

Inhibition of kinesin-5, a molecular motor protein best known for its essential role in mitosis, has notable effects on the morphology and microtubule organization of dendrites of terminally postmitotic neurons. Kinesin-5 acts as a brake that can limit the capacity of other motor proteins to influence microtubule organization and distribution.
Abstract
Kinesin-5 is a slow homotetrameric motor protein best known for its essential role in the mitotic spindle, where it limits the rate at which faster motors can move microtubules. In neurons, experimental suppression of kinesin-5 causes the axon to grow faster by increasing the mobility of microtubules in the axonal shaft and the invasion of microtubules into the growth cone. Does kinesin-5 act differently in dendrites, given that they have a population of minus end–distal microtubules not present in axons? Using rodent primary neurons in culture, we found that inhibition of kinesin-5 during various windows of time produces changes in dendritic morphology and microtubule organization. Specifically, dendrites became shorter and thinner and contained a greater proportion of minus end–distal microtubules, suggesting that kinesin-5 acting normally restrains the number of minus end–distal microtubules that are transported into dendrites. Additional data indicate that, in neurons, CDK5 is the kinase responsible for phosphorylating kinesin-5 at Thr-926, which is important for kinesin-5 to associate with microtubules. We also found that kinesin-5 associates preferentially with microtubules rich in tyrosinated tubulin. This is consistent with an observed accumulation of kinesin-5 on dendritic microtubules, as they are known to be less detyrosinated than axonal microtubules.
INTRODUCTION
During neuronal development, microtubules are regionally organized into distinct patterns. In mitotic cells, microtubules are organized by a combination of factors, and the same is true of neurons (Conde and Caceres, 2009). During cell division, various kinesin motors work in concert with cytoplasmic dynein and other factors to configure the microtubules into the biphasic spindle and then to separate the half spindles (Sharp et al., 2000). The mitotic kinesins 5, 6, and 12 all continue to be expressed in developing neurons, where they play key roles in establishing the microtubule arrays of these terminally postmitotic cells (Sharp et al., 1997; Ferhat et al., 1998; Buster et al., 2003; Liu et al., 2010). In vertebrate neurons, axonal microtubules are nearly uniform in orientation, while dendritic microtubules have a mixed orientation (Baas and Lin, 2011). Cytoplasmic dynein appears to be the principal motor for driving plus end–distal microtubules into both types of processes (Ahmad et al., 1998; Zheng et al., 2008), while kinesins 6 and 12 drive the entry of minus end–distal microtubules into dendrites and at the same time restrain the transit of plus end–distal microtubules into the axon (Lin et al., 2012).
Kinesin-5 (also called Eg5 or kif11) is a homotetrameric motor with four motor domains projected outward and hence is ideally suited to slide microtubules relative to one another (Kashina et al., 1996). However, kinesin-5 is also a very slow motor, roughly 100 times slower than cytoplasmic dynein. This means that kinesin-5 imposes a limit on how fast any other motor can slide microtubules (Saunders et al., 2007). This enables kinesin-5 to act as a brake that can effectively halt movement of microtubules by other motors. Our studies on neurons have shown that inhibition of kinesin-5 causes the axon to grow faster, to retract less, and to ignore navigational cues (Myers and Baas, 2007; Nadar et al., 2008, 2012). These effects are due to greater mobility of microtubules, as inhibition of kinesin-5 results in higher numbers of short microtubules moving within the axonal shaft and longer microtubules extending deeper into the growth cone. Given that dendritic microtubules have mixed orientation, the question arises as to whether kinesin-5 also acts as a brake in dendrites or if its forces act in a different way. We have addressed this question in the present study and in so doing have also been able to delve deeper into the mechanisms that regulate the activity and distribution of kinesin-5 in the neuron.
These studies are clinically relevant, because anti–kinesin-5 drugs have been touted for use in cancer therapy on the assumption that there is no kinesin-5 expressed in adult neurons. In fact, while its levels in adult neurons are certainly lower than during development, kinesin-5 continues to be expressed, particularly in adult neurons of the CNS (Lin et al., 2011). We have suggested that the effects of anti–kinesin-5 drugs on axons might be exploited to augment nerve regeneration after injury of the spinal cord (Haque et al., 2004; Lin et al., 2011). More recently, it has been shown that kinesin-5 is inhibited by β-amyloid, which may be a contributing factor to cognitive deficits in Alzheimer's disease (Ari et al., 2014). Given the importance of dendritic architecture for cognitive functions such as learning and memory, the impact of kinesin-5 inhibition on dendrites is an important consideration if patients are to be treated with anti–kinesin-5 drugs.
RESULTS
Kinesin-5 inhibition affects dendritic morphology
We recently established that depleting superior cervical ganglia (SCG) neurons of either of two mitotic microtubule motors, kinesin-6 (also called CHO1/MKLP1 or kif23) or kinesin-12 (also called kif15), results in dendrites becoming longer, thinner, and more axonal in character, with an accompanying shift toward a greater proportion of plus end–distal microtubules (Lin et al., 2012). We concluded that these two motors function to transport minus end–distal microtubules from the neuronal cell body into developing dendrites. We then speculated that perhaps all of the mitotic motors share this same duty, including kinesin-5 (Baas and Gonzalez-Billault, 2012). However, kinesin-6 does not enter the axon (Lin et al., 2012), while kinesin-12 behaves somewhat similarly but not identically to kinesin-5 in the axon (Liu et al., 2010), calling into question whether all three motors could act identically in any context. Moreover, whether the braking effect of kinesin-5 on axonal microtubules is likely to extend to dendrites is not obvious, because dendritic microtubules are mixed in orientation, whereas axonal microtubules are nearly uniformly oriented.
To investigate the effects of kinesin-5 depletion on dendrites, we used small interfering RNA (siRNA) to deplete kinesin-5 from SCG neurons, introducing the siRNA before plating by nucleofection (Amaxa). Neurons were plated on Matrigel-coated dishes to hasten development of dendrites and fixed at days in vitro (DIV) 5. We chose DIV 5 because it was the earliest time when bona fide dendrites were present in the cultures, as there was some concern about the siRNA-based depletion persisting beyond DIV 5. After fixation, the cultures were stained for MAP2 to demarcate dendrites (Figure 1A), and morphological measurements were made of dendrite width, length, and number, and the number of branches per dendrite was determined (Figure 1B). We found that the width of dendrites of neurons depleted of kinesin-5 (1.63 ± 0.07 μm) was significantly thinner than that of dendrites of control siRNA-treated neurons (2.42 ± 0.14 μm, p < 0.001). Dendrites of kinesin-5-depleted neurons were also significantly shorter (29.28 ± 2.01 μm) than dendrites of control siRNA-treated neurons (38.87 ± 2.96 μm, p < 0.05). There was no significant difference between control and kinesin-5–depleted neurons with respect to dendrite branching or dendrite number per cell body.
