

Abstract
β-Lactones are a privileged structural motif as enzyme inhibitors and chemical probes, particularly for the inhibition of enzymes from the serine hydrolase class. Herein, we demonstrate that cross-metathesis (CM) of α-methylene-β-lactones offers rapid access to structurally diverse, previously unexplored β-lactones. Combining this approach with competitive activity-based protein profiling (ABPP) identified lead β-lactone inhibitors/probes for several serine hydrolases, including disease-associated enzymes and enzymes of uncharacterized function. The structural diversity afforded by the α-methylene-β-lactone scaffold thus expands the landscape of serine hydrolases that can be targeted by small-molecule inhibitors and should further the functional characterization of enzymes from this class through the optimization of target-selective probes.
Keywords: β-Lactones, Serine hydrolases, Cross metathesis, α-Methylene-β-lactones, ABPP
β-Lactones are a versatile class of electrophilic small molecules that have found use as biological research probes and therapeutic agents largely due to their capacity to react with and inhibit enzymes from the serine/threonine hydrolase class.1 More recently, β-lactones have emerged as attractive probes for activity-based protein profiling (ABPP) across the larger serine hydrolase class directly in native biological systems.2 Many of the β-lactone inhibitors and probes are 3,4-disubstituted, designed around the natural product-based β-lactone, tetrahydrolipstatin (Orlistat®-Figure 1), a pancreatic lipase inhibitor used to treat obesity.3 While there are many stereoselective approaches to such β-lactones,4 we believe that cross-metathesis (CM) of α-methylene-β-lactones 1 offers rapid access to structurally diverse, previously unexplored 3,4-disubstituted-β-lactones.
Figure 1.

Examples of biologically active β-lactones.
In investigating strained heterocycles with exocyclic unsaturation, we found that α-methylene-β-lactones 1 participated in CM reactions with Type I5 alkenes, with couplings proceeding in high yields with excellent Z-stereoselectivities (Figure 2).6 This methodology is attractive for making focused libraries of β-lactones for several reasons. First, the α-alkylidene-β-lactones 4 themselves represent a new class of probes. In addition, metathesis reactions tend to be highly tolerant of a broad range of functionality, thus allowing for the preparation of diverse compounds from a single template. Moreover, it should be possible to selectively access either cis- or trans-β-lactones 5 from the α-alkylidene-β-lactones. cis-Lactones would be the expected products from hydrogenation reactions. Limited literature precedent with exocyclic α,β-unsaturated lactones suggests that 1,4-reductions can provide trans-diastereomers under the right conditions (vide infra). Most monocyclic β-lactone natural products and close analogs (see examples in Figure 1) that have been explored as drugs or probes are trans diastereomers.1d, 2b, 7 cis-β-Lactones, however, have been shown to be as potent as their trans-isomers in some cases.7b, 7c Overall, then, CM with α-methylene-β-lactones would provide a new class of β-lactone probes (α-alkylidene-β-lactones), while subsequent stereoselective reduction would lead to previously unexplored cis- or trans-β-lactones. Moreover, an opportunity to examine the potential of 2-methyleneoxetanes (see 11b/11d) in competitive ABPP is offered. We have previously shown that the 2-methyleneoxetane analog 6 (see Figure 1) of the anti-obesity drug, tetrahydrolipstatin (THL), was nearly as potent an inhibitor of porcine pancreatic lipase (PPL) as THL.8
Figure 2.

Cross-metathesis approach to β-lactone libraries.
Herein, the development of this straightforward CM approach to β-lactone probes, highlighted by a four-step synthesis of (±)-nocardiolactone (Figure 1),9 is described. Nineteen probes, including nocardiolactone, were prepared from a single α-methylene-β-lactone scaffold 9. The probes include E-and Z-α-alkylidene-β-lactones, cis- and trans-3,4-disubstituted-β-lactones, and 2-methyleneoxetanes. The utility of the probes in competitive ABPP was demonstrated by assaying these compounds in native cell (COLO205 human colon cancer cell line) and tissue (mouse brain) proteomes to assess their in vitro inhibitory activity against serine hydrolases. A combination of competitive gel- and MS-based ABPP methods identified novel β-lactone probes that target diverse members of the serine hydrolase family, including uncharacterized enzymes that lack selective inhibitors.
