

Abstract
Translation is a fundamental and highly regulated cellular process. Previously, we reported that the kinase and transcription elongation factor Ctk1 increases fidelity during translation elongation in Saccharomyces cerevisiae. Here, we show that loss of Ctk1 function also affects the initiation step of translation. Translation active extracts from Ctk1-depleted cells show impaired translation activity of capped mRNA, but not mRNA reporters containing the cricket paralysis virus (CrPV) internal ribosome entry site (IRES). Furthermore, the formation of 80S initiation complexes is decreased, which is probably due to reduced subunit joining. In addition, we determined the changes in the phosphorylation pattern of a ribosome enriched fraction after depletion of Ctk1. Thus, we provide a catalogue of phosphoproteomic changes dependent on Ctk1. Taken together, our data suggest a stimulatory function of Ctk1 in 80S formation during translation initiation.
INTRODUCTION
Translation initiation is a very intricate and highly dynamic process leading to the formation of an elongation competent 80S ribosomal complex at the start codon of the mRNA (1–4). Translation initiation starts with the association of the methionyl initiator tRNA (Met-tRNAi) to the GTP-bound eukaryotic initiation factor 2 (eIF2) to form a ternary complex (TC). In Saccharomyces cerevisiae, this TC is thought to bind another subset of initiation factors to form the multifactor complex (5), which then associates with the 40S ribosomal subunit, resulting in the formation of the 43S initiation complex. The 43S complex binds to the mRNA at its 5′ 7-methylguanosine cap in a process facilitated by eIF3, Pab1, eIF4B (B subunit of eIF4), and the multisubunit eIF4F complex to generate the 48S initiation complex (6). Within the 48S initiation complex, the 40S subunit with its associated initiation factors is thought to scan along the mRNA until it reaches the start codon. Here, correct base pairing of the initiator-tRNA Met-tRNAi with the AUG located at the ribosomal P-site causes arrest of the scanning preinitiation complex (PIC), and eIF5 promoted hydrolysis of eIF2-bound GTP and dissociation of several initiation factors. eIF5B, a GTPase, facilitates the subsequent joining of the 60S subunit, which results in an elongation competent 80S complex (1–4, 6, 7).
In addition to canonical translation initiation, a cap-independent mechanism of translation initiation exists. For example, many viruses contain special sequences in their mRNA named internal ribosome entry sites (IRESs) that also mediate cap-independent translation when canonical translation initiation is shut down. IRESs can be classified according to their dependence on translation initiation factors (reviewed in reference 8). At one extreme are the class IV IRESs exemplified by the cricket paralysis virus (CrPV) IRES that function independently of any translation initiation factors (8, 9). In addition to viruses, where they have been mainly studied, IRESs are also present in cellular mRNAs of eukaryotes. Two examples for eukaryotic IRESs are the ones present in the yeast NCE102 and GPR1 mRNAs, which require at least the initiation factor eIF4G (8, 10). The cellular role, abundance, and mode of 80S recruitment of eukaryotic IRESs remain largely enigmatic. It is assumed that the translation of cellular IRES-containing mRNAs is also favored when general translation is shut down, e.g., under stress conditions (10, 11).
Ctk1 is the kinase subunit of the carboxy-terminal domain kinase I (CTDK-I) complex. CTDK-I phosphorylates RNA polymerase II (RNAPII) on the carboxyl-terminal repeat domain (CTD) of its largest subunit, which is important for efficient transcription elongation, mRNA 3′-end processing, and transcription termination of small noncoding RNAs (12, 13). Interestingly, Ctk1 also plays a role in RNA polymerase I (RNAPI) transcription (14, 15). Previously, we showed that Ctk1—in addition to its well-described function in transcription—has a second function in translation (16). Ctk1 function increases the correct decoding of mRNA during translation elongation by phosphorylating residue S238 of Rps2, a protein of the small ribosomal subunit. Rps2 has long been known to be essential for translational accuracy (17) and is located at the beginning of the mRNA entry tunnel of the small ribosomal subunit (18).
In this study, we investigate whether Ctk1 has a second function in translation, since the dramatic decrease of translation activity upon Ctk1 depletion cannot be explained solely by the missing phosphorylation of Rps2. We show that depletion of Ctk1 leads to a translation initiation defect in vitro. In a proteomic screen for a substrate(s) of Ctk1, we identify changes in the phosphorylation patterns of numerous ribosomal proteins and proteins involved in ribosome biogenesis and translation upon Ctk1 depletion. Interestingly, formation of 80S complexes and subunit joining is decreased upon Ctk1 depletion. Taken together, Ctk1 stimulates—directly or indirectly—translation initiation.
MATERIALS AND METHODS
Yeast strains and plasmids.
All strains are derived from the Saccharomyces cerevisiae W303 wild-type (wt) strain (MATa/α ura3-1 trp1-1 his3-11,15 leu2-3,112 ade2-1 can1-100 GAL+). The GAL1::CTK1-TAP, Δctk1::HIS3, and Δrps2::HIS3 strains were described previously (16). The Δlys::KANMX6 strain was generated by exchanging the LYS1 open reading frame (ORF) with the KANMX6 cassette by homologous recombination. The GAL1::CTK1-TAP Δlys::KANMX6 strain was produced by mating the GAL1::CTK1-TAP and Δlys::KANMX6 strains.
For measuring translation activity of capped mRNA, the pSP6-Luc (Luc stands for luciferase) plasmid (19) and IRES-containing mRNAs plasmids pWG186 (capped mRNA), pWG290 (GPR1 IRES), pWG324 (NCE102 IRES), and pWG299 (CrPV IRES) (10) were used. Plasmids pRS315-RPS2 and pRS315-rps2-S238A were described previously (16). The pRS315-CTK1 plasmid was generated by cloning the CTK1 ORF with 450-bp promoter and 150-bp terminator sequences from a pUN100 library plasmid into the SmaI site of pRS315 (20). The pRS315-CTK1-D324N plasmid was generated by site-directed mutagenesis. The BSEF-RPL38, BSEF-RPL41a-22bp5′UTR, the BSEF-RPL41a-80bp5′UTR, and BSEF-PGK1 plasmids were generated by exchanging the BamHI-SacI insert of BSEF (21) with the sequence of the corresponding gene (for PGK1, the ORF was shortened to 203 bp). The length of the 5′ untranslated regions (5′UTRs) of the RPL38, RPL41, and PGK1 mRNAs was chosen by the method of Miura et al. (22).
