

Abstract
G protein–coupled receptors (GPCRs) are integral membrane proteins that represent an important class of drug targets. In particular, aminergic GPCRs interact with a significant portion of drugs currently on the market. However, most drugs that target these receptors are associated with undesirable side effects, which are due in part to promiscuous interactions with close homologs of the intended target receptors. Here, based on a systematic analysis of all 37 of the currently available high-resolution crystal structures of aminergic GPCRs, we review structural elements that contribute to and can be exploited for designing subtype-selective compounds. We describe the roles of secondary binding pockets (SBPs), as well as differences in ligand entry pathways to the orthosteric binding site, in determining selectivity. In addition, using the available crystal structures, we have identified conformational changes in the SBPs that are associated with receptor activation and explore the implications of these changes for the rational development of selective ligands with tailored efficacy.
I. Introduction
G protein–coupled receptors (GPCRs) are a large superfamily of receptors with seven transmembrane (TM) segments. They are critical in signal transduction of a wide variety of molecules, including hormones and neurotransmitters, and consequently are pharmacological targets for many diseases (Overington et al., 2006). Aminergic GPCRs, which belong to class A rhodopsin-like GPCRs, are of critical importance as they are the targets for ∼25% of current drugs (Rask-Andersen et al., 2011). However, most drugs that target these receptors have been associated with side effects, which in many cases are due to promiscuous interactions with proteins other than the intended therapeutic target(s) (Kola and Landis, 2004; Liebler and Guengerich, 2005; Besnard et al., 2012). Improving the pharmacological specificity profile of drugs, especially in the case of close receptor homologs of the primary target, is particularly challenging.
Based on their ability to recognize specific biogenic amines as their endogenous agonist, the 42 human aminergic GPCRs can be classified into subfamilies that include the adrenergic, muscarinic, dopaminergic, histaminergic, serotoninergic, and trace amine receptors. Each subfamily can be further categorized into subgroups that consist of closely-related subtypes (Fig. 1). Receptors within a particular subfamily are often expressed in different brain regions and/or peripheral tissues and can also be coupled to divergent intracellular signaling transducers, only some of which are associated with the therapeutic effect of drugs. Therefore, selective targeting of a particular receptor subtype and/or a particular signal transduction pathway is critical for elucidating mechanistic underpinnings of disease states and may also lead to improved drug design and development (Keck et al., 2014).
Fig. 1.

Alignment of the ligand-contacting residues in the 42 human aminergic receptors. An analysis of ligand-contacting residues was carried out for all 36 available ligand-bound crystal structures of aminergic receptors. The residue positions are indicated by the Ballesteros-Weinstein numbers at the top of each column (for EL2, the residues are indexed relative to the conserved Cys residue, EL2.50, which makes a disulfide bond to Cys3.25) and colored by the classification of the positions: OBS positions (see section II.A) in cyan, SBP positions (see section II.B) in yellow, and 7.42, which contacts ligand but is not in the OBS or any SBP, in gray (see Fig. 2). The OBS positions were defined as the common set of positions identified by the SASA analysis in all structures except for the agonist-bound M2R structures (PDB IDs 4MQS and 4MQT). The SASA values for each residue were calculated using the program Naccess (Hubbard and Thornton, 1993), with probe size of 1.4 Å, Z-slice of 0.001; we used a cutoff of 0.2% to identify the residues with different accessibility in the presence and absence of bound ligand. Note that the OBS includes one residue from EL2 approximately at position EL2.52—although in the H1R structure (PDB ID 3RZE) or the antagonist-bound M2R and M3R structures (PDB IDs 3UON and 4DAJ) EL2.52 does not face the OBS, the residue at EL2.49 or EL2.55 contacts ligand in a manner similar to the residues at EL2.52 in other structures. The receptors are indicated by their UniProt entry names and colored by subfamily. We define a subgroup within a subfamily, indicated by brackets, to be a set of receptors in which all the pairwise sequence identities of the TM domain are >45%. The residues that contact a ligand in any of the structures for a particular receptor are shaded in cyan, yellow, or gray as described above. The residues interacting with the positive allosteric modulator LY2119620 in the M2R structure (PDB ID 4MQT) are underlined, except for Glu172EL2.46 and Ala414EL3, which are not shown. All the amino acid sequences are human; for β1AR and M3R, however, the contact residues were identified in the crystal structures of turkey β1AR and rat M3R.
In this review, based on our analysis of all the available high-resolution crystal structures of these receptors, we discuss progress in understanding the structural basis for subtype-selective compounds within the aminergic GPCRs. Building upon our understanding of selectivity among the receptor subfamilies that we discussed previously (Shi and Javitch, 2002), we focus on specificity within individual subgroups. We highlight the difficulty in developing selective compounds targeted solely to the orthosteric binding site (OBS), as well as the role of secondary binding pockets (SBPs) and ligand entry pathways in determining receptor subtype selectivity.
II. Ligand Binding Pockets Revealed by Crystal Structures of Aminergic Receptors
A. Orthosteric Binding Site
1. Residues Forming the Orthosteric Binding Site.
Before the crystal structures of rhodopsin-like receptors became available, residues that were critical for ligand binding in aminergic GPCRs were indirectly determined by extensive mutagenesis studies, and the substituted-cysteine accessibility method was used to identify systematically the surface of the binding site crevice (reviewed in Javitch et al., 2002). Several highly conserved residues in the TM domain were found to form critical receptor-ligand interactions in the endogenous ligand binding site—specifically, Asp3.32 [superscripts denote Ballesteros-Weinstein residue numbering (Ballesteros and Weinstein, 1995)] forms a hydrogen bond with the cationic amine of the ligand; Ser5.42 and Ser5.46 interact with the meta-OH and para-OH moieties of catecholamine agonists and a cluster of aromatic residues in TM6 (Trp6.48, Phe/Tyr6.51, and Phe6.52) that interact with the aromatic moiety of the ligand (reviewed in Shi and Javitch, 2002, see references therein). In addition, residues in extracellular loop 2 (EL2) have been implicated in ligand specificity in aminergic receptors (Olah et al., 1994; Kim et al., 1996; Zhao et al., 1996; Wurch et al., 1998) and we found Ile184EL2.52, the second residue after the conserved and disulfide-bonded Cys in EL2 (defined here as CysEL2.50), to be directly involved in ligand binding to the dopamine D2 receptor (D2R) (Shi and Javitch, 2004).
In light of recent progress in GPCR structure determination, we sought to further refine our understanding of the residues forming the OBS of all aminergic receptors and thus their role in ligand selectivity at closely related receptor homologs. Thirty-seven high-resolution crystal structures have been solved for 8 of the 42 aminergic receptors [the β1- and β2-adrenergic receptors (β1AR and β2AR), dopamine D3 receptor (D3R), histamine H1 receptor (H1R), serotonin 1B and 2B receptors (5HT1BR and 5HT2BR), and muscarinic acetylcholine M2 and M3 receptors (M2R and M3R)], including several structures in active conformations and one apo-structure without a bound ligand (see Fig. 2).
Fig. 2.

Identification of SBPs in the crystal structures of aminergic receptors. The numbered (“0” to “4”) positions for each complex indicate the residues outside of the OBS (non-OBS) that interact with a specific ligand moiety—the closest distance between any heavy atoms of the residue and a ligand moiety is within the van der Waals interaction distance, which we define in this study to be the sum of the van der Waals radii plus 0.8 Å. A SBP was identified as a cluster of ≥3 non-OBS residues that interact with a small ligand moiety, which we defined as a set of ligand heavy atoms with the largest pairwise distance between any pair <4.2 Å (the largest pairwise distance within a hydroxyphenyl moiety). “0” indicates the residue is not within any SBP in that structure, “1” to “4” indicate the involvement of the residue in forming a particular SBP (“1,” “SBP237”; “2,” “SBP567”; “3,” “SBP3456”, “4,” “SBP7”). These SBPs are classified according to their location and named according to the surrounding TMs (see Fig. 4 for the representatives of “1” to “3”); thus, in a particular complex, a ligand moiety does not necessarily interact with every surrounding TM. The structures are arranged according to the conformational state of the receptor (i.e., active, intermediate-active, and inactive, which are indicated by □, ◇, and ∆, respectively) and the ligand efficacy (i.e., agonist in red, partial or biased agonist in pink, and antagonist/inverse agonist in green). The residue sets from the structures for the same receptor bound with the same or highly similar ligand are combined, e.g., the structures with PDB IDs 3P0G, 3SN6, and 4LDE are all β2AR in complex with BI-167107. The fragments bound to β1AR are categorized separately as their efficacies were unknown (colored in orange) (Christopher et al., 2013). The phosphate ion in the H1R structure (PDB ID 3RZE) is counted as an extension of the orthosteric ligand (Shimamura et al., 2011). The positions in the top row are colored if they only interact with either agonists/partial agonists (red) or antagonists/inverse agonists (green).
