

Abstract
Purpose of review
Despite the increased knowledge of osteocyte biology, the contribution of this most abundant bone cell to the development and progression of multiple myeloma in bone is practically unexplored.
Recent findings
Multiple myeloma bone disease is characterized by exacerbated bone resorption and the presence of osteolytic lesions that do not heal because of a concomitant reduction in bone formation. Osteocytes produce molecules that regulate both bone formation and resorption. Recent findings suggest that the life span of osteocytes is compromised in multiple myeloma patients with bone lesions. In addition, multiple myeloma cells affect the transcriptional profile of osteocytes by upregulating the production of pro-osteoclastogenic cytokines, stimulating osteoclast formation and activity. Further, patients with active multiple myeloma have elevated circulating levels of sclerostin, a potent inhibitor of bone formation which is specifically expressed by osteocytes in bone.
Summary
Understanding the contribution of osteocytes to the mechanisms underlying the skeletal consequences of multiple myeloma bone disease has the potential to provide important new therapeutic strategies that specifically target multiple myeloma–osteocyte interactions.
Keywords: apoptosis, osteoclasts, osteocytes, receptor activator of nuclear factor-kappa B ligand, sclerostin
INTRODUCTION
Multiple myeloma is a plasma cell malignancy characterized by the accumulation of monoclonal plasma cells in the bone marrow and the presence of osteolytic lesions [1]. Multiple myeloma has the highest incidence of bone involvement among malignant diseases. It is estimated that up to 90% of patients with multiple myeloma have evidence of osteolysis in the form of generalized osteopenia or discrete lytic lesions, and up to 60% of multiple myeloma patients develop pathologic fractures [2,3]. Skeletal manifestations, which usually involve the axial skeleton, skull and femur, are the most prominent source of pain and disability in multiple myeloma.
Under physiologic conditions, interactions between cells present in the bone marrow microenvironment result in a balanced and coupled remodeling of bone, a lifelong process responsible for bone damage repair and mineral homeostasis [4]. Bone remodeling is performed by teams of bone-resorbing osteoclasts and bone-forming osteoblasts arranged within structures known as ‘bone remodeling compartments’ (BRCs) [5–7]. In the BRC, bone surface osteoclasts and osteoblasts are physically separated from the bone marrow cavity by a canopy of flat cells (Fig. 1). Functionally, this separation has been proposed to generate a unique microenvironment that facilitates spatial and temporal ‘coupled’ osteoclast resorption and osteoblast formation, minimizing the net change in bone volume during physiological bone remodeling. Although osteoblasts and osteoclast are major participants of the bone remodeling process, osteocytes are the central regulators of bone homeostasis [8▪]. Osteocytes are the most abundant bone cells, comprising more than 95% of all bone cells, compared with 1–2% osteoclasts and 5% osteoblasts. Despite that their cell bodies lie in lacunae carved within the mineral, osteocytes extensively communicate with each other and with cells on the bone surface and in the marrow through cytoplasmic projections that run within canaliculi [9]. This osteocytic lacunar-canalicular network allows direct cell-to-cell contact between osteocytes and cells near the bone surface and also distributes secreted molecules among all bone and marrow cells as well as into blood vessels to enter the general circulation. Osteocyte-derived molecules regulate the activity of both osteoblasts and osteoclasts by releasing paracrine factors, such as sclerostin (the product of the Sost gene) or the receptor activator of nuclear factor-kappa B ligand (RANKL), to locally regulate bone formation and resorption, respectively [10–12]. In addition, emerging evidence supports the notion that osteocytes are responsible for targeting bone remodeling to specific areas in bone [13–15]. In this regard, it has been proposed that apoptotic osteocytes signal to lining cells covering the bone surface, which lift up from bone into the bone marrow and form the canopy that encloses the BRC. Osteoclast precursors are then recruited to the BRC via marrow capillaries in which they mature to osteoclasts and resorb bone, initiating bone remodeling [6].
FIGURE 1.

