Kremer L, Besra GS, Rock CO. Identification and substrate specificity of beta -ketoacyl (acyl carrier protein) synthase III (mtFabH) from Mycobacterium tuberculosis. J Biol Chem. 2000;275:28201–7. doi: 10.1074/jbc.M003241200. [DOI] [PubMed] [Google Scholar]
- 59.Surolia N, Surolia A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat Med. 2001;7:167–73. doi: 10.1038/84612. [DOI] [PubMed] [Google Scholar]
- 60.Kapoor M, Gopalakrishnapai J, Surolia N, Surolia A. Mutational analysis of the triclosan-binding region of enoyl-ACP (acyl-carrier protein) reductase from Plasmodium falciparum. Biochem J. 2004;381:735–41. doi: 10.1042/BJ20040302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.