In the present study, we carried out a systematic comparative analysis of the ligand-contacting residues in these structures. Positions at which bound ligand decreased the solvent-accessible surface area (SASA) were defined as ligand-contacting residues (Ballesteros et al., 2001; Beuming et al., 2006). Overall, we found the number of contact residues to be correlated with ligand size, such that larger ligands have more contact residues (Fig. 3). Ligand contacting residues at 12 common positions were identified in 34 of 36 ligand-bound structures. We define these positions—3.32, 3.33, 3.36, EL2.52, 5.42, 5.46, 6.48, 6.51, 6.52, 6.55, 7.39, and 7.43 (cyan in Figs. 1 and 4, A and B)—as the OBS residue positions. In the two M2R structures bound to the agonist iperoxo (PDB IDs 4MQS and 4MQT, note 4MQT is additionally bound with the allosteric modulator LY2119620), the residues at positions EL2.52, 5.42, and 6.55, which are slightly more peripheral, are not ligand-contacting, indicating that, at least for this rather small agonist, they are not essential as direct contacts. However, these residues still play a direct role for all other ligands in M2R and other receptors studied.
Fig. 3.

Statistics of ligand-contacting residues. The number of contact residues is plotted against ligand size for the 36 available ligand-receptor complexes. The color and shape of each point indicate the ligand efficacy and the receptor conformational state, respectively, as specified in Fig. 2. Although the number of contact residues is correlated with ligand size, the ligand efficacy is not correlated with ligand size.
Fig. 4.

The orthosteric binding site and secondary binding pockets. (A) All the identified ligand-contacting positions for the aminergic receptors are mapped onto a β2AR structure (PDB ID 4LDO). The 12 OBS residues are in cyan, and the SBP residues are in yellow. Three representative SBPs shown in (C–E) are indicated by red dotted circles. (B) A zoomed-in view of the OBS in the β2AR bound with adrenaline (PDB ID 4LDO). The side chains of the three highly conserved residues within the aminergic family, Asp3.32, Trp6.48, and Tyr7.43, and the conserved EL2.50-Cys3.25 disulfide bond are shown as sticks; for all other residues, sticks were drawn from the Cα to the center of mass (represented as spheres) of the heavy atoms of the side chain. The zoomed-in views of the SBPs, “SBP237” (C), “SBP567” (D), “SBP3456” (E), are shown in representative structures [PDB IDs 4AMI (C), 4IB4 (D), 3UON (E)]. The SBPs are classified according to their locations and named according to the surrounding TMs (see Fig. 2). The ligands in the respective structures are in spheres with the carbon atoms of adrenaline (B) or the moieties occupying the SBPs (C–E) colored in orange.
The OBS that we defined here is subtly different from that of Christopoulos et al. (2014) (“the binding site/s on a receptor macromolecule that is/are recognized by the endogenous agonist/s for that receptor”). Although the cavity enclosed by the OBS residues identified in this study is occupied by the endogenous agonists of aminergic receptors (e.g., all the OBS residues are in contact with the endogenous ligand adrenaline in β2AR based on our SASA analysis), our goal was to identify the entire set of residues critical for recognition of all orthosteric ligands, including full/partial agonists, antagonists, and inverse agonists.
Among the OBS residues, the triad consisting of Asp3.32, Trp6.48, and Tyr/Trp7.43 is conserved across the aminergic receptor family (Fig. 5). Whereas Trp6.48 is also conserved in many other rhodopsin-like receptors, Asp3.32 and Tyr/Trp7.43 are likely critical for recognition of aminergic ligands. A hydrogen bond between Asp3.32 and Tyr/Trp7.43, present in all available crystal structures of aminergic receptors, likely stabilizes the side chain of Asp3.32, thereby facilitating its interaction with the amine group in the ligands (Fig. 6, A and B). Similar conservation is also observed in the opioid and somatostatin receptor subfamilies of the peptide receptor family, in which the presence of an Asp at position 3.32 is correlated with a Tyr at position 7.43. Indeed, in the available crystal structures of opioid receptors, Asp3.32 always interacts simultaneously with both Tyr7.43 and a charged amine group from the ligand (Granier et al., 2012; Manglik et al., 2012; Thompson et al., 2012; Fenalti et al., 2014) (Fig. 6C), with the exception of the structure of κ-opioid receptor in complex with JDTic [(3R)-1,2,3,4-tetrahydro-7-hydroxy-N-[(1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl]-3-isoquinolinecarboxamide] (PDB ID 4DJH), in which Asp3.32 interacts with two positively charged amine groups of the ligand (Wu et al., 2012).
Fig. 5.

Conservation of ligand-contacting residues. The conservation indices (CI) of ligand-contacting residues across the aminergic receptors and within the indicated subfamilies and subgroups are calculated by the ProperTM server (Shi et al., 2001; Beuming and Weinstein, 2004). The positions with low sequence conservation are indicated by filled colors (CI values ≤ 0.25 in light blue, and those between 0.25 and 0.50 in light green). The highly conserved positions among all amine receptors with CI > 0.75 are highlighted in red. Note, at the aminergic receptor family level, except for Asp3.32, Trp6.48, Tyr/Trp7.43, other OBS positions are not more conserved than SBP positions.
Fig. 6.

The highly conserved residues in the OBS. The highly conserved OBS residues (see Fig. 5) Asp3.32, Trp6.48, Tyr/Trp7.43 (cyan) are shown as sticks in the inactive (PDB ID 2RH1) (A) and active (PDB ID 4LDO) (B) structures of β2AR. Asp3.32 forms a hydrogen bond with Tyr7.43 in all available crystal structures of aminergic receptors, suggesting that this interaction is likely to be critical for ligand-recognition of this family. This triad is similarly conserved in the opioid receptor family, as exemplified in the structure of nociceptin opioid receptor bound to a peptide-mimetic compound (PDB ID 4EA3) (C). The ligands are shown as orange sticks; the hydrogen bonds between the nitrogen atoms of the positively charged amine groups and Asp3.32 or between Asp3.32 and Tyr7.43 are indicated by dotted lines.
2. Ligand Selectivity between the Subgroups of an Individual Subfamily.
By analyzing the crystal structure of bovine rhodopsin in the context of experimental findings, we previously proposed a mechanism of structural mimicry in which the similar folds of rhodopsin and other rhodopsin-like GPCRs are formed by relatively divergent sequences (Ballesteros et al., 2001). Indeed, using the same protocol used to identify OBS residues of aminergic receptors, we analyzed the crystal structures of opioid receptors and identified a set of OBS positions highly similar to that of aminergic receptors, with the exception of the 5.46 and EL2 positions (Figs. 1 and 7). Despite these similarities in the positions of the OBS residues, differences in sequence within the common fold also contribute to structural differences that determine the selectivity for different receptor subfamilies (Ballesteros et al., 2001), and this is also likely the case for the selectivity between the subgroups of an individual subfamily.
Fig. 7.
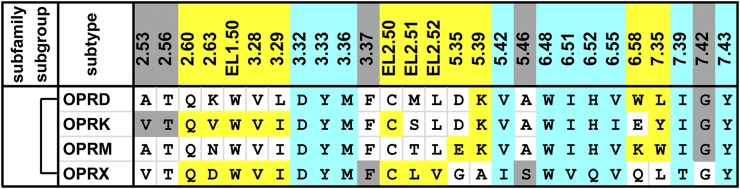
Alignment of the ligand-contacting residues in the four opioid receptors. The ligand-contacting residues were identified by SASA analysis for all five available crystal structures of opioid receptors. The residue positions are indicated by the Ballesteros-Weinstein numbers at the top of each column (for EL2, the residues are indexed relative to the conserved Cys residue, EL2.50, which makes a disulfide bond to Cys3.25) and colored by the classification of the positions: OBS positions in cyan, SBP positions in yellow, and the remaining positions in gray. The OBS and SBP positions were identified similarly to those in the aminergic receptors. Note the identified ligand-contacting residues are similar to those previously summarized (Filizola and Devi, 2013). The bracket on the left indicates high pairwise sequence identities of >60% among all four receptors. The receptors are indicated by their UniProt entry names. All the amino acid sequences are human; for the µ-opioid receptor (OPRM), the contact residues are identified in the crystal structure of mouse OPRM.
For example, the adrenergic receptor (AR) subfamily consists of three subgroups, namely α1-, α2-, and βARs, with three subtypes within each subgroup (Ahlquist, 1948; Bylund et al., 1994). These receptors are present in almost all peripheral tissues as well as the central and sympathetic nervous systems, are activated upon binding the endogenous ligands adrenaline and noradrenaline, and control heart and lung function, blood pressure, and a variety of metabolic and central nervous system functions (Ahlquist, 1948; Taira et al., 2008). The activation of αARs and βARs results in distinct, therapeutically relevant physiologic responses (vasoconstriction versus increased heart rate and contraction, respectively). Drugs have been developed that display significant selectivity between α2ARs and βARs by taking advantage of the significant divergence within the OBS of these receptors (four OBS residues differ between these two subgroups). This is highlighted by the β2AR antagonist dihydroalprenolol, which is 23,000-fold selective for β2AR over α2AAR. Mutation of just one of the four divergent OBS residues, Phe4127.39, in the human α2AAR to that in β2AR (Asn), resulted in a 3000-fold increase in binding affinity for this compound (Suryanarayana et al., 1991).
