

The localization of ECSIT into the nucleus is specifically accompanied by p65/p50 NF-κB in a TLR4-dependent manner. p65 NF-κB specifically interacts with the ubiquitinated ECSIT on the Lys-372 residue, thereby regulating NF-κB activity, NF-κB–dependent gene expression, and production of proinflammatory cytokines.
Abstract
Recent evidence shows that evolutionarily conserved signaling intermediate in Toll pathways (ECSIT) interacts with tumor necrosis factor receptor–associated factor 6 (TRAF6), is ubiquitinated, and contributes to bactericidal activity during Toll-like receptor (TLR) signaling. Here we report a new regulatory role for ECSIT in TLR4 signaling. On TLR4 stimulation, endogenous ECSIT formed a molecular complex with p65/p50 NF-κB proteins. Our biochemical studies showed that ECSIT specifically interacted with p65/p50 NF-κB proteins, which colocalized in the nucleus. Of interest, these effects were critically dependent on ubiquitination of the ECSIT lysine (K) 372 residue. K372A mutant ECSIT did not interact with p65/p50 NF-κB proteins and markedly attenuated nuclear colocalization. In addition, ECSIT-knockdown THP-1 cells could not activate NF-κB DNA-binding activities of p65 and p50, production of proinflammatory cytokines, or NF-κB–dependent gene expression in response to TLR4 stimulation. However, these activities were markedly restored by expressing the wild-type ECSIT protein but not the K372A mutant ECSIT protein. These data strongly suggest that the ubiquitination of ECSIT might have a role in the regulation of NF-κB activity in TLR4 signaling.
INTRODUCTION
Toll-like receptors (TLRs) recognize various pathogen components, referred to as pathogen-associated molecular patterns, and then initiate innate immune responses capable of acting as the first line of defense against pathogens (Medzhitov and Janeway, 2000; Akira and Hemmi, 2003; Takeuchi and Akira, 2010). TLR-mediated signaling is implicated in inflammatory and antiviral responses, as well as in dendritic cell maturation (Akira and Hemmi, 2003; Kawai and Akira, 2006; Takeuchi and Akira, 2010). Individual TLRs initially interact with different combinations of adaptor proteins and transmit downstream signaling cascades to activate various transcription factors, including nuclear factor (NF)-κB, activating protein-1, and interferon regulatory factors (IRFs; Akira and Hemmi, 2003; Ghosh and Hayden, 2008, 2012). TLR signaling pathways originate from cytoplasmic TIR domains with which TIR domain–containing adaptors, such as MyD88, TIRAP, and TRIF, are associated (Akira and Hemmi, 2003). In turn, IRAK-4, IRAK-1, and tumor necrosis factor (TNF) receptor–associated factor 6 (TRAF6) are recruited to the receptor complex. TRAF6 is a member of the TRAF family with E3 ubiquitin ligase activity and plays a key role activating IκB kinase (IKK) and mitogen-activated protein kinase, leading to activation of NF-κB (Akira et al., 2006; Uematsu and Akira, 2006; Kawai and Akira, 2011; Ghosh and Hayden, 2012)
Evolutionarily conserved signaling intermediate in Toll pathways (ECSIT) is a cytoplasmic protein that interacts specifically with the multiadaptor protein and E3 ubiquitin ligase TRAF6, which participates in Drosophila and mammalian TLR signaling pathways regulating innate immunity (Kopp et al., 1999; Moustakas and Heldin, 2003; Xiao et al., 2003; Vogel et al., 2007; West et al., 2011). A report showed that interaction with TRAF6 leads to ECSIT ubiquitination and enrichment at the mitochondrial periphery, resulting in increased mitochondrial and cellular reactive oxygen species (ROS) generation (West et al., 2011). These results strongly suggest that intracellular localization of ECSIT may be linked with its specific roles as a signaling adaptor protein in the cytoplasm (Kopp et al., 1999), a ROS regulatory protein in the mitochondria (Vogel et al., 2007; West et al., 2011; Heide et al., 2012), and a cofactor for bone morphogenic protein (BMP) signaling in the nucleus (Moustakas, 2003; Xiao, 2003). Nevertheless, nuclear localization of ECSIT and its functions in TLR signaling remain controversial and unclear. We investigated this issue in this study. Of note, our data demonstrate that localization of ECSIT in the nucleus was specifically accompanied by p65/p50 NF-κB proteins in a TLR4-dependent manner, where p65 NF-κB specifically interacted with ubiquitinated ECSIT on the Lys372 residue, thereby regulating NF-κB activity, NF-κB–dependent gene expression, and production of proinflammatory cytokines.
RESULTS
ECSIT interacts with p65/p50 NF-κB proteins after lipopolysaccharide stimulation
We first examined whether cellular localization of ECSIT changed dynamically in response to TLR4 stimulation. Subcellular fractions, including the cytosol (Cyt), nucleus (Nuc), and mitochondria (Mito), were isolated from HEK293-TLR4 cells treated or not with lipopolysaccharide (LPS), and ECSIT localization was assessed. In line with previous reports (Kopp et al., 1999; West et al., 2011), ECSIT appeared predominantly in the cytosol under resting conditions but significantly moved to the mitochondria (Figure 1A, Cyt and Mito). Of note, ECSIT markedly appeared in the nucleus in response to LPS stimulation (Figure 1A, Nuc). Moreover, these results were confirmed by confocal microscopy (Figure 1B). Previous findings indicate that ECSIT is a Toll-pathway signaling intermediate that plays a key role in activating NF-κB (Kopp et al., 1999) in the TLR4-mediated canonical NF-κB pathway. p65/p50 NF-κB translocates to the nucleus and drives NF-κB–dependent gene expression (Li and Verma, 2002; Hayden and Ghosh, 2004; Ghosh and Hayden, 2008; Häcker and Karin, 2006). Here we raise the possibility that ECSIT localization in the nucleus may be associated with NF-κB proteins.