In vitro transcription.
The IRES plasmids and the corresponding m7GpppG-capped positive control were linearized with EcI136II before being transcribed with T7 polymerase (New England BioLabs). The GpppA cap for the IRES constructs and the m7GpppG cap for the positive control (both from KEDAR, Poland) were added to the reaction mixture together with ATP, CTP, and UTP. After a 5-min incubation at 37°C, GTP was added, and DNA was transcribed for 1 h at 37°C. The template DNA was digested with DNase for 15 min at 37°C. The capped luciferase mRNA was in vitro transcribed from pSP6P (23) after its linearization with BsrBI (Fermentas). In vitro transcription was carried out with the AmpliCap high-yield message maker kit (Biozym) according to the manufacturer's directions. For generating radiolabeled mRNA, the BSEF-RPL38, BSEF-RPL41a-22bp5′UTR, BSEF-RPL41a-80bp5′UTR, and BSEF-PGK1 plasmids were linearized with HindIII before being in vitro transcribed with T3 polymerase (Roche). m7GpppG (KEDAR, Poland) together with ATP, CTP, and [α-32P]UTP were added to the reaction mixture. After a 5-min incubation at 37°C, GTP was added, and DNA was transcribed for 1 h at 37°C. The template DNA was then digested with DNase for 15 min at 37°C. Template mRNA for the toeprint assay was generated accordingly, except for the use of nonradioactive UTP. All mRNAs were purified by using the RNA MinElute kit according to the manufacturer's directions (Qiagen).
In vitro translation.
In vitro translation active extracts were prepared as described previously (16, 19). For (mock) depletion, 4-liter yeast extract-peptone-dextrose (YPD) cultures were inoculated with an overnight culture of cells grown in yeast extract-peptone-glucose (YPG). Cells were harvested after 18 h after reaching an optical density at 600 nm (OD600) of 1.0 to 1.2. In vitro translation assays were performed as described in reference 19. Approximately 300 ng of in vitro-transcribed mRNA was used for all in vitro translation assays.
Translation initiation assay.
Three A260 units of translation active extract was incubated with 6.25 μg creatine kinase (Roche) for 5 min on ice, followed by 30-min incubation with 1.25 mM EGTA, 2 mM magnesium acetate (MgAc), 76 mM KCl, 0.4 mM GTP, 1 mM ATP, 50 μM each amino acid, 12.5 mM creatine phosphate, 1 mM cycloheximide, RNase inhibitor, 35 ng of in vitro-transcribed radioactive mRNA, and where indicated, 1.25 mM 5′-guanylylimidodiphosphate (GMP-PNP) in a total volume of 120 μl at room temperature. The entire reaction mixture was loaded on a 5 to 25% sucrose gradient and centrifuged at 39,000 rpm (SW40 rotor) for 2 h. Fractions (450-μl fractions) were taken off, and 300 μl of each fraction was counted with 3 ml of scintillation cocktail (Roth) using a scintillation counter (PerkinElmer Tri-Carb 2810TR).
Toeprint assay.
Gradients with translation initiation reaction mixtures were set up as described above with the following exception. Only 1.3 mM cycloheximide and, where indicated, 0.3 mM GMP-PNP were used, and the extracts were incubated with 200 to 250 ng of in vitro-transcribed nonradioactively labeled RPL38 mRNA. Fractions (400-μl fractions) were taken off with a Teledyne ISCO gradient machine. Twenty-five microliters of each fraction was diluted 1:4 with dilution buffer (50 mM Tris-HCl [pH 7.5], 60 mM KCl, 6 mM MgCl2, 5 mM dithiothreitol [DTT], 0.5 mM cycloheximide, 0.5 mM dTTP, 0.5 mM dCTP, 0.5 mM dGTP, 5 nM dATP, and RNase inhibitor), and the reaction mixture was incubated for 105 s at 52°C, placed on ice, and incubated with 25 pmol primer (5′-GGCGGTCTTAACGTCAGCT-3′) at 37°C for 5 min. For reverse transcription, 0.5 μl [α-32P]dATP and 1 μl SuperscriptII (Invitrogen) were added, and the reaction mixture was incubated at 37°C for 15 min. SDS and EDTA were added, followed by a phenol-chloroform extraction of the reverse transcripts. The DNA was precipitated with rRNA as a coprecipitator, resuspended in denaturing formamide loading buffer, and loaded on a prerun 13% polyacrylamide denaturing sequencing gel. In the case of the 48S fraction of Ctk1-depleted cells, only half of the resuspended pellet was loaded on the gel. The gel was run at 45 W for 1 h 50 min, dried, and exposed to a phosphorimager for at least 3 days. The plate was scanned with a Storm 860 imager (Molecular Dynamics), and the bands were quantified using MultiGauge software (Fujifilm). The sequencing reaction was done according to the manufacturer's directions (USB Sequenase 2.0).
Polysome gradients.
Polysome gradients were performed as described in reference 16 except that cells were grown in minimal medium. The absorption profile was recorded with a Teledyne ISCO gradient machine.
Analysis of rRNA intermediates.
Fifty milliliters of phosphate-depleted YPD (YPD-P) was inoculated with wt or GAL1::CTK1-TAP cells growing at stationary phase in YPG. After 12 h, cells were diluted to an OD600 of 0.3 in YPD-P and grown exponentially for 6 h. Cells were labeled at an OD600 of 0.8 with 15 μCi/ml 32Pi for 5 min. Two milliliters of the cell suspensions were harvested, washed, and resuspended in buffer containing 1 M sorbitol. The suspension was incubated with 100 U Zymolyase 20T and 0.2% β-mercaptoethanol at 37°C for 5 min, centrifuged (300 rpm/1 min), and the pellet was washed before lysis according to the RNeasy standard protocol (Qiagen). The RNA was isolated using the RNeasy kit (Qiagen). RNA (1 or 3 μg) was mixed with formamide and ethidium bromide (EtBr) containing loading dye and run on a 1% denaturing formaldehyde agarose gel at 110 V for 5 h. The 25S RNA was detected with UV light, and the rRNA intermediates were analyzed with a phosphorimager (10-min exposure) and quantified with AIDA software. For a better comparison of the amounts of processed rRNA intermediates, wt signals of the lane with 1 μg total RNA and Ctk1-depleted signals of the lane with 3 μg total RNA were quantified.