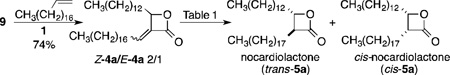
As previously noted, an attractive feature of our planned CM approach to monocyclic β-lactone analogs was that a single template could be used to access a broad range of lactones. The first important choice was the identity of the substituent at C4, since this would be a part of the initial set of analogs. THL has been shown to inhibit the thioesterase domain of fatty acid synthase,10 forming a covalent bond with an active site serine. Crystallographic studies show the C4 chain to be buried in a hydrophobic channel.11 While this channel may not be a universal motif, we and others have found that THL interacts with a variety of serine hydrolases.12 Böttcher and Sieber also took inspiration from the aliphatic chains of β-lactone natural products in the design of β-lactone ABPPs.1e In the initial series, for ease of synthesis we elected to use a simple alkyl chain at C4 and chose the chain length based on nocardiolactone, which has the same number of carbons in its alkyl chain as THL, giving us the opportunity to develop the methodology around a straightforward synthesis of (±)-nocardiolactone. Thus, the key intermediate for its synthesis and for diversification was α-methylene-β-lactone 9.
We previously reported the synthesis of α-methylene-β-lactones via lactonization of readily accessible, hydrolyzed Morita-Baylis-Hillman (MBH) adducts like 7 (Scheme 1).13 While α-methylene-β-lactone 9 was readily prepared from tetradecanal by this approach, a more direct sequence involved MBH reaction between tetradecanal and t-butyl thioacrylate, followed by mercury-mediated cyclization of 813 to 9. For nocardiolactone the remaining steps were CM and reduction.
Scheme 1.

CM between 9 and 1-nonadecene, catalyzed by Grubbs second-generation catalyst 2 under conditions we had previously developed for α-methylene-β-lactones,6 was straightforward, yielding mainly the Z-diastereomer (Z-4a). The enoate diastereomers were separable, providing two probes to evaluate. While it was anticipated that conversion to the cis-isomer of nocardiolactone could be readily achieved using hydrogenation, selective conversion to the trans-isomer was more challenging. Romo and coworkers have used epimerization of cis-3,4-disubstituted-β-lactones to access the trans-isomers,7c but the yields were relatively low (35–40%). Our preference was to avoid this extra, material depleting transformation. 1,4-Reduction of enoates 4 represented an alternative pathway that might be manipulated to provide the trans-isomers. We found no examples in the literature of 1,4-reduction of α-alkylidene-β-lactones; however, there were a number of examples involving α-alkylidene-γ-lactones.14 Although simple steric arguments would suggest that cis-diastereomers should dominate from protonation from the less hindered face of intermediate enolates, the literature cited showed that other factors play a role. Consequently, a variety of 1,4-reduction conditions were explored.
Iwasaki and coworkers had examined the use of transition metals (Co, Ni, Cu) in conjunction with NaBH4 to effect the reduction of α-alkylidene-γ-lactones with high trans-selectivity.14c They demonstrated that the protonation of the enoate was not reversible under the reaction conditions, making it the determinant of the relative stereochemistry. The high stereoselectivities were rationalized on the basis of conformational effects from 1,3-allylic strain. While our substituents were different, we reasoned that it might also be possible to use ligands to bias the outcome. Focusing first on cobalt(II) additives with NaBH4 as the reductant, CoCl2 (with MeOH as the proton source) (Table 1, entry 1) gave more cis-5a.
Table 1.
