

Abstract
The characterization of aberrant glycosylation patterns in biopsied patient samples represents a remarkable challenge for scientists and medical doctors due to the lack of specific methods for their detection. Here, we report the development of a histological method, dubbed CHoMP—Chemoenzymatic Histology of Membrane Polysaccharides—for analyzing glycosylation patterns in mammalian tissues. This method exploits a recombinant glycosyltransferase to transfer a monosaccharide analog equipped with a chemical handle to a specific cell-surface glycan target, which can then be derivatized with imaging probes using bioorthogonal click chemistry for visualization. We applied CHoMP to survey changes in expression of N-acetyllactosamine (LacNAc) in human samples from patients afflicted with lung adenocarcinoma and observed a sharp decrease in expression levels between normal and early grade tumors, suggestion a potential application of this technique in early cancer diagnosis.
Keywords: Click Chemistry, CuAAC, Gycobiology, Histology, Lung Cancer
The histological examination of biopsied tissues remains a primary method for diagnosing cancer and other pathologies in the clinical setting. For cancers, the morphological information obtained by histology often allows for the determination of tumor type and grade[1] However, in some cases, particularly for early cancer detection, the histological data provided by hematoxylin and eosin (H&E) stained tissues is insufficient to confer a proper diagnosis. In those cases, a molecular biomarker will be included when possible.[2] Currently, the most commonly used histological biomarkers for cancer diagnostics are misexpressed proteins or nucleic-acids (e.g. HER-2: human epidermal growth factor receptor-2[3] and EGFR: Epidermal Growth Factor Receptor[4]) that are detected by immunohistochemistry or in situ hybridization, respectively.
Glycans regulate cancer progression by mediating tumor proliferation, invasion, metastasis, and angiogenesis.[5] Compared to healthy tissues, tumor tissues often exhibit aberrant glycosylation patterns.[6] For example, type II N-acetyllactosamine (LacNAc; Galβ1,4GlcNAc) is barely detectable in normal mucosa, but is significantly increased in colorectal carcinoma.[7] Likewise, polysialic acid, a glycan with restricted expression, typically found attached to the protein scaffold neural cell adhesion molecule (NCAM) in the brain[8], becomes ubiquitously expressed in malignant breast, pancreatic, hematopoietic, brain, and lung tissues[9]. For these reasons, cell-surface glycans are attractive candidates in the search for cancer biomarkers. Although a few studies have reported histological analysis using glycan-targeting antibodies and lectins[10] for cancer diagnosis, these methods have well-documented limitations associated with both weak affinity and cross-reactivity.[11] Most antibodies generated against glycan epitopes are of the low-affinity IgM subtype.[12] Likewise, lectins often bind their glycan targets with affinities in the μM–mM range[13]. Plant-derived lectins, such as Concanavalin A, are particularly useful for enriching N-linked glycoproteins by binding a conserved pentasaccharide core structure[14]. However, their specificity has been contested for peripheral glycan epitopes[12b].
An example of the promiscuity of lectin binding is shown in Figure S1 and S2, in which serial sections of formalin-fixed, paraffin embedded (FFPE) mouse kidney are stained with two commercially available lectins, Erythrina cristagalli (ECA; Figure S2A) and Lycopersicon esculentum (LEA; Figure S2B). Both lectins are routinely used for the detection of type II LacNAc. However, it is known that besides binding to terminal LacNAc, ECA also targets N-acetylgalactosamine (GalNAc) and Fucα1,2Gal[15], and LEA, besides binding to internal LacNAc residues, is also known to bind high mannose-type N-glycans[16]. Therefore, it was not surprising that we observed distinct labeling patterns with these two lectins. Although both lectins labeled distal tubules, connective tissue and smooth muscle, ECA also strongly labeled nuclei of the distal tubules, whereas LEA primarily stained the glomerular basement membrane.
An alternative method for glycan detection relies on metabolic oligosaccharide engineering[11, 17], in which cells or organisms are first treated with an unnatural monosaccharide bearing a chemically reactive tag. When internalized by cells and metabolized, the modified monosaccharide is incorporated into cell-surface glycoconjugates. Then, the bioorthogonal chemical tag enables covalent conjugation with fluorescent probes for visualization[11, 18], or with affinity probes for enrichment and glycoproteomic analysis[19]. However, this method cannot be used to detect unique classes of higher order glycans, e.g. disaccharides or trisaccharides, since unnatural monosaccharide incorporation via biosynthetic pathways leads to incorporation of the unnatural substrate into many different polysaccharide glycoforms.[20] Because peripheral, higher-order glycans, rather than monosaccharides, encode information for cell-surface receptor recognition to trigger specific downstream signaling[21]; there is an unmet need to develop methods for their detection.