Western blotting.
After 18-h depletion, Ctk1- and mock-depleted cells were harvested at an OD600 of 0.5 to 1.0 and lysed via alkaline lysis with NaOH. SDS-PAGE and semidry Western blotting were performed by standard methods. The eIF2α-P51 antibody was purchased from Invitrogen. For 3-aminotriazole (3AT) treatment, 3AT was added to the yeast culture to a final concentration of 80 mM at an OD600 of 0.5. The cells were harvested at an OD600 of 1.0.
RESULTS
Translation active extracts of Ctk1-depleted cells have a reduced translation activity for a capped mRNA, but not for a CrPV internal ribosome entry site (IRES)-containing mRNA.
As reported previously, Ctk1 phosphorylates Rps2, a protein of the small ribosomal subunit, which is essential for translational accuracy and located at the beginning of the mRNA entry tunnel (17, 18). Consistently, phosphorylation of Rps2 on S238 by Ctk1 increases translational accuracy (16). However, the translation defect of an rps2-S238A (the S-to-A change at position 238 encoded by rps2) mutant is not as severe as the translation defect of Ctk1-depleted cells (16; also see below), indicating that Ctk1 has an additional role in translation. In order to identify this potential novel function of Ctk1 in translation, we assessed whether Ctk1 is important for translation initiation. The Δctk1 strain is not suited to assess a function of Ctk1 in translation, since it has a severe growth defect and shuts down translation nonspecifically as assessed by phosphorylation of eIF2α (Fig. 1). Instead, Ctk1 was depleted from cells by expressing endogenous Ctk1 under the control of the GAL1 promoter and shift of the cells to a glucose-containing medium, since this leads to almost complete depletion of Ctk1 but only a minor growth defect (16). For a control, mock-depleted cells were used. The mock-depleted cells expressed Ctk1 under the control of its endogenous promoter, resulting in continued expression of Ctk1 after the cells were shifted to glucose-containing medium.
FIG 1.

Impaired translation in Ctk1-depleted cells is not due to an overall stress defect as in Δctk1 cells. (A) Upon amino acid starvation (30) and some other stress conditions, e.g., peroxide stress (31, 32), eIF2α is phosphorylated, leading to a decrease in ternary complex (TC) formation and thus inhibition of translation initiation (30). Western blotting of Δctk1 and Ctk1- and mock-depleted extracts against total eIF2α and phosphorylated eIF2α (eIF2α-P) reveals that the phosphorylated form of eIF2α is increased in Δctk1 cells and cells treated with 3-aminotriazole (3AT), which served as the positive control. In contrast, eIF2α phosphorylation is not increased in Ctk1-depleted cells. (B and C) Consistent with an inhibition of translation initiation by deletion of Ctk1, the polysome profiles of Δctk1 cells (C) showed an increased 80S peak with a concomitant decrease in polysomes compared to wild-type (wt) cells (B). The polysome/monosome (80S) (P/M) ratio was calculated by integrating the area under the respective peaks after subtraction of the values of the baseline. Values are means ± standard errors of the means (SEM) (error bars) from three independent experiments. Changes in the P/M ratio of Δctk1 and Ctk1-depleted cells compared to wt cells are statistically significant (P < 0.05 by Student's t test). (D) 48S formation is impaired in Δctk1 cells, providing further evidence that translation is shut down nonspecifically in this strain. In contrast to the defect observed for Ctk1-depleted cells (Fig. 5F), translation initiation is affected at the stage of 48S complex formation when cells are stressed (30, 32). The incorporation of RPL38 mRNA into 48S initiation complexes analyzed by an initiation assay is increased in Ctk1-depleted extracts (Fig. 5F) and stays the same in Δctk1 extracts (D). Since 80S complex formation is also decreased in Δctk1 extracts, a decrease in 48S complex formation is probably masked. A defect in 48S complex formation is also consistent with increased mRNP formation. The nonhydrolyzable GTP analogue GMP-PNP, which prevents subunit joining and therefore leads to accumulation of 48S complexes, was added to the initiation assay for better analysis of 48S initiation complexes. The error bars represent the standard deviations from at least three independent experiments. In summary, Δctk1 cells have a defect in translation initiation most likely caused by stress that results from its severe growth impairment (see the text).
To assess a possible function of Ctk1 in translation initiation, we exploited the fact that translation of IRES-containing mRNAs can circumvent the cap- and translation initiation factor-dependent translation initiation mechanism (see the introduction). Translation of a CrPV IRES-containing mRNA does not require any translation initiation factors, whereas translation of two yeast IRESs, from the NCE102 and GPR1 genes, requires at least the initiation factor eIF4G (8, 10). We thus compared the activity of translation active extracts of Ctk1- versus mock-depleted cells to translate a capped mRNA versus the three different IRES-containing mRNAs in order to distinguish between a defect in translation initiation versus another step of translation (Fig. 2A). Translation of the capped mRNA and the two yeast IRES RNAs was reduced to 20% in extracts of Ctk1-depleted cells compared to mock-depleted wild-type (wt) cells (Fig. 2B) (16). Importantly, translation of the CrPV IRES-containing RNA was reduced to only 80% in Ctk1-depleted extracts, indicating that the translation defect of these extracts is largely circumvented by the initiation mechanism of this IRES. The fact that translation of this IRES-containing mRNA is 20% lower than in wt extracts is most likely caused by the elongation defect in Ctk1-depleted extracts (16). All three IRES-containing mRNAs were translated less efficiently than the capped mRNA (0.2% to 4% of the capped construct), but these mRNAs were still translated around 100× more efficiently in the mock-depleted extracts than in the negative control containing an ApppG cap and a stem-loop but no IRES. The three different IRES-containing mRNAs were translated with similar efficiencies with the NCE102 IRES-containing mRNA being translated best and the GPR1 IRES-containing mRNA being translated least (Fig. 2C). Since only translation initiation mediated by the CrPV IRES is independent of any translation initiation factors, this result suggests that Ctk1 mediates the correct and efficient interplay of translation initiation factors necessary to recruit translation competent 80S ribosomes to the start codon.