Optimization of stereoselectivity of 1,4-reduction of 4a.
 | ||||
|---|---|---|---|---|
| Entry | Conditions | Solvent | Temp (°C) |
trans-5a/cis-5a (yield) |
| 1 | CoCl2.6H2O (1 equiv), NaBH4 | THF:MeOH 5:1 | −5 | 1/2 |
| 2 | Co(acac)2 (1 equiv), NaBH4 | THF:MeOH 5:1 | −5 | 1.5/1 |
| 3 | Co(acac)2 (1 equiv), NaBH4 | THF:MeOH 5:1 | 5 | 4/1 (34%) opened ring product (28%) |
| 4 | Co(acac)2 (1 equiv), NaBH4 | THF:i-PrOH 5:1 | −5 | 1.85/1 |
| 5 | CoCl2(Ph3P)2 (1equiv), NaBH4 | THF:MeOH 5:1 | −5 | 2/1 |
| 6 | CoCl2(Ph3P)2 (18 mol%), NaBH4 | THF:MeOH 5:1 | −5 | 2/1 (61%) |
| 7 | CoCl2(Ph3P)2 (18 mol%), NaBH4 | THF:i-PrOH 5:1 | −5 | 1/1 |
| 8 | NiCl2.6H2O (1equiv), NaBH4 | THF:MeOH 5:1 | −5 | 1/1 |
| 9 | NiCl2(Ph3P)2 (1 equiv), NaBH4 | THF:MeOH 5:1 | −5 | 1.75/1 with smb |
| 10 | L-Selectride | THF | −78 to rt | 2.5/1 with sma,b |
| 11 | Mg0, ultrasound | MeOH | rt | Reduced opened ring product |
| 12 | Pd(Ph3P)4, ZnCl2, n-Bu3SnH | THF | rt | 1/1 with smb |
| 13 | CuCl2.2H2O, PMHS, (R)-BINAP, tBuONa | PhCH3, pentane i-PrOH | −20 | 1/1 smb |
Results were inconsistent on repetition, and material recovery was low.
sm = starting material.
The stereoselectivity improved with the use of Co(acac)2 (Entry 2), with a further improvement with a slightly more sterically hindered proton source, i-PrOH (Entry 4). Using phosphine-containing CoCl2(Ph3P)2 provided additional improvement in stereoselectivity (Entry 5). No further enhancement was seen with i-PrOH (Entry 7) or by using alternative phosphine ligands (not shown). Separating the product from phosphine related byproducts proved problematic, but this was resolved by cutting back on the additive (Entry 6). It is noteworthy that running the reaction at higher temperatures resulted in considerable quantities of methyl esters (Entry 3). Although NiCl2 initially looked more promising than the corresponding CoCl2 (Entry 8 vs 1), this was not the case with the optimal phosphine ligand (Entry 9). Copper(II) reagents gave very little conversion to product (not shown). Alternative reductants, such as L-selectride14a or Mg,14b that had been reported for the stereoselective reduction of α-alkylidene-γ-lactones (Entries 10 and 11) did not provide significant quantities of product. Although other 1,4-reduction conditions, such as those shown in Entries 1215 and 13,16 did lead to product, none of these provided higher stereoselectivities than the use of NaBH4 with catalytic CoCl2(Ph3P)2 as an additive. While we continue to explore other conditions, the optimized 1,4-reduction conditions identified thus far (Entry 6) do provide access to both cis- and trans-β-lactones 5, and these diastereomers are separable by column chromatography.
With α-methylene-β-lactone scaffold 9 in hand, the initial diversification was undertaken. Using four additional alkene partners (10b–e) that varied in chain length, with three (10c–e) incorporating a terminal aromatic moiety, five α-alkylidene-β-lactones 4 were isolated, and reduced to eight cis/trans-lactones 5 (Table 2). In addition, compounds trans-5b and trans-5d were methylenated to provide probes 11b and 11d, respectively, based on our earlier finding of the comparable activity of THL and its 2-methyleneoxetane analog in a PPL assay.8 Thus, overall, 19 β-lactone-based probes were readily assembled from a single scaffold for evaluation by competitive ABPP.
Table 2.