We have conceived a strategy for the specific labeling of complex cell-surface glycans with biophysical probes for imaging studies. This method consists of a two-step labeling process. First, a glycosyltransferase or modification enzyme is used to transfer an azide or alkyne-bearing monosaccharide or cofactor (i.e. acetyl or methyl) to the target glycans on the cell surface. These posttranslational modification enzymes are highly specific for their glycan acceptors. However, their promiscuities toward donor substrates are well documented[22]; thus, specific glycan derivatization could be achieved. Subsequently, the labeled glycans could be detected with bioorthogonal probes functionalized in a complementary fashion using the biocompatible copper-catalyzed azide-alkyne cycloaddition (CuAAC)[23], a prototypical example of bioorthogonal click chemistry[24].
Recently, we demonstrated that cell-surface type II LacNAc-containing glycans could be specifically visualized using this strategy. By using a recombinant H. pylori α1,3 fucosyltransferase (FT)[22a] to transfer a fucose residue functionalized with a “clickable” tag to the LacNAc disaccharide at the 3′OH of the GlcNAc unit from a GDP-fucose donor, the tagged LacNAc can then be detected via a click reaction with a complementary imaging probe (Figure 1)[25].
Figure 1.
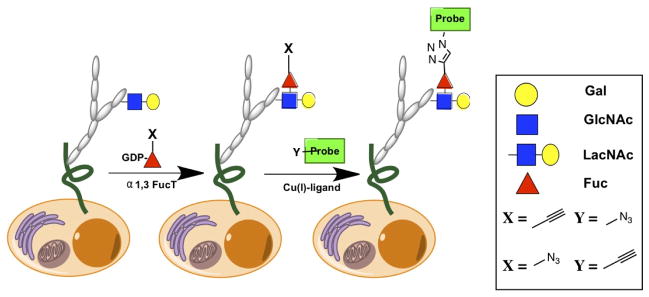
A two-step chemoenzymatic method for labeling cell-surface LacNAc.
We built upon our previous findings to develop a new method, dubbed CHoMP—Chemoenzymatic Histology of Membrane Polysaccharides— that is analogous to standard in situ hybridization and immunohistochemistry techniques. This method enables the high-throughput, cell-type specific analysis of LacNAc in heterogeneous, mammalian tissue samples. CHoMP was applied to human lung adenocarcinoma microarrays, surveying glycosylation changes accompanying disease progression. We observed a sharp decrease in LacNAc expression levels between normal and grade-one patient samples, suggesting a potential application of this technique in early cancer diagnosis. CHoMP is the first histological method for glycan detection based on chemoenzymatic approaches; this system has the benefit of high specificity towards its target glycan, and easy translation to a clinical setting. To the best of our knowledge, the Invitrogen EdU technology is the only precedent to date that exploits CuAAC in histological analysis, in an application to track DNA synthesis and cell proliferation[26].
Formalin-fixed tissue samples from 8–12 week old C57BL/6J male mice were used to develop the CHoMP method. Formalin treatment is routinely used in histology to preserve the cytoskeletal and protein structure in tissue samples. We used tissue sections from the preputial gland, an exocrine gland located in front of the genitals of mice, as a model system for method optimization due to its well-shaped structures. Direct labeling was performed and initial tests showed that glycans in both FFPE and Fixed/Frozen (FF) sections could be labeled; however, FFPE sections provided better structural definition than FF sections (Figure S2A). Therefore, all further experiments were conducted on FFPE sections. We discovered that including 0.1% Tween® 20 in the TBS buffer boosted the labeling signal dramatically and afforded significantly more consistent staining (Figure 2A and 2B). Titrations of FT and GDP-FucAl defined the optimal concentrations for each reagent: 400 μg/mL (Figure 2C) and 350 μM respectively (Figure 2D).
Figure 2.

Development of CHoMP based on direct labeling using small molecule fluorescent probes in mouse preputial gland. A) (left) FFPE labeling with 0.1% Tween-20 in TBS (right) FFPE labeling without 0.1% Tween-20 in TBS B) (left) FF labeling with 0.1% Tween-20 in TBS (right) FF labeling without 0.1% Tween-20 in TBS; C) FT titration; D) GDP-FucAl titration; E) Alexa Fluor 488-N3 titration; F) Labeling without BTTP ligand and catalyst in CuAAC; F) Time course of CuAAC. Green: LacNAc staining; Blue: DAPI nuclear staining.
The optimal concentration of Alexa Fluor 488-N3 was determined to be 12.5 μM (Figure 2E). A BTTP-Cu (I) 2:1 complex was shown to effectively accelerate the reaction, which is consistent with our previous studies on the ligand-accelerated CuAAC in vitro kinetic studies (Figure 2F). A time course of this reaction indicated that a 30–60 min incubation at room temperature provided an excellent signal-to-noise ratio (Figure 2G). A simple comparison with the cyclooctyne-based copper-free ‘click’ chemistry[27] indicated that CuAAC was a superior choice for LacNAc labeling in tissue samples due to high background produced by the fluorophore-labeled cyclooctyne (Figure S3).
