

Abstract
The hypothesis that functionally selective G protein–coupled receptor (GPCR) agonists may have enhanced therapeutic benefits has revitalized interest for many GPCR targets. In particular, although κ-opioid receptor (KOR) agonists are analgesic with a low risk of dependence and abuse, their use is limited by a propensity to induce sedation, motor incoordination, hallucinations, and dysphoria-like states. Several laboratories have produced a body of work suggesting that G protein–biased KOR agonists might be analgesic with fewer side effects. Although that has been an intriguing hypothesis, suitable KOR-selective and G protein–biased agonists have not been available to test this idea. Here we provide data using a G protein–biased agonist, RB-64 (22-thiocyanatosalvinorin A), which suggests that KOR-mediated G protein signaling induces analgesia and aversion, whereas β-arrestin-2 signaling may be associated with motor incoordination. Additionally, unlike unbiased KOR agonists, the G protein–biased ligand RB-64 does not induce sedation and does not have anhedonia-like actions, suggesting that a mechanism other than G protein signaling mediates these effects. Our findings provide the first evidence for a highly selective and G protein–biased tool compound for which many, but not all, of the negative side effects of KOR agonists can be minimized by creating G protein–biased KOR agonists.
Introduction
The term functional selectivity or biased agonism describes the ability of a G protein–coupled receptor (GPCR) ligand to selectively activate a subset of signaling cascades at a particular GPCR, as opposed to the activation of all downstream signaling cascades (e.g., G proteins, arrestins, and/or kinases). Over the past few years, the phenomenon of functional selectively has been increasingly explored, providing a novel potential for GPCR-targeted therapies with improved safety and fewer side effects (Urban et al., 2007; Allen and Roth, 2011). Currently, the potential of functionally selective drugs as biased agonists for a wide range of targets and disease states is being investigated. Thus, for instance, 1) angiotensin II receptor arrestin–biased agonists are being investigated for treating acute heart failure; 2) μ-opioid receptor G protein biased agonists have been proposed as novel analgesics; 3) δ-opioid receptor G protein–biased agonists are being considered for Parkinson disease, pain, and depression; and 4) the dopamine D2 arrestin–biased agonists are being evaluated for treating schizophrenia and related disorders (Allen et al., 2011; Pradhan et al., 2011; Whalen et al., 2011; DeWire et al., 2013; Monasky et al., 2013). Significantly, a wealth of data implies that G protein–biased KOR ligands might represent novel analgesics with lower addiction liability and fewer side effects (Bruchas et al., 2007; Tao et al., 2008; Ranganathan et al., 2012).
Although κ-opioid receptor (KOR) agonists have long been recognized to be analgesic with low abuse potential, their use can be associated with severe side effects, including dysphoria, anhedonia, and hallucinations (Pfeiffer et al., 1986; Ranganathan et al., 2012; Tejeda et al., 2013). KOR activation induces p38 mitogen-activated protein kinase (MAPK) activation in vivo, thereby mediating KOR-induced aversion in mice, but not analgesia (Bruchas et al., 2007). It has been hypothesized that p38 activation is mediated by β-arrestin-2 signaling (Bruchas and Chavkin, 2010), suggesting a therapeutic potential for G protein–biased KOR ligands as analgesics without dysphoric side effects. Until now, the only G protein–biased KOR ligand identified, 6′-GNTI (6′-guanidinonaltrindole), was not suitable for in vivo study because it does not cross the blood-brain barrier (Rives et al., 2012). Because of the lack of suitable tool compounds, however, this hypothesis has not been directly tested in vivo.
We recently reported that the centrally active salvinorin A (sal A) derivative, RB-64 (22-thiocyanatosalvinorin A; Yan et al., 2009), represents a G protein–biased KOR agonist (White et al., 2014) and is a potential tool-like compound suitable for interrogating the role of G protein signaling in mediating various behavioral responses to KOR activation. Using RB-64 in both wild-type (WT) and β-arrestin-2 KO mice, we provide evidence in favor of the hypothesis that KOR-mediated G protein signaling induces analgesic-like effects and aversion, whereas KOR-mediated β-arrestin-2 signaling induces motor incoordination. Additionally, the G protein–biased ligand RB-64 had no effect on motor coordination, sedation, or anhedonia, suggesting that these behaviors are not mediated by G protein signaling but instead, perhaps, by β-arrestin-2 signaling. Based on these results, our data support the hypothesis that G protein–biased KOR agonists might represent novel analgesics with reduced abuse potential and fewer deleterious side effects compared with unbiased agonists.
Materials and Methods
In Vitro Functional Analysis of U69593, Sal A, and RB-64
For U69593 [(+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide], sal A, and RB-64, G protein activation was measured using a previously described split-luciferase based cAMP biosensor (Glowsensor 22F; Promega, Madison, WI) (Besnard et al., 2012; White et al., 2014), whereas arrestin mobilization was measured by Tango assay exactly as previously described (White et al., 2014), with the exception that the mouse KOR was used instead of human KOR. Bias calculations were performed as previously described (White et al., 2014) (Fig. 1).
Fig. 1.

In vitro effects of agonists on signaling and internalization of the mouse KOR. Dose-response curves in HEK cells for (A) arrestin mobilization and (B) G protein signaling–induced by U69593, sal A, and RB-64. Error bars represent S.E.M. (C) Internalization of mouse KOR on stimulation with agonists (arrows indicate internalized KOR). The percent of internalization was quantified in an unbiased manner for the 15-minute study as previously described (Bhatnagar et al., 2001) as follows: control = 36 ± 9%; U69593 = 83 ± 5% (P < 0.05 versus control); salvinorin A = 82 ± 7% (P < 0.05 versus control); RB-64 = 32.7 ± 6% (NS versus control).
In Vitro Internalization Analysis
For these studies, FLAG-tagged hKOR was transiently transfected into human embryonic kidney (HEK) cells, and the day after transfection cells were plated onto polylysine-coated glass coverslips as described previously (Bhatnagar et al., 2001). After 24 hours, cells were exposed to 200 nM test compound for 0, 15, and 30 minutes and then prepared for anti-FLAG immunofluorescence confocal microscopy done essentially as previously described (Bhatnagar et al., 2001).
Animal Subjects
C57BL/6J mice and knockout (KO) mice were acquired from The Jackson Laboratory (Bar Harbor, ME), and β-arrestin-2 KO mice on a C57BL/6J background were donated by the laboratory of Robert Lefkowitz (Duke University, Durham, NC). Behavioral studies were conducted at the University of North Carolina following the National Institutes of Health’s guidelines for care and use of animals and with approved mouse protocols from the institutional animal care and use committees. An equal number of male and female subjects were age-matched: 2- to 8-month-old mice weighing between 22 and 35 g; genotypes were determined by polymerase chain reaction analysis of tail-tip digestions. WT and β-arrestin-2 KO mice from β-arrestin-2 KO heterozygous parents (C57BL/6J background) were used for all behavioral experiments. All mice were given access to food and water ad libitum.