The BRC in multiple myeloma. Osteocytes sense the need for bone resorption and send signals to lining cells, which retract from the bone surface to form the BRC, and secrete molecules that regulate osteoblast and osteoclast formation and activity. Osteoclast precursors are transported to the BRC by marrow capillaries, differentiate to mature osteoclasts and initiate bone remodeling. Osteoblasts precursors from the bone marrow or the circulation differentiate into mature, bone synthesizing cells in response to factors released from the bone matrix by resorption. In multiple myeloma, the canopy of lining cells that separate the bone marrow from the active bone remodeling areas on bone is disrupted. Multiple myeloma cells are then able to enter in close contact with osteoclasts, osteoblasts and osteocytes and modulate their function (dotted lines). BRC, bone remodeling compartment; MM, multiple myeloma; OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor-kappa B ligand.
The bone marrow microenvironment in multiple myeloma is a major contributor to tumor growth and the bone destructive process. Bone remodeling is uncoupled in multiple myeloma and is characterized by generalized osteoclasts activation, which results in exacerbated bone resorption, and by suppressed osteoblast function resulting in a reduction in bone formation [16]. As a result, lytic lesions rarely heal, even when complete remission is attained. Recent evidence demonstrates that the BRC is disrupted in multiple myeloma, supporting the notion that not only an exchange of soluble factors occurs, but also mechanisms involving direct cell-to-cell contact exist between multiple myeloma cells and bone cells (Fig. 1) [17]. The relevance of these cell-to-cell interactions to the in-vivo bone pathology in multiple myeloma is supported by in-vitro experiments and histological observations that show that extensive bone destruction occurs only in areas of the bones that are highly infiltrated with multiple myeloma cells, whereas bone resorption and formation are balanced in bones with lower tumor burden [18,19]. Interestingly, the biopsies with frequent disruptions in the BRC were also the ones that showed exacerbated resorption without matrix construction [7]. The significance of the cross-talk between multiple myeloma cells and osteoclast and osteoblasts has been a focus of recent investigations These studies found that multiple myeloma cells stimulate osteoclasts (OC) differentiation and activity through a series of potent osteoclasts effectors, such as RANKL, chemokine (C-C motif) ligand 3 (MIP-1), chemokine (C-X-C motif) ligand 12 (SDF-1), interleukin (IL)-1, IL-3, IL-6, vascular endothelial growth factor (VEGF) and tumor necrosis factor, whereas they reduce osteoblast numbers and activity through factors such as IL-7, dickkopf homolog 1 (DKK1), secreted frizzled-related protein (sFRP), VLA-4/vascular cell adhesion molecule 1 (VCAM-1), Fas ligand (FAsL), tumor necrosis factor (ligand) superfamily, member 10 (TRAIL) and hepatocyte growth factor (HGF) [20–26]. Although our knowledge of osteocyte function has markedly increased during the last few years, there is a paucity of information about the cross-talk between multiple myeloma and osteocytes and how these interactions contribute to the development and progression of multiple myeloma. In this review, we examine the current findings regarding the role of osteocytes in multiple myeloma bone disease and the potential impact of determining the consequences of osteocyte interactions with myeloma cells that might result in the identification of additional targets for therapeutic development.
OSTEOCYTES AND THE CONTROL OF OSTEOCLAST ACTIVITY
Osteocyte apoptosis is spatially, temporally and functionally linked to bone remodeling partly as a result of the osteocyte cell death [27▪]. Several studies have shown that regions containing apoptotic osteocytes are targeted for bone resorption, which results in increased bone turnover [14,28,29]. Further, in-vitro experiments demonstrated that osteocyte apoptosis not only can induce recruitment of osteoclast precursors, but also their differentiation [29,30]. Interestingly, increased osteocyte apoptosis has been shown to be induced in vivo in areas of microdamage with bone disuse, glucocorticoid administration or estrogen deficiency-induced osteoporosis. Further, exclusive induction of apoptosis of osteocytes was sufficient to increase resorption and bone loss [13]. These findings suggest that osteocytes are involved in pathological conditions involving enhanced bone resorption [14,31,32]. A recent study reported similar findings in multiple myeloma patients [33▪▪]. In this study, multiple myeloma patients with bone lesions had fewer viable osteocytes than healthy controls or multiple myeloma patients without bone lesions, partly because of increased osteocyte apoptosis. In line with previous findings in other models, these investigators found a positive correlation between the number of apoptotic osteocytes and the number of osteoclasts in the areas examined. The mechanisms underlying osteoclasts recruitment and differentiation induced by osteocyte apoptosis are not fully understood. On one hand, osteocyte death is commonly accompanied by large increases in RANKL expression, a molecule which not only promotes osteoclast differentiation, but also acts as an osteoclast precursor chemoattractant [34,35]. On the other hand, it is known that multiple myeloma cells affect the transcriptional profiles of osteocytes by upregulating the production of other osteoclastogenic cytokines such as interleukin-11 or MIP1α, thus increasing their pro-osteoclastogenic properties in an RANKL-independent fashion [33▪▪,36]. Regardless of the mechanism involved, these results suggest that osteocyte apoptosis might be an early event in multiple myeloma development and progression resulting in enhanced osteoclast migration and production. In this regard, bisphosphonates, the standard of care for multiple myeloma-induced bone disease, interfere with osteoclast function and prevent osteocyte apoptosis [37]. However, the contribution of the latter to the overall beneficial effects of these drugs on multiple myeloma bone disease remains unknown.