Next, we designed a competition experiment to verify the specificity of this new histological method. When increasing concentrations of LacNAc disaccharide were added to the solution for the enzymatic labeling, the signal produced on tissue slides could be effectively eliminated with 20 mM LacNAc (Figure 3A). By contrast, a similar treatment for tissues stained with ECA and LEA lectins failed to abolish the signal to the background level, thus highlighting the non-specific nature of the lectin-based glycan detection (Figure 3B and 3C).
Figure 3.
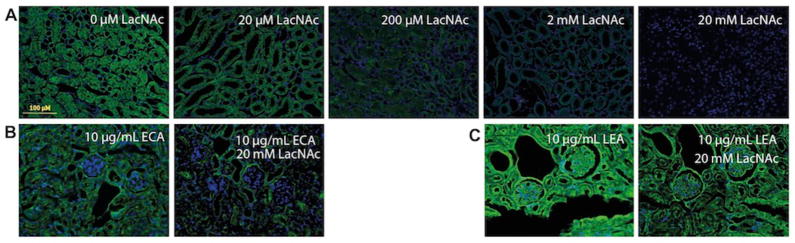
Verification of the labeling specificity using a LacNAc competition assay in FFPE kidney sections. Kidney tissue samples were labeled using A) CHoMP, B) ECA and C) LEA lectins in the presence of various concentrations of LacNAc, and imaged using a fluorescence microscope. Green: LacNAc staining; Blue: DAPI nuclear staining.
We then applied this method to screen LacNAc expression in an array of mouse tissues including the heart, liver, kidney, lung, spleen, skin, and epididymis (Figure S4), and achieved labeling across these different tissues. While labeling was achieved in all tested organs, certain tissues (i.e. spleen and lung) generated very low signal levels, rendering the reliable interpretation of the labeling patterns on different cell types difficult.
Surprisingly, when we tried to boost the output signal of this approach by labeling LacNAc using a combination of a biotinylated probe followed by streptavidin-Alexa Fluor 488, which carries multiple fluorophores attached to a streptavidin, little or no signal was detected (Figure S4). These observations prompted us to search for alternative methods to amplify the output signal for the reliable interpretation of the labeling patterns of different cell types in commonly assayed tissues.
In order to boost the output signal, we used a commercial tyramide-based system for signal amplification (TSA). We used mouse FFPE spleen and lung samples as a model system for method development (Figure S5). Tyramide binds covalently to tyrosine residues in proteins immediately proximal to the glycan target, making it ideal for visualizing the cellular and sub-cellular localization of the labeled LacNAc. By using this system, we successfully produced excellent signals in the treated tissue sections (Figure S6). The optimal conditions were determined to be 200 μg/mL FT and 1 mM GDP-FucAz. In control experiments, no signals above background were observed in the absence of the Cu(I) catalyst, GDP-FucAz, or FT (Figure S6).
The use of the ABC/TSA system revealed subtle expression patterns of LacNAc in several tissues, which were difficult to interpret using the direct labeling protocol due to the weak signals it produced. For example, the ABC/TSA system showed that, in the spleen, LacNAc-containing glycans were abundantly expressed in both the white and red pulp, in which the major cell types include red blood cells, dendritic cells, B cells, and mature T cells. Interestingly, the periarteriolar lymphoid sheaths (PALS) of the white pulp of the spleen, where naïve T cells reside, had lower LacNAc expression (Figure 4, Figure S7). This result is consistent with our previous findings, obtained by flow cytometry, which show that activated T cells bear higher levels of LacNAc as compared to their naïve counterparts[25]. This pattern, however, was not distinguishable using the direct labeling method.
Figure 4.
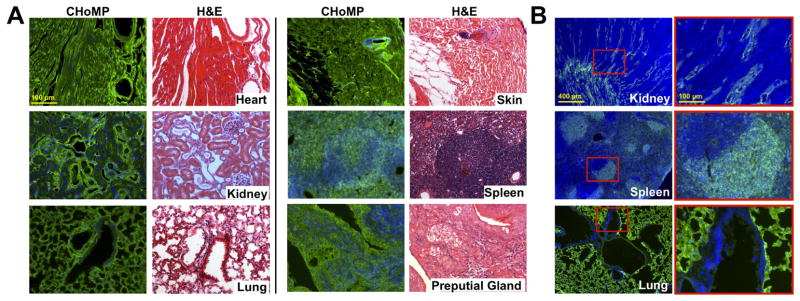
Application of CHoMP LacNAc labeling method to mouse tissue sections. (A) CHoMP LacNAc labeling method applied to 5 μm, FFPE mouse heart, kidney, lung, skin, spleen, and preputial gland tissues. (B) CHoMP method applied to 10 μm, FF mouse kidney, spleen and lung tissues. Green: LacNAc staining; Blue: DAPI nuclear staining.