FIGURE 1:
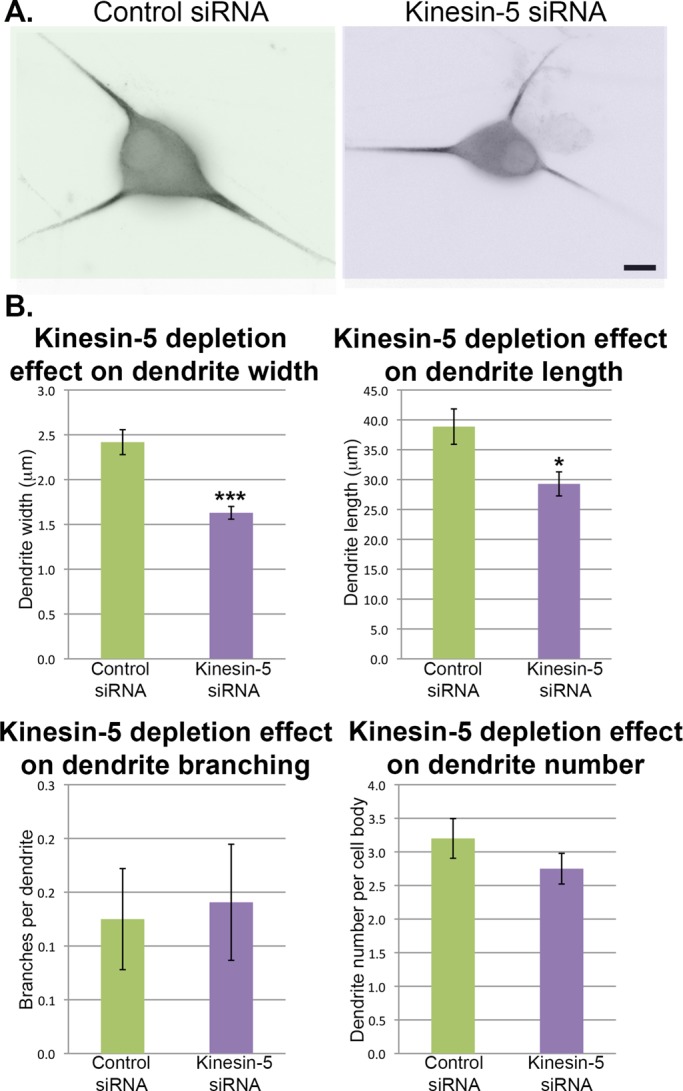
Depletion of kinesin-5 affects dendrite morphology. (A) SCG neurons were transfected with either kinesin-5 siRNA or control siRNA and then plated on Matrigel-coated coverslips. At 5 DIV, neurons were fixed and immunostained for MAP2 to visualize dendrites. Shown are examples of neurons under each experimental condition. Images are displayed using the “invert” function (see Materials and Methods). Scale bar: 10 μm. (B) Dendrite width, length, number, and branching were quantified. All quantifications are displayed as mean ± SEM. Width: dendrites of kinesin-5 siRNA-treated neurons were significantly thinner than control (Mann-Whitney U-test: 2.42 ± 0.14 vs. 1.63 ± 0.07 μm, n = 64, U = 964, p < 0.001). Length: dendrites of kinesin-5 siRNA-treated neurons were significantly shorter than control (Mann-Whitney U-test: 38.87 ± 2.96 vs. 29.28 ± 2.01 μm, n = 64, U = 1551.5, p < 0.05). Branching: there was no significant difference in branching between treatment groups (Mann-Whitney U-test: 0.13 ± 0.05 vs. 0.14 ± 0.05 branches per dendrite, n = 64, U = 2044.5, p = 0.975). Number: dendrite number was not significantly different between groups (Mann-Whitney U-test: 3.20 ± 0.30 vs. 2.75 ± 0.22 dendrites per cell body, n = 20, U = 163.5, p = 0.327). All green bars on the bar graphs correspond to control siRNA-treated neurons, while purple bars correspond to kinesin-5 siRNA-treated neurons.
In addition to the issue with the siRNA-based depletion potentially not persisting past DIV 5, another issue with the siRNA approach is that depletion begins when axons are differentiating, raising some concern that the effects on dendrites might be secondary to the effects on axons. To circumvent both of these issues, we next bath-applied monastrol, an allosteric inhibitor of kinesin-5 (Kapoor et al., 2000), over various windows of time in culture and compared the morphology of monastrol-treated neurons with that of control counterparts exposed to DMSO vehicle alone (Figure 2). Monastrol was the first kinesin-5 inhibitor identified and was named for the mono-astral spindle phenotype it induced in human osteosarcoma U2OS cells (Mayer et al., 1999; Kapoor et al., 2000; Waitzman and Rice, 2014). In previous studies, we found monastrol's effects on neurons to be entirely consistent with the effects of kinesin-5 depletion by siRNA (Myers and Baas, 2007; Nadar et al., 2008; Falnikar et al., 2011). We focused on dendrite formation and maturation stages rather than equal intervals between treatment groups. Cultures were treated for multiple days, starting on DIV 3, DIV 6, or DIV 14. The first treatment window (DIV 3–9) inhibited kinesin-5 before and during dendrite formation but after the axons had formed. This was done in order to allow the axons to form normally while kinesin-5 was still present, so we could focus strictly on dendrite-related morphological changes. The second treatment window (DIV 6–12) inhibited kinesin-5 after dendrites had formed, during their early maturation. The third treatment window (DIV 14–16) inhibited kinesin-5 in fully formed dendrites as they further matured (Figure 2A). On the last day of monastrol treatment of each of the treatment regimes, the neurons were fixed and stained for MAP2 as an unbiased indicator of dendritic identity (Figure 2B). Measurements of dendrite length, width, and number, and the number of branches per dendrite were quantified and compared with control (DMSO-treated) neurons (Figure 2C). These measurements were consistent with ones taken from kinesin-5–depleted neurons (Figure 1).