Drugs
Sal A was acquired from Apple Farms (Asheville, NC), and RB-64 was synthesized as previously reported (Yan et al., 2009). Finally, U69593 was acquired from Sigma-Aldrich (St. Louis, MO). All drugs were administered subcutaneously using 10% Tween-80 as vehicle, unless otherwise stated. A dose of 3 mg/kg RB-64 was chosen for most experiments because that was the lowest dose found to induce analgesia-like effects.
Hotplate Assay
Analgesia-like responses were measured using a hotplate analgesia meter with dimensions of 29.2 × 26.7 cm; mice were restricted to a cylinder 8.9 cm in diameter and 15.2 cm high (IITC Life Sciences, Woodland Hills, CA). Response was measured by recording the latency to lick, flutter, or splay hindpaw(s) or an attempt to jump out of the apparatus at 55°C, with a maximum cutoff time of 30 seconds. Once a response was observed or the cutoff time had elapsed, the subject was immediately removed from the hotplate and placed back in its home cage (Balter and Dykstra, 2013). The animals were acclimated to the hotplate, while cool, and a baseline analgesic response time was acquired several hours before drug treatment and testing. The analgesia-like effect was measured 10, 20, and 30 minutes after treatment administration. Animals that did not display hindpaw lick, splay, or flutter were removed from the trial. Additionally, animals that attempted to jump out of the plate or urinated on the hotplate were removed from the trial. For the vehicle-treated group, 3 of the 15 WT animals were removed and 2 of the 11 β-arrestin-2 KO mice were removed. In the sal A–treated group, none of 11 WT mice was removed and 5 of 21 β-arrestin-2 KO mice were removed. In the U69593-treated group, 2 of 10 WT mice were removed and 4 of 14 β-arrestin-2 KO were removed. Finally, in the RB-64–treated group, 1 of 10 mice was removed for both WT and β-arrestin-2 KO mice.
Conditioned Place Aversion
The conditioned place aversion (CPA) protocol was modified from Medvedev et al. (2005).
Apparatus.
Drug-naïve mice were used for each treatment condition. A three-compartment place-preference chamber was used under standardized environmental conditions using an AccuScan activity monitor system (AccuScan Instruments, Columbus, OH). The location of the subject was detected by photobeam strips, and the time spent in each compartment was recorded. The experiments were conducted in a room used only for animal behavior studies, and no other activity in the room occurred during testing. The three compartments of the chamber were separated by sliding doors, and the center compartment was 40 cm long, 30 cm deep, and 5 cm wide with white walls and plastic floor. The two larger compartments used during training were 40 cm long, 30 cm deep, and 17.5 cm wide. One compartment had white and black vertical stripes with brown, perforated paper strips on the flooring, and the other conditioning chamber had white and black vertical stripes with Diamond Dry bedding (Harlan Laboratories, Indianapolis IN) on the floor.
Pretest (Days 1–3).
During this phase, mice were placed in the center compartment and the doors were opened, so both test chambers were accessible. The location of the mice was monitored for 15 minutes during 3 days of pretest training. Mice were not considered suitable for testing if they met the following criteria during the pretest: they spent more than 25% of the time in the center compartment, they spent less than 25% of the time in one of the conditioning compartments, and/or they spent more time in the center compartment than in one of the conditioning compartments (Medvedev et al., 2005).
Conditioning Phase (Days 4–9).
Vehicle treatments were performed on days 4, 6, and 8 and were restricted to the test chamber that the particular mouse spent the least amount of time in during the pretest phase. Drug treatments were conducted on days 5, 7, and 9 and were restricted to the test chamber where the particular mouse spent more time during the pretest. The vehicle control mice received vehicle in both compartments during conditioning. Fifteen minutes after drug treatment, mice were placed in the conditioning chamber for an additional 15 minutes.
Test Phase (Day 11).
After a 1-day drug washout period, mice were placed in the center chamber and the doors were opened to allow access to all rooms. No drug or vehicle was given on this day, and the location of the mouse was measured for 15 minutes. Data were pooled over the 15-minute testing phase for each mouse, and the CPA results were analyzed for each mouse. The results were recorded as the difference in time spent in the drug-paired compartment on the test day versus pretest day.
Rotarod Performance
Balance and motor coordination were measured on an accelerating rotarod after drug treatment (Ugo-Basile, Stoelting Co., Wood Dale, IL) as previously reported (Huang et al., 2013). Briefly, the rod initially rotated at 3 rpm, gradually increasing to a maximum of 30 rpm over a 5-minute period, which was also the maximum length of the trial. Two days before the experiment, mice were trained on the apparatus in two or three trials, with a 1-minute break between trials. The latency to fall off the rod was measured by the rotarod timer. Additionally, testing was stopped for mice that rotated off the top of the rod. On testing days, each mouse first completed a drug-free trial to determine baseline performance before administration of drug. Rotarod performance was assessed 10, 20, and 30 minutes after drug administration.
Novelty-Induced Locomotion
Mouse locomotion was measured in photocell-based activity chambers under standardized environmental conditions using an AccuScan activity monitoring system (AccuScan Instruments) consisting of a 41 × 41 × 30-cm chamber with beam sensors as described earlier (Farrell et al., 2013). Distance traveled consists of horizontal movement throughout the entire chamber, and data were collected in 5-minute bins. Mice were given drug or vehicle treatments 15 minutes before being placed in the chamber, and locomotion was recorded for 1 hour. Mice were not habituated to chambers before assay.
Intracranial Self-Stimulation
Intracranial self-stimulation (ICSS) is an operant behavioral method in which mice respond for rewarding electrical stimulation of the medial forebrain bundle at the level of the lateral hypothalamus. All ICSS methods were done as previously described (Robinson et al., 2012), using the curve-shift method of ICSS (Carlezon and Chartoff, 2007). The maximum response rate (MAX) and the lowest frequency that sustained responding brain stimulation reward (BSR threshold, θ0) were compared before and after drug administration.
Statistics
The data are presented as means and S.E.M. The hotplate, CPA, rotarod, and novelty-induced locomotion studies were analyzed by GraphPad Prism statistical software (GraphPad Software, La Jolla, CA), whereas the ICSS studies were analyzed by SigmaPlot (Systat Software Inc., San Jose, CA). The hotplate data were assessed using two-way repeated measures (RM) analysis of variance (ANOVA) for within-subject effect of time, between-subject effect of treatment, and interaction between time and treatment (Fig. 2). The effect of KOR on results was determined by a one-way ANOVA of the results from KOR KO mice compared with WT mice (Fig. 2D). Additionally, the effect of genotype on baseline performance was measured by one-way ANOVA (Fig. 2E). Furthermore, we used a two-tailed t test to determine whether the analgesia-like response induced by 3 versus 10 mg/kg of sal A or RB-64 produced a more pronounced effect (Fig. 2F). Results are plotted as the percent of baseline for each mouse. The CPA data were first assessed by a Bartlett test for equal variances to determine whether the variances differ significantly, followed by a one-way ANOVA (Fig. 3A). A two-tailed t test was performed with the drug treatments that used a dimethylsulfoxide or 10% Tween-80 vehicle to determine potential ceiling effects of the 10% Tween-80 vehicle on 1 mg/kg U69593 and 3 mg/kg RB64 (Fig. 3B)
Fig. 2.
