

Abstract
Sophisticated systems for cell-cell communication enable unicellular microbes to act as multicellular entities capable of group-level behaviors that are not evident in individuals. These group behaviors influence microbe physiology, and the underlying signaling pathways are considered potential drug targets in microbial pathogens. Trypanosoma brucei is a protozoan parasite that causes substantial human suffering and economic hardship in some of the most impoverished regions of the world. T. brucei lives on host tissue surfaces during transmission through its tsetse fly vector, and cultivation on surfaces causes the parasites to assemble into multicellular communities in which individual cells coordinate their movements in response to external signals. This behavior is termed “social motility,” based on its similarities with surface-induced social motility in bacteria, and it demonstrates that trypanosomes are capable of group-level behavior. Mechanisms governing T. brucei social motility are unknown. Here we report that a subset of receptor-type adenylate cyclases (ACs) in the trypanosome flagellum regulate social motility. RNA interference-mediated knockdown of adenylate cyclase 6 (AC6), or dual knockdown of AC1 and AC2, causes a hypersocial phenotype but has no discernible effect on individual cells in suspension culture. Mutation of the AC6 catalytic domain phenocopies AC6 knockdown, demonstrating that loss of adenylate cyclase activity is responsible for the phenotype. Notably, knockdown of other ACs did not affect social motility, indicating segregation of AC functions. These studies reveal interesting parallels in systems that control social behavior in trypanosomes and bacteria and provide insight into a feature of parasite biology that may be exploited for novel intervention strategies.
INTRODUCTION
Trypanosoma brucei and other African trypanosomes are protozoan parasites that cause sleeping sickness in humans and related wasting diseases in wild and domestic animals. Sleeping sickness is recognized as one of the world's most neglected diseases, with approximately 60 million people living at risk of infection, while livestock infections account for significant economic hardship in some of the most impoverished regions of the planet (1, 2). Dedicated efforts over the last decade have reduced the human health burden, but these parasites remain a continuing threat for reemergence owing to their capacity for explosive outbreaks and their historical ability to resist eradication (3, 4). Sleeping sickness is fatal if untreated, no vaccine exists, and current treatment options are toxic, antiquated, and increasingly ineffective (5, 6). Therefore, new perspectives on trypanosome biology, transmission, and pathogenesis are urgently needed to facilitate novel intervention strategies.
T. brucei is transmitted between mammalian hosts by blood-feeding tsetse flies. Transmission through the fly requires extensive interaction with host tissue surfaces, as parasites move across and through tissues en route from the fly midgut, through the alimentary tract and mouthparts, and then into the salivary glands (7). Once in the salivary gland, parasites colonize the gland epithelial surface and complete the final stages of development into forms infectious for mammals (8). Thus, as is the case for many microbes (9), T. brucei in its natural habitat lives in intimate and continuous contact with surfaces.
Despite the ubiquity of parasite-surface interactions during T. brucei transmission, studies of these organisms are almost exclusively conducted using suspension cultures. While such studies have yielded many important insights, they overlook an important and ubiquitous feature of trypanosome biology. To overcome this gap in knowledge, we utilized semisolid agarose matrices to assess the influence of surface cultivation on parasite behavior (10). This led to the surprising discovery that rather than acting as individuals, surface-cultivated procyclic (insect-stage) trypanosomes assemble into multicellular groups that coordinate their movements across the surface (10). Initially, parasites collect into small groups, and these grow larger through recruitment of other cells. At the periphery of the inoculation site, parasites assemble in nodes of high cell density, and from there they advance outward. Movement is polarized such that cells move outward but not laterally, leading to the formation of thin projections radiating away from the center. When cells in radial projections encounter a separate group of parasites, they halt or divert their movement to avoid contact, implicating cell-cell signaling in the control of trypanosome group behavior. We termed this behavior “social motility” (SoMo), based on features shared with surface-induced social motility in bacteria (9, 11).
As is the case in bacteria (12–14), social motility in T. brucei requires cell motility and some ability of cells to sense and respond to external cues (10). Surface-induced group behaviors, such as social motility and biofilm formation, have been extensively studied in bacteria (11, 14), but molecular mechanisms that govern surface-induced group behavior in trypanosomes are completely unknown. In other microbes, collective activities of groups of cells are commonly controlled by cyclic nucleotides, which function as second messengers and as secreted signaling molecules. In Dictyostelium discoideum, for example, cyclic AMP (cAMP) acts as a chemoattractant and signaling molecule to direct surface motility of individual cells, enabling them to assemble into multicellular fruiting bodies (15). In several species of bacteria, cyclic-di-GMP (c-di-GMP) governs the transition between surface-induced swarming motility and biofilm formation (16–20). In this case, decreased intracellular c-di-GMP promotes swarming, while elevated c-di-GMP inhibits swarming and promotes biofilm formation (14, 18, 19, 21). In the yeast Candida albicans, cyclic nucleotides regulate dimorphic transitions connected to surface penetration and pathogenesis (22).
Cyclic nucleotide synthesis in T. brucei depends on a large family of receptor-type adenylate cyclases (ACs) that are localized to the flagellum membrane (23–26). Trypanosomal ACs are distinguished from mammalian ACs in that the intracellular catalytic domain is directly connected to an extracellular, putative ligand binding domain on a single polypeptide (24). As such, trypanosome ACs may be regulated by extracellular ligands directly rather than via upstream G protein-coupled receptors, as observed for mammalian ACs (24). The canonical T. brucei AC is expression site-associated gene 4 (ESAG4), a bloodstream stage-specific AC that modulates the host immune response to promote parasite survival during infection (27). Aside from ESAG4, functions for T. brucei ACs are unknown. Notably, several flagellar ACs were recently identified that are specifically upregulated in procyclic T. brucei (25), the parasite life cycle stage that undergoes social motility. Given the widespread use of cyclic nucleotides in control of microbial social behavior and the emerging role of the eukaryotic flagellum in coordinating cellular responses to external signals (28, 29), we asked whether T. brucei flagellar ACs function in social motility. Here we report that AC6, a procyclic stage-specific AC (25), is localized to the tip of the flagellum and regulates social motility. RNA interference (RNAi)-mediated knockdown of AC6 causes a hypersocial phenotype, while knockdown of several other flagellar ACs does not affect social motility. Importantly, point mutations in the AC6 catalytic domain phenocopy the knockdown, demonstrating that loss of activity, rather than loss of the protein, is responsible for the phenotype. These studies are, to our knowledge, the first to demonstrate function for trypanosomal ACs in the insect life cycle stage and extend the paradigm of cyclic nucleotide control of microbial social behavior to parasitic protozoa.