Osteocytes produce molecules that regulate osteoclast production and activity and thus bone resorption. These molecules include the previously mentioned RANKL and macrophage colony stimulating factor 1 (M-CSF), as well as the antiosteoclastogenic cytokine osteoprotegerin (OPG; the RANKL decoy receptor). M-CSF is primarily involved in stimulating the proliferation of osteoclast progenitors, whereas RANKL is essential for their differentiation [38,39]. RANKL is expressed by many different cell types, including osteoblasts and osteocytes, stromal cells, B and T lymphocytes, synovial fibroblasts and hypertrophic chondrocytes [40]. However, only mice lacking RANKL in osteocytes (not osteoblasts or stromal osteoblastic cells) exhibit osteopetrosis, as a result of decreased osteoclast numbers [11,12]. These results indicate that osteocytes are a major source of RANKL and may play a key role in the enhanced resorption displayed in multiple myeloma.
In multiple myeloma, the balance of RANKL and OPG, key determinant for osteoclast differentiation, is disrupted, promoting activation of osteoclasts differentiation and consequently bone destruction [41,42,43▪▪]. Further, increased levels of soluble RANKL in multiple myeloma patients were shown to be associated with disease activity [44]. Consistent with an important role of this molecule in multiple myeloma bone disease, RANKL neutralization delayed multiple myeloma progression in mice [45]. Further, in patients with multiple myeloma, a monoclonal antibody capable of blocking RANKL (denosumab) reduced bone turnover [46]. However, the specific origin of the elevated RANKL levels in multiple myeloma bone disease is still unclear. RANKL is expressed at both the mRNA and the protein level in multiple myeloma cells themselves purified from bone marrow aspirates of multiple myeloma patients. Further, in-vitro experiments demonstrated that multiple myeloma cells induced differentiation of functional osteoclast precursors [44,47]. In addition, multiple myeloma cells induce RANKL expression in stromal/osteoblastic cells inducing an imbalance of RANKL/OPG ratio in favor of RANKL [48], and multiple myeloma cells also upregulate RANKL in T lymphocytes [49]. These results suggest that several cells can directly contribute to the pool of RANKL in bone. Recent results reported by our laboratory demonstrated for the first time that multiple myeloma cells upregulate RANKL expression in murine osteocytic cells, supporting the idea that osteocytes also contribute to the increased levels of RANKL [50▪▪]. In addition, we also found that OPG expression is downregulated in osteocytes by multiple myeloma cells, leading to a more pro-osteoclastogenic RANKL/OPG ratio. In contrast, Giuliani et al. [33▪▪] reported that RANKL was not detected in the conditioned medium of the cocultures of human preosteocytes and multiple myeloma cells, suggesting that the contribution of osteocytes to the exacerbated bone resorption induced by multiple myeloma may also depend on other factors released by osteocytes. Further experiments are required to identify the specific role of osteocyte apoptosis and osteocytic RANKL in the early steps of multiple myeloma-induced bone disease.