FIGURE 2:
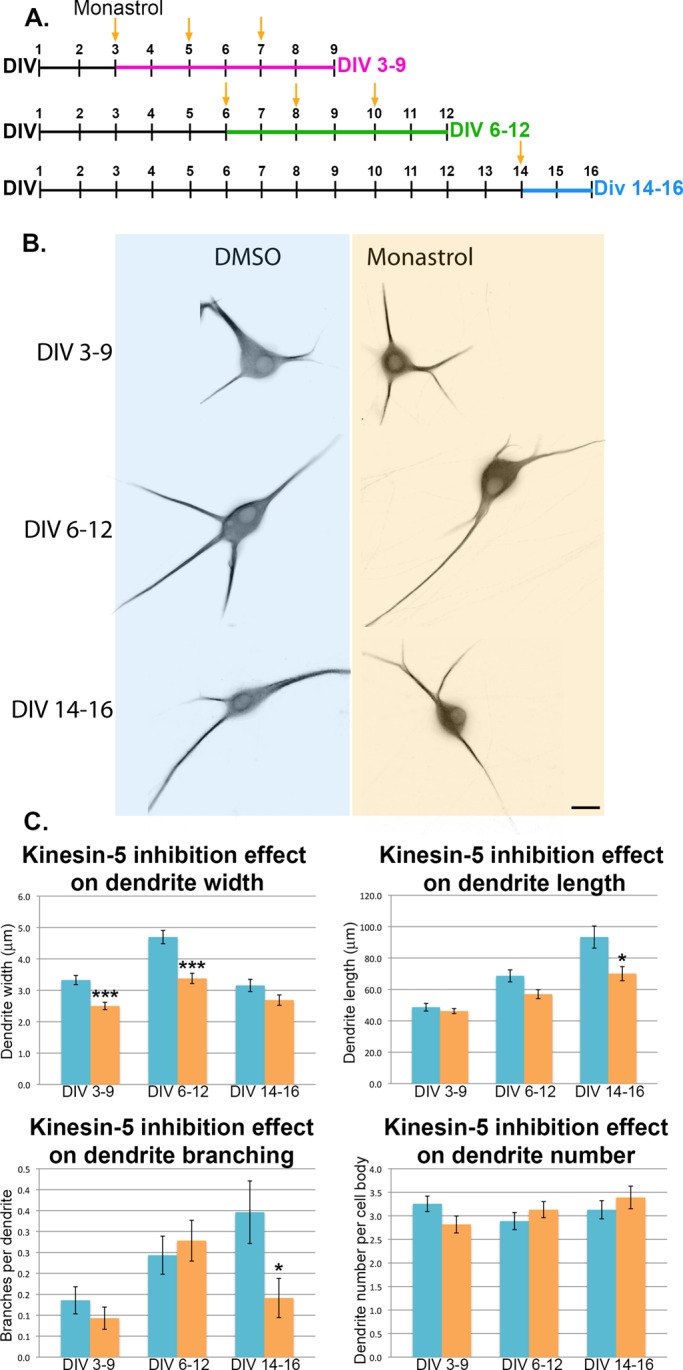
Inhibition of kinesin-5 significantly impacts dendrite morphology. (A) SCG neurons were plated on Matrigel-coated coverslips and treated with either monastrol or DMSO (as control) during dendrite development DIV 3–9, during dendrite maturation DIV 6–12, or after dendrite maturation DIV 14–16. Yellow arrows indicate monastrol (or DMSO control) application/replenishment. (B) On the last day of treatment, neurons were fixed and immunostained for MAP2 to visualize dendrites. Examples of neurons under each experimental condition are shown. Images are displayed using the “invert” function (see Materials and Methods). Scale bar: 10 μm. (C) Dendrite width, length, number, and branching were quantified. All quantifications are displayed as mean ± SEM. Width: dendrites of monastrol-treated neurons were significantly thinner in the DIV 3–9 treatment group (Mann-Whitney U-test: 3.33 ± 0.15 vs. 2.5 ± 0.11 μm, n = 140, U = 6546.5, p < 0.001) and in the DIV 6–12 treatment group (Mann-Whitney U-test: 4.70 ± 0.21 vs. 3.38 ± 0.16 μm, n = 115, U = 4333, p < 0.001), but only trending thinner dendrites were seen in the DIV 14–16 treatment group (Mann-Whitney U-test: 3.15 ± 0.20 vs. 2.69 ± 0.17 μm, n = 78, U = 2578.5, p = 0.1). Length: dendrites of monastrol-treated neurons were only trending shorter dendrites in the DIV 3–9 treatment group (48.71 ± 2.43 vs. 46.21 ± 1.56 μm, n = 140) and in the DIV 6–12 treatment group (Mann-Whitney U-test: 68.66 ± 3.82 vs. 57.02 ± 2.23 μm, n = 115, U = 5655.5, p = 0.058) and were significantly shorter in the DIV 14–16 treatment group (Mann-Whitney U-test: 93.37 ± 7.04 vs. 70.09 ± 4.49 μm, n = 78, U = 2481, p < 0.05). Branching: dendrites branched significantly less only in the monastrol-treated group DIV 14–16 (Mann-Whitney U-test: 0.35 ± 0.07 vs. 0.14 ± 0.05 branches per dendrite, n = 78, U = 2542.5, p < 0.05), with no significant difference in branching in the DIV 3–9 group (0.14 ± 0.03 vs. 0.09 ± 0.03 branches per dendrite, n = 140) or DIV 6–12 group (0.24 ± 0.05 vs. 0.28 ± 0.05 branches per dendrite, n = 115). Number: dendrite number was not significantly different in any of the groups: DIV 3–9 (3.26 ± 0.16 vs. 2.82 ± 0.18 dendrites per cell body, n = 44), DIV 6–12 (2.89 ± 0.18 vs. 3.13 ± 0.17 dendrites per cell body, n = 37), and DIV 14–16 (3.13 ± 0.19 vs. 3.39 ± 0.24 dendrites per cell body, n = 23). All blue bars on the bar graphs correspond to DMSO-treated neurons, while orange bars correspond to monastrol-treated neurons.
Kinesin-5 inhibition resulted in thinner dendrites than controls (DMSO treatment DIV 3–9: 3.33 ± 0.15 vs. monastrol treatment DIV 3–9: 2.5 ± 0.11 μm, p < 0.001; and DMSO treatment DIV 6–12: 4.70 ± 0.21 vs. monastrol treatment DIV 6–12: 3.38 ± 0.16 μm, p < 0.001; with the last treatment group trending thinner dendrites, possibly not reaching significance due to shorter monastrol treatment than other groups). This result was similar to our previously observed results on depletion from these neurons of kinesins 6 or 12 (Lin et al., 2012). However, in contrast to the effects of depleting these other motors, inhibition of kinesin-5 caused dendrites to become shorter than controls (DIV 14–16 DMSO treatment: 93.37 ± 7.04 μm vs. monastrol treatment: 70.09 ± 4.49 μm, p < 0.05; with the same trend at 6–12 DIV, but not reaching statistical significance). There was also an effect of less dendritic branching only at DIV 14–16 and no significant effect on dendrite number in any of the treatment groups. Collectively these observations indicate that kinesin-5 influences certain features of dendritic morphology, and this is true even after the neuron has achieved a mature morphology.