MATERIALS AND METHODS
Cell culture and motility assays.
Procyclic cells derived from the 2913 cell line (30) were used for all experiments. Suspension cultures were maintained using Cunningham's semidefined medium (SM) as described previously (31). For RNAi lines, 2.5 mg/ml phleomycin, was included in the medium for selection of stable transfectants, and RNAi was induced by adding 1 mg/ml tetracycline. Lines carrying pMOT vector-based sequences for rescue and hemagglutinin (HA) tagging were cultured with 1 μg/ml of puromycin. Motility in suspension cultures was assayed as previously described (31). Social motility assays were done as previously described (10). For quantitation, several independent assays were done, and the numbers of projections formed per plate were quantitated.
RNAi and quantitative reverse transcription-PCR (qRT-PCR).
RNAi target regions for the ACs were chosen by using the Trypanofan RNAit algorithm (32) and were PCR amplified using the following primers: AC1&2KD_F, TTGATGATGATGGTAGCGGA; AC1&2KD_R, ACATACACCGCCTTACTGCC; AC3KD_F, GACGGTTCTGTCCCTGTTGT; AC3KD_R, TGGCTCTGAACAGTGAATGC; AC4KD_F, AGCTTACGAGGGCTGTGAAA; AC4KD_R, AAATACACTGCCCCTTGTCG; AC5KD_F, TCTGCTTATGCAGGACGATG; AC5_R, CCTCAAAAGTCTCGAGGTGC; AC6KD_F, TGGAGCAGCAAATCTACGTG; AC6_R, TTTTCTCGGCTCTCCACTGT; AC6uKD_F, ATAAGCTTACGGGGTTCCCTCATTTAAC (restriction sites are italicized); AC6uKD_R, ATTCTAGAACAACAACAACCCCCAAAAA.
Fragments were amplified using genomic DNA from 2913 cells and ligated into the p2T7Ti-B RNAi plasmid (33). All sequences were verified by direct DNA sequencing. RNAi vectors were linearized, transfected, and selected with antibiotic as previously described (31). Clonal lines were generated by limiting dilution. Efficacy of knockdown was assessed via qRT-PCR as described previously (34) at 72 h postinduction. qRT-PCR results are reported as arithmetic means ± standard deviations from at least two independent RNA preparations, each analyzed in duplicate and normalized against the housekeeping genes glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Tb927.6.4280/Tb927.6.4300) and RPS23 (Tb10.70.7020/Tb10.70.7030). Relative gene expression was determined using the 2−ΔΔC method as previously described (35, 36). Primers used for qRT-PCR were the following: AC1_F, CGTTGACTTCACGGCTTACA; AC1_R, ACATTTCGTTCTCCCACTGC; AC2_F, GCCATGTCGTTGATTTCACA; AC2_R, CCAACCAGACCACAGACCTT; AC3_F, ACTGATGGGCGTCTTCACACAA; AC3_R, GGATGCACTTTTCTTGGGCAAC; AC4_F, CTGCGAGTGCGAGTTGGTGT; AC4_R, ACGTTCTGCGGTGCTGAGTG; AC5_F, CACATCTCAGCGCCAAAAACTG; AC5_R, TAGACCGCATAATCGCCTCACA; AC6_F, TGCAGTTAAGGTGGGTCACA; AC6_R, GATCCACCGCAGGATTAGAA; GAPDH_F, GGCTGATGTCTCTGTGGTGGA; GAPDH_R, GGCTGTCGCTGATGAAGTCG; RPS_F, AGATTGGCGTTGGAGCGAAA; RPS_R, GACCGAAACCAGAGACCAGCA.
Western blot assays and immunofluorescence.
Protein extracts were prepared and analyzed by Western blotting as described previously (37). A total of 1 × 106 cell equivalents per lane were used. Monoclonal mouse anti-HA antibody from Covance (used at 1:5,000) and monoclonal anti-β-tubulin E7 hybridoma supernatant (used at a 1:7,000 dilution) were used as primary antibodies and detected by horseradish peroxidase-coupled goat anti-mouse antibodies (Bio-Rad) at a 1:5,000 dilution. Immunofluorescence was carried out on whole cells as described previously (25), with the monoclonal anti-HA antibody HA.11 (Covance) used at a 1:250 dilution and donkey anti-mouse secondary antidbody coupled to AlexaFluor 488 (Molecular Probes) used at 1:2,500. Visualization of nucleic acid was done by staining with DAPI (4′,6-diamidino-2-phenylindole).
Generation of AC6-Ri and AC6**-Ri mutant cell lines.
AC6-Ri and AC6**-Ri cell lines were generated by expressing wild-type or mutant AC6 in the AC6 UTR knockdown line (AC6uKD) (34). To generate the AC6-Ri line, a 426-bp fragment corresponding to the 3′ terminus of the AC6 open reading frame (ORF) was cloned immediately upstream of the α/β-tubulin intergenic region in the pMOTag plasmid (39). A fragment of the AC6 3′-untranslated region (UTR) was then cloned downstream of the puromycin resistance gene in this plasmid to generate the plasmid pMOT-AC6WT426. Primers used were the following: AC6_ORF_F, ATGGTACCGAGGCATATGTGGCGGATG; AC6_ORF_R, ATGTCGACCTACTGCTTCCCCTTTTCCT; AC6_UTR_F, ATGGATCCTAACAGCAGTAGTGAATTGAAG; AC6_UTR_R, ATTCTAGAACGTGGACTTCACCTTCATC (restriction sites are italicized).
Plasmid DNA sequences were confirmed by plasmid sequencing (Genewiz, Inc.) Tagging cassettes were excised by restriction digestion, purified, and transfected into AC6uKD cells. Stable transfectants were selected, and clonal lines were generated by limiting dilution.