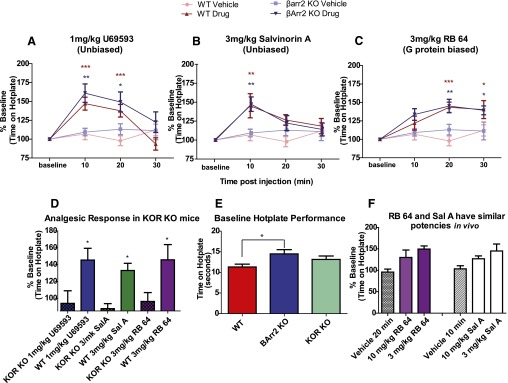
KOR agonist–induced G protein signaling causes analgesia-like effects in the hotplate assay. (A) U69593 caused an analgesia-like effect in WT (n = 8) and β-arrestin-2 KO mice (n = 10) 10 and 20 minutes after treatment. (B) Sal A produced analgesia-like effects in WT mice (n = 11) and β-arrestin-2 KO mice (n = 16) 10 minutes after treatment. (C) RB-64 induced analgesia-like effects in WT (n = 9) and β-arrestin-2 KO mice (n = 9) 20 and 30 minutes after treatment. The vehicle-treated WT mice (n = 12) and β-arrestin-2 KO mice (n = 9) showed no differences in response. (D) U69593, sal A, and RB-64 showed a KOR-selective effect when comparing KOR KO mice (n = 8 for all drug treatments) and WT mice (n = 9, 7, and 6, for U69593, sal A, and RB-64). Mice were tested 20 minutes after treatment of U69593 and sal A and 10 minutes after treatment with sal A. (E) There was an increased baseline performance in the hotplate assay for β-arrestin-2 KO mice compared with WT mice, but no difference was seen in baseline between KOR KO and WT mice (n = 12 for all genotypes). (F) A higher dose of RB-64 (n = 10) or sal A (n = 9) does not increase the analgesic response in WT mice. Data are plotted as the percent of baseline latency to respond for each animal, and baseline was established on the day of testing before drug treatment ± S.E.M. (A–D) *P < 0.05; **P < 0.01; ***P < 0.001. BArr2, β-arrestin-2.
Fig. 3.

KOR agonist–induced G protein signaling causes conditioned place aversion. (A) U69593 (1 mg/kg)-induced aversion in WT (n = 8) and β-arrestin-2 KO mice (n = 7). 1 mg/kg sal A had no effect on WT (n = 6) or β-arrestin-2 KO mice (n = 8), but 3 mg/kg sal A did cause aversion in WT (n = 9) and β-arrestin-2 KO mice (n = 6). RB-64 (1 mg/kg) did not cause aversion in WT (n = 8) or β-arrestin-2 KO mice (n = 7), but 3 mg/kg RB-64 did induce aversion in both WT (n = 8) and β-arrestin-2 KO mice (n = 7). All P values were generated in comparison with vehicle-treated WT (n = 8) or β-arrestin-2 KO mice (n = 8). (B) Using a dimethylsulfoxide (DMSO) vehicle instead of 10% Tween-80 did not cause an increased aversion in U69593-treated mice (n = 6) relative to RB-64 (n = 6). This was done to determine whether 10% Tween-80 causes a ceiling effect in U69593 mice. Using either vehicle (DMSO or 10% Tween-80), no difference is seen in the aversion induced by U69593 and 3 mg/kg RB64. Data are plotted as amount of time spent in drug-paired room during after the test compared with pretest. Data are plotted as amount of time spent in drug-paired room during posttest compared with pretest ± S.E.M. *P < 0.05; **P < 0.01. BArr2, β-arrestin-2.
Rotarod data were assessed by a two-way RM ANOVA for each treatment condition. The within-subject effect of time, the between-subject effect of genotype, and the effect of time on genotype were reported (Fig. 4, A–C). A one-way ANOVA was used to analyze any difference between baseline performances for each genotype (Fig. 4E). Results are plotted as the percent of baseline performance for each mouse. Novelty induced locomotion data were assessed by a two-way RM ANOVA to determine the effect of genotype on locomotion (Fig. 5, A–D). To determine the effect of treatment on the total distance traveled within the first 30 minutes of the assay, a one-way ANOVA was performed (Fig. 5E).
Fig. 4.

β-arrestin 2 signaling contributes to KOR agonist–induced rotarod deficit. (A) U69593 induced a rotarod deficit in both WT (n = 23) β-arrestin-2 KO mice (n = 23) but produced a larger deficit in performance for WT mice compared with β-arrestin-2 KO mice (P < 0.0003). (B) Sal A also produced a larger deficit in rotarod performance in WT mice (n = 25) than in β-arrestin-2 KO mice (n = 25) (P < 0.0006). (C) RB-64 (3 mg/kg) had no effect on performance in either WT (n = 5) or β-arrestin-2 KO mice (n = 5). (D) 3 mg/kg U69593 had a strong effect in both WT (n = 18) and β-arrestin-2 KO mice (n = 17). (E) sal A (10 mg/kg) produced a larger deficit in rotarod performance in WT mice (n = 10) than in β-arrestin-2 KO mice (n = 10). (F) Ten milligrams per kilogram RB-64 had no effect on either WT (n = 8) or β-arrestin-2 KO mice (n = 10). (G) U69593 (n = 5) and sal A (n = 5) had no effect on rotarod performance in KOR KO mice. (H) No difference was seen in rotarod baseline performances between genotypes. Data are plotted as the percent of baseline performance (A–G) or time spent on rod (H) ± S.E.M. BArr2, β-arrestin-2.
Fig. 5.

Effect of KOR agonists on novelty induced locomotion. (A) The novelty induced locomotion for vehicle-treated WT (n = 7) and β-arrestin-2 KO mice (n = 8). Both genotypes habituated and decreased activity after approximately 30 minutes. (B) U69593 did not differentially affect WT (n = 6) and β-arrestin-2 KO mice (n = 8). (C) No difference was seen in locomotion for sal A treatment between WT (n = 6) and β-arrestin-2 KO mice (n = 6). (D) RB-64 induced similar effects in both WT (n = 6) and β-arrestin-2 KO mice (n = 7). (E) The total distance traveled in the first 30 minutes (time before habituation). U69593 and sal A caused a strong decrease in activity, whereas RB-64 had no effect relative to vehicle-treated mice. Variance is represented as S.E.M. ***P < 0.001. BArr2, β-arrestin-2.