Kinesin-5 inhibition alters dendritic microtubule polarity orientation
The effect of kinesin-5 inhibition on the morphology of dendrites led us to investigate dendritic microtubule polarity orientation using the same monastrol treatment groups as in the morphological analyses, namely DIV 3–9, DIV 6–12, and DIV 14–16, along with DMSO controls for each group. One day before the end of treatment, enhanced green fluorescent protein (EGFP)-EB3 (end-binding protein 3) was expressed in the cultures using the Cellaxess electroporation device (Lin et al., 2012). When expressed as a fluorescent fusion protein, EB3 appears as fluorescent comets at the plus ends of assembling microtubules (Stepanova et al., 2003; Nadar et al., 2008) (Figure 3A). Using this method, we quantified the numbers of microtubules with plus ends assembling away from the cell body (anterograde comets) versus those assembling with minus ends away from the cell body (retrograde comets) (Baas and Lin, 2011; Figure 3B). Unlike the older hooking method for determination of microtubule polarity orientation, this method only assays microtubules that are undergoing rapid bouts of assembly during the bout of imaging. In our previous work on dendrites, we noted that this method biases toward scoring more plus end–distal microtubules within the population as compared with the hooking method, but nonetheless it is reliable in terms of revealing differences between control and experimental neurons (Lin et al., 2012). For the present studies, we scored the number of retrograde comets as a percent of total comets traveling through a line drawn perpendicularly to the proximal dendrite within 10 μm of the cell body (Figure 3C) over 60 consecutive frames made every 1 s. Measurements were made through three different portions of the proximal dendrite within 5 μm of each other.
FIGURE 3:

Inhibition of kinesin-5 significantly impacts microtubule organization in dendrites. (A) SCG neurons were plated on Matrigel-coated coverslips and treated with either monastrol or DMSO (as control) during dendrite development DIV 3–9, during dendrite maturation DIV 6–12, and after dendrite maturation DIV 14–16. The day before the last day of treatment, neurons were transfected with EGFP-EB3 using the Cellaxess device and live imaged 24 h later. Scale bar:10 μm. EB3 comets traveling anterogradely (plus end–distal microtubule) and retrogradely (minus end–distal microtubule) were quantified in the proximal dendrite. (B) Percent of retrograde comets was quantified for the three treatment groups. In all three treatment groups, percent of retrograde EB3 comets was significantly larger in monastrol-treated dendrites: DIV 3–9 (19.93 ± 2.30 vs. 29.82 ± 1.60%, n = 30, Mann-Whitney U = 250, p < 0.01), DIV 6–12 (17.68 ± 1.64 vs. 24.87 ± 2.41%, n = 30, Mann-Whitney U = 265.5, p < 0.01), and DIV 14–16 (24.66 ± 1.88 vs. 33.99 ± 2.21%, n = 30, Student's t test t(58) = −3.21, p < 0.01). Blue bars are DMSO-treated neurons and orange bars are monastrol-treated neurons (as in Figure 2). (C) Comets that crossed a line within 60 frames were included in the quantification. The measurements were made three times in the proximal dendrite over a span of 15 μm (inverted image in bottom panel A).
Inhibition of kinesin-5 resulted in significantly more minus end–distal microtubules as a percent of total microtubules traveling across the line in 60 frames in all three monastrol treatment regimes compared with their control counterparts. These results contrast with our previously reported phenotype upon depletion of kinesin-6 or kinesin-12, which resulted in fewer minus end–distal microtubules in dendrites (Lin et al., 2012), further accentuating the uniqueness of kinesin-5 compared with these other two motor proteins.
Phosphorylated kinesin-5 concentrates in dendrites during their development
We previously established that kinesin-5 is expressed in developing neurons before and after their terminal mitotic division, throughout their migratory journeys, and during the formation of axons and dendrites (Ferhat et al., 1998). Early studies from the mitosis community on kinesin-5 identified Thr-926 (or Thr-927, depending on the species) as a site that is phosphorylated during mitosis when kinesin-5 engages microtubules but not phosphorylated in interphase when kinesin-5 is diffuse and not microtubule associated (Blangy et al., 1995; Sawin and Mitchison, 1995). Later studies confirmed that phosphorylation at this site is indeed critical for kinesin-5's association with microtubules (Cahu et al., 2008). In accordance with these findings, we previously found that phosphorylation at Thr-926 is crucial for kinesin-5 to interact with microtubules in the neuron (Nadar et al., 2008, 2012). These earlier studies were conducted on neurons well before dendritic development, wherein we found kinesin-5-pThr-926 to be enriched in the transition zone of the axonal growth cone. To ascertain the distribution of kinesin-5-pThr-926 during and after dendritic differentiation, we grew SCG neuronal cultures as we did for the monastrol studies and fixed them at DIV 3, 6, 9, and 14 (Figure 4A). Average fluorescence intensity of kinesin-5-pThr-926 was recorded within a boxed region in the proximal brightest part of the dendrite and divided by the average fluorescence intensity within a boxed region of a nearby axon in the same image. We have noticed no significant variation of kinesin-5-pThr-926 distribution in axons during neuronal maturation, which leaves the ratio between dendritic and axonal differences to be explained by variations in the amount of protein in the dendrites. Background was subtracted from all measurements, and the box remained the same size throughout all images. We found that even after the processes were normalized for differences in volume, kinesin-5-pThr-926 was enriched in dendrites relative to axons during dendrite formation and maturation from DIV 3 to DIV 14, with the lowest ratio of kinesin-5Thr-926 being at DIV 3 (1.68 ± 0.18 times in dendrites compared with axons) and increasing up to 2.97 ± 0.36 times more in dendrites compared with axons at DIV 9.