To generate AC6**-Ri, the 426-bp AC6 fragment in pMOT-AC6WT426 was replaced with a fragment that was the terminal 3′ 1,332 bp of the AC6 ORF, encompassing the catalytic domain, to generate pMOT-AC6WT1332. The QuikChange mutagenesis kit (Stratagene, Inc.) was used to mutate coding sequences for conserved active site residues NMAART (amino acids 1031 to 1036), which are essential for AC catalytic activity (40). The NMART sequence was mutated to AAAAAA to generate plasmid pMOT-AC6**1332. Primers used to amplify the 1,332-bp fragment were the following: AC6_1332_F, ATGGTACCACGCGCATTAGTGTGTGGTC; AC6_1332_R, ATGTCGACCTACTGCTTCCCCTTTTCCT (restriction sites are italicized). Site-directed mutagenesis was done using the Stratagene QuikChange kit (Agilent) and primers MUT_F, GGATATGACTATTACGGTCAAACGGCAGCCGCGGCTGCCGCCGCGGAGAGCATTGCGAA, and MUT_R, TTCGCAATGCTCTCCGCGGCGGCAGCCGCGGCTGCCGTTTGACCGTAATAGTCATATCC. Plasmid sequence verification and generation of stably transfected clonal lines were done as described above.
Epitope tagging.
Cell lines carrying HA-tagged versions of AC6 and AC6** were generated by in situ tagging with the pMOTag-HA plasmid, as described previously (31). The 3′ end of the wild-type AC6 ORF was amplified with genomic DNA from 2913 cells using the following primers: F, ATGGTACCGAGGCATATTGTGGCGGATG, and R, ATCTCGAGCTGCTTCCCCTTTTCCTCC (restriction sites are italicized).
The AC** catalytic domain mutant fragment was PCR amplified from the pMOT-AC6**1332 plasmid by using the following primers: F, ATGGTACCACGCGCATTAGTGTGTGGTC; R, ATCTCGAGCTGCTTCCCCTTTTCCTCC (restriction sites are italicized).
Amplified fragments were cloned upstream of the HA tag in pMOTag-HA. All sequences were confirmed by direct DNA sequencing (GeneWiz, Inc.), and tagging cassettes were excised and used to generate stably transfected clonal lines as described above.
cAMP ELISA.
Total cellular cyclic AMP levels were measured using the cyclic-AMP Direct enzyme-linked immunosorbent assay (ELISA) kit (EnzoLife Sciences) essentially as described previously (41). A total of 4 × 107 cells were harvested, washed in phosphate-buffered saline (PBS), resuspended into a hypotonic lysis buffer (1 mM HEPES, 1 mM EDTA, 1× SigmaFAST protease inhibitors), and left on ice for 10 min. Samples were then passed through a 25 (± 3/8)-gauge needle 10 times before the addition of 1 M HCl to give a final concentration of 0.1 M HCl. Cells were spun at 14,000 × g at 4°C for 10 min, and supernatant fractions were saved for analysis. Aliquots of 100 μl of supernatant corresponding to 2.5 × 107 cells were used per well. The ELISA was done following the manufacturer's suggested protocols, and output was read at 405 nM and analyzed using a MasterPlex2010 system (MiraiBio Group of Hitachi America). A best-fit curve using 5PL logistics was utilized. All values are averages of independent biological replicates that were assayed in technical duplicates.
Northern blot assays.
Northern blot assays were done similar to assays previously described (42), except that digoxigenin (DIG) was used for detection rather than 32P-labeling. A probe unique to the AC6 ORF was generated with primers F, TGTGCTTTTGTTTGGTGCTC, and R, AGTAGTTCGGGTCCGTGATG, and suspended into DIG-EasyHyb buffer (Roche). This corresponds to a 305-bp region that is AC6-specific according to RNAit (32) and NCBI-BLAST. Blots were visualized using the DIG nucleic acid detection kit (Roche). As a loading control, total RNA was visualized by UV shadowing (43).
RESULTS
A subset of insect stage-specific ACs regulates social motility without impacting individual cell motility in suspension culture.
Cyclic AMP production in T. brucei is catalyzed by a large family of receptor-type ACs whose biological functions are mostly unknown (44). We recently identified a group of 6 ACs in the flagellum of procyclic T. brucei and found that 5 of these are specifically upregulated in procyclic-stage cells (25). To test whether any of these ACs function in social motility, we generated gene-specific, tetracycline-inducible RNAi knockdowns by targeting unique regions of each gene's open reading frame. The open reading frames of AC1 and AC2 are 89% identical, with a single region of high divergence near the 3′ end that distinguishes between the two genes. We therefore generated a single RNAi construct for simultaneous knockdown of both AC1 and AC2, allowing us to reserve unique regions for assessing expression of each gene individually. qRT-PCR demonstrated that the targeted mRNA was significantly reduced following tetraycycline induction of RNAi (see Fig. S2 in the supplemental material). Parasite growth rates in suspension culture were not affected in any of the AC knockdowns (see Fig. S2). This differs from bloodstream-form T. brucei, where knockdown of ESAG4 causes morphological defects and is lethal (48). Knockdown of AC3, -5 or -6 and dual knockdown of AC1 and -2 did not impact motility of individual cells in suspension culture, while AC4 knockdown parasites showed reduced motility (see Fig. S3 in the supplemental material).
We next examined social motility in each of the AC knockdowns. T. brucei social motility is characterized by the formation of multicellular communities that grow through recruitment of nearby cells (45). Collective motility of parasites outward produces projections that radiate away from the site of inoculation (10). There were no apparent differences in the social motilities of AC3, 4 or -5 knockdowns compared to the wild type (Fig. 1). However, social motility in the AC1 and -2 dual knockdown and the AC6 knockdown differed from that of wild-type cells (Fig. 1). The general pattern was the same, with projections extending outward and tending to spiral in a clockwise direction, but the number of projections formed was greater in the knockdowns and projections were more closely spaced. We termed this behavior “hypersocial,” by analogy to hyperswarming observed in bacterial diguanylate cyclase mutants (14, 16, 19). The hypersocial phenotype of the AC1 and -2 dual knockdown and AC6 knockdown cell lines was dependent on tetracycline-induced RNAi (Fig. 1).
FIG 1.