ICSS data were assessed by a two-way RM ANOVA to compare the effects of drug and dose (Fig. 6). The lowest frequency that sustained responding (BSR threshold) andMAX values were represented as the percent of the predrug baseline for each mouse. A two-way RM ANOVA was performed to assess the effects of treatments on the average rate-frequency curves. For all data sets assessed with two-way RM ANOVA and one-way ANOVA, when significant (P < 0.05), all post-hoc analyses were by Bonferroni corrected pairwise comparisons. To satisfy assumptions for parametric analysis using a one-way ANOVA, data were evaluated for equal variances using the Bartlett test.
Fig. 6.

A G protein–biased KOR agonist does not display anhedonia-like effects. (A) The response for different frequencies of brain stimulation reward by C57BL/6J mice treated with 1 mg/kg drug. All treatments showed a rightward shift in the average rate-frequency curves compared with vehicle; U69593 had the largest effect, followed by sal A, and then RB-64 (n = 13 for all conditions). (B) Dose-response relationship for the effects of U69593, sal A, and RB-64 on the BSR. Results are presented as mean percentages of preinjection baseline during the four 15-minute postinjection response series ± S.E.M. *Significance (P < 0.05) of drug versus vehicle. #Significance (P < 0.05) of drug compared with another drug treatment. (C) Dose-response relationship for the effects of U69593, sal A, and RB-64 on the maximum response rate (MAX) in C57BL/6J mice. Results are presented as mean percentage of preinjection baseline during the four 15-minute postinjection response periods ± S.E.M. *Significance (P < 0.05) versus vehicle. #Significance (P < 0.05) versus a separate drug treatment. n = 13 for all treatment conditions.
Results
RB-64 Is an Extremely G Protein–Biased Ligand at the Mouse KOR.
RB-64 was originally identified as a G protein–biased ligand from a screen using the human KOR (White et al., 2014). Because prior studies have indicated that mouse and human KOR may differ substantially with regard to patterns of functional selectivity (Schattauer et al., 2012), it was essential to evaluate the degree of bias for RB-64 at the mouse KOR before in vivo studies. In an in vitro assay system, we showed that RB-64 is nearly inactive at the arrestin pathway and represents an exceptionally G protein–biased agonist at the mouse KOR (bias factor = 96) (Fig. 1; Table 1). Importantly, U69593 is unbiased (bias factor = 3) relative to sal A, which is a potent and selective balanced KOR agonist (Roth et al., 2002) (Fig. 1; Table 1). Given that RB-64 represents an extremely potent and selective G protein–biased mouse KOR agonist with pronounced in vivo central nervous system activity in mice (Yan et al., 2009), we elected to compare its actions with those induced by the chemically similar but unbiased agonist sal A and with a chemically dissimilar unbiased agonist U69593.
TABLE 1.
Potencies and efficacies of ligands for mouse κ-opioid receptor in vitro
| Drug | G Protein Activation | Arrestin Mobilization | Bias Factor |
|---|---|---|---|
| nM | |||
| Salvinorin A | EC50 = 4.73 (−8.33 ± 0.04) | EC50 = 10.5 (−7.98 ± 0.05) | 1 |
| Emax = 101 | Emax = 93.6 | ||
| U69593 | EC50 = 3.68 (−8.43 ± 0.04) | EC50 = 25.4 (−7.59 ± 0.03) | 3 G protein |
| Emax = 99.7 | Emax = 100 | ||
| RB-64 | EC50 = 5.22 (−7.92 ± 0.05) | EC50 = 1130 (−5.94 ± 0.03) | 96 G protein |
| Emax = 99.0 | Emax = 126 | ||
In addition, we examined the effects of 200 nM U69593, sal A, and RB-64 on KOR internalization in HEK cells. Although U69593 and sal A induced robust internalization at 15 and 30 minutes, RB-64 induced no internalization at 15 minutes and modest internalization at 30 minutes (Fig. 1C), consistent with its G protein bias at this dose.
KOR-Mediated G Protein Signaling Induces Analgesia-Like Effects.
In the first set of assays, we examined the analgesic actions of our three test KOR agonists in a hotplate analgesic assay in both WT and β-arrestin-2 KO mice. As can be seen in Fig. 2A, all three compounds were analgesic in both WT and β-arrestin-2 KO mice. Specifically, there was an interaction of time and U69593 treatment compared with vehicle-treated mice in WT [F(3,54) = 11.93, P < 0.0001] and β-arrestin-2 KO mice [F(3,51) = 4.25, P = 0.0093]. WT mice showed an analgesic effect at 10 and 20 minutes after U69593 treatment (P < 0.001), and β-arrestin-2 KO mice displayed an analgesic effect 10 minutes (P < 0.01) and 20 minutes (P < 0.05) after U69593 treatment (Fig. 2A). Additionally, there was an interaction of sal A treatment and time for both WT [F(3,63) = 3.51, P = 0.0201] and β-arrestin-2 KO mice [F(3,69) = 3.14, P = 0.0309]. The analgesia-like effect of sal A occurred 10 minutes posttreatment of both WT and β-arrestin-2 KO mice (P < 0.01) (Fig. 1B). Furthermore, there was an interaction of time and RB-64 treatment of both WT [F(3,57) = 5.81, P = 0.0016] and β-arrestin-2 KO mice [F(3,48) = 2.96, P = 0.0417]. The analgesia-like effects of 3 mg/kg RB-64 occurred 20 (P < 0.001, WT mice; and P < 0.01, β-arrestin-2 KO mice) and 30 minutes after treatment (P < 0.05, for both WT and β-arrestin-2 KO mice) (Fig. 2C). There was no difference in analgesia-like responses between genotypes for any drug conditions (Fig. 2, A–C).
To determine whether the KOR mediated the analgesia-like effects of the tested drugs, KOR KO and WT mice were treated with 1 mg/kg U69593, 3 mg/kg sal A, or 3 mg/kg RB-64, but there was no analgesic effect in KOR KO mice compared with WT mice for all drug treatments [F(5,40) = 4.734, P = 0.0017] (Fig. 2D). Furthermore, there was no effect of genotype on analgesia-like effects of vehicle-treated mice [F(1,48) = 0.37, P = 0.5526] (Fig. 2, A–C). However, genotype did effect baseline performance [F(2,33) = 3.438, P = 0.044] (Fig. 2E). β-Arrestin-2 KO mice have a higher baseline than WT mice (P < 0.05), but KOR KO mice and WT mice do not differ (P > 0.05). To confirm that sal A and RB-64 have similar potencies in vivo, we tested the analgesia-like response at a higher dose of both sal A and RB-64 and found no significant difference among the 3 mg/kg dose versus the 10 mg/kg dose for either drug (P > 0.05) (Fig. 2F).
The G Protein–Biased KOR Agonist Induces Aversion in a Conditioned Place Aversion Assay.