Using this method, we analyzed LacNAc expression patterns in several other FFPE tissues, including the mouse heart, kidney, lung, and skin (Table 1; Figure 4, Figure S7). The high signal-to-noise ratio obtained in all of these tissue types highlights the broad utility of this method. We found that LacNAc was ubiquitously expressed in heart tissue. Interestingly, the kidney showed a distinct pattern of labeling in the glomeruli and in the distal tubules but not in the proximal tubules. These tubules are both lined with cuboidal epithelial cells, however they differ by the presence of a brush border found on the proximal tubules, suggesting a function for the LacNAc pattern observed. We also observed interesting labeling patterns in lung tissues, in which strong labeling was observed in the pulmonary alveoli, and little to no staining was detected in the cells lining the bronchiole, where air interacts directly with the cells.
Table 1.
Cell type specific labelling in mouse tissues using CHoMP method
| High Fluorescence Intensity | Low Fluorescence Intensity | |
|---|---|---|
| Heart | Cardiac myocytes, connective tissue, endothelium | |
| Kidney | Glomerulus, distal tubules | Connective tissues |
| Lung | Pulmonary alveolus | Smooth muscle |
| Skin | Keratinocytes, adipose, matrix stem cells, fibroblasts | Connective tissue |
| Spleen | Activated T cells in white pulp, central arteriole | Red pulp, naïve T cells in PALS of white pulp |
| Preputial Gland | Sebaceous secretory cells, acini |
The signal amplification system also enabled us to examine LacNAc labeling patterns in frozen tissue sections (10 micron, Figure 3B). We observed similar patterns of labeling in the FF mouse spleen, kidney, and lung tissues as compared to those observed in the FFPE tissues. Although FFPE tissues are generally preferred for their finer resolution, it is desirable to have a complementary method for FF tissues as well, especially for studies where CHoMP may be combined with in situ hybridization, which may require the use of frozen samples.
Importantly, we discovered that the CHoMP method could be combined with antibody-based IHC to enable cell-type specific glycan analysis. As shown in Figure 5, we applied CHoMP to label type II LacNAc in an array of human lung adenocarcinoma tissue samples. Then, the labeled samples were stained with an antibody against pan-cytokeratin, an epithelial-cell specific marker, and probed with an Alexa Fluor 555-labeled secondary antibody. The combination of these two techniques allowed for the analysis of epithelial-cell specific glycans.
Figure 5.
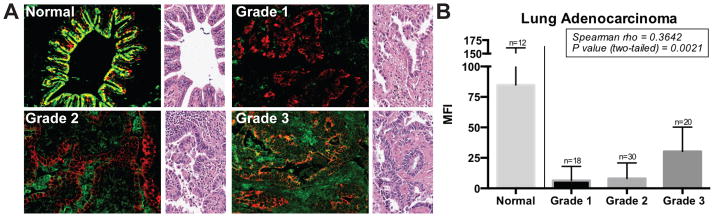
Analysis of the LacNAc level on a lung adenocarcinoma microarray. (A) LacNAc (green) is overlayed with Pan-cytokeratin (red) to delineate epithelial cells in normal lung, and grades 1 to 3 of lung adenocarcinoma patient samples. H&E stained, representative region from the same tissue sample is shown on the right of each image. (B) Mean fluorescence intensity (MFI) for LacNAc labeling in regions of epithelial cells is quantified, and normalized to the MFI of normal cores. A total of 6–8 discrete foci from each sample were examined microscopically at 40X magnification per core. Two trends were observed: (1) Normal lung tissue has significantly higher LacNAc expression than all adenocarcinoma samples (Mann-Whitney, two-tailed; p < 0.0001). (2) LacNAc expression increased as a function of grade (Spearman; p < 0.0021).
Lung cancer is the leading cause of cancer mortality, and adenocarcinomas account for nearly 40% of all the cases. These patients have a poor prognosis, with only about 15% of those diagnosed surviving five years[28]. Therefore, it is critical to develop early detection methods that may allow for curative surgery. Unfortunately, even state-of-the-art methods for early detection such as low-dose computed tomography and chest radiography, have shown a limited impact on reducing lung cancer mortality[29]. These methods suffer from high false positive rates (27.3% of subjects screened had a positive result of which only 3.6% were cancer, implying >96% detected nodules are benign). These results can lead to unnecessary surgery for benign nodules (about 15–30% of surgeries removed only benign nodules)[29]. Recent studies have reported that many lung cancer tissues express aberrant glycan epitopes, such as the Thomsen-Friedenreich antigen (Galβ1–3GalNAcα1-Ser/Thr)[30] and polysialic acid[31], as detected by low-affinity IgM antibodies and lectins. These studies suggest the potential of using aberrant glycan epitopes as biomarkers for cancer detection and evaluation. However, to date, no analysis has been conducted to evaluate the feasibility of using LacNAc as a biomarker for early detection of lung cancer.