FIGURE 1:

ECSIT interacts with p65/p50 NF-κB proteins after LPS stimulation. (A) HEK293-TLR4 cells were treated or not with 100 ng/ml LPS for 45 min, and then cytosol (Cyt), nuclear (Nuc), and mitochondrial (Mito) fractions were isolated, followed by immunoblot (IB) with antibodies to ECSIT, GAPDH, PCNA, and GRIM19. (B) HEK293-TLR4 cells were treated or not with100 ng/ml LPS for 45 min, and a confocal microscopy analysis was performed. (C) HEK293T cells were transiently transfected with Flag-ECSIT or p65-expression vector, as indicated, and then the immunoprecipitation (IP) assay was performed with anti-Flag antibodies, followed by IB with antibodies to anti-p65 or anti-Flag. (D) HEK293T cells were transiently transfected with Flag-ECSIT or p50-expressing vector, as indicated, and the IP assay was performed with anti-Flag antibodies, followed by IB with antibody to anti-p50 or anti-Flag. (E) HEK293T cells were transiently transfected with Flag-ECSIT wt or Flag-ECSIT truncated mutants, along with the p65-expressing vector, as indicated, and the IP assay was performed with anti-Flag antibodies, followed by IB with antibody to anti-Flag or anti-p65. (F) HEK293T cells were transiently transfected with Flag-ECSIT wt or Flag-ECSIT truncated mutants, along with p50-expressing vector, as indicated, and the IP assay was performed with anti-Flag antibodies, followed by IB with antibody to anti-Flag or anti-p50. (G) HEK293T cells were transiently transfected with p65-expressing vector, p50-expressiong vector, different concentrations of Flag-ECSIT wt, or different concentrations Flag-ECSIT 1-257, as indicated, and the IP assay was performed with anti-p65 antibody, followed by IB with antibodies to anti-Flag, anti-p50, and anti-p65.
To investigate this possibility, we examined whether ECSIT interacted with the NF-κB proteins. An immunoprecipitation (IP) assay in HEK293T cells expressing Flag-ECSIT and the p65 or p50 protein revealed that Flag-ECSIT proteins precipitated markedly with the p65 or p50 protein (Figure 1, C, p65, and D, p50). To identify the nature of the molecular interaction, we determined the ECSIT interaction site of p65 or p50. As shown in Figure 1, E and F, only wild-type (wt) ECSIT, but not other truncated ECIST mutants, precipitated p65 or p50. To verify these results, we transiently transfected HEK293T cells with Flag-ECSIT wt, the Flag-ECSIT 1–257 truncated mutant lacking the p65/p50, p65, and p50 binding-site vectors, as indicated, and performed the IP assay with anti-p65 antibodies. As expected, p65 coprecipitated markedly with p50 and Flag-ECSIT wt, whereas no significant interaction was seen with Flag-ECSIT 1–257 (Figure 1G), strongly suggesting that ECSIT interacts with p65 and p50 through its C-terminal region.
To confirm these results in an endogenous system, we examined whether the ECSIT/p65/p50 complex forms after LPS stimulation. We treated human monocytic THP-1 cells with or without LPS and isolated cellular complexes by gel filtration column chromatography (Noguchi et al., 2005). Endogenous ECSIT moved specifically to higher–molecular mass fractions (i.e., fractions 14–16) compared with those without stimulation (Figure 2A, IB: ECSIT), suggesting that ECSIT movement might be inducible in response to LPS stimulation. Of interest, the p65 and p50 NF-κB proteins appeared significantly in fractions 14–16 in response to LPS stimulation (Figure 2A, IB: p65 and IB: p50). Therefore we wondered whether ECSIT interacted with the p65/p50 proteins in the complex. As shown in Figure 2B, endogenous ECSIT coprecipitated markedly with the p65 and p50 NF-κB proteins in fractions 14–16. In addition, a direct interaction between ECSIT and p65 was confirmed by histidine (His)-tagged p65 pull-down assay (Figure 2C). These results suggest that ECSIT interacts with the p65/p50 NF-κB proteins after LPS stimulation.
FIGURE 2:

ECSIT endogenously forms the ECSIT/p65/p50 complex and interacts with the p65/p50 NF-κB proteins. (A) THP1 cells were treated for 60 min or not with LPS. The cell extracts were prepared and fractionated through a Superose6 10/300 GL column. Each fraction (40 μl) was analyzed by immunoblot (IB) with the indicated antibody to anti-ECSIT, anti-p65, or anti-p50. Apparent molecular mass was evaluated after column calibration with standard proteins: thyroglobulin (669 kDa), ferritin (400 kDa), catalase (232 kDa), and aldolase (158 kDa). The elution positions of these proteins are indicated at the top. (B) Endogenous immunoprecipitation (IP) of ECSIT from fractions 14–16 prepared from THP-1 cells, as described in A, followed by IB with antibodies to anti-ECSIT, anti-p65, or anti-p50. (C) The His-tagged p65 pull-down assay was performed with whole-cell lysates prepared from THP-1 cells treated or not with LPS (100 ng/ml) for 45 min, and then IB with antibody to anti-ECSIT or anti-p65 was performed.