FIG 2.

Ctk1 stimulates translation initiation. (A) Firefly luciferase (F-luc) reporter constructs used to assess initiation-independent translation contain a nonphysiological ApppG cap followed by an inhibitory stem-loop and an IRES. IRESs used correspond to the CrPV IRES or yeast IRESs of the GPR1 or NCE102 gene. A corresponding reporter that carried an m7G cap and lacked the inhibitory stem-loop as well as the IRES was used as a control for canonical translation. (B) Loss of Ctk1 causes decreased translation activity of capped mRNA, but not of CrPV IRES-containing mRNA. Translation activity of translation active extracts from mock- and Ctk1-depleted cells for the reporters shown in panel A was analyzed. For each RNA construct, the activity of the mock-depleted extracts was set at 100%. The values for mock-depleted and Ctk1-depleted cells are significantly different (P < 0.05 by Student's t test) as indicated by the brackets and asterisks. (C) Absolute values of luciferase activity of mock- and Ctk1-depleted extracts for the three IRES-containing reporters. The error bars represent the standard deviations from at least three independent experiments. Luciferase activity is shown in arbitrary units (AU). (D and E) Initiation is slightly but significantly impaired in Ctk1-depleted cells in vivo. The 80S peak in polysome profiles of Ctk1-depleted cells (E) is increased compared to mock-depleted cells (D), reflecting a translation initiation defect. The polysome/monosome (80S) (P/M) ratio was calculated by integrating the area under the respective peaks after subtraction of the values of the baseline. Values are means ± SEM from three independent experiments. Changes in the P/M ratio of Ctk1 compared to mock-depleted cells are statistically significant (P < 0.05 by Student's t test).
To assess whether translation initiation by depletion of Ctk1 is also impaired in vivo, we analyzed polysome profiles of mock- and Ctk1-depleted cells. Consistent with impaired translation initiation, the 80S peak increases, whereas the polysome peaks decrease when Ctk1 is depleted (Fig. 2D and E). The decrease in the polysome/monosome (P/M) ratio of Ctk1- versus mock-depleted cells is small but significant (Fig. 2D and E). The surprisingly small effect on the P/M ratio and the lack of halfmers in gradients (also see below) of Ctk1-depleted cells is probably due to the fact that in vitro translation experiments are more sensitive to perturbation than the polysome gradients that reflect an in vivo situation. Alternatively, the in vitro translation defect could also be due to a missing or defective translation factor or ribosome that can be compensated for in the in vivo situation. Taken together, in addition to its function in translation elongation, Ctk1 stimulates translation initiation.
Ctk1 depletion causes reduced phosphorylation of proteins involved in ribosome biogenesis and translation.
Since Ctk1 is a serine/threonine kinase, we determined whether the Ctk1 kinase activity is needed for efficient translation. Translation active extracts from a strain depleted for wt Ctk1 but expressing the kinase dead Ctk1 mutant encoded by ctk1-D324N (24) (Fig. 3A) showed the same reduction in overall translation efficiency as the Ctk1 depletion (Fig. 3B). Thus, the kinase activity of Ctk1 is essential for Ckt1's function in translation. Furthermore, a different substrate than Rps2 is important for full translational activity, since the rps2-S238A mutant does not exhibit a translation defect in vitro (Fig. 3C).
FIG 3.

Ctk1's kinase activity is required for its function in translation. Translation active extracts were prepared from cells expressing CTK1 from the GAL1 promoter and containing a plasmid encoding either wt Ctk1, the kinase dead Ctk1 mutant Ctk1-D324N (24), or an empty plasmid. After the cells were shifted to glucose-containing medium, only the CTK1 copy on the plasmid was expressed. (A) Cellular levels of the Ctk1 wt protein and the D324N mutant are similar. Ctk1 depl., Ctk1 depletion; α-Ctk1, anti-Ctk1 antibody. (B) Translation of a luciferase reporter mRNA is reduced to 30% in Ctk1-depleted extracts as well as in extracts expressing Ctk1-D324N after Ctk1 depletion. The values for mock- and Ctk1-depleted and mock-depleted and Ctk1-D324N cells are significantly different (*, P < 0.05, and **, P < 0.01, by Student's t test). (C) The missing phosphorylation of Rps2 on S238 in Ctk1-depleted cells is not responsible for the strong translation defect in Ctk1-depleted cells. In contrast to translation fidelity (16), the overall translation rate of extracts of cells expressing a wild-type Rps2 or Rps2-S238A is the same as that measured by a luciferase reporter assay.
We thus wanted to identify Ctk1's substrate(s) important for translation initiation. To this end, we employed a proteomic approach to analyze the phosphorylation status of ribosomes and ribosome-associated proteins in mock- and Ctk1-depleted cells using stable isotope labeling with amino acids in cell culture (SILAC) and subsequent mass spectrometry (see Fig. S1 in the supplemental material). Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) analysis of these samples showed a multitude of changes in the phosphorylation pattern upon Ctk1 depletion (see Table S1 in the supplemental material). Interestingly, a number of phosphorylation sites are less phosphorylated in Ctk1-depleted cells. The proteins listed in Table S2 were selected based on their known function in translation or ribosome biogenesis and tested for direct phosphorylation by Ctk1 in in vitro kinase assays with purified CTDK-I complex and the candidate substrate. Three of the 23 candidate substrates tested were directly phosphorylated by Ctk1, namely, Sda1, Mrs6, and Ltv1 (data not shown). However, serine-to-alanine mutations at the site identified in the mass spectrometric analysis in these three proteins showed unchanged phosphorylation by Ctk1 in vitro (data not shown), indicating that the decreased phosphorylation of this site upon Ctk1 loss is likely not due to a direct phosphorylation by this kinase. However, even though we have been unable to identify a direct target of Ctk1, we provide a catalogue of phosphoproteomic changes upon Ctk1 depletion, i.e., not obscured by unspecific changes caused by the growth defect of Δctk1 cells, that might provide a valuable resource for other researchers interested in Ctk1 function.