To evaluate whether type II LacNAc expression patterns are altered in lung adenocarcinomas, we acquired a lung adenocarcinoma microarray from US Biomax for our study. This microarray comprised 68 samples from lung adenocarcinoma patients across grades and 12 samples from healthy humans. We applied the CHoMP method to label type II LacNAc on the microarray. As shown in Figure 5A, LacNAc was visible across many cell types in both the normal and cancerous tissue samples. We quantified LacNAc expression in epithelial (by co-staining with a specific epithelial cell marker based upon the antibody against pan-cytokeratin). Data produced from a Mann-Whitney, 2-tailed ranks sum analysis (median of normals = 84.88, median of grade 1 adenocarcinomas = 6.57 p < 0.0001) indicated that there were significant differences in LacNAc expression in lung epithelium between normal and grade 1 adenocarcinoma patients. In particular, we observed a 13-fold decrease in LacNAc expression from normal lung to grade 1 adenocarcinoma. We also observed a significant trend across tumor grades (p = 0.0021) using a Spearman correlation of the MFIs (Figure 5), where the levels of LacNAc increased slightly as the tumor grade progressed, up to roughly 40% of the LacNAc level of healthy tissues.
In this work, we developed a method for the histological detection of the LacNAc epitope using a two-step, chemoenzymatic labeling approach, dubbed CHoMP, which showed greater sensitivity and specificity as compared to traditional lectin-based methods. We applied this method to human tumor microarray screening and obtained a positive correlation between LacNAc expression patters with tumor grade in human lung adenocarcinoma patient samples. Strikingly, we observed a 13-fold decrease in LacNAc expression in grade 1-lung adenocarcinoma patient samples as compared to that of healthy humans, suggesting that the LacNAc epitope may be used as an early detection marker for lung cancer.
In healthy tissues, LacNAc acts as a substrate for galectins, lectins that bind galactose-containing glycans, to mediate inter- and intra-cellular signaling. Dysregulation of galectin-glycan interactions have been previously reported for many types of cancer[32]. Although the mechanism for aberrant LacNAc expression in lung adenocarcinomas remains obscure, a previous study discovered that metastatic human lung adenocarcinoma cells over express α1,3-fucosyltransferase, the enzyme responsible for capping LacNAc moieties with α1,3-linked fucose residues[33]. Likewise, over expression of the sialyl Lewis X antigen was found in the sera of lung adenocarcinoma patients—this epitope would also block LacNAc residues from being labeled by our approach[34]. Future studies using a larger sample set will be performed to elucidate the mechanism(s) underlying our observations.
It is worth noting that while the optimal conditions for direct labeling and signal amplification determined here could be applied to all tissue types tested, the signal intensity and signal-to-noise ratios varied in samples derived from different organs. For example, to obtain 90% signal saturation during image acquisition, heart tissue sections required a 1.2 sec exposure time, in contrast to the 1.7 sec used for the lung tissue sample analysis. Enzyme activities may also vary between different batches. Thus, for specific studies, both the enzyme and donor substrate concentrations should be titrated to obtain the optimal signal for the target tissue samples.
CHoMP represents the first example of a chemoenzymatic protocol for the detection of glycans on histological samples. The development of new enzymes that can be used to expand the CHoMP method would provide the ability to detect multiple glycans on tissue samples. Such expansion may allow for the creation of unique glycodes to mark disease progression with high specificity and sensitivity. These methods may readily be applied in clinical settings for cancer diagnostics and prognostics.
Experimental Section
Tissue Preparation and Processing
All studies were carried out under a protocol approved by the Institutional Animal Care And Use Committee at the Albert Einstein College of Medicine. Male, 8–12 week old C57BL/6J mice (Jackson Labs) were kept under isoflurane before and during organ perfusion with 50 mL of Tris-HCL buffered Saline (TBS, pH 7.4). Following perfusion with TBS, the mice were perfused with 50 mL of 4% paraformaldehyde (PFA) in TBS. Tissues were harvested and immersed in 4% PFA/TBS for 24h at 4°C. For frozen sectioning, the tissues were mounted in Tissue-Tek OCT Compound, then frozen in a dry-ice/ethanol bath in a cryomold (Tissue-Tek) then either stored at −80°C or sectioned to 10 μm slices with a Leica cryostat and mounted ion Superfrost Plus glass slides. For paraffin sectioning, the tissues were paraffin embedded by the Histology and Comparative Pathology facility at the Albert Einstien College of Medicine, then sectioned on a Leica microtome at 5 μm.
Direct CHoMP Labeling
For paraffin slides, the tissues were deparaffinized and rehydrated according to standard protocols. Briefly, the slides were sequentially immerged in coplin jars for 5 min in 40 mL: 2x Histo-Clear II (National Diagnostics), 2x Ethanol, 2x 70% Ethanol, 2x 50% Ethanol and d.H2O. Note: after deparaffinization and rehydration, the methods for frozen and paraffin sections are the same. A hydrophobic barrier was drawn around the tissue samples on the slides using a PAP pen. Tissues were immersed in 50 mL of TBS + 0.1% Tween-20 (TBST) for 10 minutes. For the enzymatic reaction, slides were placed in a humidified chamber and 50–500 μl of enzyme solution were added (volume depending on tissue area). The enzyme solution contained: 400 μg/mL of α1,3 fucosyltransferase, 350 μM GDP-FucAl, and 5 mM MgCl2 in TBST. The slides were incubated for 1h at 37°C. The slides were then washed 3 times in 50 mL of TBST for 5 min in coplin jars. The slides were again placed in a humidified chamber for the ‘click’ reaction. A solution containing 12.5 μM Alexa-488-azide, 75 μM CuSO4·5H2O premixed with 150 μM BTTP ligand, 2.5 mM sodium ascorbate in TBST was added to the slides, and these were then incubated for 30 min at RT. Following 5x 3-min washes in TBST, the slides were rinsed with d.H2O and mounted with Prolong anti-fade gold with DAPI (Invitrogen).