REGULATION OF BONE FORMATION BY OSTEOCYTES
Osteocyte-derived molecules also regulate the activity of osteoblasts. Only osteocytes in bone secrete sclerostin, the product of the Sost gene, a potent inhibitor of bone formation and the first discovered and long-sought molecular mediator between osteocytes and bone surface cells [51]. Sclerostin antagonizes canonical Wnt signaling by binding to Wnt coreceptors LRP-4, LRP-5, and LRP-6, thus inhibiting wnt-dependent osteoblast differentiation and osteoblast survival [10,52–54]. Functional mutations of the Sost gene cause abnormal skeletal phenotypes in humans, characterized by high bone mineral density [55,56]. Likewise, Sost KO mice have a high bone mass phenotype characterized by increased bone formation [57]. Further, several clinical studies have shown that circulating sclerostin levels increase with age, suggesting that the increased production of sclerostin leads to the impairment of bone formation associated with aging [58]. In multiple myeloma, sclerostin levels are elevated in the serum or the plasma of patients, and correlate with the extent of bone disease and other adverse myeloma features [59,60,61▪▪]. Several recent studies reported that myeloma cells produce sclerostin or induce sclerostin expression in myeloma patients [62,63]; however, the specific contribution of sclerostin derived from osteocytes has not been explored. Giuliani et al. [33▪▪] did not find significant differences in sclerostin expression by osteocytes in multiple myeloma patients when compared with healthy controls, suggesting that higher sclerostin levels could be secreted directly by multiple myeloma cells rather than by osteocytes. However, we found that multiple myeloma cells promote upregulation of Sost mRNA levels in osteocytes, decreasing Wnt signaling and downregulating Wnt target genes such as OPG [50▪▪]. In contrast, we did not find Sost mRNA transcripts in the multiple myeloma cell line JJN3. Furthermore, we cannot exclude the possibility that apoptotic osteocytes in multiple myeloma contribute to the increased sclerostin levels, as it has been reported that proapoptotic factors may induce sclerostin expression in osteocytes [64]. These results suggest the possibility that osteocyte-derived sclerostin contributes to the inhibition of the bone forming activity of osteoblasts in multiple myeloma bone disease. In addition to its role as a potent inhibitor of bone formation, recent findings suggest that sclerostin may also have a catabolic action, promoting osteoclast formation and activity, in an RANKL-dependent manner [65]. Although more experiments are needed to determine the mechanisms leading to the elevated levels of sclerostin in multiple myeloma bone disease and their consequences, Wnt proteins and their inhibitors offer a potential for the development of novel anabolic therapeutics in multiple myeloma.
CONCLUSION
The potential role of osteocytes in multiple myeloma is underexplored. The studies reviewed in this article suggest that osteocytes may be responsible for many of the deleterious effects present in multiple myeloma bone disease (Fig. 2). Osteocytes are an important source of RANKL and sclerostin in bone, and the levels of these molecules are altered in multiple myeloma. The source of these molecules and the mechanisms of their aberrant production, as well as their roles in tumor growth, osteoclastogenesis and bone formation in multiple myeloma bone lesions still remain obscure. Findings demonstrating that multiple myeloma triggers osteocyte apoptosis, followed by increased osteoclast recruitment, bone destruction and poor bone quality, provide a new direction of mechanistic research that has not yet been explored. In addition, the knowledge that multiple myeloma cells regulate sclerostin, as well as Wnt signaling in osteocytes, raises the possibility of using currently available tools that target these pathways as new therapeutic approaches for multiple myeloma, and eventually other cancers targeting bone.
FIGURE 2.