We next tested all three KOR agonists in a commonly used assay of aversion—namely, the conditioned place aversion assay. As can be seen all three KOR agonists cause an aversion-like response that can be observed in both WT and β-arrestin-2 KO mice (F(11,78) = 3.241, P = 0.0011) (Fig. 3A). In WT mice, there was an aversive effect of 1 mg/kg U69593 (P < 0.05), 3 mg/kg sal A (P < 0.01), and 3 mg/kg RB-64 (P < 0.05), but no aversive effect of the lower tested doses of sal A (1 mg/kg; P > 0.05) or RB-64 (1 mg/kg; P > 0.05). Similarly, deleting β-arrestin-2 did not significantly alter the aversion-like action of 1 mg/kg U69593 (P < 0.05), 3 mg/kg sal A (P < 0.05), or 3 mg/kg RB-64 (P < 0.05). Similarly, there was no aversion-like action of the lower tested dose of sal A (1 mg/kg; P > 0.05) or RB-64 (1 mg/kg; P > 0.05). Furthermore, there was no difference in aversion-like effects between WT and β-arrestin 2 KO mice for any of the treatment conditions (vehicle, 1 mg/kg U69593, 1 mg/kg sal A, 3 mg/kg sal A, 1 mg/kg RB-64, or 3 mg/kg RB-64) (P > 0.05, for all conditions).
Because we used a vehicle (10% Tween-80) that could alter the aversive response to the test agents, we also examined the effect of 1 mg/kg U69593 or 3 mg/kg RB-64 dissolved in dimethylsulfoxide instead of 10% Tween-80. For these studies, the test drugs were microinjected into WT mice using a Hamilton syringe (Hamilton, Reno, NV) in a total volume of 1 μl for each milligram of mouse weight. Using this alternative vehicle, we again observed no significant difference in aversion-like actions of U69593 and RB-64 [t(10) = 1.237, P > 0.2442] (Fig. 3B).
The G Protein–Biased Agonist RB-64 Does Not Impair Rotarod Performance.
The effect of opioids on rotarod performance has long been used to evaluate potential sedative or coordination-impairing actions of KOR agonists (Kieffer, 1999). Here we found that the tested unbiased KOR agonists caused a strong deficit in rotarod performance and that this effect was greater in WT mice than in β-arrestin-2 KO mice (Fig. 4). There was a greater performance deficit in WT mice than β-arrestin-2 KO mice for both 1 mg/kg U69593 treatment [F(1,88)=15.38, P < 0.003] and 3 mg/kg sal A treatment [F(1,96) = 13.69, P < 0.0006] (Fig. 4, A and B). Significantly, 3 mg/kg RB-64 had no effect on rotarod performance in either genotype [F(1,24)=0.14, P = 0.7124] (Fig. 4C).
We also examined the effect of higher doses of U69593, sal A, and RB-64 on rotarod performance in WT and β-arrestin-2 KO mice. No difference was seen in the performance of the WT and β-arrestin-2 KO mice treated with 3 mg/kg U69593 [F(1,66) = 0.26, P = 0.615] (Fig. 4D); however, in both genotypes, a strong deficit in performance was observed. We found that 10 mg/kg sal A induced a larger deficit in performance in WT mice than β-arrestin-2 KO mice [F(1,34) = 11.65, P = 0.0033] (Fig. 4E). Furthermore, 10 mg/kg RB-64 induced no effect on rotarod performance in either genotype [F(1,32) = 0.05, P = 0.8288] (Fig. 4F).
To ensure that the effect of these drugs was due to the KOR, we tested them in KOR KO mice and found that KOR KO animals show no deficit in rotarod performance when treated with 1 mg/kg U69593 or 3 mg/kg sal A (Fig. 4G). For both drug treatments, all KOR KO animals performed at 100% baseline for all time points tested. Additionally, there was no difference in baseline performance among genotypes [F(2,53) = 0.6736, P = 0.5142] (Fig. 4H).
The G Protein–Biased KOR Agonist RB-64 Does Not Affect Novelty-Induced Locomotion.
The KOR agonists are also well known to induce sedation and to decrease novelty-induced locomotion (Kieffer, 1999). We therefore next evaluated our three KOR agonists in a novelty-induced locomotion paradigm as described previously (Allen et al., 2011; Besnard et al., 2012) (Fig. 5). Here we pooled the data for the first 30 minutes of each treatment condition to compare the effects of all treatments and genotypes on total distance traveled (Fig. 5) and found that the treatments had different effects on locomotion [F(7,49)=21.97, P < 0.0001]. In both WT and β-arrestin-2 KO mice, 1 mg/kg U69593 caused a large decrease in distance traveled (P < 0.001, for both WT and β-arrestin-2 KO mice), and 3 mg/kg sal A similarly decreased distance traveled (P < 0.001, for both WT and β-arrestin-2 KO mice), but there was no effect of 3 mg/kg RB-64 on distance traveled (P > 0.05, for both WT and β-arrestin-2 KO mice). Additionally, there was no difference in distance traveled between WT and β-arrestin-2 KO mice for any treatment condition.
A G Protein–Biased KOR Agonist Does Not Display Anhedonia-Like Effects.
The KOR agonists are well known to induce dysphoria and an anhedonic-like response in humans and mice (Potter et al., 2011; Ranganathan et al., 2012). For these studies, the anhedonia-like effects of sal A, RB-64, or U69593 were tested in C57BL/6J mice using the curve-shift method of ICSS (Carlezon and Chartoff, 2007). The mice responded in a frequency-dependent manner for BSR, as shown by the average baseline rate-frequency curves (Fig. 6A). There was an interaction between frequency and drug treatment [F(42,672) = 12.98, P < 0.0001]; all three drugs caused a rightward shift in average rate-frequency curves at a dose of 1 mg/kg where U69593 is the most potent, followed by sal A and RB-64 (Fig. 6A).
After a daily baseline measurement, the effects of each drug (0.01–1.0 mg/kg) on the minimum frequency that maintained responding (BSR threshold; Fig. 6B), and maximum operant response rate (MAX; Fig. 6C) during four 15-minute response periods post-treatment were measured (Fig. 6). A significant interaction was revealed between drug and dose on BSR threshold during the first posttreatment period [F(8,96) = 2.439, P = 0.019], the second post-treatment period [F(8.96) = 8.776, P < 0.001], the third post-treatment period [F(8,96) = 6.111, P < 0.0001], and the fourth post-treatment period [F(8,96) = 2.499, P = 0.016]. For the first post-treatment period, 1 mg/kg sal A and U69593 elevated BSR threshold compared with vehicle (P < 0.001 for both). For the second post-treatment period, 0.3 U69593, 1.0 mg/kg U69593, and 1.0 mg/kg sal A elevated BSR threshold compared with vehicle (P < 0.001 for each). There was a differential effect of treatment at 1.0 mg/kg dose: U69593 versus RB-64 (P < 0.001), U69593 versus sal A (P = 0.016), and sal A versus RB-64 (P < 0.001). During the third and fourth post-treatment periods, 1.0 mg/kg U69593 significantly elevated BSR thresholds compared with vehicle (P < 0.001). Additionally, U69593 differed from sal A and RB64 during the third (P < 0.001) and fourth post-treatment periods (P = 0.005 and P = 0.01 for sal A and RB-64, respectively). All doses of RB-64 had no significant effect on BSR (Fig. 6B).