CHoMP procedure with amplification
For paraffin slides, the tissues were deparaffinized and rehydrated according to standard protocols (as above). A hydrophobic barrier was drawn around the tissue samples on the slides using a PAP pen. Tissues were immersed in 50 mL of TBS + 0.1% Tween-20 (TBST) for 10 minutes. For the enzymatic reaction, slides were placed in a humidified chamber and 50–500 μl of enzyme solution were added (volume depending on tissue area). The enzyme solution contained: 200 μg/mL of α1,3 fucosyltransferase, 1 mM GDP-FucAz, and 5 mM MgCl2 in TBST. The slides were incubated for 1h at 37°C. The slides were then washed 3 times in 50 mL of TBST in coplin jars. The slides were again placed in a humidified chamber face-up and an alkyne-tagged, biotin-probe was then ‘clicked’ to the FucAz (100 μM biotin-alkyne, 75 μM CuSO4·5H2O premixed with 150 μM BTTP ligand, 2.5 mM sodium ascorbate) in TBST for 30 min at RT. Following three washes in TBST, the tissues were blocked for 10 min in 0.3% hydrogen peroxide diluted in TBS in coplin jars at RT, and washed 3X in TBST to remove the H2O2. The slides were then placed in a humidified chamber and incubated with Neutravidin-HRP (1:100 in TBST) for 1h at RT, then subsequently washed 3X with TBST in coplin jars. Finally, the slides were placed in a humidified chamber and incubated with TSA-Plus FITC reagent according to manufacturers protocol (1:50 dilution for 10 min at RT, protected from light), then washed 3X for 5 min each in TBST in coplin jars and mounted with Prolong anti-fade gold with DAPI (Invitrogen).
Direct labeling with DIFO
The tissues were deparaffinized and rehydrated according to standard protocols (as above). A hydrophobic barrier was drawn around the tissue samples on the slides using a PAP pen. Tissues were immersed in 50 mL of TBST for 10 minutes. For the enzymatic reaction, slides were placed in a humidified chamber and 50–500 μl of enzyme solution were added (volume depending on tissue area). The enzyme solution contained: 200 μg/mL of α1,3 fucosyltransferase, 1 mM GDP-fucose-azide, and 5 mM MgCl2 in TBST. The slides were incubated for 1h at 37°C. The slides were then washed 3 times in 50 mL of TBST in coplin jars. The slides were again placed in a humidified chamber face-up and a difluorinated cyclooctyne-Alexa 488 conjugate (DIFO-488)[35] was then ‘clicked’ to the azido-fucose (10 μM DIFO) in TBST for 30 min at RT. The slides were then washed 3X for 5 min each in TBST in coplin jars and mounted with Prolong anti-fade gold with DAPI (Invitrogen).
Lectin Staining
Lectin staining was done according to manufacturers protocol using biotinylated ECA and LEA from EY laboratories. In summary, after paraffin removal (as above), slides were incubated with 10 μg/mL lectin in TBS for 1h at RT. Following three washes in TBST, the tissues were blocked for 10 min in 0.3% hydrogen peroxide diluted in TBS in coplin jars at RT, and washed 3X in TBST to remove the H2O2. The slides were then placed in a humidified chamber and incubated with Neutravidin-HRP (1:100 in TBST) for 1h at RT, then subsequently washed 3X with TBST in coplin jars. Finally, the slides were placed in a humidified chamber and incubated with TSA-Plus FITC reagent according to manufacturers protocol (1:50 dilution for 10 min at RT, protected from light), then washed 3X for 5 min each in TBST in coplin jars and mounted with Prolong anti-fade gold with DAPI (Invitrogen).
Cytokeratin counterstaining for TMAs
CHoMP labeling (ABC/TSA) was performed according to protocol above without mounting. Slides were subjected to antigen retrieval after CHoMP labeling with Vector unmasking solution in a coplin jar heated to 95 degrees for 20 min. Coplin jar was removed and allowed to cool at RT for 30 min. Slides were laid flat in a humidified chamber and blocked with 5% horse serum with 2% BSA in TBST for 30 min at RT. Slides were then incubated with mouse anti-cytokeratin (Sigma) diluted in serum block (1:100) for 1h at RT. Slides were rinsed 3X in TBST in a coplin jar, then laid flat in a humidified chamber. The slides were incubated with goat anti-mouse IgG Alexa 555 diluted in serum block 1:250 for 1h at RT. The slides were then washed 3X for 5 min each in TBST in coplin jars and mounted with Prolong anti-fade gold with DAPI (Invitrogen).