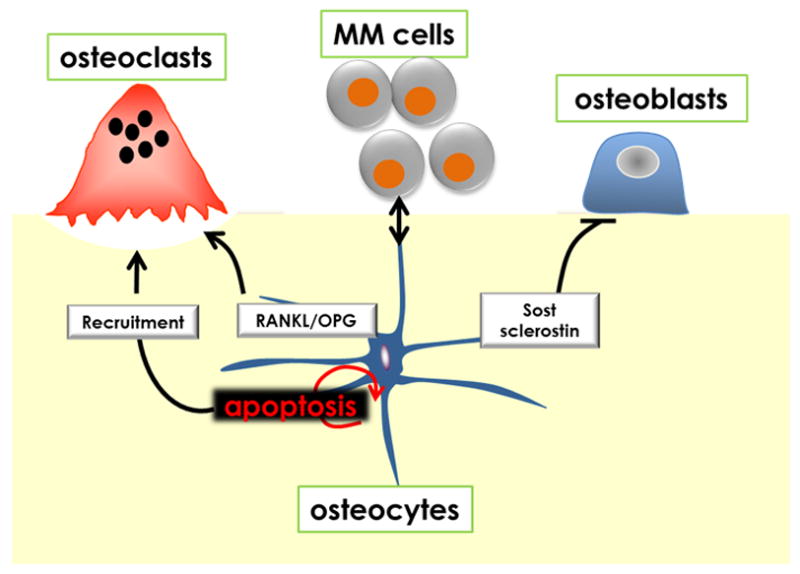
Potential role of osteocytes in multiple myeloma bone disease. Interactions between osteocytes and multiple myeloma cells within the BRC modulate critical signaling pathways and mediators responsible for bone resorption and bone formation. Osteocyte apoptosis is markedly increased in multiple myeloma patients with bone lesions and results in targeted osteoclast recruitment. The RANKL/OPG ratio in osteocytes is also elevated by multiple myeloma cells because of a combination of downregulation of OPG and upregulation of RANKL expression potentially increasing osteoclast differentiation. In addition, increased Sost/Sclerostin in osteocytes induced by multiple myeloma could contribute to the decreased bone formation that characterizes multiple myeloma-induced bone disease. MM, multiple myeloma; OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor-kappa B ligand.
KEY POINTS.
Osteocytes are key regulators of bone remodelling, and their role in multiple myeloma bone disease is largely unknown.
Osteocyte viability is decreased in multiple myeloma patients with bone lesions.
Crosstalk between multiple myeloma cells and osteocytes induces profound changes in osteocytic gene expression.
Understanding multiple myeloma–osteocyte interactions has the potential to develop new therapeutic strategies.
Acknowledgments
This work was supported in part by the National Institutes of Health Grants, VA Merit Review (Indiana-CTSI) and The Gideon and Sevgi Rodan Fellowship (International Bone and Mineral Society).
Footnotes
Conflicts of interest
The authors have no conflicts of interest to declare.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
- 1.Roodman GD. Pathogenesis of myeloma bone disease. Leukemia. 2009;23:435–441. doi: 10.1038/leu.2008.336. [DOI] [PubMed] [Google Scholar]
- 2.Roodman GD. Pathogenesis of myeloma bone disease. Blood Cells Mol Dis. 2004;32:290–292. doi: 10.1016/j.bcmd.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg AJ, Rajkumar SV, Therneau TM, et al. Relationship between initial clinical presentation and the molecular cytogenetic classification of myeloma. Leukemia. 2014;28:398–403. doi: 10.1038/leu.2013.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen MR, Burr DB. Bone modeling and remodeling. In: Burr D, Allen M, editors. Basic and Applied Bone Biology. 1. San Diego, United States: Elsevier; 2014. pp. 75–90. [Google Scholar]
- 5.Hauge EM, Qvesel D, Eriksen EF, et al. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. J Bone Miner Res. 2001;16:1575–1582. doi: 10.1359/jbmr.2001.16.9.1575. [DOI] [PubMed] [Google Scholar]
- 6.Eriksen EF, Eghbali-Fatourechi GZ, Khosla S. Remodeling and vascular spaces in bone. J Bone Miner Res. 2007;22:1–6. doi: 10.1359/jbmr.060910. [DOI] [PubMed] [Google Scholar]
- 7.Andersen TL, Sondergaard TE, Skorzynska KE, et al. A physical mechanism for coupling bone resorption and formation in adult human bone. Am J Pathol. 2009;174:239–247. doi: 10.2353/ajpath.2009.080627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8▪.Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int. 2013;94:25–34. doi: 10.1007/s00223-013-9774-y. This article provides a detailed review of osteocytes in bone remodeling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–238. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poole KE, Van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–1844. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- 11.Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–1234. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 12.Xiong J, Onal M, Jilka RL, et al. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–1241. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tatsumi S, Ishii K, Amizuka N, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–475. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Aguirre JI, Plotkin LI, Stewart SA, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Min Res. 2006;21:605–615. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 15.Bellido T. Osteocyte apoptosis induces bone resorption and impairs the skeletal response to weightlessness. BoneKEy-Osteovision. 2007;4:252–256. [Google Scholar]