Dopamine receptors (DRs) are classified into two subgroups, D1-like and D2-like, based on sequence, G protein coupling, and pharmacology (Lachowicz and Sibley, 1997). D1-like DRs are quite divergent from the D2-like DRs, and in fact are closer in sequence to the βARs (∼43% versus ∼39% sequence identity between D1-like DRs and βARs and between D1-like and D2-like DRs, respectively) (Fredriksson et al., 2003). Although both D1-like and D2-like DRs share dopamine as their endogenous ligand, 6 of 12 OBS positions are divergent between these two subgroups (Fig. 1). Not surprisingly, agonists and antagonists that display a high degree of D1- or D2-like selectivity have been developed (Beaulieu and Gainetdinov, 2011).
Histamine H1R and H3R/H4R, which are classified into two subgroups, are relatively divergent (<∼30% sequence identity in TM domain and only 4 or 5 of 12 OBS residues conserved), and the development of subgroup-selective compounds may be of great clinical significance (Kiss et al., 2008). Structure-based virtual high-throughput screening (VHTS) of ∼100,000 fragment-like compounds against the H1R crystal structure led to the identification of subgroup-selective compounds with up to 1000-fold selectivity for H1R over H3R/H4R (de Graaf et al., 2011), again illustrating effective differentiation between subgroups with divergent OBS residues.
3. The Difficulty of Developing Subtype-Selective Ligands Targeting the Orthosteric Binding Site.
The development of subtype-selective drugs is one approach to optimizing therapeutic efficacy while diminishing untoward side effects. However, among the subtypes within a receptor subgroup, the sequence similarity in the OBS can be very high, and this has made the development of subtype-selective drugs for some aminergic GPCRs extremely challenging (Congreve et al., 2011). There are 11 subgroups of closely related aminergic receptors (defined as >45% sequence identity in the TM domain; see Fig. 1). Within these subgroups, the 12 OBS residues described above are completely conserved between two or more subtypes: M1,3,4,5Rs; α2B,CARs; β1–3ARs; D1,5Rs; D2,3Rs; 5HT1B,DRs; and 5HT2B,CRs (Fig. 1).
Despite the high conservation of the OBS, subtype-specific compounds have been developed for some receptors through extensive medicinal chemistry campaigns. For example, within the D2-like dopamine receptor subgroup, D3R- or D4R-selective ligands (see below) and to a lesser degree D2R-preferential ligands have been discovered. However, within the D1-like dopamine receptor subgroup, separation of D1R- and D5R-selective ligands using traditional medicinal chemistry efforts has thus far been unsuccessful (Giorgioni et al., 2008), as has been the case with other subgroups as well.
Crystal structures of receptors from the same subgroups are now available for two subfamilies of the aminergic receptors, thus enabling comparison of conformational differences in the OBS of these receptors. The structures of β1AR and β2AR in inactive conformations reveal that, between the two subtypes, the size and shape of the OBS are almost identical (RMSDheavy atoms [root-mean-square deviation] < 0.8 Å). Similarly, the structures of 3-quinuclidinyl-benzilate-bound M2R (PDB ID 3UON) and tiotropium-bound M3R (PDB ID 4DAJ) show high structural similarity in the completely conserved OBS (RMSDheavy atoms < 0.9 Å), although there are minor rearrangements of the Tyr7.39 side chains, likely caused by the different residues at position 2.61 (Haga et al., 2012; Kruse et al., 2012).
The difficulty of developing subtype-selective orthosteric ligands also has been reflected in VHTS efforts to identify subtype-selective compounds for closely related subtype receptors, such as the D3R and D2R. VHTS of over 3 million “lead-like” compounds from the ZINC library was performed against the D3R structure, and the identified hits were tested for binding at D3R and its close homolog D2R (Carlsson et al., 2011). This study was not designed to address specificity, and not surprisingly, the hit compounds showed little selectivity for D3R over D2R, despite having relatively high affinity for D3R (Carlsson et al., 2011).
In a separate study, a counter-screening strategy failed to identify M2R/M3R subtype-selective compounds (Kruse et al., 2013b). In this study, over 3 million “lead-like” and fragment compounds from the ZINC library were screened against the OBS of both the M2R and M3R structures, and the top-ranked M3R hits with the largest rank difference between the M3R and M2R were selected to identify novel M3R-selective compounds. The experimentally measured binding selectivity of the selected compounds, however, was only modest (M2R/M3R Ki < 6-fold) (Kruse et al., 2013b). This example highlights the challenge in targeting only the highly conserved OBS for subtype specificity. Curiously, although the virtual screening was against the antagonist bound M3R and M2R structures, the screening identified a M3R-selective partial agonist with low micromolar EC50, which was devoid of activity at M2R. Intriguingly, this compound had extremely low binding affinity at both M2R and M3R (Ki > 100 μM), highlighting the complexity of designing selective ligands with tailored efficacy (see below).
Thus, an approach that is focused solely on the near-identical OBS within a subfamily of aminergic GPCR is likely to fail in identifying highly selective compounds. In contrast, more recent efforts to develop subtype-selective drugs have focused on targeting concomitantly both the OBS and SBPs that are more divergent in sequence and structure than the OBS (Jones et al., 2008; Langmead et al., 2008; Chien et al., 2010; Newman et al., 2012a; Michino et al., 2013).
B. Secondary Binding Pockets Revealed by Crystal Structures of Aminergic Receptors
An SBP is a part of the ligand binding site that extends beyond the OBS. Because the residues at the peripheral regions of the OBS are less conserved within individual subgroups (Fig. 5), these regions can be exploited to achieve subtype selectivity. In our analysis of the available aminergic receptor crystal structures, we define an SBP as a cavity consisting of three or more ligand-contacting residues outside of the OBS that are in direct contact with a defined ligand moiety (see Fig. 2). In this way, we avoid the complexity of identifying and defining cavities based solely on residue position and side chain orientation (Nayal and Honig, 2006). However, due to the difficulty in identifying such cavities with the SASA approach described above, we applied a distance-based approach to identify the residues that interact with a specific ligand moiety—the closest distance between any heavy atoms of the residue and a ligand moiety is within the van der Waals interaction distance, which we define in this study to be the sum of the van der Waals radii plus 0.8 Å. We found ligand-contacting residues forming SBPs in 30 out of 36 ligand-bound crystal structures of aminergic receptors (Fig. 2). Except for position 7.42, all the other ligand-contacting positions outside the OBS are part of a SBP in at least one structure (Figs. 1 and 2). This method led us to identify several distinct SBPs, and we describe below several examples that appear to be of functional importance.
In the β1AR structure that is in complex with the agonist bucindolol (PDB ID 4AMI), the aromatic substituent at the amine end of bucindolol occupies a SBP formed by TMs 2, 3, 7, EL1, and EL2 (SBP237) and contacts residues Trp1173.28, Cys199EL2.50, Asp200EL2.51, Phe3257.35, and Val3267.36 (Fig. 4C). These contacts in the SBP, especially those with EL2 residues, have been proposed to result in biased agonism by increasing the probability of conformational changes that promote the binding of arrestin more efficiently than G protein (Warne et al., 2012).
In the structures of 5HT2BR bound to ergotamine (PDB IDs 4IB4 and 4NC3), because of an inward shift of TM5, the phenyl moiety of ergotamine occupies a SBP located near the top of TMs 5, 6, and 7 (SBP567) and interacts with residues Thr210EL2.53, Lys211EL2.54, Met2185.39, Leu3476.58, Val3486.59, and Leu3627.35 (Fig. 4D). In the structure of 5HT1BR bound to ergotamine (PDB ID 4IAR), the phenyl moiety does not interact with this SBP because of the lack of similar rearrangements at the extracellular tip of TM5. The additional interaction with the SBP at the tip of TM5 in 5HT2BR has been proposed to contribute to β-arrestin biased signaling in 5HT2BR by stabilizing a conformation of the receptor that selectively interferes with G protein signaling (Wacker et al., 2013; Wang et al., 2013).
In the inactive structure of M2R bound to the antagonist 3-quinuclidinyl-benzilate (PDB ID 3UON), one of the phenyl moieties of 3-quinuclidinyl-benzilate points toward a SBP formed by TMs 3, 4, 5, 6 (SBP3456) and contacts residues Asn1083.37, Val1113.40, Trp1554.57, and Phe1955.47 (Haga et al., 2012) (Fig. 4E). Similar occupancies of the SBP3456, which is located more intracellularly than the OBS, are also observed in the inactive M3R structure bound to the antagonist tiotropium (PDB ID 4DAJ) and the active M2R structures bound to the agonist iperoxo (PDB IDs 4MQS and 4MQT). In the two inactive M2R and M3R structures, the moieties occupying SBP3456 could help to stabilize the side chain of Trp6.48 in the OBS in a parallel orientation relative to lipid bilayer, in contrast to the relatively perpendicular orientation in the active M2R structures and other crystal structures of aminergic receptors.
As of yet, none of these SBPs have been targeted rationally for the development of selective compounds. However, in our recent studies (Newman et al., 2012a; Michino et al., 2013), we extensively characterized the role of the SBPs in dopamine D3R/D2R-subtype selectivity (see below).
Interestingly, using a similar analysis to identify the SBP residues in opioid receptors, we found the positions forming SBPs in opioid receptors to be very similar to those of aminergic receptors, except for the positions from TM2, i.e., positions 2.53, 2.56, 2.60, and 2.63 in opioid receptors versus 2.61, 2.64, and 2.65 in aminergic receptors (Figs. 1 and 7). Thus, it appears that there is a face shift in the helix orientations of TM2 in the opioid receptors compared with those in aminergic receptors. Although the functional significance of this divergence is not clear, it argues that structural mimicry is less conserved beyond the OBS for different rhodopsin-like receptor families.