Image analysis
Photomicrograms were acquired on Zeiss AxioObserver digital microscope and processed using ImageJ software. The acquisitions paramenters were set to obtain 80% signal saturation in each fluorescent channel. Every image was obtained as a pair, including a -E control, with the same acquisition parameters. Image processing involved lowering the MFI of the negative controls to eliminate all signal, and the subtracting the same value for each sample to obtain enzyme dependent signals.
Statistical analysis
For TMA analysis, LacNAc labeling was quantified in epithelial cell regions (cytokeratin positive regions), and normalized to the average mean fluorescence intensity (MFI) of the normal cores. Nonparametric analysis was performed on data sets and Spearman correlation was calculated using Prism 6 software.
TMA Description
TMA LC2085a (US Biomax, Rockville, MD) high-density tissue array with normal lung from autopsy, cancer adjacent tissue, and multiple types of lung adenocarcinoma (grades 1–3, representing male and female patients of all ages).
Supplementary Material
Acknowledgments
This work was supported partially by the Albert Einstein College of Medicine and the National Institutes of Health to P.W. (R01GM093282). We thank the Histology and Comparative Pathology facility at the Albert Einstein College of Medicine, and Rani S. Sellers, D.V.M., Ph.D. for her assistance in guiding our pathological analysis.
References
- 1.Wick MR. Diagnostic Histochemistry. Cambridge University Press; New York: 2008. [Google Scholar]
- 2.Smits AJ, Kummer JA, de Bruin PC, Bol M, van den Tweel JG, Seldenrijk KA, Willems SM, Offerhaus GJ, de Weger RA, van Diest PJ, Vink A. Mod Pathol. 2014;27:168–174. doi: 10.1038/modpathol.2013.134. [DOI] [PubMed] [Google Scholar]
- 3.Ridolfi RL, Jamehdor MR, Arber JM. Mod Pathol. 2000;13:866–873. doi: 10.1038/modpathol.3880154. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Garcia E, Gomez-Martin C, Angulo B, Conde E, Suarez-Gauthier A, Adrados M, Perna C, Rodriguez-Peralto JL, Hidalgo M, Lopez-Rios F. Histopathology. 2011;59:8–17. doi: 10.1111/j.1365-2559.2011.03894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuster MM, Esko JD. Nat Rev Cancer. 2005;5:526–542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 6.a Hakomori S. Adv Cancer Res. 1989;52:257–331. doi: 10.1016/s0065-230x(08)60215-8. [DOI] [PubMed] [Google Scholar]; b Dube DH, Bertozzi CR. Nat Rev Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 7.Ichikawa T, Nakayama J, Sakura N, Hashimoto T, Fukuda M, Fukuda MN, Taki T. J Histochem Cytochem. 1999;47:1593–1602. doi: 10.1177/002215549904701211. [DOI] [PubMed] [Google Scholar]
- 8.Rutishauser U, Landmesser L. Trends Neurosci. 1996;19:422–427. doi: 10.1016/0166-2236(96)10041-2. [DOI] [PubMed] [Google Scholar]
- 9.Zhang S, Cordon-Cardo C, Zhang HS, Reuter VE, Adluri S, Hamilton WB, Lloyd KO, Livingston PO. Int J Cancer. 1997;73:42–49. doi: 10.1002/(sici)1097-0215(19970926)73:1<42::aid-ijc8>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 10.Adamczyk B, Tharmalingam T, Rudd PM. Biochim Biophys Acta. 2012;1820:1347–1353. doi: 10.1016/j.bbagen.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Laughlin ST, Bertozzi CR. Proc Natl Acad Sci U S A. 2009;106:12–17. doi: 10.1073/pnas.0811481106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Stray SJ, Cummings RD, Air GM. Glycobiology. 2000;10:649–658. doi: 10.1093/glycob/10.7.649. [DOI] [PubMed] [Google Scholar]; b Manimala JC, Roach TA, Li Z, Gildersleeve JC. Angew Chem Int Ed Engl. 2006;45:3607–3610. doi: 10.1002/anie.200600591. [DOI] [PubMed] [Google Scholar]
- 13.a aIP, Goldstein RD. The Lectins: Properties, Functions, and Applications in Biology and Medicine. Academic Press; 1986. [Google Scholar]; b Debray H, Decout D, Strecker G, Spik G, Montreuil J. Eur J Biochem. 1981;117:41–55. doi: 10.1111/j.1432-1033.1981.tb06300.x. [DOI] [PubMed] [Google Scholar]
- 14.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 15.Lis H, Sharon N. Methods Enzymol. 1987;138:544–551. doi: 10.1016/0076-6879(87)38049-8. [DOI] [PubMed] [Google Scholar]
- 16.Oguri S. Glycoconj J. 2005;22:453–461. doi: 10.1007/s10719-005-5329-4. [DOI] [PubMed] [Google Scholar]