Social motility is regulated by a subset of procyclic stage-specific adenylate cyclases. (A) Gene-specific knockdowns targeting the indicated ACs were assayed for social motility in comparison with 2913 control cells (ctrl) on plates containing tetracycline to induce RNAi. (B) Social motility assay results are shown for the indicated knockdowns, maintained in the absence or presence of tetracycline (Tet) to induce RNAi. (C) Quantitation of the number of projections formed for wild-type cells (WT) or the indicated knockdown lines, maintained in the absence or presence of tetracycline. Mean values and standard errors are shown. P values (unpaired t test) are shown. Sample sizes: WT –Tet, n = 12; WT +Tet, n = 7; AC1 and -2KD –Tet, n = 24; AC1 and -2KD +Tet, n = 20; AC6KD –Tet, n = 16; AC6KD +Tet, n = 23; AC5KD –Tet, n = 36; AC5KD +Tet, n = 36.
Restoring AC6 expression in AC6 uKD cells rescues the hypersocial phenotype.
Our results implicate a subset of T. brucei ACs in trypanosome social motility. As AC1 and 2 are 89% identical at the nucleotide level, distinguishing their specific contributions poses challenges and we therefore focused our analyses on AC6. To assess the specific role of AC6, we asked whether the hypersocial phenotype could be rescued by restoring expression of the wild-type protein in the knockdown cell line. To achieve this, we generated a knockdown line targeting the AC6 3′ UTR, “AC6-uKD,” because this enables one to test for rescue under RNAi-inducing conditions by expressing an AC6 transgene with an alternate 3′ UTR that is immune to RNAi (34, 38). Tetracycline induction of AC6-uKD cells effectively reduced AC6 expression, without affecting growth or individual cell motility in suspension culture (see Fig. S4 in the supplemental material). Expression of other ACs was unaffected (see Fig. S5 in the supplemental material). Importantly, the AC6-uKD phenocopied the open reading frame knockdown, exhibiting an RNAi-dependent hypersocial phenotype that was qualitatively and quantitatively indistinguishable from that observed for the open reading frame knockdown (Fig. 2). These results established a background in which an AC6 transgene can be expressed while the endogenous gene is knocked down.
FIG 2.

Social motility is modulated by AC6 catalytic activity. (A) Social motility assays on AC6-UTR-knockdown cells (AC6 uKD), AC6-RNAi-immune cells (AC6-Ri), or the RNAi-immune catalytic domain mutant cells (AC6**-Ri). Cells were maintained in the absence or presence of tetracycline (Tet) to induce RNAi. Schematics at the top of the panel illustrate the region targeted for RNAi. RNAi targets the 3′ UTR of the endogenous AC6 gene, while transgenes are immune to RNAi, owing to the presence of an alternate UTR. (B) Northern blot probed with an AC6-specific probe (top). RNA was prepared from bloodstream cells (BSF), procyclic 2913 control cells (PCF), AC6-uKD cells, AC6-Ri cells, or AC6**-Ri cells maintained with or without Tet as indicated. The position of the endogenous AC6 mRNA (open arrowhead) and transgene mRNA (closed arrowhead) are indicated. Total RNA was visualized by UV illumination of the blot (bottom). (C) Quantitation of the number of projections formed for the indicated cell lines maintained in the absence or presence of Tet. Mean values and standard errors are shown. P values (unpaired t test) are indicated. Sample sizes: AC6uKD –Tet, n = 90; AC6uKD +Tet, n = 90; AC6-Ri –Tet, n = 34; AC6-Ri +Tet, n = 34; AC6**-Ri –Tet, n = 30; AC6**-Ri +Tet, n = 30.
We next replaced the 3′ UTR of one AC6 allele with the tubulin 3′ UTR. We refer to this cell line as “AC6-RNAi-immune” (AC6-Ri). Northern blotting demonstrated that AC6 mRNA was upregulated in procyclic cells (Fig. 2), consistent with qRT-PCR results (25). A single AC6 mRNA was observed in control and AC6-uKD cells, while AC6-Ri cells expressed two mRNAs, corresponding to endogenous and RNAi-immune transcripts (Fig. 2). Abundance of the endogenous transcript was substantially reduced upon RNAi induction, while the RNAi-immune transcript was unaffected (Fig. 2). Thus, AC6-Ri cells maintain AC6 expression following knockdown of the endogenous gene. Growth and individual cell motility in suspension culture were unaffected in AC6-Ri cells with or without RNAi induction (see Fig. S6 in the supplemental material). Maintenance of AC6 expression under RNAi-induced conditions rescued the hypersocial phenotype (Fig. 2), demonstrating that the hypersocial phenotype is a consequence of AC6-specific knockdown.
RNAi alone does not distinguish between phenotypes caused by loss of protein activity, versus indirect consequences arising from loss of the target protein (38). For example, while the AC6 knockdown phenotype might be a direct result of impaired cAMP output, it might alternatively be due to changes in potential interaction partners of AC6 caused by loss of the protein. To distinguish between these possibilities, we asked if blocking AC catalytic function without altering protein levels altered social motility. The T. brucei AC active site contains a highly conserved NMAART domain that is required for catalytic activity (40). We therefore mutated the NMAART domain to all alanines to generate a catalytically inactive version, termed AC6**, and introduced an RNAi-immune copy of AC6** into the AC6 uKD line. This cell line is referred to as AC6**-Ri. Following tetracycline induction, endogenous AC6 mRNA levels were substantially reduced, while AC6**-Ri mRNA levels were unaffected. Growth and motility in suspension culture were unaffected in AC6**-Ri cells (see Fig. S6 in the supplemental material), but expression of the AC6** catalytic mutant failed to compensate for loss of endogenous AC6 (Fig. 2).
It is possible that the phenotype of AC6**-Ri mutants might be due to altered expression levels and/or localization of the mutant protein. To test this, we compared expression and localization of wild-type AC6 to that of AC6**. For this, we used in situ tagging (39) to place a hemagglutinin (HA) epitope at the C terminus of the wild-type and mutant proteins. Western blot analysis showed a single band of approximately equal abundance in AC6-HA and AC6**-HA cells (Fig. 3). Immunofluorescence revealed that wild-type AC6-HA localized to the flagellum, but interestingly, was restricted to the flagellar tip (Fig. 3). Interestingly, tip localization for AC6 was predicted previously based on sequences within a C-terminal domain that is required for flagellum targeting (25). Importantly, the AC6** catalytic mutant gave a flagellum tip distribution that was indistinguishable from that of the wild type (Fig. 3), indicating that the hypersocial phenotype of AC6**-Ri cells is a consequence of lost activity and is not caused by altered expression or mislocalization.