Preparation of α-alkylidene-β-lactones and their corresponding β-lactones
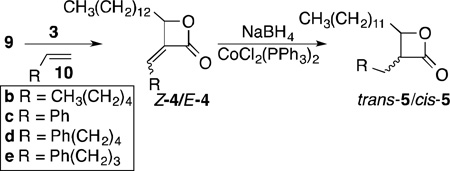 | |||
| Entry | R | Alkylidene lactone Z-4/E-4, (yield) |
β-Lactone trans-5/cis-5, (yield) |
| 1 | 10b; CH3(CH2)4 | 3.3/1 (97%) | 1. 3 /1 (44%) |
| 2 | 10c; Ph | Z-only (40%) | 2/1 (39%) |
| 3 | 10d; Ph(CH2)4 | Z-only (68%) | 2/1 (32%) |
| 4 | 10e; Ph(CH2)3 | Z-only (51%) | 1. 4 /1 (46%) |
The β-lactone probe set was screened against the membrane proteomes of mouse brain and the human COLO205 colon cancer cell line by competitive gel-based ABPP. Briefly, proteomes (1mg/mL) were incubated with β-lactone compounds at 25 µM for 30 minutes at 37 °C and then treated with the serine hydrolase-directed activity-based probe fluorophosphonate (FP)-rhodamine17 (2 µM, 45 min, 37 °C) and analyzed by SDS-PAGE and in-gel fluorescence scanning. Proteomes were also treated with DMSO and THL (25 µM) as controls. The β-lactone probes partially or fully inhibited activities of several serine hydrolase targets in the mouse brain membrane proteome (Figure 3A), including established targets of this inhibitor class, such as fatty acid synthase (FASN)1a, 18 and lysophospholipase 1 and 2 (LYPLA1, LYPLA2),2d, 19 and poorly characterized enzymes like ABHD10 and ABHD16A. These enzyme assignments were based on previous studies where gel-based ABPP experiments of mouse membrane proteome were related to mass spectrometry-based identifications of enzymes.12b, 20 Profiles of the human COLO205 colon cancer membrane proteome also revealed a diverse set of serine hydrolase targets for the β-lactone probes (Figure 3B).
Figure 3.

Competitive ABPP screening and identification of the serine hydrolase targets of β-lactone probe set. Competitive gel-based ABPP in mouse brain membrane proteome (A) and COLO205 colon cancer cell membrane proteome (B) for the β-lactone probe set. (C) ABPP-SILAC analysis of the COLO205 membrane proteome treated with trans-5c.
Among the β-lactones, trans-5c exhibited one of the broadest reactivity profiles across the detected serine hydrolases (Figure 3A, 3B) and was therefore selected for a more in-depth, mass spectrometry (MS)-based analysis of the landscape of serine hydrolases susceptible to inhibition by the β-lactone probes. COLO205 cells were cultured in media with isotopically heavy or light amino acids and their membrane proteomes isolated (1mg/mL) and treated with DMSO or trans-5c (25 µM, 30 min, 37 °C), respectively. The heavy and light proteomes were then incubated with FP-biotin (10 µM, 45 min, 37 °C),21 combined, and biotinylated proteins enriched and analyzed using avidin chromatography and multidimensional liquid chromatography (LC)-MS using an LTQ-Orbitrap instrument, where serine hydrolase activities were identified and quantified based on tryptic peptide MS2 and MS1 data, respectively.22 Enriched serine hydrolases that showed substantially greater (≥ 3-fold) MS1 signals in DMSO (heavy) versus trans-5c (light)-treated proteomes were considered targets of the trans-5c probe (Figure 3C). Eight serine hydrolases satisfied this criterion, with the most potently inhibited targets being ABHD16A, PNPLA4, FAAH, ABHD2, CES2, and ABHD12 (all inhibited > 90%). A second set of enzymes that included FASN and ABHD3 were inhibited from 70–90%. Among these enzymes, only FAAH and FASN, to our knowledge, are well-annotated in terms of biological functions and have selective inhibitors suitable for cellular or in vivo pharmacology studies. Our findings thus suggest that α-alkylidene-β-lactones could serve as useful starting points for developing selective inhibitors that target a diverse range of poorly characterized serine hydrolases. We have successfully tested this premise in a separate study, where we identified an α-alkylidene-β-lactone inhibitor and structurally related inactive control probe, that facilitated the functional characterization of the membrane-bound enzyme ABHD16A.23
In summary, a diverse set of 19 β-lactones has been readily prepared from a single α-methylene-β-lactone scaffold 9 by a straightforward sequence involving cross-metathesis and subsequent reduction. A combination of competitive gel- and MS-based ABPP experiments identified individual α-alkylidene-β-lactones and their reduced β-lactone counterparts that target a diverse array of serine hydrolases, including disease-relevant and uncharacterized enzymes that lack selective inhibitors. Thus, the utility of CM with α-methylene-β-lactones to access novel biologically active motifs and substitution patterns has been demonstrated. Coupling these tools with ABPP amplifies the significance of this approach for discovering new small-molecule inhibitors as chemical probes of cell biology. We are currently working to identify conditions for 1,4-reductions of the α-alkylidene-β-lactones that provide greater trans-selectivity and on the enantioselective synthesis of the α-methylene-β-lactones. In addition, efforts to optimize lead inhibitors discovered in this study are underway, as exemplified in the development of an inhibitor and accompanying inactive control probe of ABHD16A that have been used for functional characterization of this enzyme.23
Supplementary Material
Scheme 2.