- 16.Roodman GD. Targeting the bone microenvironment in multiple myeloma. J Bone Miner Metab. 2010;28:244–250. doi: 10.1007/s00774-009-0154-7. [DOI] [PubMed] [Google Scholar]
- 17.Andersen TL, Soe K, Sondergaard TE, et al. Myeloma cell-induced disruption of bone remodelling compartments leads to osteolytic lesions and generation of osteoclast-myeloma hybrid cells. Br J Haematol. 2010;148:551–561. doi: 10.1111/j.1365-2141.2009.07980.x. [DOI] [PubMed] [Google Scholar]
- 18.Taube T, Beneton MNC, McCloskey EV, et al. Abnormal bone remodelling in patients with myelomatosis and normal biochemical indices of bone resorption. Eur J Haematol. 1992;49:192–198. doi: 10.1111/j.1600-0609.1992.tb00046.x. [DOI] [PubMed] [Google Scholar]
- 19.Bataille R, Chappard D, Basle M. Excessive bone resorption in human plasmacytomas: direct induction by tumour cells in vivo. Br J Haematol. 1995;90:721–724. doi: 10.1111/j.1365-2141.1995.tb05609.x. [DOI] [PubMed] [Google Scholar]
- 20.Silvestris F, Cafforio P, Tucci M, et al. Upregulation of osteoblast apoptosis by malignant plasma cells: a role in myeloma bone disease. Br J Haematol. 2003;122:39–52. doi: 10.1046/j.1365-2141.2003.04374.x. [DOI] [PubMed] [Google Scholar]
- 21.Gunn WG, Conley A, Deininger L, et al. A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: a potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells. 2006;24:986–991. doi: 10.1634/stemcells.2005-0220. [DOI] [PubMed] [Google Scholar]
- 22.Choi SJ, Oba T, Callander NS, et al. AML-1A and AML-1B regulation of MIP-1alpha expression in multiple myeloma. Blood. 2003;101:3778–3783. doi: 10.1182/blood-2002-08-2641. [DOI] [PubMed] [Google Scholar]
- 23.Silbermann R, Bolzoni M, Storti P, et al. Bone marrow monocyte/macrophage derived activin a mediates the osteoclastogenic effect of IL-3 in multiple myeloma. Leukemia. 2014;28:951–954. doi: 10.1038/leu.2013.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehrlich LA, Roodman GD. The role of immune cells and inflammatory cytokines in Paget’s disease and multiple myeloma. Immunol Rev. 2005;208:252–266. doi: 10.1111/j.0105-2896.2005.00323.x. [DOI] [PubMed] [Google Scholar]
- 25.Giuliani N, Colla S, Morandi F, et al. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood. 2005;106:2472–2483. doi: 10.1182/blood-2004-12-4986. [DOI] [PubMed] [Google Scholar]
- 26.Giuliani N, Colla S, Rizzoli V. New insight in the mechanism of osteoclast activation and formation in multiple myeloma: focus on the receptor activator of NF-kappaB ligand [RANKL] Exp Hematol. 2004;32:685–691. doi: 10.1016/j.exphem.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 27▪.Plotkin LI. Apoptotic osteocytes and the control of targeted bone resorption. Curr Osteoporos Rep. 2014;12:121–126. doi: 10.1007/s11914-014-0194-3. This article reviews targeted bone remodeling by osteocytes apoptosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cardoso L, Herman BC, Verborgt O, et al. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res. 2009;24:597–605. doi: 10.1359/JBMR.081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noble B. Microdamage and apoptosis. Eur J Morphol. 2005;42:91–98. doi: 10.1080/09243860500096248. [DOI] [PubMed] [Google Scholar]
- 30.O’Brien FJ, Brennan O, Kennedy OD, et al. Microcracks in cortical bone: how do they affect bone biology? Curr Osteoporos Rep. 2005;3:39–45. doi: 10.1007/s11914-005-0002-1. [DOI] [PubMed] [Google Scholar]