Loss of Ctk1 causes a decrease in 80S initiation complex formation.
Since the phosphorylation status of many proteins involved in ribosome biogenesis changed due to Ctk1 depletion (see Table S1 in the supplemental material), we assessed whether defective ribosome biogenesis could be the cause of the translation initiation defect in Ctk1-depleted extracts. Consistent with previous studies (14), the RNAPI transcription rate was decreased in Ctk1-depleted cells (Fig. 4A). Thus, the decreased amount of rRNA and consequently ribosomes could cause the decrease in translation initiation. On the other hand, this seems unlikely, as the levels of Rps8 and Rpl6 and thus most likely both ribosomal subunits are not decreased in extracts of Ctk1-depleted cells (25). In addition, as rRNA processing is not affected by Ctk1 depletion (Fig. 4B), this is also most likely not the reason for the translation initiation defect.
FIG 4.
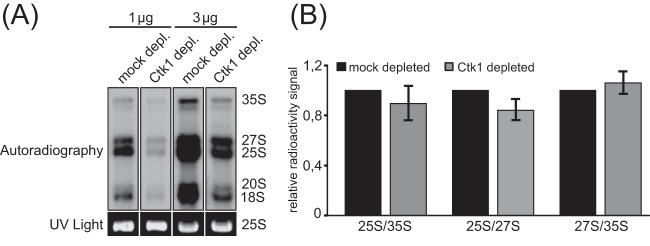
rRNA processing is not affected by loss of Ctk1 function. Since the phosphorylation state of many proteins involved in ribosome biogenesis changed due to Ctk1 depletion (see Table S2 in the supplemental material), we analyzed whether defective ribosome biogenesis could be the cause of the translation initiation defect in Ctk1-depleted extracts. To test whether rRNA processing is affected upon Ctk1 depletion, we performed pulse-chase experiments with mock- and Ctk1-depleted cells. (A) Consistent with previous studies showing an involvement of Ctk1 in RNAPI transcription (14, 15), the intensity of radiography signals was decreased upon Ctk1 depletion, but the ratios of RNA intermediates (25S/35S, 25S/27S, and 27S/35S) did not change significantly upon Ctk1 depletion. (B) Quantification of the signals shown in panel A. The error bars represent the standard deviations from at least three independent experiments.
In order to corroborate that Ctk1 indeed stimulates translation initiation and to determine specifically which step in translation initiation is affected, we next compared the incorporation of radiolabeled mRNA into translation initiation complexes in extracts of Ctk1- and mock-depleted cells. To do this, we adapted the initiation assay described by Beckmann and colleagues (21) to yeast (Fig. 5A). Four yeast transcripts were used for this analysis: RPL38 mRNA, RPL41 mRNA with two different lengths of the 5′UTR (22 bp and 80 bp), and PGK1 mRNA with a shortened open reading frame (ORF). These mRNAs were selected based on their high expression, lack of introns, and—in the case of the mRNAs coding for the ribosomal proteins—short ORFs necessary for a good resolution of the initiation complexes on sucrose density gradients. All mRNAs contain a 3′UTR of approximately 140 bp and a poly(A) tail of 71 adenines. To characterize these four novel reporter mRNAs, their incorporation into initiation complexes was first analyzed in wt extracts under conditions in which translation initiation occurs but elongation is blocked by the addition of cycloheximide. The resulting translation initiation complexes were separated on a sucrose density gradient followed by scintillation counting of the gradient fractions in order to assess the distribution of the radioactively labeled mRNA (Fig. 5A). All four mRNAs were mostly incorporated into 80S initiation complexes with a smaller portion present in the 48S initiation complex or in the light, ribosome-free fractions.
FIG 5.

In extracts of Ctk1-depleted cells, formation of 80S initiation complexes is decreased. (A) Incorporation of PGK1, RPL38, RPL41 with 80-bp 5′UTR (RPL41 80 bp), and RPL41 with 22 bp 5′UTR (RPL41 22 bp) into initiation complexes was analyzed in initiation reactions of translation active extracts. The fractions containing mRNPs, 48S, and 80S complexes are indicated. (B to E) In comparison to mock-depleted cells, Ctk1 depletion caused a decrease in incorporation of RPL38 mRNA (B), PGK1 mRNA (C), RPL41 mRNA with 80-bp 5′UTR (D), and RPL41 mRNA with 22 bp 5′UTR (E) into 80S initiation complexes. 80S formation was decreased to 68% ± 4% for the RPL38 mRNA, to 54% ± 16% for the RPL41 (22-bp) mRNA, to 68% ± 8% for the RPL41 (80-bp) mRNA and to 70% ± 4% for the PGK1 mRNA. Free mRNPs and, to a lesser extent, mRNAs bound to 48S initiation complexes are increased upon Ctk1 depletion with all mRNAs tested. Differences in the 80S peak area between Ctk1- and mock-depleted cells are statistically significant (P < 0.05 by Student's t test). (F) The incorporation of RPL38 mRNA into 48S initiation complexes is increased in Ctk1-depleted extracts. The experiment was performed as described above for panels A to E except for the addition of the nonhydrolyzable GTP analogue GMP-PNP, which prevents subunit joining and therefore leads to accumulation of 48S complexes. Differences in the 48S peak area between Ctk1- and mock-depleted cells are statistically significant (P < 0.05 by Student's t test). Each error bar represents the standard deviation of each fraction from at least three independent experiments.