ECSIT K372 ubiquitination is essential for the interaction and translocation of p65/p50 NF-κB to the nucleus
ECSIT was identified previously as a cytoplasmic protein interacting specifically with TRAF6 (Kopp et al., 1999). A report showed that TRAF6 is trafficked into periphagosomal mitochondria after TLR4 stimulation, where it interacts with and ubiquitinates ECSIT (West et al., 2011). Therefore we asked whether ECSIT ubiquitination would be affected by the interaction and translocation of p65/p50 NF-κB proteins to the nucleus. Overexpressed ECSIT proteins appeared markedly in both the cytosol and nucleus fractions and were significantly ubiquitinated by LPS stimulation (Figure 3A, IB: Flag). As expected, translocation of p65 NF-κB to the nucleus increased significantly after LPS stimulation compared with that without LPS treatment and was markedly enhanced by ECSIT overexpression (Figure 3A, IB: p65), suggesting that ubiquitinated ECSIT might be affected by translocation of p65 NF-κB to the nucleus in response to LPS stimulation.
FIGURE 3:

Ubiquitination of ECSIT is critical for the interaction with and translocation of p65 NF-κB to the nucleus. (A) HEK293-TLR4 cells were transfected with Flag-ECSIT and treated or not with 100 ng/ml LPS for 45 min. The cytosolic and nuclear fractions were prepared from the cells, and immunoblot (IB) with antibodies to anti-Flag, anti-p65, anti-PDI, and anti-PCNA was performed. (B) HEK293T cells were transfected with mock or Flag-ECSIT vector and immunoprecipitated (IP) with anti-Flag antibody, followed by IB with anti-Flag antibody. The immunoprecipitants were reacted in a K63-Ub reaction kit and treated with or without CYLD enzymes (left). The reactants were mixed with lysates prepared from HEK293T cells transfected with mock or p65- or p50-expressing vector, as indicated (right), and then the mixtures were centrifuged and washed three times with lysis buffer, and Western blotting with antibodies to anti-Flag, anti-p65, or anti-p50 was performed. (C) HEK293T cells were transiently transfected with Flag-ECSIT wt or Flag-ECSIT truncated mutants, along with the HA-Ub vector, as indicated, and then the IP assay was performed with anti-Flag antibodies, followed by IB with antibodies to anti-Flag or anti-Ub. (D) Comparison of sequences of wt ECSIT and K372A ECSIT mutant.
Thus HEK293T cells were transfected with mock or Flag-ECSIT vector, and Flag-ECSIT proteins were prepared by immunoprecipitation with anti-Flag antibodies. The K63- ubiquitin (Ub) reaction for polyubiquitination to Flag-ECSIT proteins was performed on the precipitates, followed by CYLD enzyme treatment (Figure 3B, left). The p65 and p50 vectors were independently transfected into HEK293T cells, and p65/p50 lysates were prepared (Figure 3B, right). To determine whether ECSIT ubiquitination is critical for the interaction with p65/p50 NF-κBs, we mixed ubiquitinated Flag-ECSIT or deubiquitinated Flag-ECSIT protein pretreated with the CYLD enzyme with p65/p50 lysates, respectively, and evaluated the interaction between ECSIT and p65/p50 NF-κB proteins. The polyubiquitinated ECSIT coprecipitated markedly with p65 and p50, whereas the interactions were dramatically attenuated after CYLD treatment (Figure 3B, right, bottom), strongly suggesting that ECSIT ubiquitination is critical for the interaction with p65/p50 NF-κB proteins. On the basis of these results, we sought to identify the ECSIT ubiquitination site. Flag-ECSIT wt or the Flag-ECSIT truncated mutants were cotransfected with a HA-Ub vector and then immunoprecipitated with anti-Flag antibodies. The wt ECSIT proteins were massively ubiquitinated, whereas no significant results were detected for the other ECSIT truncated mutants (Figure 3C), indicating that the site might be in the ECSIT 300–431 region if ECSIT is ubiquitinated. When we looked up the ECSIT amino acid sequence, only one lysine residue, K372, was found at ECSIT 300–431 (Figure 3D). Therefore we strongly considered the K372 lysine as an ECSIT-specific ubiquitination site and generated a K372A mutant ECSIT (Figure 3D)
Next we examined whether ECSIT ubiquitination occurred at the ECSIT K372 position and whether it is essential for the interaction and translocation of p65/p50 NF-κB to the nucleus. Flag-ECSIT wt or the Flag-ECSIT K372A mutant was transfected into HEK293T cells along with the HA-Ub vector. Flag-ECSIT wt was markedly ubiquitinated, whereas no significant change was seen in the Flag-ECSIT K372A mutant (Figure 4A), strongly demonstrating that ECSIT ubiquitination occurred at the K372 position. We then examined whether K372 ubiquitination directly affected not only the interaction between ECSIT and p65/p50 NF-κB proteins, but also translocation of p65/p50 NF-κBs to the nucleus. As shown in Figure 4B, Flag-ECSIT wt markedly coprecipitated with p65 or p50, whereas the interactions were severely attenuated in the Flag-ECSIT K372A mutant, indicating that ECSIT K372 ubiquitination is critical for the interaction with p65/p50 NF-κB proteins. Furthermore, nuclear localization of endogenous p65/p50 NF-κB proteins increased significantly after LPS stimulation and was markedly enhanced by overexpressing ECSIT (Figure 4C, Flag-ECSIT wt), whereas marked attenuation was detected when the K372A mutant ECSIT was overexpressed (Figure 4C, Flag-ECSIT K372A). These results strongly suggest that ECSIT K372 ubiquitination is critical for the interaction and colocalization of p65/p50 NF-κB to the nucleus.