- 31.Kennedy OD, Herman BC, Laudier DM, et al. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone. 2012;50:1115–1122. doi: 10.1016/j.bone.2012.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brennan O, Kennedy OD, Lee TC, et al. Effects of estrogen deficiency and bisphosphonate therapy on osteocyte viability and microdamage accumulation in an ovine model of osteoporosis. J Orthop Res. 2011;29:419–424. doi: 10.1002/jor.21229. [DOI] [PubMed] [Google Scholar]
- 33▪▪.Giuliani N, Ferretti M, Bolzoni M, et al. Increased osteocyte death in multiple myeloma patients: role in myeloma-induced osteoclast formation. Leukemia. 2012;26:1391–1401. doi: 10.1038/leu.2011.381. This article reports increased osteocyte apoptosis in multiple myeloma patients with bone lesions. In addition, the authors explored some of the consequences and mechanisms involved in multiple myeloma-induced osteocyte apoptosis. [DOI] [PubMed] [Google Scholar]
- 34.Al-Dujaili SA, Lau E, Al-Dujaili H, et al. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J Cell Biochem. 2011;112:2412–2423. doi: 10.1002/jcb.23164. [DOI] [PubMed] [Google Scholar]
- 35.Bivi N, Condon KW, Allen MR, et al. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J Bone Min Res. 2012;27:374–389. doi: 10.1002/jbmr.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisenberger S, Ackermann K, Voggenreiter G, et al. Metastases and multiple myeloma generate distinct transcriptional footprints in osteocytes in vivo. J Pathol. 2008;214:617–626. doi: 10.1002/path.2322. [DOI] [PubMed] [Google Scholar]
- 37.Bellido T, Plotkin LI. Novel actions of bisphosphonates in bone: preservation of osteoblast and osteocyte viability. Bone. 2011;49:50–55. doi: 10.1016/j.bone.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kong YY, Yoshida H, Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida H, Hayashi S-I, Kunisada T, et al. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- 40.O’Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone. 2013;54:258–263. doi: 10.1016/j.bone.2012.08.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pearse RN, Sordillo EM, Yaccoby S, et al. Multiple myeloma disrupts the TRANCE/osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc Natl Acad Sci U S A. 2001;98:11581–11586. doi: 10.1073/pnas.201394498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giuliani N, Bataille R, Mancini C, et al. Myeloma cells induce imbalance in the osteoprotegerin/osteoprotegerin ligand system in the human bone marrow environment. Blood. 2001;98:3527–3533. doi: 10.1182/blood.v98.13.3527. [DOI] [PubMed] [Google Scholar]
- 43▪▪.Schmiedel BJ, Scheible CA, Nuebling T, et al. RANKL expression, function, and therapeutic targeting in multiple myeloma and chronic lymphocytic leukemia. Cancer Res. 2013;73:683–694. doi: 10.1158/0008-5472.CAN-12-2280. This article reviews RANKL biology in multiple myeloma. [DOI] [PubMed] [Google Scholar]
- 44.Jakob C, Goerke A, Terpos E, et al. Serum levels of total-RANKL in multiple myeloma. Clin Lymphoma Myeloma. 2009;9:430–435. doi: 10.3816/CLM.2009.n.085. [DOI] [PubMed] [Google Scholar]
- 45.Sordillo EM, Pearse RN. RANK-Fc: a therapeutic antagonist for RANK-L in myeloma. Cancer. 2003;97(Suppl 3):802–812. doi: 10.1002/cncr.11134. [DOI] [PubMed] [Google Scholar]
- 46.Vij R, Horvath N, Spencer A, et al. An open-label, phase 2 trial of denosumab in the treatment of relapsed or plateau-phase multiple myeloma. Am J Hematol. 2009;84:650–656. doi: 10.1002/ajh.21509. [DOI] [PubMed] [Google Scholar]
- 47.Farrugia AN, Atkins GJ, To LB, et al. Receptor activator of nuclear factor-kappaB ligand expression by human myeloma cells mediates osteoclast formation in vitro and correlates with bone destruction in vivo. Cancer Res. 2003;63:5438–5445. [PubMed] [Google Scholar]
- 48.Michigami T, Shimizu N, Williams PJ, et al. Cell-cell contact between marrow stromalcells and myeloma cells via VCAM-1 and alpha[4]beta[1]-integrin enhances production of osteoclast-stimulating activity. Blood. 2000;96:1953–1960. [PubMed] [Google Scholar]