FIG 3.

AC6 is localized to the tip of the trypanosome flagellum. (A) Western blot of protein extracts from 2913 cells and from cells expressing wild-type HA-tagged AC6 (AC6HA) or catalytically inactive AC6 (AC6**HA). Blots were probed with anti-HA antibody, or anti-tubulin antibody as a loading control. (B) Control cells (2913) or cells expressing HA-tagged wild-type AC6 (AC6HA) or the catalytic domain mutant (AC6**HA) were subjected to immunofluorescence with anti-HA antibody (green) and DAPI (blue). Bar, 2 μm.
We next asked whether knockdown of AC6, or expression of AC6**, affected total cellular cAMP levels (see Fig. S7 in the supplemental material). Although there was a trend toward reduced total cellular cAMP following RNAi induction in AC6uKD and AC6**-Ri cells, the changes were not statistically significant. Given the discrete localization of AC6 to a tiny subdomain of the parasite surface and the fact that this is just one of several ACs expressed in this life cycle stage, it is not surprising that RNAi did not cause gross perturbations of total cellular cAMP.
DISCUSSION
Recognition of social behavior and cell-cell communication as ubiquitous properties of bacteria (9, 13) transformed our view of microbiology and microbial pathogenesis, but protozoan parasites have not generally been considered in this context. Recently, we showed that the paradigm of microbial social behavior applies to the protozoan parasite T. brucei when cultivated on surfaces to mimic their natural environment (10). Here we provide the first molecular dissection of mechanisms underlying social motility in trypanosomes, and our results suggest that cAMP signaling systems in the flagellum control T. brucei social behavior. As discussed below, our studies reveal intriguing parallels in systems used by trypanosomes and bacteria for controlling surface-induced social behavior. Moreover, by discovering an accessible readout for receptor-type adenylate cyclase function in T. brucei, our findings provide a foundation for dissecting structure-function relationships among these enigmatic proteins and, by extension, cAMP signaling in trypanosomes.
Several independent analyses point to a specific role for cAMP produced by AC6 in social motility. First, a hypersocial phenotype was observed in two independent AC6 knockdown lines targeting distinct regions of the AC6 mRNA, while expression of other ACs was unaffected. Second, the phenotype was rescued by expression of wild-type AC6, but not by a catalytic domain mutant. Thus, normal social motility depends on AC6 activity, and the mutant phenotype is not due to off-target effects of knockdown.
cAMP is implicated in the T. brucei transmission cycle as well as pathogenesis, and elevated cAMP is lethal for bloodstream-form cells, making cAMP pathways a target of drug discovery efforts (46). Pathways of cAMP signaling in these organisms remain poorly characterized, however, and lack familiar features found in mammalian cells (44). One outstanding question is why trypanosomes have so many AC genes, ∼75 compared to ∼10 in mammals, for example (47). Some T. brucei ACs may substitute for others (48), suggesting some redundancy or overlap in function. However, the presence of ACs that are specific to different life cycle stages and that exhibit distinct subcellular localizations (24, 25) argues against strictly redundant functions for these proteins. The idea of distinct functions for individual ACs is also supported by the fact that expression of AC1 to AC5 is unaltered in AC6 knockdowns but does not accommodate for loss of AC6. Likewise, normal expression of AC6 does not accommodate for loss of AC1 and -2. At least two possibilities may explain the inability of AC6 to compensate for loss of AC1 and -2 or vice versa. One possibility is that normal social motility requires interaction between different AC isoforms, such that each isoform must be present to form an active complex. It is also possible that rather than direct interaction, independent signaling through each is required, and different isoforms function as coincidence detection modules.
Simultaneous expression of several ACs in a single cell presents a conundrum if each AC controls a different pathway, because cAMP output must be segregated to avoid interference. In mammalian cells, this problem is mitigated via a microdomain model, in which spatial separation of cAMP signaling output enables different ACs to control distinct cellular responses (49). Such a model has been suggested for T. brucei (25, 50) and is supported by our results. In this model (Fig. 4), social motility is regulated by local levels of intraflagellar cAMP specifically derived from an AC6 microdomain at the flagellum tip. When levels are high, social motility is attenuated, and when levels are low social motility ensues. In wild-type cells (Fig. 4A), AC6 activity is downregulated in response to a specific signal, e.g., the presence of an inhibitor or loss of an activator, resulting in reduced local cAMP levels and initiation of social motility. Upon constitutive inactivation of AC6, e.g., through knockdown or expression of a catalytic mutant (Fig. 4B), a signal-independent decrease in local cAMP within the AC6 microdomain results in a precocious, hypersocial phenotype. A key tenet of this model is that a cAMP-specific phosphodiesterase localized throughout the flagellum provides a diffusion barrier to shield the AC6 tip domain from cAMP generated by other flagellar ACs (50). As such, the model predicts that if local cAMP levels were constitutively elevated through loss of the diffusion barrier, social motility would be blocked. In support of this model, in separate work we found that genetic or pharmacological inhibition of flagellum-localized cAMP-specific phosphodiesterase leads to elevated cAMP and a complete block of social motility (M. Oberholzer and K. M. Hill, submitted for publication).
FIG 4.