FIGURE 4:

K372 ubiquitination of ECSIT is essential for the interaction and translocation of p65/p50 NF-κB into the nucleus. (A) HEK293T cells were transfected with Flag-ECSIT wt or the Flag-ECSIT K372A mutant, along with the HA-Ub vector. Immunoprecipitation (IP) with anti-Flag antibodies was performed, followed by immunoblot (IB) with antibodies to anti-Flag or anti-Ub. (B) HEK293T cells were transfected with mock, Flag-ECSIT wt, Flag-ECSIT K372A mutant, or p65- or p50-expressing vectors, as indicated. IP with anti-Flag antibody was performed, followed by IB with antibodies to anti-Flag, anti-p65, or anti-p50. (C) THP-1 cells were transfected with Flag-ECSIT wt or Flag-ECSIT K372A mutant vector and treated with LPS for the times indicated, cytosolic and nuclear fractions were isolated from cells, and a Western blot analysis was performed with antibody to anti-p65, anti-p50, anti-Flag, anti-PDI, or anti-PCNA.
ECSIT K372 ubiquitination is functionally involved in TLR4-mediated signaling to activate NF-κB
On the basis of the foregoing results, we examined whether ECSIT K372 ubiquitination was functionally involved in TLR4 signaling leading to NF-κB activation. The NF-κB reporter assay in HEK293-TLR4 cells revealed that Flag-ECSIT wt markedly enhanced NF-κB reporter activity in a dose-dependent manner, whereas no significant increase was seen in the Flag-ECSIT K372A mutant (Figure 5A). Moreover, consistent results were obtained for p65 or p50 DNA–binding activities (Figure 5, B, p65, and C, p50). To functionally verify these results, we generated control THP-1 (ctrl THP-1) or ECSIT-knockdown THP-1 (ECSITKD THP-1) cells using lentiviral particles containing control or short hairpin RNA (shRNA)–targeted human ECSIT, respectively (Figure 5D). ECSIT knockdown resulted in decreased NF-κB activity in response to LPS stimulation (Figure 5E, lane 2 vs. lane 16). NF-κB activities were markedly enhanced or restored in ctrl or ECSITKD THP-1 cells based on ECSIT expression (Figure 5E, lanes 6–8 in ctrl; lanes 20–22 in ECSITKD THP-1). In contrast, no significant changes were seen in K372A mutant ECSIT expression (Figure 5E, ECSIT K372A). Similar results were observed for p65 and p50 DNA–binding activities (Figure 5, F, p65, and G, p50). Moreover, ctrl THP-1 cells showed markedly higher secretion of interleukin-6 (IL-6) and IL-1β than that by ECSIT-knockdown THP-1 cells in response to LPS stimulation (Figure 5, H, IL-6, and I, IL-1β). IL-6 and IL-1β levels were markedly enhanced in ctrl THP-1 cells when subjected to overexpression of wt ECSIT and significantly restored in ECSIT-knockdown THP-1 cells, whereas no significant changes were seen in either ctrl THP-1 or ECSIT-knockdown THP-1 cells after overexpression of the Flag-tagged ECSIT K372A mutant (Figure 5, H, IL-6, and I, IL-1β). These results strongly suggest that ubiquitination of ECSIT on the 372 lysine residue is functionally important for activating NF-κB during TLR4-mediated signaling.
FIGURE 5:

K372 ubiquitination of ECSIT is functionally involved in activating NF-κB induced by LPS stimulation. (A) NF-κB reporter assay in HEK293-TLR4 cells transfected with mock, different concentrations of wt ECSIT, or K372A mutant ECSIT in the presence or absence of LPS. All error bars represent SDs of the mean from triplicate samples. (B, C) p65 or p50 DNA–binding assay in HEK293-TLR4 cells transfected with the indicated vectors, mock, wt ECSIT, or K372A mutant ECSIT, in the presence or absence of LPS. All error bars represent SDs of the mean from triplicate samples. (D) THP-1 cells were infected with lentiviral particles containing control lentiviral particles or shRNA-targeted human ECSIT lentiviral particles according to the manufacturer's protocol. Control THP-1 (Ctrl) or ECSIT-knockdown THP-1 (ECSITKD THP-1) were cultured in puromycin-containing medium (4 μg/ml) for 2 wk to select stable clones, and immunoblots (IB) with antibodies to anti-ECSIT or anti-GAPDH were performed to evaluate knockdown efficacy. (E) NF-κB reporter assay in control (ctrl) or ECSITKD THP-1 cells transfected with mock, different concentrations of wt ECSIT, or K372A mutant ECSIT in the presence or absence of LPS. All error bars represent SDs of the mean from triplicate samples. (F, G) p65 or p50 DNA–binding assay in ctrl or ECSITKD THP-1 cells transfected with the indicated vectors, mock, wt ECSIT, or K372A mutant ECSIT in the presence or absence of LPS. All error bars represent SDs of the mean from triplicate samples. (H, I) Ctrl or ECSITKD THP-1 cells were transfected with mock, wt ECSIT, or K372A mutant ECSIT, treated with or without LPS, and the levels of IL-6 (H) and IL-1β (I) were measured by ELISA. All error bars represent SDs of the mean from triplicate samples. *p < 0.05 and **p < 0.01.
TLR4 signaling activates p65/p50 NF-κB components primarily dependent on the canonical IKK complex (Oeckinghaus and Ghosh, 2009; Hyden and Ghosh, 2011; Ghosh and Hyden, 2012). Because ECSIT K372 ubiquitination was specifically involved in activating the p65/p50 NF-κB proteins induced by LPS, we asked whether the regulatory mechanism was dependent on IKKs. Thus mock, wt ECSIT, or K372A mutant ECSIT vectors were transiently transfected into wt mouse embryonic fibroblasts (MEFs) or NEMO−/− MEF cells and treated with or without LPS, and p65 or p50 DNA–binding activity was measured. As shown in Figure 6, A and B, p65 and p50 DNA–binding activities were significantly enhanced by LPS and were markedly augmented by overexpressing wt ECSIT, whereas no significant increase was seen by overexpressing the K372A mutant ECSIT. Of interest, neither wt ECSIT nor K372A mutant ECSIT increased p65 or p50 DNA–binding activity after TLR4 stimulation in NEMO−/− MEFs (Figure 6, C, p65, and D, p50). These results strongly demonstrate that the ECSIT regulatory mechanism is critically dependent on the canonical IKK complex.