Acknowledgments
This manuscript is based upon work partially supported by the National Science Foundation (AH) under Grant Nos. CHE-0111522 and CHE-1048717, the US National Institutes of Health (DA033760) (BFC) and the 9th Irving S. Sigal Postdoctoral Fellowship, American Chemical Society (SSK). Preliminary synthetic studies by Anisa Barolli and Thomas Farrell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Notes
The authors declare competing financial interests. BFC is a founder and advisor to Abide Therapeutics, a biotechnology company interested in developing serine hydrolase inhibitors and therapeutics
Supplementary Material
Experimental procedures, spectral characterization for all new compounds. This material is free of charge via the Internet at http://bmcl.
References
- 1.For leading references and reviews see: Flavin R, Peluso S, Nguyen PL, Loda M. Future Oncol. 6. 2010:551. doi: 10.2217/fon.10.11. Lam KS, Lloyd GK, Neuteboom STC, Palladino MA, Sethna KM, Spear MA, Potts BC. From natural products to clinical trials: NPI-0052 (salinosporamide A), a marine actinomycete-derived anticancer agent. Royal Society of Chemistry; 2010. pp. 355–373. Kluge AF, Petter RC. Curr. Opin. Chem. Biol. 2010;14:421. doi: 10.1016/j.cbpa.2010.03.035. Richardson RD, Ma G, Oyola Y, Zancanella M, Knowles LM, Cieplak P, Romo D, Smith JW. J. Med. Chem. 2008;51:5285. doi: 10.1021/jm800321h. Böttcher T, Sieber SA. Angew. Chem. Int. Ed. 2008;47:4600. doi: 10.1002/anie.200705768.
- 2.For leading references and reviews see: Lajkiewicz NJ, Cognetta AB, Niphakis MJ, Cravatt BF, Porco JA. J. Am. Chem. Soc. 2014;136:2659. doi: 10.1021/ja412431g. Böttcher T, Sieber SA. MedChemComm. 2012;3:408. Yang P-Y, Liu K, Zhang C, Chen GYJ, Shen Y, Ngai MH, Lear MJ, Yao SQ. Chem. - Asian J. 2011;6:2762. doi: 10.1002/asia.201100306. Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, Wetzel S, Renner S, Gerauer M, Schoelermann B, Rusch M, Kramer JW, Rauh D, Coates GW, Brunsveld L, Bastiaens PIH, Waldmann H. Nat. Chem. Biol. 2010;6:449. doi: 10.1038/nchembio.362. Wang Z, Gu C, Colby T, Shindo T, Balamurugan R, Waldmann H, Kaiser M, van der Hoorn RAL. Nat. Chem. Biol. 2008;4:557. doi: 10.1038/nchembio.104.