To assess the translation initiation defect in Ctk1-depleted cells, initiation assays were performed with extracts from Ctk1- and mock-depleted cells. Strikingly, the incorporation of mRNA into 80S ribosomes is lower in Ctk1-depleted extracts than in mock-depleted extracts. The level of mRNA incorporated in 80S complexes is reduced to 54 and 70% compared to mock-depleted extracts as measured by integration of the 80S peak area for each of the four mRNAs (Fig. 5B to E). Importantly, the decrease in 80S formation is statistically significant (Fig. 5B to E). This defect does not seem to be specific for a certain mRNA, since it occurred with all four reporters (Fig. 5B to E). In addition, the amount of mRNAs in the messenger ribonucleoprotein particle (mRNP) and—at least for the RPL38 mRNA (see below)—the 48S PIC fractions is increased, suggesting a defect in 48S PIC and 80S initiation complex formation (Fig. 5B to E). In order to corroborate that formation of 48S complexes is increased in Ctk1-depleted extracts, an initiation assay was performed in the presence of GMP-PNP, which prevents subunit joining, leading to an accumulation of 48S PICs and thus rendering an increase of 48S PICs of Ctk1- versus mock-depleted extracts more visible. The significant increase in 48S PICs in Ctk1-depleted extracts (as determined by comparison of the 48S peak areas and application of Student's t test; P < 0.05) suggests that a step in translation initiation after the formation of 48S PICs is affected upon Ctk1 depletion (Fig. 5F). In contrast to the defect observed for Ctk1-depleted cells, 48S complex formation is decreased in Δctk1 cells as the peak for mRNPs is increased, the 80S peak is decreased, and the 48S peak is basically unaffected (Fig. 1D). Thus, translation initiation is affected at the stage of 48S complex formation in Δctk1 cells as indicated by the increased phosphorylation of eIF2α (Fig. 1A), corroborating that the initiation block in these cells is due to stress. Taken together, 80S formation is defective in Ctk1-depleted extracts.
40S subunits accumulate at the start codon in extracts of Ctk1-depleted cells.
A decreased conversion of 48S PICs to 80 initiation complexes in Ctk1-depleted extracts could be caused either by decreased scanning of the 5′UTR by the small ribosomal subunit or by decreased 60S subunit joining at the start codon. To distinguish between these two possibilities, we determined the position of the 40S subunit on the RPL38 mRNA in Ctk1-depleted extracts by a toeprint assay. Translation reactions were performed with unlabeled RPL38 mRNA, the proteins were separated on sucrose density gradients, and fractions containing 48S initiation complexes were pooled (Fig. 6A to C). Reverse transcription with a primer annealing to the ORF in the direction of the cap renders transcription products whose lengths depend on the position of the 40S subunit on the mRNA (“toeprint”). Thus, 40S subunits positioned at the start codon yield a distinct “short” transcription product, whereas scanning 40S subunits yield a “long” transcription product, since loosely bound 40S subunits are either displaced from the mRNA during the reverse transcription reaction or are not detected, since scanning is a very fast process, rendering scanning intermediates difficult to detect. Importantly, there was a clear toeprint signal for 40S subunits positioned at the start codon in Ctk1-depleted extracts in contrast to mock-depleted extracts, indicating that 40S subunits accumulate at the start codon (Fig. 6D). The toeprint for the long transcript, on the other hand, was about the same in both extracts (Fig. 6D). Thus, the ratio of the short transcript to the long transcript, i.e., the fraction of mRNAs with 40S subunits at the AUG codon, increases 1.3-fold when Ctk1 is missing (Fig. 6E). This indicates a subunit joining defect caused by the lack of Ctk1. The fact that this defect is not visible in the absorption profiles (Fig. 6A to C) might be due to the fact that in the toeprint assay, only newly formed 48S complexes are assessed, whereas on the gradient, all ribosomal complexes are visible. As a positive control for a subunit joining defect, GMP-PNP, a nonhydrolyzable GTP analogue, which blocks subunit joining, was added to the mock-depleted reaction mixture. As expected, this treatment led—as in the case of Ctk1-depleted cells—to an increased toeprint signal for the short transcripts corresponding to 40S positioned at the start codon (Fig. 6D). The ratio of short to long transcripts in GMP-PNP-treated extracts increased 1.6-fold (Fig. 6E). Thus, 40S initiation complexes accumulate to a significant degree at the start codon in Ctk1-depleted and GMP-PNP-treated extracts. These results suggest that the reduced formation of 80S initiation complexes in Ctk1-depleted extracts might be due to a ribosomal subunit joining defect.
FIG 6.

Loss of Ctk1 function causes 40S subunits to accumulate at the start codon of the RPL38 mRNA in vitro. (A to C) Gradient fractions (in gray) of 48S and 80S initiation complexes were taken from mock-depleted (A), Ctk1-depleted (B), and GMP-PNP-treated mock-depleted extracts (C). (D) The 40S subunit accumulates at the start codon of 48S initiation complexes in Ctk1-depleted extracts. Toeprints of fractions taken from panels A to C were determined by primer extension and analysis on a sequencing gel. The short toeprint is produced when the 40S subunit is located at the start codon (AUG), whereas the long transcript is generated when the primer is extended to the cap of the mRNA. The plasmid DNA of the template for the mRNA used was sequenced to determine the position on the gel. (E) Quantification of the ratios of the short transcript versus the long transcript of the 48S initiation complexes shown in panel D. The signal for the short transcript is significantly increased in Ctk1-depleted extracts in comparison to mock-depleted extracts. The error bars represent the standard deviations from three independent experiments. The values for mock- and Ctk1-depleted and mock-depleted and GMP-PNP-treated cells are significantly different (*, P < 0.05, and **, P < 0.01, by Student's t test).