C. Allosteric Ligand Binding Pockets
The significant sequence divergence in the SBPs of aminergic GPCRs (Fig. 5) makes these cavities an enticing target for the development of more specific pharmacological agents. It is important, therefore, to consider the relationship between the SBPs that we identified for extended ligands and allosteric sites in aminergic GPCRs that have been targeted to achieve improved selectivity profiles (Valant et al., 2012; Wootten et al., 2013). The first and only high-resolution structure of a GPCR in complex with an allosteric ligand was recently reported; in M2R, the M2R/M4R-selective positive allosteric modulator LY2119620 (3-amino-5-chloro-N-cyclopropyl-4-methyl-6-[2-(4-methylpiperazin-1-yl)-2-oxoethoxy]thieno[2,3-b]pyridine-2-carboxamide), cocrystallized with an agonist bound in the OBS, was bound in a more extracellular location >10 Å away from the OBS (Kruse et al., 2013a). Intriguingly, except for Glu172EL2.46 and Ala414EL3, all residues in contact with LY2119620 have already been shown to interact with the extended portions of orthosteric ligands in various receptors (Fig. 1). Thus, the binding site of LY2119620 consists of two OBS residues Tyr4267.39 and Phe181EL2.55 and spans two SBPs in the extracellular vestibule identified by our analysis (Fig. 2), including one at the interface of TMs 2 and 7 and another enclosed by TM6, TM7, and EL2. Furthermore, the binding site overlaps significantly with those identified for several allosteric modulators of M2R by long-timescale molecular dynamics (MD) simulations (Dror et al., 2013). Similarly in this region of M1R, Tyr179EL2.51, and Trp4007.35 were found be critical for the binding of the selective positive allosteric modulator benzylquinolone carboxylic acid, as well as its modulation of the efficacy of carbachol (Ma et al., 2009; Abdul-Ridha et al., 2014). The interactions of receptor with these allosteric modulators involve several nonconserved SBP residues, which may account for the high selectivity of some allosteric compounds for specific subtypes of muscarinic receptors.
For example, the presence of positively charged Lys5237.32 in the extracellular vestibule of M3R was implicated as the critical determinant of M2R over M3R selectivity for currently available allosteric modulators of these receptors (Dror et al., 2013), and the introduction of acidic or basic residues at this position increases or decreases, respectively, the affinity of the allosteric modulator gallamine for several muscarinic receptors (Gnagey et al., 1999). Recent characterization of two M1R-selective agonists, TBPB (1-(1′-(2-methylbenzyl)-1,4′-bipiperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one) and 77-LH-28-1 (1-[3-(4-butyl-1-piperidinyl)propyl]-3,4-dihydro-2(1H)-quinolinone), indicates that the crevice formed by TMs 2, 3, and 7 is likely involved in binding the allosteric components of these compounds and may contribute to the selectivity of these agonists (Keov et al., 2014).
III. Mechanisms for Achieving Subtype Selectivity
A. Improving Selectivity by Targeting Secondary Binding Pockets with Extended Ligands.
Because allosteric compounds do not bind the OBS, developing such compounds with high affinity has been challenging, especially for certain aminergic receptors, such as D2-like receptors (Hoare et al., 2000; Raghavan et al., 2009). Another strategy to improve subtype-selectivity is to extend the orthosteric or primary pharmacophore (PP) of a ligand to take advantage of the divergence in the SBPs. In particular, a novel strategy is to link orthosteric and allosteric pharmacophores to create so-called “bitopic” ligands (Lane et al., 2013), which has been successfully applied to improve ligand affinity and selectivity in muscarinic acetylcholine receptors (Leach et al., 2012). Moreover, a SBP that has not yet been successfully targeted by an allosteric ligand can still be targeted by an extended moiety from the PP to gain selectivity, such as in the case of selective compounds for individual D2-like receptors described below. Although such compounds may not be formally bitopic as currently defined, because their secondary pharmacophore (SP) is not in itself a documented allosteric compound, they nonetheless bind and function in a bitopic manner (Christopoulos et al., 2014).
The subtypes within the D2-like subgroup are highly homologous. All 12 OBS residues are identical between D2R and D3R and only one differs in D4R. Overall, D2R and D3R are more closely related to each other than either is to D4R (78% identity between D2R and D3R and ∼50% identity between D4R and D2R/D3R in the TM domain). Although it is generally thought that antagonism of the D2R is essential for therapeutic efficacy of all antipsychotic drugs, these agents block both D3R and D2R. A significant difficulty in determining the relevance for antipsychotic efficacy of signaling processes downstream of specific dopamine receptors stems not only from limitations in modeling the diseases, e.g., in rodents, but just as importantly from a lack of appropriate pharmacological tools to selectively target the different receptors (Beaulieu and Gainetdinov, 2011). Furthermore, the finding of increased D3R expression in response to drugs of abuse, e.g., in the brains of cocaine-associated fatalities (Staley and Mash, 1996) and more recently in cocaine and methamphetamine abusers (Boileau et al., 2012; Matuskey et al., 2014; Payer et al., 2014) using the D3-preferential PET ligand [11C]PHNO [(+)-4-propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b][1,4]oxazin-9-ol], has prompted efforts toward the development of selective D3R antagonists for the treatment of drug addiction (Heidbreder and Newman, 2010; Newman et al., 2012b). The D4R has lower levels of expression in the brain compared with D2R and D3R but also was implicated recently in relapse to stimulant abuse (Di Ciano et al., 2014).
Early studies aimed at understanding ligand binding specificity at the D2-like subgroup focused on the selectivity between the relatively more divergent D2R and D4R (Simpson et al., 1999; Kortagere et al., 2004). From the accessible positions identified in D2R using substituted-cysteine accessibility method (Javitch et al., 2002), nonconserved residues in D2R were mutated either individually or in combination with the aligned residues in the D4R. A combined substitution of 4 to 6 residues in TMs 2, 3, and 7 was found to be sufficient to switch the affinity of the receptors for several chemically distinct D4R-selective antagonists. For example, the D2R affinity of the D4R-selective compound, CPPMA (chlorophenylpiperazinyl methylazaindole) (D2R/D4R Ki > 1000-fold), could be increased by three orders of magnitude to D4R-like affinity with a combination of mutations at residues Val912.61, Phe1103.28, Val1113.29, and Tyr4087.35, and it was hypothesized, prior to the availability of any high resolution GPCR structure, that these residues form a divergent pocket into which the ligand extends (Simpson et al., 1999).
The high-resolution structure of D3R (Chien et al., 2010) has invigorated interest in understanding the structural basis of subtype-selectivity between the closely related D3R and D2R. The D3R structure revealed a binding site located in the upper-half of the TM domain, in which the nonselective D2-like dopamine receptors antagonist eticlopride is bound. Of the 18 eticlopride contact residues in the D3R structure, 17 are identical in the D2R and one is similar (Fig. 1), highlighting the challenge in creating subtype-selective agents.
Previously, extensive medicinal chemistry efforts have led to the discovery of a class of 4-phenylpiperazine derivatives that is highly selective for D3R over D2R (e.g., compound R-22 [(R)-N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)-1H-indole-2-carboxamide] has a >100-fold selectivity for D3R over D2R) (Boeckler and Gmeiner, 2006; Newman et al., 2009). These D3R-selective compounds are characterized by a 4-phenylpiperazine scaffold and an aryl amide moiety connected by a four-carbon linking chain (Heidbreder and Newman, 2010). Based on previous studies (Ehrlich et al., 2009; Newman et al., 2009), we hypothesized that the 4-phenylpiperazine, the PP of R-22, would bind within the OBS of both D3R and D2R (Chien et al., 2010). This orientation of the PP positions the arylamide, the SP of R-22, in the SBP237 (see section II.B), which can accommodate the SP in several distinct binding modes. Given that the OBS is completely conserved between D3R and D2R, the substantial difference between the SBP in these receptors suggests that the different interactions of ligands with the SBP is responsible for D3R over D2R selectivity.
Indeed, our results from docking and subsequent MD simulations of D3R and D2R in complex with the D3R-selective 4-phenylpiperazine compound, R-22, showed that the PP moiety makes extensive interactions with the conserved OBS in both D3R and D2R. Although the SP moiety of R-22 in D3R packs against a hydrophobic pocket formed by residues Val862.61, Leu892.64, Gly94EL1.48, Phe1063.28, and Cys181EL2.50 [we define this part of the SBP237 to be Ptm23, to indicate the TMs (2 and 3) that form contact with the SP], the SP in D2R binds at the interface of TMs 1, 2, and 7 (Newman et al., 2012a). Interestingly, the Ptm23 residues overlap with the positions of the residues identified previously as responsible for D2R versus D4R selectivity (see above) (Simpson et al., 1999; Ehrlich et al., 2009).