FIGURE 6:
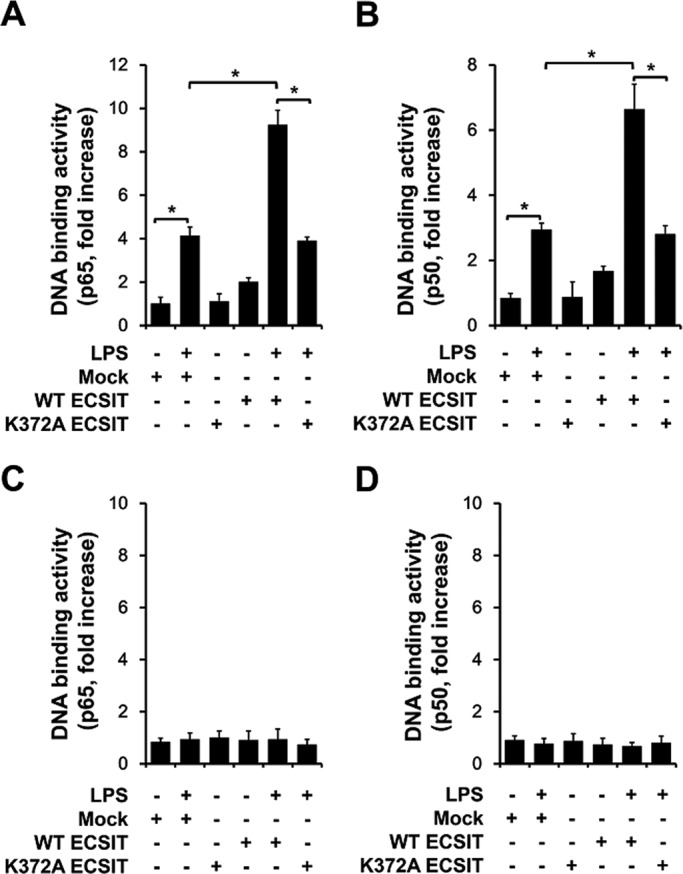
ECSIT is involved in the IKK-dependent canonical pathway. (A, B) Wild-type MEF cells were transfected with mock, wt ECSIT, or K372A ECSIT mutant vector, as indicated. After 38 h, the cells were treated for 60 min in the presence or absence of 100 ng/ml LPS, and then the p65 or p50 DNA–binding assay was performed. All error bars represent SDs of the mean from triplicate samples. *p < 0.05. (C, D) NEMO−/− MEF cells were transfected with mock, wt ECSIT, or K372A ECSIT mutant vector, as indicated. After 38 h, the cells were treated for 60 min in the presence or absence of 100 ng/ml LPS, and then the p65 or p50 DNA–binding assay was performed. All error bars represent SDs of the mean from triplicate samples.
ECSIT K372 ubiquitination is critical for expression of NF-κB–dependent genes induced by LPS
We finally examined whether the ECSIT regulatory mechanism is involved in gene expression induced by LPS. A microarray analysis was performed to evaluate ECSIT-related gene expression profiles using ctrl THP-1 and ECSITKD THP-1 cells. After TLR4 stimulation for different lengths of time, genes related to TLR4-mediated signaling were significantly up-regulated in ctrl THP-1 cells, whereas these profiles were significantly reduced in ECSITKD THP-1 cells (Supplemental Figure S1, A and B). ECSIT deficiency was found to have resulted in a marked down-regulation of gene expression when the results between ctrl and ECSITKD THP-1 cells were compared (Supplemental Figure S1C). In line with previous results (Xiao, 2003; West, 2011), ECSIT deficiency also led to down-regulation of genes related to mitochondrial electron transfer (Supplemental Figure S2) and mesoderm development (Supplemental Figure S3).
To determine whether K372A mutant ECSIT affected the NF-κB–dependent gene expression induced by TLR4 stimulation, we transfected ctrl or ECSITKD THP-1 cells with mock, wt ECSIT, or K372A mutant ECSIT, treated with or without LPS, and examined them by quantitative reverse transcription-PCR (qRT-PCR) to assess the expression of four NF-κB–dependent genes, Il-1β, CD44, IRF7, and NF-κB2, which have a consensus κB-binding site. As shown in Figure 7, A– D, induction of wt ECSIT in ctrl THP-1 cells resulted in marked increases in expression of these genes in the presence of LPS, and their expression was significantly restored in ECSITKD THP-1 cells compared with that in ECSITKD THP-1 cells. In contrast, no significant changes in expression were observed after inducing the K372A mutant ECSIT in ctrl and ECSITKD THP-1 cells (Figure 7, A, Il-1β; B, CD44; C, IRF7; and D, NF-κB2). These results strongly suggest that K372 ubiquitination of ECSIT plays a pivotal role in regulating NF-κB–dependent gene expression induced by TLR4 stimulation.
FIGURE 7:

K372A mutant ECSIT impairs NF-κB–dependent gene expression induced by TLR4 stimulation. (A–D) Control (ctrl) or ECSITKD THP-1 cells were transfected with mock, wt ECSIT, or K372A mutant ECSIT vector, as indicated. After 38 h, cells were treated for 7 h with or without LPS (100 ng/ml), and then qRT-PCR analysis with specific primers to IL-1β (A), CD44 (B), IRF7 (C), and NF-κB2 (D) was performed. All error bars represent SDs of the mean from triplicate samples. *p < 0.05 and **p < 0.01.