DISCUSSION
Previously, we showed that the transcription factor Ctk1 functions in translation elongation by enhancing translational accuracy through phosphorylation of the ribosomal protein Rps2 (16). In this study, we present a second function of Ctk1 in translation. We show that loss of Ctk1 also affects translation initiation. Specifically, depletion of Ctk1 impairs formation of 80S initiation complexes in vitro. 80S formation is a multistep process, and it remains to be determined which of these steps is stimulated by Ctk1—either directly or indirectly. However, initiation factors are likely to play a role because translation of the CrPV IRES-containing reporter RNA, which does not depend on any translation initiation factors, was not significantly affected in cells lacking Ctk1 (Fig. 2). This is consistent with the finding that the levels of the ribosomal proteins Rps8 and Rpl6 and thus most likely the levels of ribosomal subunits are not affected in extracts of Ctk1-depleted cells (16, 25). In addition, the translation elongation defect in extracts of Ctk1-depleted cells can be rescued by adding purified CTDK-I complex back, indicating that the ribosomes in these extracts are functional (16). Ctk1 could, for instance, phosphorylate an initiation factor and modulate its recruitment to or its dissociation from initiation complexes. Alternatively, Ctk1 could enhance subunit joining by phosphorylation of ribosomal proteins, which in turn could enhance directly the binding of the small subunit to the large subunit or the association of translation factors with or their dissociation from the initiation complex. We previously showed that Ctk1 consistently associates with ribosomal subunits, monosomes, and polysomes (16). In an attempt to show that Ctk1 directly functions in translation initiation, purified CTDK-I complex was added to the translation active extracts of Ctk1-depleted cells prior to the luciferase or initiation assay. Unfortunately, we could not observe a “rescue” of the translation activity by the purified complex, and there could be many possible reasons for this. Thus, Ctk1 could also affect translation initiation indirectly. Since RNAPI and RNAPII transcription is decreased upon loss of Ctk1 function, the amount of specific ribosomal components other than Rps8 or Rpl6 (see above) (16, 25) and/or translation factors might be compromised. Moreover, the integrity and/or phosphorylation status of these factors could be affected, since the phosphorylation status of numerous proteins changed upon depletion of Ctk1 (see Table S1 in the supplemental material). Even though a major rRNA processing defect could be excluded (Fig. 4), ribosome biogenesis could be impaired in a more subtle way, leading to a translation initiation defect.
Using an unbiased SILAC approach, we identified a multitude of phosphosites on ribosomal proteins and proteins involved in ribosome biogenesis and/or translation that are less phosphorylated in vivo upon Ctk1 depletion (see Tables S1 and S2 in the supplemental material). Unfortunately, however, we were not able to identify a direct substrate of Ctk1 with a function in translation initiation, and there are many possible explanations for this. Thus, the mechanism by which Ctk1 stimulates translation initiation remains to be determined. Another aspect that remains to be analyzed is the question whether Ctk1 is important for global translation or for translation of a specific subset of mRNAs. The fact that the novel translation initiation defect presented here was observed with various different reporter mRNAs (luciferase, RPL38, RPL41, PGK1, two yeast IRESs) favors global translation control by Ctk1.
Bodenmiller and colleagues recently conducted an extensive phosphoproteomic analysis determining the impact of a multitude of kinase deletions on the phosphoproteome (26). They showed that of all the kinases tested, deletion of Ctk1 caused the most dramatic change in the phosphoproteome. This is consistent with the results of our study and shows that Ctk1 function is crucial for maintaining homoeostasis of the phosphoproteome. The changes in the phosphoproteome observed by Bodenmiller and colleagues are—with the exception of only a few phosphosites—different from the changes that we determined in this study. Besides technical differences, the main reason for this is most likely the fact that we used cells depleted of Ctk1 instead of a Δctk1 strain as Bodenmiller et al. did; the Δctk1 strain has a severe growth defect that is expected to affect the phosphoproteome also nonspecifically. Thus, our study focusing solely on the function of Ctk1 most likely represents a more adequate view of phosphorylation changes caused by the lack of Ctk1, since any influence of slow growth on the phosphoproteome as expected for the Δctk1 strain is absent. Thus, we provide a valuable catalogue of phosphosite changes on ribosomes and ribosome-associated proteins when Ctk1 activity is strongly reduced.
Ctk1 is believed to be highly conserved with three potential human homologues: CDK12 or CDK13 (27) and CDK9, the kinase subunit of the positive transcription elongation factor b (P-TEFb) (28). Thus, it is of great interest whether its human homologue(s) also influences translation initiation. Interestingly, one of the potential mammalian homologues—CDK9—was reported to shuttle between the nucleus and cytoplasm (29) and to associate with polysomes (16). Thus, CDK9 might also function in translation. In addition, it remains to be elucidated whether the newly identified homologues of Ctk1, CDK12 and CDK13 (27), also function in translation.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Wendy Gilbert for the IRES-containing plasmids, to Adolfo Saiardi for the pGEX4T-2-Srp40 plasmid, and Tom Dever for the eIF2α antibody. We thank Karsten Beckmann (U3 Pharma) for suggestions and technical help with initiation and toeprint assays as well as the BSEF plasmid. Furthermore, we are thankful to Roland Beckmann, Daniel Wilson, Klaus Förstemann, and Ralf-Peter Jansen for help and discussions. We thank Achim Dickmanns, Viter Márquez, and Helena Dickinson for critically reading the manuscript.
This work was supported by a grant from the European Research Council (ERC) and Sonderforschungsbereich SFB646 (K.S.), Sonderforschungsbereich SFB684 (D.E.), and the Novo Nordisk Foundation Center for Protein Research (B.S. and J.V.O.). B.C. was supported by a Ph.D. fellowship of the Boehringer Ingelheim Fonds, K.M.B. was supported by a fellowship of the Chemischen Industrie, and K.B. was supported by a fellowship of the Deutsche José Carreras-Stiftung e.V.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00106-14.