FIGURE 4:

Kinesin-5 phosphorylation during neuronal development. (A) SCG neurons were plated on Matrigel-coated coverslips and then fixed at different time points in development: DIV 3, 6, 9, or 14. Neurons were immunostained for kinesin-5-pThr-926. Images are displayed using the “invert” function (see Materials and Methods). Scale bar: 10 μm. At day 3, “proto-dendrites” were considered dendrites for these analyses. Fluorescence intensity was quantified within a boxed region at the brightest region in the dendrite and compared with the same-sized box over the axonal region within the same image. All quantifications displayed are mean ± SEM, and measurements were normalized for volume differences between dendrites and axons and background fluorescence. There was a statistically significant difference between groups as determined by one-way analysis of variance (F(3,56) = 6.95, p < 0.001). A Tukey post hoc test revealed that the ratio of kinesin-5-pThr-926 in dendrites significantly increased (p < 0.01) from DIV 3 (1.68 ± 0.18) to DIV 6 (2.87 ± 0.22). There were no statistically significant differences between DIV 6 and DIV 9 (p = 0.99). However, the ratio significantly decreased (p < 0.05) from DIV 9 (2.97 ± 0.36) to DIV 14 (1.84 ± 0.22). (B) SCG neurons were plated on PDL-coated glass coverslips and fixed at 24 h. A subgroup of dishes was treated with laminin for the entire 24-h period. Neurons were stained for kinesin-5-pThr-926 and β3-tubulin. Fluorescence intensity was measured within a boxed region around the middle of the axon, background subtracted, and normalized against the tubulin immunofluorescence. A Mann-Whitney U-test revealed that laminin treatment resulted in a significantly lower amount of kinesin-5-pThr-926 (0.38 ± 0.02, n = 60) compared with no treatment (0.51 ± 0.03, n = 60) (U = 1154, p < 0.01). (C) Cortical neurons were plated on PLL-coated plastic dishes for 48–96 h. Left two bands, neurons were collected using NP40 lysis buffer and treated with Lambda PP as per manufacturer's instructions or with control buffer. Middle bands, cortical neurons were treated for 24 h with either DMSO control (0.1%), roscovitine (50 μM), p38 MAPK inhibitor SB203580 (10 μM), or GSK3β inhibitor SB216763 (10 μM). Right three bands, cortical neurons were electroporated with either control, GSK3β, or CDK5 siRNA at the time of plating and collected at 48 h. Protein samples were run on a Western blot and probed for kinesin-5-pThr-926 and GAPDH as internal control. Measurements of band intensity were made using ImageJ, normalized against GAPDH, and ratioed against the control band. Control is designated as 1.0, roscovitine treatment resulted in 0.68 ± 0.06 fraction of control, SB203580 (1.09 ± 0.15), and SB216763 (1.03 ± 0.16) (n = 3 for all samples). One sample t test revealed significant difference between control and roscovitine treatment (t = −4.8, p < 0.05). Control siRNA was also designated as 1.0; CDK5 siRNA resulted in 0.28 ± 0.11 fraction of control (n = 3) and GSK3β siRNA (1.09 ± 0.41, n = 4). One sample t test revealed significant difference between control and CDK5 siRNA (t = −6.1, p < 0.05).
Phosphorylation of kinesin-5 is responsive to signaling molecules
Kinesin-5 would only be useful as a brake if it were regulated by signaling cascades relevant to axonal and dendritic development and key milestones in the life of the neuron, such as cessation of migration (Falnikar et al., 2011). We previously reported that when axonal growth cones encounter signals to turn, kinesin-5 becomes preferentially phosphorylated on the side of the growth cone opposite the direction of the turn (Nadar et al., 2012). To fortify the case for kinesin-5 being regulated by signaling molecules, we compared the levels of kinesin-5-pThr-926 in axons of neurons treated with laminin with the levels in axons of control neurons (i.e., not treated with laminin). The axons of sympathetic neurons are known to grow dramatically faster when exposed to laminin, a major extracellular matrix component of the basal lamina (He et al., 2005). Axons of neurons depleted of kinesin-5 or treated with monastrol grow faster than they otherwise would (Haque et al., 2004; Myers and Baas, 2007), and hence it is appealing to speculate that laminin might function through a signaling pathway to dephosphorylate kinesin-5 at Thr-926. To investigate this, we plated neurons on poly-d-lysine (PDL), with a subgroup treated with 10 μM laminin for 24 h. Indeed, we found that the laminin-treated neurons had less kinesin-5-pThr-926 per unit area of axon normalized to tubulin levels than the control neurons, as assessed by quantitative immunofluorescence (Figure 4B; PDL kinesin-5-pThr-926 levels: 0.51 ± 0.03 vs. laminin kinesin-5-pThr-926 levels: 0.38 ± 0.02, p < 001).
Cyclin-dependent kinase 5 (CDK5) is the kinase that phosphorylates kinesin-5 in neurons
Kinesin-5's function in mitosis is regulated by phosphorylation at Thr927 in human cells and Thr-926 in rat cells through the action of cyclin-dependent kinase 1 (CDK1). Such phosphorylation controls association of the motor with the spindle and hence its localization (Blangy et al., 1995; Cahu et al., 2008; Ferenz et al., 2010; Nadar et al., 2012). In previously published studies from our laboratory, we reported that, in axonal development, phosphorylation at this site is crucial for kinesin-5 to concentrate on growth cone microtubules and for it to elicit its function in regulating axonal growth rate and navigation (Nadar et al., 2012). In these earlier studies, a phospho-mutant that cannot be phosphorylated at this site acted as a dominant negative, duplicating the axonal phenotype of either monastrol or siRNA. Before the present study, we had not yet ascertained the kinase that phosphorylates kinesin-5 at Thr-926 in neurons, as terminally postmitotic cells do not express CDK1 in the same manner as dividing cells. We have speculated that CDK5 is the relevant kinase, and we have endeavored to investigate this matter here.
We first validated that our antibody developed to be phospho-specific actually does recognize only the phosphorylated form of kinesin-5. For this, cortical neurons were collected into NP40 lysis buffer and treated with Lambda protein phosphatase (Lambda PP; New England BioLabs) per the manufacturer's instructions. Lambda PP can be used to release phosphate groups from phosphorylated serine, threonine, and tyrosine residues in proteins and was used to show that the antibody recognizes the control buffer–treated lysate, but not the phosphatase-treated lysate (left two bands in Figure 4C).
Cortical neurons were then treated with either DMSO control, roscovitine (50 μM), p38 mitogen-activated protein kinase (p38 MAPK) inhibitor SB203580 (10 μM), or glycogen synthase kinase 3β (GSK3β) inhibitor SB216763 (10 μM) to evaluate which of these potential kinases phosphorylates kinesin-5 at Thr-926. Roscovitine is a selective inhibitor of CDKs, including 1, 2, and 5. We found that a 24-h treatment of cortical neurons with roscovitine resulted in a significant decrease of phosphorylated kinesin-5 compared with control, which was not observed with a p38 MAPK inhibitor or with a GSK3β inhibitor (middle four bands in Figure 4C). We then introduced siRNA for CDK5 or GSK3β (or control siRNA) into cortical neurons at the time of plating, which effectively depleted the relevant proteins over the next 48 h, as we have previously seen. CDK5 depletion significantly reduced the amount of kinesin-5-pThr-926 in the neurons compared with control, while GSK3β siRNA did not (right three bands in Figure 4C). Collectively these results indicate that CDK5 is the kinase that phosphorylates kinesin-5 at Thr-926 in neurons.