Based on our simulation results, we hypothesized that the ability of the SP to adopt a favorable binding pose in Ptm23 of D3R is dependent on local receptor conformational flexibility. In particular, the flexibility of TM2 is likely determined by both the conserved Pro2.59, which acts as a hinge, and by the length and configuration of EL1, which modulates the degree of bending at the proline kink. EL1 in D3R is one residue longer than in D2R: D3R has two Gly residues in EL1 (93GGV95), whereas D2R has only one Gly residue (98GE99). The extra Gly94 in D3R both interacts directly with the SP of R-22 and renders EL1 sufficiently flexible so as to allow the TM2 rearrangement necessary to accommodate optimally the SP of R-22 (Michino et al., 2013). This is consistent with the dramatic loss of affinity of R-22 for D3R when this residue is deleted and the large increase of affinity of R-22 when a Gly is inserted into EL1 of D2R (Michino et al., 2013). In addition, computational hydration site predictions suggest that the Ptm23 subpocket has higher binding affinity in D3R than in D2R, which is optimally exploited by the R-22 binding mode in the SBP of D3R.
Thus, our combined computational and experimental results point to the difference in the conformations of the SBP between D3R and D2R and identify TM2 and EL1 as the critical determinants for D3R over D2R selectivity (Michino et al., 2013). The D3R-selective ligands exploit interactions at amino acid residues in both the OBS and SBP in a bivalent manner, resulting in subtype selectivity in binding.
A recent study of SB269652 (1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide), an allosteric modulator of D2R, used molecular modeling and simulations in combination with site-directed mutagenesis to show that a SBP between TM2 and TM7 extensively interacts with the linker and indole moieties of SB269652 that extend from the PP bound in the OBS (Lane et al., 2014). Functional studies suggested that the allosteric effect is manifest through a dimeric interface between two D2R protomers. Interestingly, the TM1-TM1 dimer interface of D2R (Guo et al., 2008), which is close to the binding cavity for the indole moiety of SB269652, includes the extracellular portion of TM1, which is divergent even between highly homologous D3R and D2R (Shi et al., 2001; Chien et al., 2010). Thus, such a novel mechanism of allostery opens a new venue to achieve selectivity by taking advantage of the divergence in dimer interfaces, which very likely exist in other subfamilies of aminergic receptors.
To our knowledge, no other studies of aminergic receptors have specifically addressed the structural basis of ligand selectivity as being due to interactions with a divergent SBP. Nevertheless, several studies have implied that interactions with allosteric sites contribute to the selectivity of bitopic ligands (see section II.C), such as the M1R-selective agonists, TBPB, and 77-LH-28-1, mentioned above—their interactions with the crevice formed by TMs 2, 3, and 7 may contribute to their selectivity (Keov et al., 2014). Given the overall similar shapes and sizes of binding pockets among the aminergic receptors, we propose that the extension of ligands into the divergent SBP may be a common exploitable scheme to achieve subtype selectivity for other subfamilies of aminergic receptors.
Interestingly, and conversely, modifications to the SP (aryl amide) that binds in the SBP (as well as the PP that binds to the OBS) in the 4-phenylpiperazine class of drugs have been used to develop multifunctional atypical antipsychotics (Butini et al., 2009; Brindisi et al., 2014). In these studies, using structure-based drug design, compounds with a defined “balance” of affinities and efficacies (e.g., potent D3R antagonism with 5HT1AR partial agonism and 5HT2AR antagonism and low affinities for both D2R and 5HT2CR) have been discovered. The lead compound 5bb showed promising antipsychotic actions in animal models, devoid of D2R side effects that render classic and atypical D2R antagonists therapeutically suboptimal. Hence, clearly defining the structural components of the SBP, in particular, can be used to design novel therapeutically superior drug molecules, with multiple and defined activities.
B. The Role of Extracellular Loop 2 and Ligand Entry/Exit Pathways in Ligand Selectivity.
Whereas the sides and the bottom of the OBS are formed by positions in the TM domain, EL2 forms a “lid” enclosing the OBS with at least one residue in direct contact with the ligands in all the structures of amine receptors (Fig. 1). Despite the conserved role of this lid, the overall structure of EL2 varies greatly between receptors due to differences in the length and physicochemical properties of the composite residues, especially in the region N-terminal to CysEL2.50, which is highly divergent even among the most homologous subtypes. For example, EL2 was suggested to play a role in D2R/D3R-selectivity based on receptor chimera experiments, in which replacing EL2 in the D2R with that of D3R improved binding affinity of a D3R-selective 4-phenylpiperazine compound (Newman et al., 2009; Banala et al., 2011).
The divergence of EL2 may affect its dynamics as well, and consequently the access of ligand to the OBS and the egress of the bound ligand from the OBS. In a recent study, it was found that the compound tiotropium more easily dissociates from the OBS of M2R than M3R due to higher mobility of EL2 in M2R (Kruse et al., 2012). This may explain the clinically important “kinetic selectivity” of this drug (Barnes, 2000) for M3R over M2R, despite its similar equilibrium binding affinities for both receptors.
βARs are also significantly divergent in their extracellular loops—how may the divergency play a role in ligand selectivity? Several β1AR and β2AR structures are now available that are bound with a variety of ligands (Kobilka, 2011; Katritch et al., 2012; Venkatakrishnan et al., 2013), and although there are no currently available structures of β1AR bound with β1AR-selective ligands, there are several β2AR structures bound with β2AR-selective ligands [including ICI-118551 [(±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol], salbutamol, and timolol; 500-, 30-, and 20-fold selective for β2AR over β1AR, respectively (Baker, 2005)]. Despite the availability of these structures, the mechanistic basis of selectivity of currently available subtype-selective βAR compounds remains poorly understood.
To investigate the contribution of static ligand binding modes to the selectivity of βAR compounds, we can make use of predicted ligand poses. Retrospective docking benchmarks (Beuming and Sherman, 2012) have shown that docking of most βAR ligands into the crystal structures can replicate the binding modes identified by these structures with very high confidence (typical RMSD values <1.0 Å), even when crystal structures with other cocrystalized ligands are used. Thus, the availability of accurately predicted binding modes for several specific ligands for which no crystal structure is available makes it possible to assess the basis of β1AR/β2AR selectivity through an analysis of predicted contacts formed in the binding site.
Based on both the crystal structures and docking studies, in very few cases, selective binding to either β1AR or β2AR can be rationalized by specific interactions formed between ligands and a divergent binding residue. One such case is that of the compound salmeterol, which is bound with ∼1500 higher affinity by β2AR than by β1AR. In its predicted binding mode, salmeterol can interact with β2AR via aromatic interactions with the side chains of Tyr3087.35 and His2966.58, which are Phe and Lys, respectively, in β1AR. Indeed, it has been shown that a mutation of Tyr3087.35 to Ala reduces binding of this compound to β2AR by more than a 100-fold (Isogaya et al., 1998), and the H296K mutation of β2AR reduces the affinity by 18-fold (Baker et al., 2015). Conversely, chimeras of β1AR with either TM7 alone or both TM7 and TM2 replaced by those of β2AR showed 8- and 88-fold increased affinities for this compound, respectively; interestingly, the chimera with only TM2 replaced showed a 5-fold decreased affinity (Isogaya et al., 1998). Thus, it is likely that the orientation of position 7.35 is affected by TM2, similar to our finding in studying D3R versus D2R selectivity that divergence in a peripheral region can affect the functional role of residues that are directly involved in ligand binding (see above). However, unlike salmeterol and the extended D3R-selective compounds discussed above, most existing βAR-selective compounds are relatively small and do not appear to possess two distinct pharmacophores.
In a number of β2AR structures, Tyr3087.35 forms a hydrogen bond with the conserved Asn2936.55. It is likely that the presence or absence of the 7.35–6.55 hydrogen bond has a strong effect on the rotameric preferences of both residues and that this in part mediates the selectivity of a number of selective compounds. Indeed, structures of the β1AR and β2AR reveal a spectrum of conformations of these residues depending on the ligand bound, suggesting that the conformational properties of these residues may affect the specificity of a subset of compounds, including the β2AR-specific compound timolol, which forms a direct hydrogen bond with Asn2936.55 (but not Tyr3087.35) (PDB ID 3D4S).
However, in addition to these examples of roles for the divergence of 7.35 or its interaction with 6.55, there are also several examples of highly selective compounds that only interact with residues that are identical in both β1AR and β2AR. For example, the highly β2AR-selective compound ICI-118551 (Bilski et al., 1983) forms neither aromatic nor hydrogen bond interactions with the Tyr7.35–Asn6.55 pair. Comparing the crystal structures of β2AR complexed with ICI-118551 (PDB ID 3NY8) and β1AR complexed with cyanopindolol (PDB ID 2VT4), ICI-118551 and cyanopindolol have very similar binding modes, but ICI-118551 is significantly more selective than pindolol (500- versus 2-fold selectivity, respectively) for β2AR over β1AR (Hoffmann et al., 2004; Baker, 2005, 2010). Thus, additional elements beyond the completely conserved contact residues must also play an important role in determining selectivity.
One such element in the extracellular region is the salt bridge formed between Asp192EL2.51 and Lys3057.32 in β2AR (Bokoch et al., 2010) that is absent in β1AR or β3AR, in which the corresponding residues are two aspartic acids in β1AR and alanine and glycine in β3AR. It has been found that K305D mutation of β2AR reduces the affinity of salmeterol by more than 30-fold, possibly due to an effect on the overall size of and access to the binding site in the absence of the salt bridge (Baker et al., 2015). Interestingly, in β1AR there is a salt bridge formed between Asp184/Asp186 in EL2 and Arg317 in EL3 that may play a functionally similar role but result in divergent loop conformations and dynamics.