- 3.(a) Nelson RH, Miles JM. Expert Opin. Pharmacother. 2005;6:2483. doi: 10.1517/14656566.6.14.2483. [DOI] [PubMed] [Google Scholar]; (b) McNeely W, Benfield P. Drugs. 1998;56:241. doi: 10.2165/00003495-199856020-00007. [DOI] [PubMed] [Google Scholar]
- 4.For leading references and reviews see: Mulzer M, Tiegs BJ, Wang Y, Coates GW, O'Doherty GA. J. Am. Chem. Soc. 2014;136:10814. doi: 10.1021/ja505639u. Mulzer M, Ellis WC, Lobkovsky EB, Coates GW. Chem. Sci. 2014;5:1928. Chen S, Mondal M, Ibrahim AA, Wheeler KA, Kerrigan NJ. J. Org. Chem. 2014;79:4920. doi: 10.1021/jo500486e. Zhao C, Mitchell TA, Vallakati R, Pérez LM, Romo D. J. Am. Chem. Soc. 2012;134:3084. doi: 10.1021/ja209163w. Weise CF, Pischl MC, Pfaltz A, Schneider C. J. Org. Chem. 2012;77:1477. doi: 10.1021/jo202330b. Meier P, Broghammer F, Latendorf K, Rauhut G, Peters R. Molecules. 2012;17:7121. doi: 10.3390/molecules17067121. Marqués-López E, Christmann M. Angew. Chem. Int. Ed. 2012;51:8696. doi: 10.1002/anie.201204026. Purohit VC, Matla AS, Romo D. Heterocycles. 2008;76:949. Schneider C. Angew. Chem. Int Ed. 2002;41:744. doi: 10.1002/1521-3773(20020301)41:5<744::aid-anie744>3.0.co;2-v. Yang HW, Romo D. Tetrahedron. 1999;55:6403.
- 5.Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH. J. Am. Chem. Soc. 2003;125:11360. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 6.Raju R, Howell AR. Org. Lett. 2006;88:2139. doi: 10.1021/ol060624v. [DOI] [PubMed] [Google Scholar]
- 7.(a) Weinandy F, Lorenz-Baath K, Korotkov VS, Böttcher T, Sethi S, Chakraborty T, Sieber SA. ChemMedChem. 2014;9:710. doi: 10.1002/cmdc.201300325. [DOI] [PubMed] [Google Scholar]; (b) Ponzano S, Bertozzi F, Mengatto L, Dionisi M, Armirotti A, Romeo E, Berteotti A, Fiorelli C, Tarozzo G, Reggiani A, Duranti A, Tarzia G, Mor M, Cavalli A, Piomelli D, Bandiera T. J. Med. Chem. 2013;56:6917. doi: 10.1021/jm400739u. [DOI] [PubMed] [Google Scholar]; (c) Purohit VC, Richardson RD, Smith JW, Romo D. J. Org. Chem. 2006;71:4549. doi: 10.1021/jo060392d. [DOI] [PubMed] [Google Scholar]
- 8.Dollinger LM, Howell AR. Bioorg. Med. Chem. Lett. 1998;8:977. doi: 10.1016/s0960-894x(98)00140-1. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kumaraswamy G, Markondaiah B. Tetrahedron. 2008;64:5861–5865. [Google Scholar]; (b) Sun Y-P, Wu Y. Synlett. 2005:1477. [Google Scholar]; (c) Mikami Y, Yazawa Y, Ritzau M, Gräfeb U. Nat. Prod. Lett. 1999;13:277. [Google Scholar]
- 10.Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Cancer Res. 2004;64:2070. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- 11.Pemble CWIV, Johnson LC, Kridel SJ, Lowther WT. Nat. Struct. Mol. Biol. 2007;14:704. doi: 10.1038/nsmb1265. [DOI] [PubMed] [Google Scholar]
- 12.(a) Baggelaar MP, Janssen FJ, van Esbroeck ACM, den Dulk H, Allara M, Hoogendoorn S, McGuire R, Florea BI, Meeuwenoord N, van den Elst H, van der Marel GA, Brouwer J, Di Marzo V, Overkleeft HS, van der Stelt M. Angew. Chem., Int. Ed. 2013;52:12081. doi: 10.1002/anie.201306295. [DOI] [PubMed] [Google Scholar]; (b) Hoover HS, Blankman JL, Niessen S, Cravatt BF. Bioorg. Med. Chem. Lett. 2008;18:5838. doi: 10.1016/j.bmcl.2008.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez I, Andrews AE, Emch JD, Ndakala AJ, Wang J, Howell AR. Org. Lett. 2003;5:399. doi: 10.1021/ol020202v. [DOI] [PubMed] [Google Scholar]