DISCUSSION
We showed that ECSIT interacts with p65/p50 NF-κB proteins and colocalizes to the nucleus after LPS stimulation, eventually leading to activation of NF-κB proteins. Biochemical studies revealed that ECSIT ubiquitination specifically occurs at the 372 lysine and that this is critical for both interaction and translocation of p65/p50 NF-κB proteins to the nucleus. The ECSIT K372A mutant failed to activate the NF-κB proteins, thereby causing severely attenuated production of proinflammatory cytokines and the expression of NF-κB–dependent genes induced after TLR4 stimulation. These findings suggest that ECSIT might play a key role regulating NF-κB activity during TLR4 signaling.
ECSIT had been identified as a multifunctional protein, as it participates in mammalian TLR signaling pathways regulating innate immunity (Kopp et al., 1999), regulates bactericidal activity through mitochondrial ROS generation in response to TLR stimulation (West et al., 2011), and is involved in BMP signaling in the nucleus (Xiao et al., 2003), strongly suggesting that intracellular localization of ECSIT is linked with its specific roles. We found that ECSIT formed a high–mass molecular complex after TLR4 stimulation, with which p65/p50 NF-κB proteins were specifically associated. Therefore we hypothesized that ECSIT activates NF-κB induced by TLR4 stimulation, although we could not rule out that other proteins may be associated with the ECSIT complex and involved in TLR4 signaling. We showed that ECSIT specifically interacts with p65/p50 NF-κB proteins through the ECSIT 300–341 region and regulates activation of NF-κB proteins. Furthermore, the ECSIT/p65/p50 complex specifically translocated to the nucleus in response to TLR4 stimulation. Of note, these biochemical and molecular functions are critically dependent on ubiquitination of the ECSIT 372 lysine residue. The ECSIT K372A mutant markedly failed to activate NF-κB, produce proinflammatory cytokines, or express NF-κB–dependent genes. These results indicate that ECSIT acts as a key regulator during TLR4 signaling leading to activation of NF-κB.
In summary, ECSIT interacts and forms a signal complex including TRAF6 and TAK1 in which TAK1 is activated and, in turn, activates IKKs (Figure 8). ECSIT is also simultaneously ubiquitinated by TRAF6 (West et al., 2011). Ubiquitinated ECSIT at the K372 residue further interacts with p65/p50 NF-κB recently dissociated from IκB-α. The ECSIT/p65/p50 complex eventually translocates to the nucleus and is involved in p65/p50 NF-κB–dependent gene expression. Several reports showed that ECSIT plays an essential role in signaling networks gone awry in Alzheimer's disease (AD; Mattson and Meffert, 2006; Okun et al., 2009; Soler-López et al., 2012) and cellular oncogenesis (Xiao, 2003). We believe that our results will contribute to the understanding of ECSIT-related signals, including TLRs, BMP, and transforming growth factor-β signaling, and the pathogenesis of ECSIT-related diseases such as AD.
FIGURE 8:

Model detailing the roles of ECSIT in TLR4-mediated signaling. On TLR4 stimulation, ECSIT forms the TAK1/TRAF6 complex, in which TAK1 is activated and ECSIT is simultaneously ubiquitinated on K372 by TRAF6. Activated TAK1 activates the IKKs complex through phosphorylation, leading to IκB-α degradation through phosphorylation and polyubiquitination. The dissociated p65/p50 NF-κB proteins from IκB-α interact with K372-ubiquitinated ECSIT and translocates to the nucleus, leading to the induction of p65/p50-dependent gene expression.
MATERIALS AND METHODS
Cells
HEK293T, wt MEFs, and NEMO−/− MEFs were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (Irrinki et al., 2011; Kim et al., 2012a, b; Supplemental Materials and Methods). HEK293-TLR4 cells were purchased from InvivoGen (San Diego, CA; 293-htlr4md2cd14) and cultured according to the manufacturer's guidelines. THP-1 cells were maintained in RPMI medium (Invitrogen, Carlsbad, CA) containing 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 5 × 10−5 M β-mercaptoethanol.
Knockdown by lentiviral particles
THP-1 cells were infected with lentiviral particles containing shRNA targeted human ECSIT (sc-77224-V; Santa Cruz Biotechnology, Santa Cruz, CA) or control lentiviral particles (sc-108080; Santa Cruz Biotechnology) according to the manufacturer's protocol. Control THP-1 (Ctrl) and ECSIT-knockdown THP-1 (ECSITKD THP-1) cells were cultured in puromycin-containing medium (4 μg/ml) for 2 wk to select stable clones and then maintained in RPMI medium containing 4 μg/ml puromycin, 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 5 × 10−5 M β-mercaptoethanol (Kim et al., 2012b, 2014).
Plasmids and antibodies
The Flag-ECSIT 1–100, Flag-ECSIT 1–200, Flag-ECSIT 1–257, Flag-ECSIT 1–300, and Flag-ECSIT 257–431 truncated mutants were generated by PCR using Flag-ECSIT wt as a template and inserted into pcDNA3. The Flag-tagged ECSIT K372A mutant was generated using the MORPH plasmid DNA mutagenesis kit (5 Prime → 3 Prime, Boulder, CO). The p65 and p50 expression vectors were purchased from Invitrogen. We used anti-ECSIT (Santa Cruz Biotechnology), anti-p65 (Cell Signaling Technology, Danvers, MA), anti-p50 (Cell Signaling Technology), anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Cell Signaling Technology), anti–proliferating cell nuclear antigen (PCNA; Santa Cruz Biotechnology), anti-GRIM19 (Abcam, Cambridge, MA), anti-Flag (Abcam), anti-Myc (Cell Signaling Technology), anti-hemagglutinin (HA; Cell Signaling Technology), anti-ubiquitin (Abcam), and anti-PDI (Abcam) antibodies.