Kinesin-5 preferentially interacts with microtubules not enriched in detyrosinated tubulin
What is the mechanism by which phosphorylated kinesin-5 concentrates in dendrites? There is precedent for proteins to be compartmentalized in neurons by greater phosphorylation of the protein in dendrites relative to axons. For example, tau is phosphorylated at sites that cause it to detach from microtubules, and a principal reason why tau does not accumulate in dendrites is that it tends to be phosphorylated in dendrites at these sites (Binder et al., 1985; Kosik and Finch, 1987; Papasozomenos and Binder, 1987). While it seems reasonable that kinesin-5 accumulates in dendrites at least in part because of greater phosphorylation, we wondered whether there might be additional factors. Recent studies have established that many microtubule-related proteins interact differently with microtubules depending on their levels of posttranslationally modified tubulin subunits. Posttranslational modifications of tubulin impose a kind of code onto microtubules, and proteins that associate with the microtubule lattice can then read this code. Not all but many microtubule-related proteins have a preferred affinity for microtubules that are richer or poorer in tubulin subunits that bear a particular posttranslational modification (Palazzo et al., 2003; Hammond et al., 2010; Badding and Dean, 2013; Sirajuddin et al., 2014). One of the first discovered and best studied of these modifications is the enzymatic removal of the C-terminal tyrosine residue from the α-tubulin subunit (Gundersen et al., 1987). This modification, called detyrosination, occurs only after tubulin has incorporated into a microtubule. When tubulin subunits are released into the soluble pool due to microtubule depolymerization, the α-tubulin component of the subunit is rapidly retyrosinated by an enzyme called tubulin tyrosine ligase (TTL). Another modification, called acetylation, has a cycle similarly linked to polymerization and depolymerization of the microtubule; this cycle involves the enzymatic addition of an acetyl moiety to Lys-40 of α-tubulin after incorporation of the tubulin subunit into the microtubule, with enzymatic deacetylation occurring rapidly after the tubulin is released by microtubule depolymerization (Maruta et al., 1986). Detyrosinated and acetylated subunits gradually accumulate in microtubules over time and hence are markers for stable microtubules or stable regions of otherwise labile microtubules (Baas and Black, 1990; Baas et al., 1991; Brown et al., 1993). It is well established that dendritic microtubules are less rich in detyrosinated and acetylated tubulin than axonal microtubules (Baas et al., 1991; Kollins et al., 2009), so if kinesin-5-pThr-926 is distributed in neurons according to the tubulin code, a preference for microtubules rich in tyrosinated or unacetylated subunits would be predicted.
To ascertain whether kinesin-5 has a preference for microtubules of a particular composition, we began with CHO cells, which have flattened regions well suited for resolving individual microtubules by fluorescence microscopy. In most interphase cells, ectopically expressed kinesin-5 remains diffuse rather than binding microtubules (because it tends only be phosphorylated at Thr-926 during mitosis), but we found that CHO cells are unusual and advantageous in this regard, because most of the ectopically expressed kinesin-5 becomes microtubule-associated, presumably because it becomes phosphorylated. Two days (48 h) after transfecting EGFP-kinesin-5 into the cells, we fixed and immunostained for either tyrosinated, detyrosinated, or acetylated tubulin. (No antibody exists that is specific to unacetylated tubulin.) ImageJ was used to quantify colocalization of the EGFP-kinesin-5 with staining for these antibodies with Pearson correlation coefficient (r), in which perfect positive correlation has a value of 1.0. The quantification shows that EGFP-kinesin-5 strongly colocalizes with microtubules rich in tyrosinated tubulin (r = 0.75 ± 0.02) compared with acetylated (r = 0.59 ± 0.03) and detyrosinated tubulin (r = 0.46 ± 0.04) (Figure 5). While some richly acetylated microtubules were bound by EGFP-kinesin-5, this was the case for virtually none of the richly detyrosinated microtubules, thus fortifying the conclusion that it is lack of detyrosination not the lack of acetylation to which the kinesin-5 is attracted. Such a preference has previously been reported for kinesin-13 (microtubule-depolymerizing kinesin) and KIF3C (a kinesin-2 family member) (Peris et al., 2009; Gumy et al., 2013). Kinesin-1 (conventional kinesin, also known as kif5), on the other hand, associates preferentially with microtubules rich in detyrosinated tubulin (Kreitzer et al., 1999; Dunn et al., 2008; Konishi and Setou, 2009).