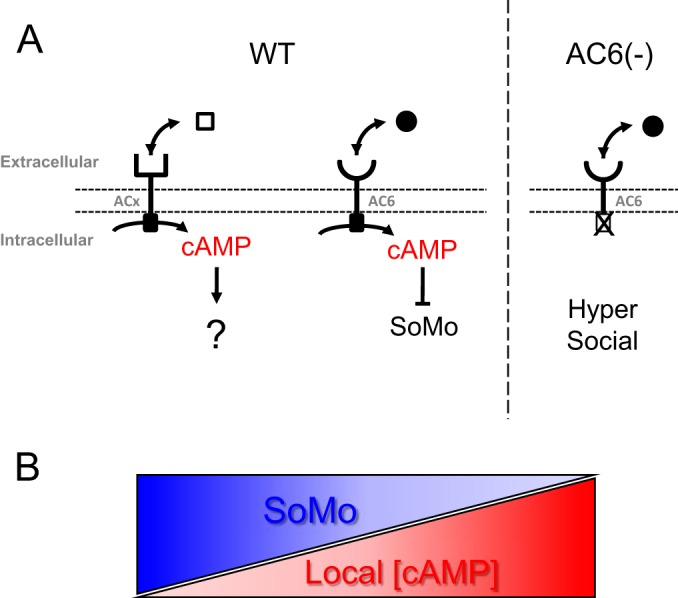
Model for cAMP microdomain regulation of social motility. (A) Schematic model for AC6-dependent control of social motility in wild-type cells (WT) and AC6 knockdown or catalytic mutant cells [AC6(−)]. AC6 is one of several ACs in the trypanosome flagellum. The model posits that ACs recognize different ligands, depending on their divergent extracellular domains, and that ligand binding regulates AC activity. cAMP produced specifically by AC6 acts to inhibit social motility, and when AC6 activity is reduced through ligand-mediated regulation (WT), this results in reduced cAMP and activation of social motility. Constitutive inactivation of AC6, e.g., through knockdown or expression of a catalytic mutant [AC6(−)], causes a signal-independent decrease in local cAMP, resulting in a precocious, hypersocial phenotype. (B) Reciprocal relationship between social motility (SoMo) and cAMP concentration.
Our findings extend the concept of cyclic nucleotides as regulators of microbial social behaviors to parasitic protozoa and reveal parallels with c-di-GMP regulation of bacterial swarming motility. Two specific examples are the SadC/BifA system in Pseudomonas aeruginosa and the ScrABC system in Vibrio parahaemolyticus (14, 19). Pseudomonas and Vibrio species undergo surface-dependent swarming motility. In P. aeruginosa, swarming is regulated by the combined activities of SadC, a diguanylate cyclase (DC) that synthesizes c-di-GMP, and BifA, a phosphodiesterase (PDE) that specifically degrades c-di-GMP (19). Knockout of the cyclase reduces intracellular c-di-GMP levels and causes hyperswarming, while knockout of the phosphodiesterases results in elevated c-di-GMP levels and blocks swarming (19). In V. parahaemolyticus, swarming motility is regulated by the ScrABC system. Here, the ScrC protein is a c-di-GMP cyclase and phosphodiesterase that is regulated by the periplasmic binding protein ScrB (14). The ligand for ScrB is an extracellular “S-signal” that accumulates in a cell density-dependent fashion and binds ScrB. Once bound to its ligand, ScrB neutralizes the c-di-GMP cyclase activity of ScrC and simultaneously activates its phosphodiesterase activity. This causes intracellular c-di-GMP levels to drop and stimulates swarming. At low density, in the absence of S-signal, the cyclase activity of ScrC predominates, resulting in elevated c-di-GMP and inhibition of swarming. Thus, in both bacteria and trypanosomes, the extent of social motility on surfaces is inversely proportional to levels of intracellular cyclic nucleotide, as controlled by reciprocal regulation of cyclase and phosphodiesterase activities. In bacteria, an expanded repertoire of DC genes is common and makes it possible to use c-di-GMP signaling for responding to a wide variety of signals (16, 17).
In addition to pathway architecture, the domain structure of ScrC in Vibrio spp. is similar to that of trypanosome ACs, having a surface exposed ligand binding domain coupled via a transmembrane segment to a cytoplasmic nucleotide cyclase domain (14, 44). Ligand binding to trypanosome ACs has not been demonstrated, but the AC extracellular domain bears homology to bacterial periplasmic binding proteins (52), such as ScrB. Bacterial periplasmic binding proteins bind a variety of small-molecule ligands and control chemotaxis and swarming motility (14, 53), raising the intriguing possibility that small-molecule ligands might control social motility through AC6 in T. brucei. In bacteria, where social motility has been well-studied, this behavior facilitates rapid colonization of surfaces, antibiotic resistance, and efficient use of nutrients (9, 54, 55). Although the in vivo correlates of social motility in T. brucei are not yet known, similar activities would be beneficial to parasites in the tsetse fly (10). More broadly, studies of bacterial surface-associated group behaviors have provided tremendous insights into signaling mechanisms used by these organisms (13, 14). Likewise, our findings present an important advance by providing the first demonstrated requirement for ACs in procyclic-form parasites and establishing a foundation for dissecting cAMP signaling systems in these organisms in a readily accessible in vitro assay.
Supplementary Material
ADDENDUM IN PROOF
Recent work has shown that T. brucei social motility is a property of a distinct life cycle stage within the fly transmission stage (S. Imhof, S. Knüsel, K. Gunasekera, X. L. Vu, and I. Roditi, PLoS Pathog 10:e1004493, 2014, http://dx.doi.org/10.1371/journal.ppat.1004493).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00217-14.