In addition, it has been postulated that selectivity at βARs can also arise from different initial transient interactions between the ligand and receptor in an extracellular vestibule, which influences ligand association rate. However, it has been difficult using direct experimental approaches to identify transient ligand binding sites involved in the specificity of βAR ligands. Thus, several groups have sought to characterize these interactions along the ligand entrance pathway using MD simulations (Wang and Duan, 2009; Dror et al., 2011; Gonzalez et al., 2011; Selvam et al., 2012), and a number of common transient binding sites near EL2, EL3, and TM7 have been identified. The most computationally rigorous study, carried out by Dror and colleagues (2011), implicated a pocket proximal to the OBS in an extracellular vestibule enclosed by EL2, EL3, and TMs 5–7 and proposed that transient interactions with divergent residues in this pocket could affect the specific binding of ligands. In particular, the authors proposed that these interactions are associated with an energetic barrier related to the partial dehydration of both the ligand and the extracellular vestibule. Selvam and colleagues (2012) further characterized this entrance pathway and identified specificity-determining interactions from the simulated unbinding of the selective ligands esmolol in β1AR and ICI-118551 in β2AR. They suggested that the nonconserved residue Lys3476.58 in the β1AR was a key transient interaction for esmolol, whereas ICI-118551 formed mostly hydrophobic and aromatic interactions along the entrance pathway in β2AR. Interestingly, other groups have computationally identified a second ligand entrance site closer to TM2 (Wang and Duan, 2009; Gonzalez et al., 2011)—the existence of multiple entry routes suggests that the preference for one route over another for different ligands in different receptors may also contribute to ligand selectivity.
C. Conformational Changes within Secondary Binding Pockets during Receptor Activation.
The development of superior subtype-selective drugs will require a better understanding of the molecular determinants not only of selectivity but also of efficacy. Among the available crystal structures of aminergic receptors, an active conformational state stabilized by G protein or a G protein–mimetic nanobody has been captured for β2AR (Rasmussen et al., 2011a,b; Ring et al., 2013) and M2R (Kruse et al., 2013a). Relative to the inactive states of these receptors, the largest conformational changes in the active state are on the intracellular side, including a >10 Å outward movement of the intracellular end of TM6 that results in dissociation of the TM3–TM6 ionic lock as well as smaller rearrangements of TM5 and the NPXXY motif of TM7 that form the Tyr5.58–Tyr7.53 interaction (Rasmussen et al., 2011b; Kruse et al., 2013a).
Smaller conformational changes, characterized by various degrees of contraction of the ligand binding crevice (including both the OBS and SBP) have been observed in active state structures of M2R and β2AR, as well as in agonist-bound structures of β1AR (Warne et al., 2011; Kruse et al., 2013a; Ring et al., 2013). Although larger conformational changes in this region are seen in the M2R active structure compared with that of β2AR, both show similar trends: the extracellular portions of TM6 and TM7 move toward the center of the OBS, and a bulge near position 5.46 of TM5 protrudes into the OBS (Fig. 8, A and B). Compared with the OBS residues, some SBP residues in β2AR and M2R display only subtle changes in the active structures, e.g., those in TM2 and TM3. However, substantially larger conformational changes are seen in SBP residues 5.38, 5.39, 6.58, 6.59, 7.32, 7.35, and 7.36 (Fig. 8, C and D). Interestingly, based on our analysis of the crystal structures of aminergic receptors available so far, some SBP residues appear to uniquely contact agonists/partial agonists (see top row of Fig. 2), which include the aforementioned positions 6.58, 6.59, 7.32, and 7.36. Given the sequence divergence of these SBP residues (e.g., positions 6.58, 6.59, 7.32, 7.35, and 7.36 are KADFV in β1AR and HVKYI in β2AR), agonist-induced conformational rearrangements in the SBP may differ among receptor subtypes, creating an opportunity for the rational development of both subtype-selective agonists and inverse agonists by taking advantage of the divergence at these positions between active and inactive states.
Fig. 8.

Rearrangements of binding site residues in the conformational transition from the inactive state to the active state. The rearrangements in the OBS (A and B) and SBPs (C and D) between the inactive and active structures of β2AR (PDB IDs 2RH1 and 4LDE/4LDO) (A and C) and M2R (PDB IDs 3UON and 4MQT) (B and D) are shown. The color coding of residues is the same as in Fig. 1. For simplicity, EL2 is not shown in (C and D). The agonists bound in the active-state structures are represented as cyan surfaces to indicate the space occupied by the OBS. For clarity, only the backbones of the inactive-state structures are shown in gray cartoon representation, whereas the active-state structures are superimposed to the corresponding inactive-state structures by the Cα atoms of 70 TM residue positions that undergo the smallest changes between the inactive and active states. To select these positions, we ranked the distance changes of the corresponding Cα atoms in the inactive and active structures, after aligning them by TM residues using the iterative-fit “align” command in PyMOL (version 1.3r1; Schrödinger LLC, New York, NY). This ranking has been carried out for both β2AR and M2R structures pairs; the 70 highest ranked positions were selected after averaging the ranks from both receptor comparisons and ensuring that at least two positions from each TM were included. The ligand-binding site residues are represented as sticks, drawn from the Cα atom to the COM of the side chain heavy atoms of the residue (for Gly, only the Cα atom is shown). The red arrows indicate the conformational changes from the inactive to the active state for the COM of each residue. In (C), two arrows are drawn for each of the positions 6.58 and 6.59, showing the differing conformational changes to the BI-167107–bound active-state (PDB ID 4LDE) compared with the adrenaline-bound active-state (PDB ID 4LDO) for β2AR.
Interestingly, different agonists can stabilize divergent configurations of the SBP residues of β2AR, e.g., the side chain of His2966.58 is oriented more toward the OBS in the adrenaline-bound structure (PDB ID 4LDO) than in the BI-167107 (8-[(1R)-2-{[1,1-dimethyl-2-(2-methylphenyl)ethyl]amino}-1-hydroxyethyl]-5-hydroxy-2H-1,4-benzoxazin-3(4H)-one)–bound structure (PDB ID 4LDE) and forms an extended polar interaction network that is propagated up to EL3 (shown by two arrows for position 6.58 in Fig. 8C). Thus, in addition to the divergence in residue identity, the conformational dynamics around the SBP residues may also be exploited in developing subtype-selective agents with tailored efficacy.
IV. Conclusions
Unlike for receptors from different subfamilies or subgroups, the OBS of receptors within the same subgroup can be highly homologous or even identical, and thus the development of subtype-selective compounds has been extremely challenging. With recent progress in aminergic GPCR structure determination and molecular modeling, the basis of subtype selectivity is beginning to be elucidated. Divergent extracellular regions can be exploited for the development of 1) selective compounds that bind concomitantly to the OBS and SBP, 2) allosteric modulators that bind only to SBPs, or 3) compounds that differ in their interactions along entry/exit pathways to the OBS of different receptor subtypes. Although there are opportunities to achieve selectivity with all three strategies, practically, it would be easier to achieve rationalized optimizations of existing lead compounds for strategy 1 and possibly strategy 2; to fully understand the structural basis for the ligand selectivity that results from strategy 3 will require significant effort from both the experimental and computational perspectives, but it nonetheless has the potential to address more difficult selectivity issues.
Significant challenges remain, however, in the development of subtype-selective compounds. Although currently available subtype-selective compounds (e.g., the D3R over D2R-selective compound R-22) can be used as tools to identify targetable SBPs, the identification of SBPs in receptors for which there are no available leads to selective compounds remains challenging. In addition, although characterization of binding modes of SBP-targeting compounds can reveal the identity and conformational spectrum of SBPs, further efforts are required for the development of subtype-selective allosteric modulators with sufficient affinity and in vivo activity for therapeutic development.
Given that crystal structures provide information on the binding modes of relatively rigid compounds, characterization of binding modes of selective compounds that are bivalent and flexible, determination of ligand entry/exit pathways of existing selective compounds, and in silico screening for selective compounds that take advantage of these features must rely heavily on computational analysis, in particular MD simulations that take protein dynamics into consideration.
Abbreviations
- 77-LH-28-1
1-[3-(4-butyl-1-piperidinyl)propyl]-3,4-dihydro-2(1H)-quinolinone
- AR
adrenergic receptor
- BI-167107
8-[(1R)-2-{[1,1-dimethyl-2-(2-methylphenyl)ethyl]amino}-1-hydroxyethyl]-5-hydroxy-2H-1,4-benzoxazin-3(4H)-one
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- D4R
dopamine D4 receptor
- DR
dopamine receptor
- EL2
extracellular loop 2
- GPCR
G protein–coupled receptor
- H1R
histamine H1 receptor
- 5HT1BR
serotonin 1B receptor
- 5HT2BR
serotonin 2B receptor
- ICI-118551
(±)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol
- JDTic
(3R)-1,2,3,4-tetrahydro-7-hydroxy-N-[(1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl]-3-isoquinolinecarboxamide
- LY2119620
3-amino-5-chloro-N-cyclopropyl-4-methyl-6-[2-(4-methylpiperazin-1-yl)-2-oxoethoxy]thieno[2,3-b]pyridine-2-carboxamide
- M2R
muscarinic acetylcholine M2 receptor
- M3R
muscarinic acetylcholine M3 receptor
- MD
molecular dynamics
- OBS
orthosteric binding site
- PHNO
(+)-4-propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b][1,4]oxazin-9-ol
- PP
primary pharmacophore
- R-22
(R)-N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)-1H-indole-2-carboxamide
- RMSD
root-mean-square deviation
- SASA
solvent-accessible surface area
- SB269652
1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide
- SBP
secondary binding pocket
- SP
secondary pharmacophore
- TBPB
1-(1′-(2-methylbenzyl)-1,4′-bipiperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one
- TM
transmembrane
- VHTS
virtual high-throughput screening
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Michino, Beuming, Donthamsetti, Newman, Javitch, Shi.