REFERENCES
- 1.Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lorsch JR, Dever TE. 2010. Molecular view of 43 S complex formation and start site selection in eukaryotic translation initiation. J Biol Chem 285:21203–21207. doi: 10.1074/jbc.R110.119743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson RJ, Hellen CU, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinnebusch AG, Dever TE, Asano K. 2007. Mechanism of translation initiation in the yeast Saccharomyces cerevisiae, p 225–268. In Mathews MB, Sonenberg N, Hershey JWB (ed), Translational control in biology and medicine, vol 48 Cold Spring Harbor Monograph Series 48 Cold Spring Harbor Laboratory Press, New York, NY. [Google Scholar]
- 5.Asano K, Clayton J, Shalev A, Hinnebusch AG. 2000. A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5, and initiator tRNA(Met) is an important translation initiation intermediate in vivo. Genes Dev 14:2534–2546. doi: 10.1101/gad.831800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinnebusch AG, Lorsch JR. 2012. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol 4:a011544. doi: 10.1101/cshperspect.a011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aitken CE, Lorsch JR. 2012. A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol 19:568–576. doi: 10.1038/nsmb.2303. [DOI] [PubMed] [Google Scholar]
- 8.Hellen CU. 2009. IRES-induced conformational changes in the ribosome and the mechanism of translation initiation by internal ribosomal entry. Biochim Biophys Acta 1789:558–570. doi: 10.1016/j.bbagrm.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez IS, Bai XC, Murshudov G, Scheres SH, Ramakrishnan V. 2014. Initiation of translation by cricket paralysis virus IRES requires its translocation in the ribosome. Cell 157:823–831. doi: 10.1016/j.cell.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilbert WV, Zhou K, Butler TK, Doudna JA. 2007. Cap-independent translation is required for starvation-induced differentiation in yeast. Science 317:1224–1227. doi: 10.1126/science.1144467. [DOI] [PubMed] [Google Scholar]
- 11.Gilbert WV. 2010. Alternative ways to think about cellular internal ribosome entry. J Biol Chem 285:29033–29038. doi: 10.1074/jbc.R110.150532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buratowski S. 2009. Progression through the RNA polymerase II CTD cycle. Mol Cell 36:541–546. doi: 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lenstra TL, Tudek A, Clauder S, Xu Z, Pachis ST, van Leenen D, Kemmeren P, Steinmetz LM, Libri D, Holstege FC. 2013. The role of Ctk1 kinase in termination of small non-coding RNAs. PLoS One 8:e80495. doi: 10.1371/journal.pone.0080495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouchoux C, Hautbergue G, Grenetier S, Carles C, Riva M, Goguel V. 2004. CTD kinase I is involved in RNA polymerase I transcription. Nucleic Acids Res 32:5851–5860. doi: 10.1093/nar/gkh927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grenetier S, Bouchoux C, Goguel V. 2006. CTD kinase I is required for the integrity of the rDNA tandem array. Nucleic Acids Res 34:4996–5006. doi: 10.1093/nar/gkl493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rother S, Strasser K. 2007. The RNA polymerase II CTD kinase Ctk1 functions in translation elongation. Genes Dev 21:1409–1421. doi: 10.1101/gad.428407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eustice DC, Wakem LP, Wilhelm JM, Sherman F. 1986. Altered 40 S ribosomal subunits in omnipotent suppressors of yeast. J Mol Biol 188:207–214. doi: 10.1016/0022-2836(86)90305-0. [DOI] [PubMed] [Google Scholar]
- 18.Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. 2011. The structure of the eukaryotic ribosome at 3.0 A resolution. Science 334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 19.Altmann M, Trachsel H. 1997. Translation initiation factor-dependent extracts from yeast Saccharomyces cerevisiae. Methods 11:343–352. doi: 10.1006/meth.1996.0432. [DOI] [PubMed] [Google Scholar]
- 20.Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beckmann K, Grskovic M, Gebauer F, Hentze MW. 2005. A dual inhibitory mechanism restricts msl-2 mRNA translation for dosage compensation in Drosophila. Cell 122:529–540. doi: 10.1016/j.cell.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 22.Miura F, Kawaguchi N, Sese J, Toyoda A, Hattori M, Morishita S, Ito T. 2006. A large-scale full-length cDNA analysis to explore the budding yeast transcriptome. Proc Natl Acad Sci U S A 103:17846–17851. doi: 10.1073/pnas.0605645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verge V, Vonlanthen M, Masson JM, Trachsel H, Altmann M. 2004. Localization of a promoter in the putative internal ribosome entry site of the Saccharomyces cerevisiae TIF4631 gene. RNA 10:277–286. doi: 10.1261/rna.5910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn SH, Keogh MC, Buratowski S. 2009. Ctk1 promotes dissociation of basal transcription factors from elongating RNA polymerase II. EMBO J 28:205–212. doi: 10.1038/emboj.2008.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rother S. 2007. Functional analysis of the RNA polymerase II C-terminal domain kinase Ctk1 in the yeast Saccharomyces cerevisiae. Ph.D. thesis Ludwig Maximilians University, Munich, Germany. [Google Scholar]
- 26.Bodenmiller B, Wanka S, Kraft C, Urban J, Campbell D, Pedrioli PG, Gerrits B, Picotti P, Lam H, Vitek O, Brusniak MY, Roschitzki B, Zhang C, Shokat KM, Schlapbach R, Colman-Lerner A, Nolan GP, Nesvizhskii AI, Peter M, Loewith R, von Mering C, Aebersold R. 2010. Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci Signal 3:rs4. doi: 10.1126/scisignal.2001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, Adelman K, Lis JT, Greenleaf AL. 2010. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev 24:2303–2316. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garriga J, Grana X. 2004. Cellular control of gene expression by T-type cyclin/CDK9 complexes. Gene 337:15–23. doi: 10.1016/j.gene.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 29.Napolitano G, Licciardo P, Carbone R, Majello B, Lania L. 2002. CDK9 has the intrinsic property to shuttle between nucleus and cytoplasm, and enhanced expression of cyclin T1 promotes its nuclear localization. J Cell Physiol 192:209–215. doi: 10.1002/jcp.10130. [DOI] [PubMed] [Google Scholar]
- 30.Hinnebusch AG. 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- 31.Mascarenhas C, Edwards-Ingram LC, Zeef L, Shenton D, Ashe MP, Grant CM. 2008. Gcn4 is required for the response to peroxide stress in the yeast Saccharomyces cerevisiae. Mol Biol Cell 19:2995–3007. doi: 10.1091/mbc.E07-11-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altmann M, Linder P. 2010. Power of yeast for analysis of eukaryotic translation initiation. J Biol Chem 285:31907–31912. doi: 10.1074/jbc.R110.144196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.