- 17.a Springer GF. Science. 1984;224:1198–1206. doi: 10.1126/science.6729450. [DOI] [PubMed] [Google Scholar]; b Dube DH, Bertozzi CR. Curr Opin Chem Biol. 2003;7:616–625. doi: 10.1016/j.cbpa.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Rouhanifard SH, Nordstrom LU, Zheng T, Wu P. Chem Soc Rev. 2013;42:4284–4296. doi: 10.1039/c2cs35416k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a Hanson SR, Hsu TL, Weerapana E, Kishikawa K, Simon GM, Cravatt BF, Wong CH. J Am Chem Soc. 2007;129:7266–7267. doi: 10.1021/ja0724083. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jiang H, Zheng T, Lopez-Aguilar A, Feng L, Kopp F, Marlow FL, Wu P. Bioconjug Chem. 2014;25:698–706. doi: 10.1021/bc400502d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaubard JL, Krishnamurthy C, Yi W, Smith DF, Hsieh-Wilson LC. J Am Chem Soc. 2012;134:4489–4492. doi: 10.1021/ja211312u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakada H, Inoue M, Tanaka N, Numata Y, Kitagawa H, Fukui S, Yamashina I. Biochem Biophys Res Commun. 1991;179:762–767. doi: 10.1016/0006-291x(91)91882-d. [DOI] [PubMed] [Google Scholar]
- 22.a Wang W, Hu T, Frantom PA, Zheng T, Gerwe B, Del Amo DS, Garret S, Seidel RD, 3rd, Wu P. Proc Natl Acad Sci U S A. 2009;106:16096–16101. doi: 10.1073/pnas.0908248106. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Varki AC, RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. Essentials of Glycobiology. 2. Cold Spring Harbor Press; New York: 2008. [PubMed] [Google Scholar]
- 23.a Soriano del Amo D, Wang W, Besanceney C, Zheng T, He Y, Gerwe B, Seidel RD, 3rd, Wu P. Carbohydr Res. 2010;345:1107–1113. doi: 10.1016/j.carres.2010.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Besanceney-Webler C, Jiang H, Zheng T, Feng L, Soriano del Amo D, Wang W, Klivansky LM, Marlow FL, Liu Y, Wu P. Angew Chem Int Ed Engl. 2011;50:8051–8056. doi: 10.1002/anie.201101817. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; d Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 24.Baskin JM, Bertozzi CR. QSAR & Combinatorial Science. 2007;26:1211–1219. [Google Scholar]
- 25.Zheng T, Jiang H, Gros M, del Amo DS, Sundaram S, Lauvau G, Marlow F, Liu Y, Stanley P, Wu P. Angew Chem Int Ed Engl. 2011;50:4113–4118. doi: 10.1002/anie.201100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavanagh BL, Walker T, Norazit A, Meedeniya AC. Molecules. 2011;16:7980–7993. doi: 10.3390/molecules16097980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a Jewett JC, Sletten EM, Bertozzi CR. J Am Chem Soc. 2010;132:3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ning X, Guo J, Wolfert MA, Boons GJ. Angew Chem Int Ed Engl. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.N. C. Institute. 2013 [Google Scholar]
- 29.a Church TR, Black WC, Aberle DR, Berg CD, Clingan KL, Duan F, Fagerstrom RM, Gareen IF, Gierada DS, Jones GC, Mahon I, Marcus PM, Sicks JD, Jain A, Baum S. N Engl J Med. 2013;368:1980–1991. doi: 10.1056/NEJMoa1209120. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Aberle DR, DeMello S, Berg CD, Black WC, Brewer B, Church TR, Clingan KL, Duan F, Fagerstrom RM, Gareen IF, Gatsonis CA, Gierada DS, Jain A, Jones GC, Mahon I, Marcus PM, Rathmell JM, Sicks J. N Engl J Med. 2013;369:920–931. doi: 10.1056/NEJMoa1208962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takanami I. Oncol Rep. 1999;6:341–344. doi: 10.3892/or.6.2.341. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka F, Otake Y, Nakagawa T, Kawano Y, Miyahara R, Li M, Yanagihara K, Nakayama J, Fujimoto I, Ikenaka K, Wada H. Cancer Res. 2000;60:3072–3080. [PubMed] [Google Scholar]
- 32.Liu FT, Rabinovich GA. Nat Rev Cancer. 2005;5:29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- 33.Martin-Satue M, de Castellarnau C, Blanco J. Br J Cancer. 1999;80:1169–1174. doi: 10.1038/sj.bjc.6690482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kannagi R, Fukushi Y, Tachikawa T, Noda A, Shin S, Shigeta K, Hiraiwa N, Fukuda Y, Inamoto T, Hakomori S, et al. Cancer Res. 1986;46:2619–2626. [PubMed] [Google Scholar]
- 35.Codelli JA, Baskin JM, Agard NJ, Bertozzi CR. J Am Chem Soc. 2008;130:11486–11493. doi: 10.1021/ja803086r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.