- 49.Giuliani N, Colla S, Sala R, et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand [RANKL] in T lymphocytes: a potential role in multiple myeloma bone disease. Blood. 2002;100:4615–4621. doi: 10.1182/blood-2002-04-1121. [DOI] [PubMed] [Google Scholar]
- 50▪▪.Delgado-Calle J, Bellido T, Roodman GD. Direct cell-to-cell interactions between osteocytes and multiple myeloma [MM] cells upregulate Sost and downregulate OPG expression in osteocytes: evidence for osteocytic contributions to MM-induced bone disease. Blood. 2013;122:3140. This study reports for the first time direct regulation of RANKL, OPG and Sost in osteocytes by multiple myeloma MM cells. [Google Scholar]
- 51.Weivoda MM, Oursler MJ. Developments in sclerostin biology: regulation of gene expression, mechanisms of action, and physiological functions. Curr Osteoporos Rep. 2014;12:107–114. doi: 10.1007/s11914-014-0188-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Bezooijen RL, Roelen BA, Visser A, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–814. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sutherland MK, Geoghegan JC, Yu C, et al. Sclerostin promotes the apoptosis of human osteoblastic cells: a novel regulation of bone formation. Bone. 2004;35:828–835. doi: 10.1016/j.bone.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 54.Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 55.Wergedal JE, Veskovic K, Hellan M, et al. Patients with Van Buchem disease, an osteosclerotic genetic disease, have elevated bone formation markers, higher bone density, and greater derived polar moment of inertia than normal. J Clin Endocrinol Metab. 2003;88:5778–5783. doi: 10.1210/jc.2003-030201. [DOI] [PubMed] [Google Scholar]
- 56.Balemans W, Patel N, Ebeling M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X, Niu Q-T, Sun N, et al. Mice lacking sclerostin have increased bone formation and bone strength. Calcif Tissue Int. 2007;80(Suppl 1):S24. [Google Scholar]
- 58.Ke HZ, Richards WG, Li X, et al. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr Rev. 2012;33:747–783. doi: 10.1210/er.2011-1060. [DOI] [PubMed] [Google Scholar]
- 59.Terpos E, Christoulas D, Katodritou E, et al. Elevated circulating sclerostin correlates with advanced disease features and abnormal bone remodeling in symptomatic myeloma: reduction postbortezomib monotherapy. Int J Cancer. 2012;131:1466–1471. doi: 10.1002/ijc.27342. [DOI] [PubMed] [Google Scholar]
- 60.Brunetti G, Oranger A, Mori G, et al. Sclerostin is overexpressed by plasma cells from multiple myeloma patients. Ann N Y Acad Sci. 2011;1237:19–23. doi: 10.1111/j.1749-6632.2011.06196.x. [DOI] [PubMed] [Google Scholar]
- 61▪▪.Wang XT, He YC, Zhou SY, et al. Bone marrow plasma macrophage inflammatory protein protein-1 alpha [MIP-1 alpha] and sclerostin in multiple myeloma: relationship with bone disease and clinical characteristics. Leuk Res. 2014;38:525–531. doi: 10.1016/j.leukres.2014.02.010. This article reports elevated sclerostin levels in the serum of multiple myeloma patients and the potential relationship with multiple myeloma-induced bone disease. [DOI] [PubMed] [Google Scholar]
- 62.Colucci S, Brunetti G, Oranger A, et al. Myeloma cells suppress osteoblasts through sclerostin secretion. Blood Cancer J. 2011;1:e27. doi: 10.1038/bcj.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Habibi H, Abroun S, Hajifathali A, et al. Osteogenic inhibition in multiple myeloma. Cell J. 2013;15:266–271. [PMC free article] [PubMed] [Google Scholar]
- 64.Mabilleau G, Mieczkowska A, Edmonds ME. Thiazolidinediones induce osteocyte apoptosis and increase sclerostin expression. Diabet Med. 2010;27:925–932. doi: 10.1111/j.1464-5491.2010.03048.x. [DOI] [PubMed] [Google Scholar]
- 65.Wijenayaka AR, Kogawa M, Lim HP, et al. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One. 2011;6:e25900. doi: 10.1371/journal.pone.0025900. [DOI] [PMC free article] [PubMed] [Google Scholar]