Immunoprecipitation and immunoblot assays
Total protein was resolved on a 10% polyacrylamide gel and transferred to polyvinylidene fluoride membranes, as described previously (Kim et al., 2012a, b, 2014). Cells were transfected with appropriated vectors, as indicated in each figure, and the immunoprecipitation assay was performed as described previously (Kim et al., 2012a, b, 2014), followed by immunoblot with specific antibodies.
Gel filtration chromatography
THP-1 cells were treated or not with 100 ng/ml LPS (Sigma-Aldrich, St. Louis, MO) for 45 min. The S-100 fraction was prepared and loaded onto a Superose6 10/300 GL column (Noguchi et al., 2005). Samples were analyzed by Western blotting using the indicated antibodies.
His-tagged pull-down assay
The His-tagged p65 pull-down assay was performed as per the manufacturer's protocol (21277; Thermo Scientific, Rockford, IL). Briefly, His-tagged p65 was immobilized as bait, and protein lysates were bound to the control as prey or immobilized on a His-tagged p65 column. The interaction with ECSIT was analyzed by Western blotting with the indicated antibodies.
Cellular fractionation
HEK293-TLR4 cells were treated with or without LPS (100 ng/ml) for 45–60 min. Cytosol, mitochondria, and nucleus fractions were isolated using a Cell Fractionation Kit (ab109719; Abcam) according to the manufacturer's protocol. ECSIT localization was determined by immunoblotting analysis with anti-ECSIT antibody (Abcam). THP-1 cells were transiently transfected with Flag-tagged ECSIT wt or Flag-tagged K372A expression vector. At 36 h posttransfection, the cells were treated or not with LPS (100 ng/ml) for different times. The cytosol and nucleus fractions were isolated using a Cell Fractionation Kit (Abcam). p65, p50, and Flag-ECSIT wt or Flag-ECSIT K372A localization was detected by Western blotting with anti-p65 (Cell Signaling Technology), anti-p50 (Cell Signaling Technology), and anti-Flag (Abcam) antibodies.
Confocal microscopy
HEK293T cells were grown on coverslips and stimulated or not with LPS (100 ng/ml) for 45–60 min. After washing, the cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) for 5 min, blocked with PBS containing 10% FBS for 30 min, stained with anti-ECSIT antibody for 60 min, and stained with secondary antibodies for 60 min. Nuclei were stained with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich), and coverslips were mounted with Prolong Gold antifade reagent (Molecular Probes, Sunnyvale, CA). Cells were imaged on a Zeiss LSM 710 laser-scanning confocal microscope (Carl Zeiss, Jena, Germany).
Ubiquitination assay
Flag-ECSIT wt, the Flag-ECSIT truncated mutant, or HA-ubiquitin was transfected into HEK293T cells. Immunoprecipitated complexes were separated by SDS–PAGE and probed with anti-ubiquitin antibody. Flag-ECSIT wt, Flag-K372A ECSIT mutant, HA-ubiquitin, or mock plasmids were transfected into HEK293T cells. The cells were treated or not with LPS (100 ng/ml) for 45 min after 36 h. Immunoprecipitated complexes were separated by SDS–PAGE and probed with anti-ubiquitin antibody.
Deubiquitination assay
HEK293T cells were transfected with mock or Flag-ECSIT vector. At 36 h posttransfection, the cells were lysed in lysis buffer containing 150 mM NaCl, 20 mM Tris-HCl, pH 7.5, 10 mM EDTA, 1% Triton X-100, 1% deoxycholate, 1.5% aprotinin, and 1 mM phenylmethylsulfonyl fluoride, and Flag-ECSIT proteins were prepared by immunoprecipitation with anti-Flag antibodies. The K63 Ub-reaction for polyubiquitination to Flag-ECSIT proteins was performed on the precipitates, followed by treatment with or without CYLD enzyme treatment (Xia et al., 2009). The p65 and p50 vectors were independently transfected into HEK293T cells, and p65/p50 lysates were prepared. Ubiquitinated Flag-ECSIT or deubiquitinated Flag-ECSIT protein was mixed with p65/p50 lysates. The samples were pulled down with an anti-Flag antibody, and the immunoprecipitated complexes were separated by SDS–PAGE and probed with anti-Flag, anti-p65, or anti-p50 antibody.
Enzyme-linked immunosorbent assay
Cell culture supernatants were collected and assayed for cytokines. Cytokine production was measured with human IL-1β and IL-6 enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN) according to the manufacturer's protocol (Kim et al., 2012a, b; 2014).
p65/p50 DNA-binding assay
HEK293T cells, ctrl THP-1, ECSITKD THP-1, wt MEFs, or NEMO−/− MEFs were transiently transfected with different vectors, as indicated in the figures, using the Lipofectamine LRX (Invitrogen) or the Neon transfection system (Invitrogen). At 36 h posttransfection, the cells were treated or not with LPS (100 ng/ml) for 60 min. Nuclear proteins were prepared with a CelLytic NuCLEAR Extraction kit (Sigma-Aldrich) in accordance with the manufacturer's protocol. p65 and p50 transcription factor activities were determined with a TransAM NF-κB transcription factor assay kit (Active Motif, Carlsbad, CA) according to the manufacturer's instructions.