Footnotes
This work was supported in part by the National Institutes of Health National Institute on Drug Abuse [Grants K05-DA022413 (to J.A.J.) and R00-DA023694 (to L.S.)]; the National Institutes of Health National Institute of Mental Health [Grant R01-MH54137 (to J.A.J.)]; Lieber Center for Schizophrenia Research and Training; and the Intramural Research Program of the National Institutes of Health [National Institute on Drug Abuse] (to A.H.N.).
References
- Abdul-Ridha A, Lane JR, Mistry SN, Lopez L, Sexton PM, Scammells PJ, Christopoulos A, Canals M. (2014) Mechanistic insights into allosteric structure-function relationships at the M1 muscarinic acetylcholine receptor. J Biol Chem 89:33701–33711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlquist RP. (1948) A study of the adrenotropic receptors. Am J Physiol 153:586–600. [DOI] [PubMed] [Google Scholar]
- Baker JG. (2005) The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br J Pharmacol 144:317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG. (2010) The selectivity of beta-adrenoceptor agonists at human beta1-, beta2- and beta3-adrenoceptors. Br J Pharmacol 160:1048–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Proudman RG, Hill SJ. (2015) Salmeterol's extreme beta2-selectivity is due to residues in both extracellular loops and transmembrane domains. Mol Pharmacol 87:103–120. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Shi L, Javitch JA. (2001) Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol 60:1–19. [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci 25:366–428. [Google Scholar]
- Banala AK, Levy BA, Khatri SS, Furman CA, Roof RA, Mishra Y, Griffin SA, Sibley DR, Luedtke RR, Newman AH. (2011) N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J Med Chem 54:3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. (2000) The pharmacological properties of tiotropium. Chest 117(2, Suppl)63S–66S. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63:182–217. [DOI] [PubMed] [Google Scholar]
- Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, et al. (2012) Automated design of ligands to polypharmacological profiles. Nature 492:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuming T, Sherman W. (2012) Current assessment of docking into GPCR crystal structures and homology models: successes, challenges, and guidelines. J Chem Inf Model 52:3263–3277. [DOI] [PubMed] [Google Scholar]
- Beuming T, Shi L, Javitch JA, Weinstein H. (2006) A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol Pharmacol 70:1630–1642. [DOI] [PubMed] [Google Scholar]
- Beuming T, Weinstein H. (2004) A knowledge-based scale for the analysis and prediction of buried and exposed faces of transmembrane domain proteins. Bioinformatics 20:1822–1835. [DOI] [PubMed] [Google Scholar]
- Bilski AJ, Halliday SE, Fitzgerald JD, Wale JL. (1983) The pharmacology of a beta 2-selective adrenoceptor antagonist (ICI 118,551). J Cardiovasc Pharmacol 5:430–437. [DOI] [PubMed] [Google Scholar]
- Boeckler F, Gmeiner P. (2006) The structural evolution of dopamine D3 receptor ligands: structure-activity relationships and selected neuropharmacological aspects. Pharmacol Ther 112:281–333. [DOI] [PubMed] [Google Scholar]
- Boileau I, Payer D, Houle S, Behzadi A, Rusjan PM, Tong J, Wilkins D, Selby P, George TP, Zack M, et al. (2012) Higher binding of the dopamine D3 receptor-preferring ligand [11C]-(+)-propyl-hexahydro-naphtho-oxazin in methamphetamine polydrug users: a positron emission tomography study. J Neurosci 32:1353–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, Choi HJ, Thian FS, Kobilka TS, et al. (2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brindisi M, Butini S, Franceschini S, Brogi S, Trotta F, Ros S, Cagnotto A, Salmona M, Casagni A, Andreassi M, et al. (2014) Targeting dopamine D and serotonin 5-HT and 5-HT receptors for developing effective antipsychotics: synthesis, biological characterization, and behavioral studies. J Med Chem 57:9578–9597. [DOI] [PubMed] [Google Scholar]
- Butini S, Gemma S, Campiani G, Franceschini S, Trotta F, Borriello M, Ceres N, Ros S, Coccone SS, Bernetti M, et al. (2009) Discovery of a new class of potential multifunctional atypical antipsychotic agents targeting dopamine D3 and serotonin 5-HT1A and 5-HT2A receptors: design, synthesis, and effects on behavior. J Med Chem 52:151–169. [DOI] [PubMed] [Google Scholar]
- Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, Molinoff PB, Ruffolo RR, Jr, Trendelenburg U. (1994) International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev 46:121–136. [PubMed] [Google Scholar]
- Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. (2011) Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol 7:769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, et al. (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330:1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher JA, Brown J, Doré AS, Errey JC, Koglin M, Marshall FH, Myszka DG, Rich RL, Tate CG, Tehan B, et al. (2013) Biophysical fragment screening of the β1-adrenergic receptor: identification of high affinity arylpiperazine leads using structure-based drug design. J Med Chem 56:3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA, Olsen RW, Peters JA, Neubig RR, Pin JP, et al. (2014) International union of basic and clinical pharmacology. XC. multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev 66:918–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M, Langmead CJ, Mason JS, Marshall FH. (2011) Progress in structure based drug design for G protein-coupled receptors. J Med Chem 54:4283–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf C, Kooistra AJ, Vischer HF, Katritch V, Kuijer M, Shiroishi M, Iwata S, Shimamura T, Stevens RC, de Esch IJ, et al. (2011) Crystal structure-based virtual screening for fragment-like ligands of the human histamine H(1) receptor. J Med Chem 54:8195–8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Ciano P, Grandy DK, Le Foll B. (2014) Dopamine D4 receptors in psychostimulant addiction. Adv Pharmacol 69:301–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, et al. (2013) Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503:295–299. [DOI] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE. (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci USA 108:13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich K, Götz A, Bollinger S, Tschammer N, Bettinetti L, Härterich S, Hübner H, Lanig H, Gmeiner P. (2009) Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J Med Chem 52:4923–4935. [DOI] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. (2014) Molecular control of δ-opioid receptor signalling. Nature 506:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filizola M, Devi LA. (2013) Grand opening of structure-guided design for novel opioids. Trends Pharmacol Sci 34:6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. (2003) The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63:1256–1272. [DOI] [PubMed] [Google Scholar]
- Giorgioni G, Piergentili A, Ruggieri S, Quaglia W. (2008) Dopamine D5 receptors: a challenge to medicinal chemists. Mini Rev Med Chem 8:976–995. [DOI] [PubMed] [Google Scholar]
- Gnagey AL, Seidenberg M, Ellis J. (1999) Site-directed mutagenesis reveals two epitopes involved in the subtype selectivity of the allosteric interactions of gallamine at muscarinic acetylcholine receptors. Mol Pharmacol 56:1245–1253. [DOI] [PubMed] [Google Scholar]
- González A, Perez-Acle T, Pardo L, Deupi X. (2011) Molecular basis of ligand dissociation in β-adrenergic receptors. PLoS ONE 6:e23815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. (2012) Structure of the δ-opioid receptor bound to naltrindole. Nature 485:400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Urizar E, Kralikova M, Mobarec JC, Shi L, Filizola M, Javitch JA. (2008) Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J 27:2293–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, et al. (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482:547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidbreder CA, Newman AH. (2010) Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann N Y Acad Sci 1187:4–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare SR, Coldwell MC, Armstrong D, Strange PG. (2000) Regulation of human D(1), d(2(long)), d(2(short)), D(3) and D(4) dopamine receptors by amiloride and amiloride analogues. Br J Pharmacol 130:1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Leitz MR, Oberdorf-Maass S, Lohse MJ, Klotz KN. (2004) Comparative pharmacology of human beta-adrenergic receptor subtypes—characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch Pharmacol 369:151–159. [DOI] [PubMed] [Google Scholar]
- Hubbard SJ, Thornton JM. (1993) NACCESS, Department of Biochemistry and Molecular Biology, University College London, UK. [Google Scholar]
- Isogaya M, Yamagiwa Y, Fujita S, Sugimoto Y, Nagao T, Kurose H. (1998) Identification of a key amino acid of the beta2-adrenergic receptor for high affinity binding of salmeterol. Mol Pharmacol 54:616–622. [PubMed] [Google Scholar]
- Javitch JA, Shi L, Liapakis G. (2002) Use of the substituted cysteine accessibility method to study the structure and function of G protein-coupled receptors. Methods Enzymol 343:137–156. [DOI] [PubMed] [Google Scholar]
- Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR, et al. (2008) Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J Nneurosci 28:10422–10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci 33:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck TM, Burzynski C, Shi L, Newman AH. (2014) Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv Pharmacol 69:267–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keov P, López L, Devine SM, Valant C, Lane JR, Scammells PJ, Sexton PM, Christopoulos A. (2014) Molecular mechanisms of bitopic ligand engagement with the M1 muscarinic acetylcholine receptor. J Biol Chem 289:23817–23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jiang Q, Glashofer M, Yehle S, Wess J, Jacobson KA. (1996) Glutamate residues in the second extracellular loop of the human A2a adenosine receptor are required for ligand recognition. Mol Pharmacol 49:683–691. [PMC free article] [PubMed] [Google Scholar]