A significant interaction between drug and dose on MAX was revealed at the second post-treatment period [F(8,96) = 14.317, P < 0.001], the third post-treatment period [F(8,96) = 18.426, P < 0.001], and the fourth post-treatment period [F(8,96) = 7.896, P < 0.001]. During the second post-treatment period, 0.3 mg/kg U69593, 1.0 mg/kg U69593, or 1.0 mg/kg sal A significantly decreased MAX compared with vehicle (P = 0.001 for each). Furthermore, the drugs caused differential effects at the 0.3 mg/kg dose between U69593 and sal A (P < 0.001), and U69593 and RB-64 (P < 0.001), but not sal A and RB-64. Additionally, the drugs had differential effects at a 1 mg/kg dose between all pairs of drugs: U69593 and sal A (P < 0.001), U69593 and RB-64 (P < 0.001), and sal A and RB-64 (P = 0.027). During the third post-treatment period, only U69593 (both 0.3 and 1 mg/kg) decreased MAX compared with vehicle, sal A, and RB-64 (P < 0.001 for each). During the fourth post-treatment period, only 1.0 mg/kg U69593 decreased MAX compared with vehicle, sal A, and RB-64 (P < 0.001 for each) (Fig. 6C).
Discussion
The major finding of this study is that the selective G protein–biased KOR agonist RB-64 is analgesic with fewer of the prototypical side effects associated with unbiased KOR agonists. Prototypical KOR agonists are well known to be effective analgesics with low dependence and abuse potential (Tao et al., 2008; Wee and Koob, 2010; Prevatt-Smith et al., 2011). Unfortunately, KOR agonists can cause dysphoria, psychotomimesis, hallucinations, and anhedonia-like effects (Pfeiffer et al., 1986; Ebner et al., 2010; Potter et al., 2011; Ranganathan et al., 2012). Our findings are consistent with the hypothesis that creating G protein–biased KOR agonists represents a novel approach for creating analgesics with fewer side effects.
Elegant prior studies have provided evidence that KOR-mediated p38 MAPK signaling regulates dysphoria and that G protein signaling regulates analgesia (Bruchas et al., 2007). Before the current study, the role of β-arrestin-2 signaling for the actions of KOR agonists has not been directly tested in vivo. To investigate further which KOR signaling pathways contribute to specific behaviors, we examined WT and β-arrestin-2 KO mice in a number of behavioral paradigms with a G protein–biased ligand (RB-64) and two unbiased ligands (U69593 and sal A). If an expected KOR-induced response is absent in β-arrestin-2 KO mice, β-arrestin-2 signaling may be involved in mediating that response. Additionally, if RB-64 fails to induce a response similar to the unbiased ligands U69593 and sal A, that would suggest that RB-64 has a biased signaling profile in vivo.
We previously showed that RB-64 is a G protein–biased, human KOR ligand using in vitro signaling assay, and here we show a similar effect in activation of the mouse KOR (Fig. 1; Table 1). Additionally, we observed an altered KOR internalization on RB-64 stimulation relative to the unbiased KOR ligands U69593 and sal A (Fig. 1C). In the future, an in-depth investigation of downstream signaling events activated by a diverse set of biased and unbiased ligands will provide major insights into what contributes to behavior beyond G proteins, arrestin, and p38 MAPK.
The analgesia-like effects of U69593 and sal A shown here are similar to those of previous reports (Waddell and Holtzman, 1999; Ansonoff et al., 2006). This study shows no statistical difference in analgesia-like responses between WT and β-arrestin-2 KO mice treated with KOR agonists (Fig. 2, A–C). However, we did observe a significant difference in baseline analgesia between WT and β-arrestin-2 KO mice, consistent with previous reports (Fig. 2E) (Bohn et al., 2002). Additionally, the G protein–biased ligand (RB-64) showed a significant and long-lasting analgesia-like effect (Fig. 2C). This long-lasting effect may be due to the lack of β-arrestin-2 recruitment, and therefore altered receptor desensitization, but it could also be due to differential pharmacokinetics of the test agents. The transient effect of sal A was expected because it is known that sal A has a very short half-life in vivo owing to plasma esterase activity (Cunningham et al., 2011). Importantly, the analgesia-like effect of U69593, sal A, and RB-64 was shown to be specific to KOR and not due to an off-target effect because there was no analgesic response in KOR KO mice treated with U69593, sal A, or RB-64 (Fig. 2D). These studies suggest that G protein–signaling mediates KOR-induced analgesia-like effects. The analgesic studies are consistent with recent hints that a G protein–biased KOR agonist of a different chemotype can induce analgesia-like effects (Zhou et al., 2013).
To determine whether β-arrestin-2 signaling mediates KOR-mediated aversion, as previously hypothesized (Bruchas and Chavkin, 2010), we examined the effects of U69593, sal A, and RB-64 in both WT and β-arrestin-2 KO mice in the conditioned place aversion behavioral paradigm. U69593 and sal A have previously been shown to induce aversion in this paradigm (Shippenberg and Herz, 1986; McLaughlin et al., 2003; Zhang et al., 2005; Tejeda et al., 2013; Sufka et al., 2014). We found that U69593, sal A, and RB-64 all caused a similar degree of aversion-like activity in both WT and β-arrestin-2 KO mice (Fig. 3A). This finding suggests that G protein signaling, not β-arrestin-2 signaling, mediates the aversion-like effect measured in this assay. It is important to note that CPA is a measure of conditioned learning, and the avoidance observed in this assay could be caused by actions of the drugs not directly associated with an aversive sensation. Additionally, previous reports link the activation of p38 MAPK to KOR-induced aversion that was hypothesized to be mediated by β-arrestin-2. Our results suggest that β-arrestin-2 is not the only factor involved in mediated KOR dependent aversion. Here p38 MAPK may be activated by an alternative signaling node or the aversion may be induced by a p38 MAPK-independent manner. Further investigation of the global signaling events activated by KOR biased and unbiased ligands will be illuminating.
To explore further the specific KOR-mediated signaling cascades that mediate its actions on reward and various hedonic states, we performed ICSS experiments to determine the effect of G protein–biased ligands on motivation and brain reward circuitry. Previous reports have shown a negative effect of KOR agonists on BSR, implying that treatment with KOR agonists reduces the hedonic value of brain stimulation reward (Ebner et al., 2010; Negus et al., 2010). Because RB-64 appears biased in vivo, we treated WT mice with U69593, sal A, or RB-64. Interestingly, U69593 and sal A reduce an animal’s motivation to work for brain stimulation reward, whereas RB-64 lacks this effect (Fig. 6). These data are consistent with the original premise that β-arrestin-2 may mediate some of the negative side effects of KOR agonism; however, it is now clear that other signaling components may also mediate KOR-induced negative side effects.