REFERENCES
- 1.Hill KL. 2003. Mechanism and biology of trypanosome cell motility. Eukaryot Cell 2:200–208. doi: 10.1128/EC.2.2.200-208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Legros D, Ollivier G, Gastellu-Etchegorry M, Paquet C, Burri C, Jannin J, Buscher P. 2002. Treatment of human African trypanosomiasis: present situation and needs for research and development. Lancet Infect Dis 2:437–440. doi: 10.1016/S1473-3099(02)00321-3. [DOI] [PubMed] [Google Scholar]
- 3.Ekwanzala M, Pepin J, Khonde N, Molisho S, Bruneel H, De Wals P. 1996. In the heart of darkness: sleeping sickness in Zaire. Lancet 348:1427–1430. doi: 10.1016/S0140-6736(96)06088-6. [DOI] [PubMed] [Google Scholar]
- 4.Franco JR, Simarro PP, Diarra A, Jannin JG. 2014. Epidemiology of human African trypanosomiasis. Clin Epidemiol 6:257–275. doi: 10.2147/CLEP.S39728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alsford S, Eckert S, Baker N, Glover L, Sanchez-Flores A, Leung KF, Turner DJ, Field MC, Berriman M, Horn D. 2012. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 482:232–236. doi: 10.1038/nature10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stuart K, Brun R, Croft S, Fairlamb A, Gurtler RE, McKerrow J, Reed S, Tarleton R. 2008. Kinetoplastids: related protozoan pathogens, different diseases. J Clin Invest 118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Den Abbeele J, Claes Y, van Bockstaele D, Le Ray D, Coosemans M. 1999. Trypanosoma brucei spp. development in the tsetse fly: characterization of the post-mesocyclic stages in the foregut and proboscis. Parasitology 118:469–478. doi: 10.1017/S0031182099004217. [DOI] [PubMed] [Google Scholar]
- 8.Vickerman K, Tetley L, Hendry KA, Turner CM. 1988. Biology of African trypanosomes in the tsetse fly. Biol Cell 64:109–119. doi: 10.1016/0248-4900(88)90070-6. [DOI] [PubMed] [Google Scholar]
- 9.Harshey RM. 2003. Bacterial motility on a surface: many ways to a common goal. Annu Rev Microbiol 57:249–273. doi: 10.1146/annurev.micro.57.030502.091014. [DOI] [PubMed] [Google Scholar]
- 10.Oberholzer M, Lopez MA, McLelland BT, Hill KL. 2010. Social motility in African trypanosomes. PLoS Pathog 6:e1000739. doi: 10.1371/journal.ppat.1000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaiser D. 2003. Coupling cell movement to multicellular development in myxobacteria. Nat Rev Microbiol 1:45–54. doi: 10.1038/nrmicro733. [DOI] [PubMed] [Google Scholar]
- 12.Gibiansky ML, Conrad JC, Jin F, Gordon VD, Motto DA, Mathewson MA, Stopka WG, Zelasko DC, Shrout JD, Wong GC. 2010. Bacteria use type IV pili to walk upright and detach from surfaces. Science 330:197. doi: 10.1126/science.1194238. [DOI] [PubMed] [Google Scholar]
- 13.Bassler BL, Losick R. 2006. Bacterially speaking. Cell 125:237–246. doi: 10.1016/j.cell.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Trimble MJ, McCarter LL. 2011. bis-(3′-5′)-cyclic dimeric GMP-linked quorum sensing controls swarming in Vibrio parahaemolyticus. Proc Natl Acad Sci U S A 108:18079–18084. doi: 10.1073/pnas.1113790108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Firtel RA, Meili R. 2000. Dictyostelium: a model for regulated cell movement during morphogenesis. Curr Opin Genet Dev 10:421–427. doi: 10.1016/S0959-437X(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 16.Merritt JH, Ha DG, Cowles KN, Lu W, Morales DK, Rabinowitz J, Gitai Z, O'Toole GA. 2010. Specific control of Pseudomonas aeruginosa surface-associated behaviors by two c-di-GMP diguanylate cyclases. mBio 1(4):e00183-10. doi: 10.1128/mBio.00183-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srivastava D, Waters CM. 2012. A tangled web: regulatory connections between quorum sensing and cyclic Di-GMP. J Bacteriol 194:4485–4493. doi: 10.1128/JB.00379-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuchma SL, Brothers KM, Merritt JH, Liberati NT, Ausubel FM, O'Toole GA. 2007. BifA, a cyclic-di-GMP phosphodiesterase, inversely regulates biofilm formation and swarming motility by Pseudomonas aeruginosa PA14. J Bacteriol 189:8165–8178. doi: 10.1128/JB.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merritt JH, Brothers KM, Kuchma SL, O'Toole GA. 2007. SadC reciprocally influences biofilm formation and swarming motility via modulation of exopolysaccharide production and flagellar function. J Bacteriol 189:8154–8164. doi: 10.1128/JB.00585-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyd CD, O'Toole GA. 2012. Second messenger regulation of biofilm formation: breakthroughs in understanding c-di-GMP effector systems. Annu Rev Cell Dev Biol 28:439–462. doi: 10.1146/annurev-cellbio-101011-155705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simm R, Morr M, Kader A, Nimtz M, Romling U. 2004. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol Microbiol 53:1123–1134. doi: 10.1111/j.1365-2958.2004.04206.x. [DOI] [PubMed] [Google Scholar]
- 22.Klengel T, Liang WJ, Chaloupka J, Ruoff C, Schroppel K, Naglik JR, Eckert SE, Mogensen EG, Haynes K, Tuite MF, Levin LR, Buck J, Muhlschlegel FA. 2005. Fungal adenylyl cyclase integrates CO2 sensing with cAMP signaling and virulence. Curr Biol 15:2021–2026. doi: 10.1016/j.cub.2005.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gould MK, Bachmaier S, Ali JA, Alsford S, Tagoe DN, Munday JC, Schnaufer AC, Horn D, Boshart M, de Koning HP. 2013. Cyclic AMP effectors in African trypanosomes revealed by genome-scale RNA interference library screening for resistance to the phosphodiesterase inhibitor CpdA. Antimicrob Agents Chemother 57:4882–4893. doi: 10.1128/AAC.00508-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paindavoine P, Rolin S, Van Assel S, Geuskens M, Jauniaux JC, Dinsart C, Huet G, Pays E. 1992. A gene from the variant surface glycoprotein expression site encodes one of several transmembrane adenylate cyclases located on the flagellum of Trypanosoma brucei. Mol Cell Biol 12:1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saada EA, Kabututu ZP, Lopez M, Shimogawa MM, Langousis G, Oberholzer M, Riestra A, Jonsson Z, Wohlschlegel JA, Hill KL. 2014. Insect stage-specific receptor adenylate cyclases are localized to distinct subdomains of the Trypanosoma brucei flagellar membrane. Eukaryot Cell 13:1064–1076. doi: 10.1128/EC.00019-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naula C, Schaub R, Leech V, Melville S, Seebeck T. 2001. Spontaneous dimerization and leucine-zipper induced activation of the recombinant catalytic domain of a new adenylyl cyclase of Trypanosoma brucei, GRESAG4.4B. Mol Biochem Parasitol 112:19–28. doi: 10.1016/S0166-6851(00)00338-8. [DOI] [PubMed] [Google Scholar]