FIGURE 5:
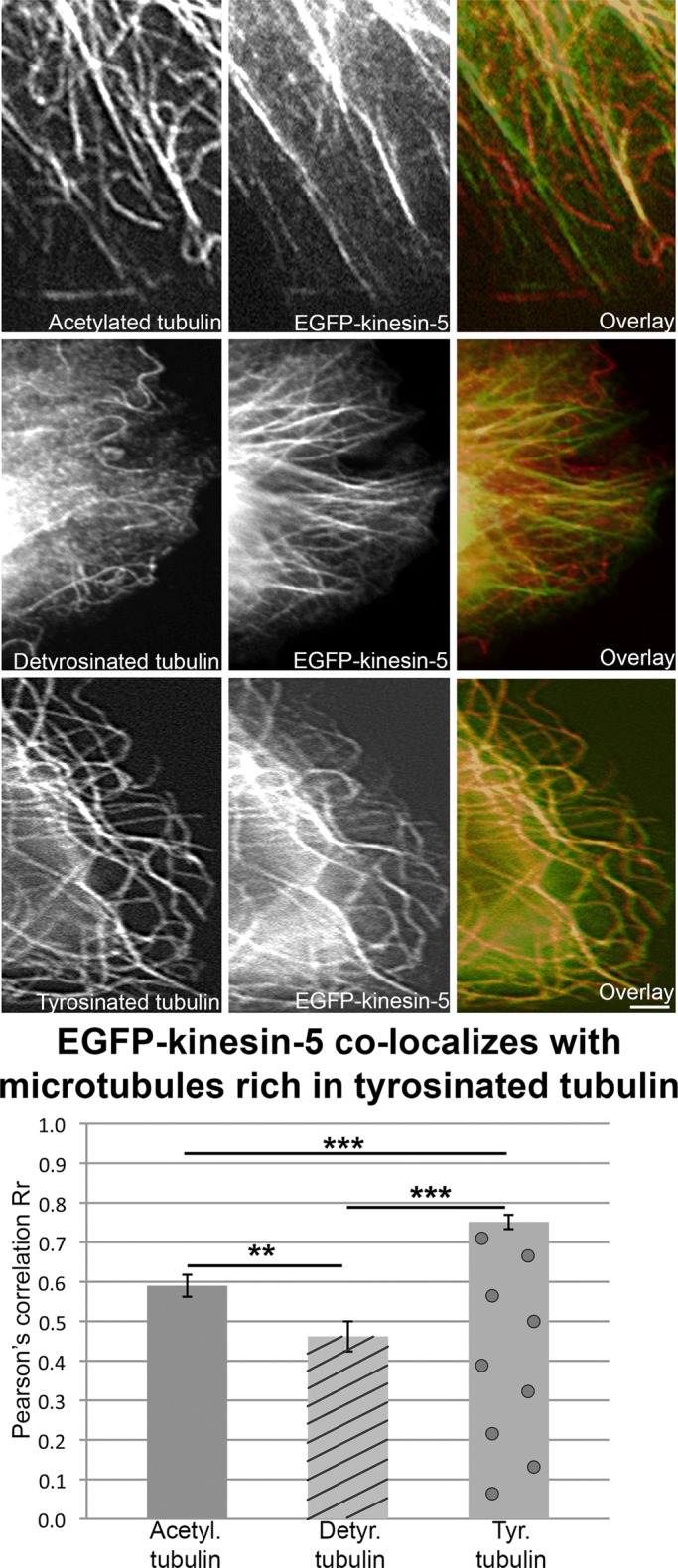
EGFP-kinesin-5 ectopically expressed in CHO cells preferentially interacts with microtubules rich in tyrosinated tubulin. CHO cells were transfected with EGFP-kinesin-5 and plated on glass coverslips. After 48 h, the cells were fixed and immunostained for either acetylated, detyrosinated, or tyrosinated tubulin. Scale bar: 10 μm. For ascertaining the degree of colocalization, Pearson r correlations were quantified between EGFP-kinesin-5 and the different types of tubulin, using WCIF-ImageJ. There was a statistically significant difference between groups as determined by a Kruskal-Wallis test (H(2) = 45.81, p < 0.001). A Mann-Whitney U-test determined that EGFP-kinesin-5 colocalized with microtubules rich in tyrosinated tubulin (n = 65, 0.75 ± 0.02) significantly more than microtubules rich in acetylated tubulin (n = 65, 0.59 ± 0.03) (U = 1065, p < 0.001) or with microtubules rich in detyrosinated tubulin (n = 65, 0.46 ± 0.04) (U = 780, p < 0.001). EGFP-kinesin-5 also colocalized significantly more with microtubules rich in acetylated tubulin than with detyrosinated tubulin (U = 1552.5, p < 0.01).
To confirm this finding in studies on endogenous kinesin-5 in neurons, we used siRNA to deplete TTL from the SCG cultures, introducing the siRNA by nucleofection (Amaxa) before plating. By depleting the enzyme that restores the tyrosine on soluble tubulin, we were able to markedly diminish the levels of tyrosinated tubulin throughout all compartments of the neuron (Figure 6A, top panels). Neurons were plated on Matrigel-coated dishes to hasten development of dendrites and fixed at DIV 5. Images of neurons were acquired with a Zeiss Pascal system (see Materials and Methods), and the ratio function of the software was used to obtain ratio images of tyrosinated tubulin to β3-tubulin from double-stained cultures (Figure 6A). As previously reported, TTL depletion caused morphological changes in the neuron (Erck et al., 2005), which were intriguingly similar to those that we have documented here with kinesin-5 inhibition. Furthermore, we quantified fluorescence intensity of the kinesin-5 signal in the brightest portion of the dendrite and compared it with the signal in axons as in the developmental studies shown in Figure 4. In the case of total kinesin-5, the dendrite-to-axon ratio remained roughly 1:1 in cultures treated with either control siRNA or TTL siRNA. However, in the case of kinesin-5-pThr-926, the dendrite-to-axon ratio decreased from 2.43 ± 0.16 in control siRNA to 1.11 ± 0.08 in TTL siRNA (Figure 6B). Thus, without an axon–dendrite difference in microtubule tyrosinated tubulin levels, the phosphorylated form of kinesin-5 could not properly compartmentalize (Figure 6C).
FIGURE 6:

Endogenous kinesin-5 in neurons preferentially interacts with microtubules rich in tyrosinated tubulin. (A) SCG neurons were transfected via nucleofection at the time of plating with control or TTL siRNA. Cultures were fixed and immunostained for either tyrosinated tubulin and β3-tubulin or only total kinesin-5 or only kinesin-5-pThr-926. Top panels are ratio images showing decrease in tyrosinated tubulin (ratioed against β3-tubulin) in neurons depleted of TTL with siRNA. Middle and bottom panels are fire-scale images (generated using ImageJ) depicting levels of total kinesin-5 or kinesin-5-pThr-926 in dendrites of neurons treated with TTL siRNA. Scale bar:10 μm. (B) Fluorescence intensity was quantified within a boxed region at the brightest region in the dendrite and compared with the same-sized box over an axonal region within the same image. All quantifications displayed are mean ± SEM, and measurements were normalized for volume differences between dendrites and axons and background fluorescence. Phosphorylated kinesin-5 ratio in dendrite compared with axon significantly decreased from neurons treated with control siRNA (n = 43, 2.43 ± 0.16) to TTL-depleted neurons (n = 43, 1.11 ± 0.08) (Mann-Whitney U = 193, p < 0.001). Dendrite-to-axon ratios of total kinesin-5 are not significantly different between control siRNA (n = 43, 0.91 ± 0.04) and TTL-depleted neurons (n = 43, 0.91 ± 0.05) (U = 867, p = 0.62). (C) Schematic showing redistribution of phosphorylated kinesin-5 from its normal concentration in dendrites relative to axons to spreading more evenly throughout the neuron upon diminution of tyrosinated tubulin in all compartments of the neuron driven by TTL depletion.