To explore further how G protein and β-arrestin-2 signaling contribute to KOR-mediated behavioral effects, we measured the performance of WT and β-arrestin-2 KO mice in the rotarod assay after treatment with U69593, sal A, and RB-64. This assay measures balance and motor coordination and has long been used to assess the potential liability of central nervous system–active drugs. Both 1 mg/kg U69593 and 3 mg/kg sal A caused a stronger deficit in rotarod performance in WT mice than in β-arrestin-2 KO mice, whereas 3 mg/kg RB-64 had absolutely no effect on performance in either genotype (Fig. 4, A–C). Additionally, we found that at a higher dose of 3 mg/kg U69593, the deficit in rotarod performance is strong in both genotypes (Fig. 4D). At all doses of sal A tested, there was a significant difference in performance between genotypes (Fig. 4E), but we expect that at higher doses, both genotypes would perform equally poorly as seen with 3 mg/kg U69593. Interestingly, even with a dose of 10 mg/kg RB-64, we observed little effect on rotarod performance and no difference between genotypes (Fig. 4F). These findings suggest that β-arrestin-2 signaling is important for the KOR-induced deficit in rotarod performance. Furthermore, the complete lack of an effect of RB-64 on rotarod performance suggests that there could be some β-arrestin-1 compensation in the β-arrestin-2 KO mice, leading to the slight performance deficit observed when mice were treated with U69593 and sal A. KOR KO mice were treated with U69593 and sal A and showed no impairment in performance, suggesting that the effects of U69593 and sal A are mediated through the KOR. Taken together, these data suggest that β-arrestin-2 signaling contributes to KOR-induced rotarod deficit and strongly suggest that RB-64 is an extremely G protein–biased ligand in vivo.
The effect of these drugs on the rotarod assay could be due to a number of factors: sedation, altered motor coordination, and/or a cognitive disruption. To understand more clearly why RB-64 does not affect rotarod performance but U69593 and sal A cause an impairment in performance, we tested the locomotor-attenuating actions of these drugs in the novelty-induced locomotion assay. We observed decreased locomotion in both WT and β-arrestin-2 KO mice treated with U69593 or sal A, but not RB-64 (Fig. 5). Although both WT and β-arrestin-2 KO mice showed a strong suppression of locomotion, the G-protein–biased ligand did not cause this effect. It is possible that, in this behavioral paradigm, there is compensation for the absence of β-arrestin-2 by β-arrestin-1.
Despite a lack of effect of RB-64 on novelty-induced locomotion, the rotarod assay, and MAX and BSR thresholds, we conclude that RB-64 is active in the brain because of the strong effects observed in the hotplate and CPA paradigms. Further support comes from our prior studies showing that RB-64 has potent effects on prepulse inhibition (Yan et al., 2009). We attempted to quantify RB-64 levels in the brain using light chromatography–tandem mass spectrometry; however, the collection conditions used for this analysis interfered with the detection of RB-64. A more thorough investigation with additional G protein–biased, β-arrestin biased and, perhaps, p38 MAPK biased KOR selective ligands paired with a full pharmacokinetic evaluation will clarify the role of specific signaling cascades in KOR-dependent behaviors. Furthermore, RB-64 was originally synthesized to covalently bind KORs (Yan et al., 2009) and in preliminary studies (F. Yan and B. L. Roth, unpublished observations), we found that RB-64, when administered in vivo, apparently does not irreversibly interact with KOR.
In summary, our findings suggest that KOR-mediated G protein signaling induces analgesia-like effects and aversion-like actions in the CPA assay, whereas KOR-mediated β-arrestin-2 signaling mediates motor incoordination. Given the lack of effect of the G protein–biased ligand RB-64 on sedation and anhedonia, our data are consistent with the hypothesis that β-arrestin-2 activity is essential for the sedative and anhedonia-like actions of KOR agonists. Based on these results, our studies imply that G protein–biased KOR agonists may represent novel analgesics with fewer side effects compared with unbiased agonists.
Acknowledgments
The authors thank Dr. Wesley Kroeze and Reid Olsen for critically reading this manuscript.
Abbreviations
- 6′-GNTI
6′-guanidinonaltrindole
- ANOVA
analysis of variance
- BSR
brain stimulation reward threshold
- CPA
conditioned place aversion
- GPCR
G protein–coupled receptor
- ICSS
intracranial self-stimulation
- KO
knockout
- KOR
κ-opioid receptor
- MAPK
mitogen-activated protein kinase
- RB-64
22-thiocyanatosalvinorin A
- RM
repeated measure
- sal A
salvinorin A
- U69593
(+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide
- WT
wild type
Authorship Contributions
Participated in research design: White, Roth, Robinson.
Conducted experiments: White, Robinson, DiBerto, Zhu, Roth.
Contributed new reagents or analytic tools: Nichols, Polepally, Zjawiony.
Performed data analysis: White, Robinson, DiBerto.
Wrote or contributed to the writing of the manuscript: White, Roth.
Footnotes
This research was supported by the National Institutes of Health National Institute on Drug Abuse [Grants R01-DA017204 and P01-DA035764]; and the National Institutes of Health National Institute of Mental Health Psychoactive Drug Screening Program.
A preliminary account of this work was previously presented at the following meeting: White KL, Vardy E, and Roth BL (2013) Utilizing functionally selective ligands to probe specific signaling pathways of the kappa opioid receptor. 2013 Kappa Therapeutics Meeting; 2013 Apr 24–27; Cambridge, MA.