Luciferase reporter assay
HEK293-TLR4 cells, ctrl THP-1, or ECSITKD THP-1 cells were transiently transfected with different vectors, as indicated in the figures, using the Lipofectamine LRX (Invitrogen) or Neon transfection system (Invitrogen), together with the pBIIx-luc NF-κB-dependent reporter construct and the Renilla luciferase vector (Promega, Madison, WI). At 36 h posttransfection, the cells were treated or not with LPS (100 ng/ml) for 6 h and lysed, and luciferase activity was measured using a dual luciferase assay kit (Promega).
qRT-PCR
Total RNA was isolated and cDNA was synthesized following the manufacturer's protocol (Qiagen, Valencia, CA). All primers were purchased from Qiagen: IRF7 (PPH02014E), CD44 (PPH 00114A), NF-κB2 (PPH 00782E), and IL-1β (PPH 00171B). The qRT-PCR analysis was performed using Roter-GeneQ (Qiagen) according to the manufacturers' protocol.
Statistical analysis
In vitro data are presented as means ± SDs from triplicate samples. Data were analyzed with the Mann–Whitney test. In vivo data are presented as Tukey's box-and-whisker plots and analyzed with the Mann–Whitney test. p < 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank the Hyewa Forum members, namely, Doo Hyun Chung, Jun Chang, You-Me Kim, Eun Sook Hwang, Eui-Cheol Shin, Seung-Hyo Lee, Heung kyu Lee, and Sang-Jun Ha, for helpful discussions. This study was supported by the Mid-career Researcher Program through a National Research Foundation Grant (NRF-2012R1A2A2A01005659) and by a grant from the Korea Healthcare Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (A111636).
Abbreviations used:
- ECSIT
evolutionarily conserved signaling intermediate in Toll pathways
- IKK
IκB kinase
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- TRAF6
tumor necrosis factor receptor–associated factor.
Footnotes
*These authors contributed equally to this work.
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-08-1277) on October 29, 2014.
REFERENCES
- Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85:85–95. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Hayden MS. Celebrating 25 years of NF-κB research. Immunol Rev. 2012;246:5–13. doi: 10.1111/j.1600-065X.2012.01111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. NF-κB in immunobiology. Cell Res. 2011;21:223–244. doi: 10.1038/cr.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Heide H, Bleier L, Steger M, Ackermann J, Dröse S, Schwamb B, Zörnig M, Reichert AS, Koch I, Wittig I, Brandt U. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012;16:538–549. doi: 10.1016/j.cmet.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Irrinki KM, Mallilankaraman K, Thapa RJ, Chandramoorthy HC, Smith FJ, Jog NR, Gandhirajan RK, Kelsen SG, Houser SR, May MJ, et al. Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol Cell Biol. 2011;31:3745–3758. doi: 10.1128/MCB.05303-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Kim SY, Baik KH, Baek KH, Chah KH, Kim KA, Moon G, Jung E, Kim ST, Shim JH, Greenblatt MB, et al. S6K1 negatively regulates TAK1 activity in the toll-like receptor signaling pathway. Mol Cell Biol. 2014;34:510–521. doi: 10.1128/MCB.01225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Jeong S, Jung E, Baik KH, Chang MH, Kim SA, Shim JH, Chun E, Lee KY. AMP-activated protein kinase-α1 as an activating kinase of TGF-β-activated kinase 1 has a key role in inflammatory signals. Cell Death Dis. 2012b;3:e357. doi: 10.1038/cddis.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Shim JH, Chun E, Lee KY. Reciprocal inhibition between the transforming growth factor-β-activated kinase 1 (TAK1) and apoptosis signal-regulating kinase 1 (ASK1) mitogen-activated protein kinase kinase kinases and its suppression by TAK1-binding protein 2 (TAB2), an adapter protein for TAK1. J Biol Chem. 2012a;287:3381–3391. doi: 10.1074/jbc.M111.317875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp E, Medzhitov R, Carothers J, Xiao C, Douglas I, Janeway CA, Ghosh S. ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev. 1999;13:2059–2071. doi: 10.1101/gad.13.16.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-kappa B regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway C A. Innate immunity. N Engl J Med. 2000;343:338–344. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Ecsit-ement on the crossroads of Toll and BMP signal transduction. Genes Dev. 2003;17:2855–2859. doi: 10.1101/gad.1161403. [DOI] [PubMed] [Google Scholar]
- Noguchi T, Takeda K, Matsuzawa A, Saegusa K, Nakano H, Gohda J, Inoue J, Ichijo H. Recruitment of tumor necrosis factor receptor-associated factor family proteins to apoptosis signal-regulating kinase 1 signalosome is essential for oxidative stress-induced cell death. J Biol Chem. 2005;280:37033–37040. doi: 10.1074/jbc.M506771200. [DOI] [PubMed] [Google Scholar]
- Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. 2009;59:278–292. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-López M, Badiola N, Zanzoni A, Aloy P. Towards Alzheimer's root cause: ECSIT as an integrating hub between oxidative stress, inflammation and mitochondrial dysfunction. Hypothetical role of the adapter protein ECSIT in familial and sporadic Alzheimer's disease pathogenesis. Bioessays. 2012;34:532–541. doi: 10.1002/bies.201100193. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Akira S. Toll-like receptors and innate immunity. J Mol Med (Berl) 2006;84:712–725. doi: 10.1007/s00109-006-0084-y. [DOI] [PubMed] [Google Scholar]
- Vogel RO, Janssen RJ, van den Brand MA, Dieteren CE, Verkaart S, Koopman WJ, Willems PH, Pluk W, van den Heuvel LP, Smeitink JA, Nijtmans LG. Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev. 2007;21:615–624. doi: 10.1101/gad.408407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Shim JH, Klüppel M, Zhang SS, Dong C, Flavell RA, Fu XY, Wrana JL, Hogan BL, Ghosh S. Ecsit is required for Bmp signaling and mesoderm formation during mouse embryogenesis. Genes Dev. 2003;17:2933–2949. doi: 10.1101/gad.1145603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.