- Kiss R, Kiss B, Könczöl A, Szalai F, Jelinek I, László V, Noszál B, Falus A, Keseru GM. (2008) Discovery of novel human histamine H4 receptor ligands by large-scale structure-based virtual screening. J Med Chem 51:3145–3153. [DOI] [PubMed] [Google Scholar]
- Kobilka BK. (2011) Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci 32:213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola I, Landis J. (2004) Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3:711–715. [DOI] [PubMed] [Google Scholar]
- Kortagere S, Gmeiner P, Weinstein H, Schetz JA. (2004) Certain 1,4-disubstituted aromatic piperidines and piperazines with extreme selectivity for the dopamine D4 receptor interact with a common receptor microdomain. Mol Pharmacol 66:1491–1499. [DOI] [PubMed] [Google Scholar]
- Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, et al. (2012) Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482:552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hübner H, Pardon E, Valant C, Sexton PM, et al. (2013a) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504:101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Weiss DR, Rossi M, Hu J, Hu K, Eitel K, Gmeiner P, Wess J, Kobilka BK, Shoichet BK. (2013b) Muscarinic receptors as model targets and antitargets for structure-based ligand discovery. Mol Pharmacol 84:528–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachowicz JE, Sibley DR. (1997) Molecular characteristics of mammalian dopamine receptors. Pharmacol Toxicol 81:105–113. [DOI] [PubMed] [Google Scholar]
- Lane JR, Donthamsetti P, Shonberg J, Draper-Joyce CJ, Dentry S, Michino M, Shi L, López L, Scammells PJ, Capuano B, et al. (2014) A new mechanism of allostery in a G protein-coupled receptor dimer. Nat Chem Biol 10:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JR, Sexton PM, Christopoulos A. (2013) Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol Sci 34:59–66. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Austin NE, Branch CL, Brown JT, Buchanan KA, Davies CH, Forbes IT, Fry VA, Hagan JJ, Herdon HJ, et al. (2008) Characterization of a CNS penetrant, selective M1 muscarinic receptor agonist, 77-LH-28-1. Br J Pharmacol 154:1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Simms J, Sexton PM, Christopoulos A. (2012) Structure-function studies of muscarinic acetylcholine receptors. Handb Exp Pharmacol 208:29–48. [DOI] [PubMed] [Google Scholar]
- Liebler DC, Guengerich FP. (2005) Elucidating mechanisms of drug-induced toxicity. Nat Rev Drug Discov 4:410–420. [DOI] [PubMed] [Google Scholar]
- Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, et al. (2009) Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci USA 106:15950–15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. (2012) Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 485:321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matuskey D, Gallezot JD, Pittman B, Williams W, Wanyiri J, Gaiser E, Lee DE, Hannestad J, Lim K, Zheng MQ, et al. (2014) Dopamine D₃ receptor alterations in cocaine-dependent humans imaged with [¹¹C](+)PHNO. Drug Alcohol Depend 139:100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michino M, Donthamsetti P, Beuming T, Banala A, Duan L, Roux T, Han Y, Trinquet E, Newman AH, Javitch JA, et al. (2013) A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Mol Pharmacol 84:854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayal M, Honig B. (2006) On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins 63:892–906. [DOI] [PubMed] [Google Scholar]
- Newman AH, Beuming T, Banala AK, Donthamsetti P, Pongetti K, LaBounty A, Levy B, Cao J, Michino M, Luedtke RR, et al. (2012a) Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J Med Chem 55:6689–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AH, Blaylock BL, Nader MA, Bergman J, Sibley DR, Skolnick P. (2012b) Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem Pharmacol 84:882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, Ho D, Luedtke RR. (2009) N-(4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J Med Chem 52:2559–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah ME, Jacobson KA, Stiles GL. (1994) Role of the second extracellular loop of adenosine receptors in agonist and antagonist binding. Analysis of chimeric A1/A3 adenosine receptors. J Biol Chem 269:24692–24698. [PMC free article] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. (2006) How many drug targets are there? Nat Rev Drug Discov 5:993–996. [DOI] [PubMed] [Google Scholar]
- Payer D, Balasubramaniam G, Boileau I. (2014) What is the role of the D3 receptor in addiction? A mini review of PET studies with [(11)C]-(+)-PHNO. Prog Neuropsychopharmacol Biol Psychiatry 52:4–8. [DOI] [PubMed] [Google Scholar]
- Raghavan B, Skoblenick KJ, Bhagwanth S, Argintaru N, Mishra RK, Johnson RL. (2009) Allosteric modulation of the dopamine D2 receptor by Pro-Leu-Gly-NH2 peptidomimetics constrained in either a polyproline II helix or a type II beta-turn conformation. J Med Chem 52:2043–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Andersen M, Almén MS, Schiöth HB. (2011) Trends in the exploitation of novel drug targets. Nat Rev Drug Discov 10:579–590. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, et al. (2011a) Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 469:175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011b) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK. (2013) Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502:575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvam B, Wereszczynski J, Tikhonova IG. (2012) Comparison of dynamics of extracellular accesses to the β(1) and β(2) adrenoceptors binding sites uncovers the potential of kinetic basis of antagonist selectivity. Chem Biol Drug Des 80:215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Javitch JA. (2002) The binding site of aminergic G protein-coupled receptors: the transmembrane segments and second extracellular loop. Annu Rev Pharmacol Toxicol 42:437–467. [DOI] [PubMed] [Google Scholar]
- Shi L, Javitch JA. (2004) The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci USA 101:440–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Simpson MM, Ballesteros JA, Javitch JA. (2001) The first transmembrane segment of the dopamine D2 receptor: accessibility in the binding-site crevice and position in the transmembrane bundle. Biochemistry 40:12339–12348. [DOI] [PubMed] [Google Scholar]
- Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, et al. (2011) Structure of the human histamine H1 receptor complex with doxepin. Nature 475:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson MM, Ballesteros JA, Chiappa V, Chen J, Suehiro M, Hartman DS, Godel T, Snyder LA, Sakmar TP, Javitch JA. (1999) Dopamine D4/D2 receptor selectivity is determined by A divergent aromatic microdomain contained within the second, third, and seventh membrane-spanning segments. Mol Pharmacol 56:1116–1126. [DOI] [PubMed] [Google Scholar]
- Staley JK, Mash DC. (1996) Adaptive increase in D3 dopamine receptors in the brain reward circuits of human cocaine fatalities. J Neurosci 16:6100–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryanarayana S, Daunt DA, Von Zastrow M, Kobilka BK. (1991) A point mutation in the seventh hydrophobic domain of the alpha 2 adrenergic receptor increases its affinity for a family of beta receptor antagonists. J Biol Chem 266:15488–15492. [PubMed] [Google Scholar]
- Taira CA, Carranza A, Mayer M, Di Verniero C, Opezzo JA, Höcht C. (2008) Therapeutic implications of beta-adrenergic receptor pharmacodynamic properties. Curr Clin Pharmacol 3:174–184. [DOI] [PubMed] [Google Scholar]
- Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, et al. (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485:395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valant C, Robert Lane J, Sexton PM, Christopoulos A. (2012) The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu Rev Pharmacol Toxicol 52:153–178. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494:185–194. [DOI] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. (2013) Structural features for functional selectivity at serotonin receptors. Science 340:615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, et al. (2013) Structural basis for molecular recognition at serotonin receptors. Science 340:610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Duan Y. (2009) Ligand entry and exit pathways in the beta2-adrenergic receptor. J Mol Biol 392:1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Edwards PC, Leslie AG, Tate CG. (2012) Crystal structures of a stabilized β1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure 20:841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Moukhametzianov R, Baker JG, Nehmé R, Edwards PC, Leslie AG, Schertler GF, Tate CG. (2011) The structural basis for agonist and partial agonist action on a β(1)-adrenergic receptor. Nature 469:241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D, Christopoulos A, Sexton PM. (2013) Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov 12:630–644. [DOI] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, et al. (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurch T, Colpaert FC, Pauwels PJ. (1998) Chimeric receptor analysis of the ketanserin binding site in the human 5-Hydroxytryptamine1D receptor: importance of the second extracellular loop and fifth transmembrane domain in antagonist binding. Mol Pharmacol 54:1088–1096. [DOI] [PubMed] [Google Scholar]
- Zhao MM, Hwa J, Perez DM. (1996) Identification of critical extracellular loop residues involved in alpha 1-adrenergic receptor subtype-selective antagonist binding. Mol Pharmacol 50:1118–1126. [PubMed] [Google Scholar]