- 14.(a) He M, Lei A, Zhang X. Tetrahedron Lett. 2005;46:1823–1826. [Google Scholar]; (b) Kim K, Okamoto S, Takayama Y, Sato F. Tetrahedron Lett. 2002;43:4237. [Google Scholar]; (c) Moritani Y, Fukushima C, Miyagishima T, Ohmizu H, Iwasaki T. Bull. Chem. Soc. Jpn. 1996;69:2281. doi: 10.1021/jo9601932. [DOI] [PubMed] [Google Scholar]
- 15.Matsuda Y, Endo Y, Saikawa Y, Nakata M. J. Org. Chem. 2011;76:6258. doi: 10.1021/jo2010186. [DOI] [PubMed] [Google Scholar]
- 16.Hughes G, Kimura M, Buchwald SL. J. Am. Chem. Soc. 2003;125:11253. doi: 10.1021/ja0351692. [DOI] [PubMed] [Google Scholar]
- 17.Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Proteomics. 2001;1:1067. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 18.(a) Agostini M, Almeida LY, Bastos DC, Ortega RM, Moreira FS, Seguin F, Zecchin KG, Raposo HF, Oliveira HCF, Amoedo ND, Salo T, Coletta RD, Graner E. Mol. Cancer Ther. 2014;13:585. doi: 10.1158/1535-7163.MCT-12-1136. [DOI] [PubMed] [Google Scholar]; (b) Chuang H-Y, Chang Y-F, Hwang J-J. Biomed. Pharmacother. 2011;65:286. doi: 10.1016/j.biopha.2011.02.016. [DOI] [PubMed] [Google Scholar]; (c) Kant S, Kumar A, Singh SM. Biochim. Biophys. Acta, Gen. Subj. 2014;1840:294. doi: 10.1016/j.bbagen.2013.09.020. [DOI] [PubMed] [Google Scholar]
- 19.(a) Xu J, Hedberg C, Dekker FJ, Li Q, Haigis KM, Hwang E, Waldmann H, Shannon K. Blood. 2012;119:1032. doi: 10.1182/blood-2011-06-358960. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rusch M, Zimmermann TJ, Bürger M, Dekker FJ, Goermer K, Triola G, Brockmeyer A, Janning P, Böttcher T, Sieber SA, Vetter IR, Hedberg C, Waldmann H. Angew. Chem., Int. Ed. 2011;50:9838. doi: 10.1002/anie.201102967. [DOI] [PubMed] [Google Scholar]; (c) Hedberg C, Dekker FJ, Rusch M, Renner S, Wetzel S, Vartak N, Gerding-Reimers C, Bon RS, Bastiaens PIH, Waldmann H. Angew. Chem., Int. Ed. 2011;50:9832. doi: 10.1002/anie.201102965. [DOI] [PubMed] [Google Scholar]
- 20.(a) Zuhl AM, Mohr JT, Bachovchin DA, Niessen S, Hsu K-L, Berlin JM, Dochnahl M, Lopez-Alberca MP, Fu GC, Cravatt BF. J. Am. Chem. Soc. 2012;134:5068. doi: 10.1021/ja300799t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Blankman JL, Simon GM, Cravatt BF. Chem. Biol. 2007;14:1347. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Patricelli MP, Cravatt BF. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14694. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adibekian A, Martin BR, Wang C, Hsu K-L, Bachovchin DA, Niessen S, Hoover H, Cravatt BF. Nat. Chem. Biol. 2011;7:469. doi: 10.1038/nchembio.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamat SS, Camara K, Parsons WH, Chen D-H, Dix MM, Bird TD, Howell AR, Cravatt BF. manuscript submitted. 2014 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.