References
- Allen JA, Roth BL. (2011) Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol 51:117–144. [DOI] [PubMed] [Google Scholar]
- Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, et al. (2011) Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci USA 108:18488–18493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansonoff MA, Zhang J, Czyzyk T, Rothman RB, Stewart J, Xu H, Zjwiony J, Siebert DJ, Yang F, Roth BL, et al. (2006) Antinociceptive and hypothermic effects of Salvinorin A are abolished in a novel strain of kappa-opioid receptor-1 knockout mice. J Pharmacol Exp Ther 318:641–648. [DOI] [PubMed] [Google Scholar]
- Balter RE, Dykstra LA. (2013) Thermal sensitivity as a measure of spontaneous morphine withdrawal in mice. J Pharmacol Toxicol Methods 67:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, et al. (2012) Automated design of ligands to polypharmacological profiles. Nature 492:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar A, Willins DL, Gray JA, Woods J, Benovic JL, Roth BL. (2001) The dynamin-dependent, arrestin-independent internalization of 5-hydroxytryptamine 2A (5-HT2A) serotonin receptors reveals differential sorting of arrestins and 5-HT2A receptors during endocytosis. J Biol Chem 276:8269–8277. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Caron MG. (2002) Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J Neurosci 22:10494–10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Chavkin C. (2010) Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl) 210:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. (2007) Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci 27:11614–11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Chartoff EH. (2007) Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nat Protoc 2:2987–2995. [DOI] [PubMed] [Google Scholar]
- Cunningham CW, Rothman RB, Prisinzano TE. (2011) Neuropharmacology of the naturally occurring kappa-opioid hallucinogen salvinorin A. Pharmacol Rev 63:316–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, et al. (2013) A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther 344:708–717. [DOI] [PubMed] [Google Scholar]
- Ebner SR, Roitman MF, Potter DN, Rachlin AB, Chartoff EH. (2010) Depressive-like effects of the kappa opioid receptor agonist salvinorin A are associated with decreased phasic dopamine release in the nucleus accumbens. Psychopharmacology (Berl) 210:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell MS, Pei Y, Wan Y, Yadav PN, Daigle TL, Urban DJ, Lee HM, Sciaky N, Simmons A, Nonneman RJ, et al. (2013) A Gαs DREADD mouse for selective modulation of cAMP production in striatopallidal neurons. Neuropsychopharmacology 38:854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HS, Burns AJ, Nonneman RJ, Baker LK, Riddick NV, Nikolova VD, Riday TT, Yashiro K, Philpot BD, Moy SS. (2013) Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav Brain Res 243:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer BL. (1999) Opioids: first lessons from knockout mice. Trends Pharmacol Sci 20:19–26. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. (2003) Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci 23:5674–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev IO, Gainetdinov RR, Sotnikova TD, Bohn LM, Caron MG, Dykstra LA. (2005) Characterization of conditioned place preference to cocaine in congenic dopamine transporter knockout female mice. Psychopharmacology (Berl) 180:408–413. [DOI] [PubMed] [Google Scholar]
- Monasky MM, Taglieri DM, Henze M, Warren CM, Utter MS, Soergel DG, Violin JD, Solaro RJ. (2013) The β-arrestin-biased ligand TRV120023 inhibits angiotensin II-induced cardiac hypertrophy while preserving enhanced myofilament response to calcium. Am J Physiol Heart Circ Physiol 305:H856–H866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Morrissey EM, Rosenberg M, Cheng K, Rice KC. (2010) Effects of kappa opioids in an assay of pain-depressed intracranial self-stimulation in rats. Psychopharmacology (Berl) 210:149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. (1986) Psychotomimesis mediated by kappa opiate receptors. Science 233:774–776. [DOI] [PubMed] [Google Scholar]
- Potter DN, Damez-Werno D, Carlezon WA, Jr, Cohen BM, Chartoff EH. (2011) Repeated exposure to the κ-opioid receptor agonist salvinorin A modulates extracellular signal-regulated kinase and reward sensitivity. Biol Psychiatry 70:744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan AA, Befort K, Nozaki C, Gavériaux-Ruff C, Kieffer BL. (2011) The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci 32:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevatt-Smith KM, Lovell KM, Simpson DS, Day VW, Douglas JT, Bosch P, Dersch CM, Rothman RB, Kivell B, Prisinzano TE. (2011) Potential drug abuse therapeutics derived from the hallucinogenic natural product salvinorin A. Medchemcomm 2:1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan M, Schnakenberg A, Skosnik PD, Cohen BM, Pittman B, Sewell RA, D’Souza DC. (2012) Dose-related behavioral, subjective, endocrine, and psychophysiological effects of the κ opioid agonist Salvinorin A in humans. Biol Psychiatry 72:871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rives L-M, Rossillo M, Liu-Chen LY, Javitch JA. (2012) 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased kappa-opioid receptor agonist that inhibits arrestin recruitment. J Biol Chem 287:27050–27054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JE, Fish EW, Krouse MC, Thorsell A, Heilig M, Malanga CJ. (2012) Potentiation of brain stimulation reward by morphine: effects of neurokinin-1 receptor antagonism. Psychopharmacology (Berl) 220:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. (2002) Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci USA 99:11934–11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattauer SS, Miyatake M, Shankar H, Zietz C, Levin JR, Liu-Chen LY, Gurevich VV, Rieder MJ, Chavkin C. (2012) Ligand directed signaling differences between rodent and human κ-opioid receptors. J Biol Chem 287:41595–41607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippenberg TS, Herz A. (1986) Differential effects of mu and kappa opioid systems on motivational processes. NIDA Res Monogr 75:563–566. [PubMed] [Google Scholar]
- Sufka KJ, Loria MJ, Lewellyn K, Zjawiony JK, Ali Z, Abe N, Khan IA. (2014) The effect of Salvia divinorum and Mitragyna speciosa extracts, fraction and major constituents on place aversion and place preference in rats. J Ethnopharmacol 151:361–364. [DOI] [PubMed] [Google Scholar]
- Tao YM, Li QL, Zhang CF, Xu XJ, Chen J, Ju YW, Chi ZQ, Long YQ, Liu JG. (2008) LPK-26, a novel kappa-opioid receptor agonist with potent antinociceptive effects and low dependence potential. Eur J Pharmacol 584:306–311. [DOI] [PubMed] [Google Scholar]
- Tejeda HA, Counotte DS, Oh E, Ramamoorthy S, Schultz-Kuszak KN, Bäckman CM, Chefer V, O’Donnell P, Shippenberg TS. (2013) Prefrontal cortical kappa-opioid receptor modulation of local neurotransmission and conditioned place aversion. Neuropsychopharmacology 38:1770–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, et al. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320:1–13. [DOI] [PubMed] [Google Scholar]
- Waddell AB, Holtzman SG. (1999) Modulation of cocaine-induced antinociception by opioid-receptor agonists. Pharmacol Biochem Behav 62:247–253. [DOI] [PubMed] [Google Scholar]
- Wee S, Koob GF. (2010) The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology (Berl) 210:121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen EJ, Rajagopal S, Lefkowitz RJ. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol Med 17:126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Scopton AP, Rives ML, Bikbulatov RV, Polepally PR, Brown PJ, Kenakin T, Javitch JA, Zjawiony JK, Roth BL. (2014) Identification of novel functionally selective κ-opioid receptor scaffolds. Mol Pharmacol 85:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Bikbulatov RV, Mocanu V, Dicheva N, Parker CE, Wetsel WC, Mosier PD, Westkaemper RB, Allen JA, Zjawiony JK, et al. (2009) Structure-based design, synthesis, and biochemical and pharmacological characterization of novel salvinorin A analogues as active state probes of the kappa-opioid receptor. Biochemistry 48:6898–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Butelman ER, Schlussman SD, Ho A, Kreek MJ. (2005) Effects of the plant-derived hallucinogen salvinorin A on basal dopamine levels in the caudate putamen and in a conditioned place aversion assay in mice: agonist actions at kappa opioid receptors. Psychopharmacology (Berl) 179:551–558. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F, et al. (2013) Development of functionally selective, small molecule agonists at kappa opioid receptors. J Biol Chem 288:36703–36716. [DOI] [PMC free article] [PubMed] [Google Scholar]