- 27.Salmon D, Vanwalleghem G, Morias Y, Denoeud J, Krumbholz C, Lhomme F, Bachmaier S, Kador M, Gossmann J, Dias FB, De Muylder G, Uzureau P, Magez S, Moser M, De Baetselier P, Van Den Abbeele J, Beschin A, Boshart M, Pays E. 2012. Adenylate cyclases of Trypanosoma brucei inhibit the innate immune response of the host. Science 337:463–466. doi: 10.1126/science.1222753. [DOI] [PubMed] [Google Scholar]
- 28.Gerdes JM, Davis EE, Katsanis N. 2009. The vertebrate primary cilium in development, homeostasis, and disease. Cell 137:32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langousis G, Hill KL. 2014. Motility and more: the flagellum of Trypanosoma brucei. Nat Rev Microbiol 12:505–518. doi: 10.1038/nrmicro3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wirtz E, Leal S, Ochatt C, Cross GA. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol 99:89–101. doi: 10.1016/S0166-6851(99)00002-X. [DOI] [PubMed] [Google Scholar]
- 31.Oberholzer M, Lopez MA, Ralston KS, Hill KL. 2009. Approaches for functional analysis of flagellar proteins in African trypanosomes. Methods Cell Biol 93:21–57. doi: 10.1016/S0091-679X(08)93002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Redmond S, Vadivelu J, Field MC. 2003. RNAit: an automated web-based tool for the selection of RNAi targets in Trypanosoma brucei. Mol Biochem Parasitol 128:115–118. doi: 10.1016/S0166-6851(03)00045-8. [DOI] [PubMed] [Google Scholar]
- 33.LaCount DJ, Barrett B, Donelson JE. 2002. Trypanosoma brucei FLA1 is required for flagellum attachment and cytokinesis. J Biol Chem 277:17580–17588. doi: 10.1074/jbc.M200873200. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen HT, Sandhu J, Langousis G, Hill KL. 2013. CMF22 is a broadly conserved axonemal protein and is required for propulsive motility in Trypanosoma brucei. Eukaryot Cell 12:1202–1213. doi: 10.1128/EC.00068-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kabututu ZP, Thayer M, Melehani JH, Hill KL. 2010. CMF70 is a subunit of the dynein regulatory complex. J Cell Sci 123:3587–3595. doi: 10.1242/jcs.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Ralston KS, Lerner AG, Diener DR, Hill KL. 2006. Flagellar motility contributes to cytokinesis in Trypanosoma brucei and is modulated by an evolutionarily conserved dynein regulatory system. Eukaryot Cell 5:696–711. doi: 10.1128/EC.5.4.696-711.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ralston KS, Kisalu NK, Hill KL. 2011. Structure-function analysis of dynein light chain 1 identifies viable motility mutants in bloodstream-form Trypanosoma brucei. Eukaryot Cell 10:884–894. doi: 10.1128/EC.00298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oberholzer M, Morand S, Kunz S, Seebeck T. 2006. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol Biochem Parasitol 145:117–120. doi: 10.1016/j.molbiopara.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 40.Bieger B, Essen LO. 2001. Structural analysis of adenylate cyclases from Trypanosoma brucei in their monomeric state. EMBO J 20:433–445. doi: 10.1093/emboj/20.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zoraghi R, Kunz S, Gong K, Seebeck T. 2001. Characterization of TbPDE2A, a novel cyclic nucleotide-specific phosphodiesterase from the protozoan parasite Trypanosoma brucei. J Biol Chem 276:11559–11566. doi: 10.1074/jbc.M005419200. [DOI] [PubMed] [Google Scholar]
- 42.Merchant S, Hill K, Howe G. 1991. Dynamic interplay between two copper-titrating components in the transcriptional regulation of Cyt C6. EMBO J 10:1383–1389. (Erratum, 10:2320.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thurston SJ, Saffer JD. 1989. Ultraviolet shadowing nucleic acids on nylon membranes. Anal Biochem 178:41–42. doi: 10.1016/0003-2697(89)90353-9. [DOI] [PubMed] [Google Scholar]
- 44.Gould MK, de Koning HP. 2011. Cyclic-nucleotide signalling in protozoa. FEMS Microbiol Rev 35:515–541. doi: 10.1111/j.1574-6976.2010.00262.x. [DOI] [PubMed] [Google Scholar]
- 45.Lopez MA, Nguyen HT, Oberholzer M, Hill KL. 2011. Social parasites. Curr Opin Microbiol 14:642–648. doi: 10.1016/j.mib.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Koning HP, Gould MK, Sterk GJ, Tenor H, Kunz S, Luginbuehl E, Seebeck T. 2012. Pharmacological validation of Trypanosoma brucei phosphodiesterases as novel drug targets. J Infect Dis 206:229–237. doi: 10.1093/infdis/jir857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanoune J, Defer N. 2001. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol 41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- 48.Salmon D, Bachmaier S, Krumbholz C, Kador M, Gossmann JA, Uzureau P, Pays E, Boshart M. 2012. Cytokinesis of Trypanosoma brucei bloodstream forms depends on expression of adenylyl cyclases of the ESAG4 or ESAG4-like subfamily. Mol Microbiol 84:225–242. doi: 10.1111/j.1365-2958.2012.08013.x. [DOI] [PubMed] [Google Scholar]
- 49.Baillie GS, Houslay MD. 2005. Arrestin times for compartmentalised cAMP signalling and phosphodiesterase-4 enzymes. Curr Opin Cell Biol 17:129–134. doi: 10.1016/j.ceb.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 50.Oberholzer M, Bregy P, Marti G, Minca M, Peier M, Seebeck T. 2007. Trypanosomes and mammalian sperm: one of a kind? Trends Parasitol 23:71–77. doi: 10.1016/j.pt.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 51.Oberholzer M, Marti G, Baresic M, Kunz S, Hemphill A, Seebeck T. 2007. The Trypanosoma brucei cAMP phosphodiesterases TbrPDEB1 and TbrPDEB2: flagellar enzymes that are essential for parasite virulence. FASEB J 21:720–731. doi: 10.1096/fj.06-6818com. [DOI] [PubMed] [Google Scholar]
- 52.Emes RD, Yang Z. 2008. Duplicated paralogous genes subject to positive selection in the genome of Trypanosoma brucei. PLoS One 3:e2295. doi: 10.1371/journal.pone.0002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tam R, Saier MH Jr. 1993. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol Rev 57:320–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butler MT, Wang Q, Harshey RM. 2010. Cell density and mobility protect swarming bacteria against antibiotics. Proc Natl Acad Sci U S A 107:3776–3781. doi: 10.1073/pnas.0910934107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Velicer GJ, Vos M. 2009. Sociobiology of the myxobacteria. Annu Rev Microbiol 63:599–623. doi: 10.1146/annurev.micro.091208.073158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.