DISCUSSION
It was a decade ago when we first reported that monastrol treatment causes axons to grow faster (Haque et al., 2004). Since then, we have found that depleting kinesin-5 with siRNA similarly causes axons to grow faster (Myers and Baas, 2007) and that inhibiting or depleting kinesin-5 causes growth cones to fail to turn in response to guidance cues (Nadar et al., 2008, 2012). We then reported that inhibition or depletion of kinesin-5 causes migratory neurons to move faster (Falnikar et al., 2011). In this paper, we report that dendritic morphology is also notably affected by kinesin-5 inhibition, with dendrites mainly getting thinner in response to kinesin-5 inhibition but also getting shorter at one phase of development. In vertebrate dendrites, the microtubules have a nonuniform (antiparallel) orientation, especially in their more proximal regions. We have puzzled over the years about the mechanism of kinesin-5's effects on neurons. The simplest view is that, because it is a homotetramer, kinesin-5 would drive antiparallel microtubules apart and presumably transit to the plus ends of parallel microtubules without affecting their movement. However, growing evidence indicates that kinesin-5's mode of action is more complex and can affect parallel microtubules in the spindle as well as antiparallel microtubules (Kapitein et al., 2005; van den Wildenberg et al., 2008).
As with our previous observations, our present observations are consistent with kinesin-5 acting as a brake on microtubules. Releasing the brake results in greater mobility of microtubules and less restraint of the invasion of microtubules from one region of the neuron to another. For example, the growth cone fails to turn when kinesin-5 is inhibited, because the microtubules no longer show a preference for invading one side of the cone versus the other but rather invade all regions with the same vigor (Nadar et al., 2008). In the leading process of the migratory neuron, as in the axon of a postmigratory neuron, inhibiting kinesin-5 results in a notable increase in the number of short microtubules in transit (Myers and Baas, 2007; Falnikar et al., 2011). Available evidence suggests that, in these situations, cytoplasmic dynein is the principal motor whose forces are attenuated by the kinesin-5 brake. In the case of dendrites, however, to explain our results, kinesin-5 would have to impose a brake relevant to the motor proteins that transport minus end–distal microtubules into dendrites, as a greater proportion of microtubules of this orientation are transported into dendrites when kinesin-5 is inhibited. We posit that kinesin-5 acts as a brake on the forces generated by virtually any other microtubule-based motor that pushes or pulls on microtubules to orient them, transport them, or control their distribution. This is irrespective of the polarity orientation of the microtubules being affected.
Phosphorylation would appear to be the simplest means by which kinesin-5 is regulated and distributed, as the Thr-926–phosphorylated form is microtubule-associated, whereas the unphosphorylated form is freely diffusible and does not associate with microtubules. In typical dividing cells, kinesin-5 is not phosphorylated at this site during interphase and then becomes phosphorylated at this site by CDK1, as the cell transitions into prophase (Blangy et al., 1995; Sawin and Mitchison, 1995). Our present results confirm our previous speculation (Haque et al., 2004) that the relevant kinase for phosphorylating kinesin-5 at Thr-926 in neurons is CDK5. In developing axons, the greatest concentration of kinesin-5 lies in the transition zone of the growth cone, and this is because kinesin-5 is most prominently phosphorylated and hence most predominantly microtubule bound in this region (Ferhat et al., 1998; Nadar et al., 2008, 2012). Asymmetries in the distribution of kinesin-5 in the transition zone cause the growth cone to turn in a direction opposite to the side where kinesin-5 is enriched, presumably as a result of signaling cascades that either phosphorylate or dephosphorylate kinesin-5 at Thr-926. In addition, as we have shown here, treatment with laminin lowers Thr-926 phosphorylation of kinesin-5 in the axon, which is consistent with laminin's effects on axonal growth rate being at least partly related to attenuation of the kinesin-5 brake on microtubules. Our results on dendrites show that kinesin-5 is more heavily phosphorylated at Thr-926 in dendrites as opposed to axons. Just as the relevant kinase and phosphatases in neurons are strong candidates for modulating axonal navigation via kinesin-5 phosphorylation, it seems reasonable to posit that such regulators of kinesin-5 phosphorylation could be powerful contributors to how dendritic shape responds to environmental factors.
Differences in phosphorylation between axons and dendrites are not sufficient to explain kinesin-5's enrichment in dendrites, as the enrichment of kinesin-5-pThr-926 in dendrites is lost when differences in the tyrosination status of axonal and dendritic microtubules are experimentally reduced. Rather, it appears that kinesin-5-pThr-926 distributes according to the “tubulin code,” wherein many but not all microtubule-related proteins have a preferential affinity for microtubules richer or poorer in certain posttranslational modifications. Our results indicate that kinesin-5-pThr-926 associates preferentially with microtubules rich in tyrosinated tubulin, which is consistent with the enrichment of kinesin-5-pThr-926 in two regions of the neuron known to contain microtubules that are especially rich in tyrosinated tubulin—the dendrite and the axonal growth cone (Baas et al., 1991; Ahmad et al., 1993).
Our studies indicate that neurons have at least two levels of control over kinesin-5, as to where and when it will impose its braking effects on microtubule movements—CDK5-regulated phosphorylation and preference for tyrosinated microtubules. Such a brake would be especially important during development, because the brake is apparently responsive to signaling cascades that modulate such things as axonal growth rates, navigation of the growth cone, speed and cessation of neuronal migration, and the shape of dendrites. However, especially in the case of dendrites, the functions of kinesin-5 may also be important beyond development. Dendritic arbors are known to remain dynamic throughout childhood and even into adulthood, with such dynamics being crucial for learning and memory (Wu et al., 1999; McAllister, 2000; Tronel et al., 2010, 2012). Thus a potential role for kinesin-5 in modulating dendritic architecture throughout the life of the neuron is a provocative idea. We envision a scenario wherein small bursts of kinesin-5 expression or phosphorylation may be crucial for the fine-tuning of dendritic shape underlying cognitive challenges. This idea is reinforced by recent studies showing that β-amyloid inhibits kinesin-5 and that this may be a contributing factor in neurodegeneration during Alzheimer's disease. Kinesin-5 activity is absent from amyloid precursor protein/presenilin-1 (APP/PS) transgenic mice brain and in neurons directly treated with β-amyloid. Furthermore, anti–kinesin-5 drugs have been shown to inhibit long-term potentiation in a manner entirely similar to β-amyloid in a cellular model of N-methyl-d-aspartate (NMDA)-dependent learning and memory (Ari et al., 2014). Our results highlight the need for clinicians to be watchful for potential structural and cognitive deficits that may arise in the brains of patients treated with contemporary anti–kinesin-5 drugs.
MATERIALS AND METHODS
Cell culture
Cultures of neonatal rat sympathetic neurons from the SCG were used in the present studies for consistency with a large body of previous work from our laboratory on the roles of mitotic motor proteins in organizing axonal and dendritic microtubules (Myers and Baas, 2007; Nadar et al., 2008; Liu et al., 2010; Lin et al., 2011). While not actually brain neurons, these cultures have been used extensively and are recognized as a valid experimental model for the effects of drugs, toxins, and growth factors on brain neurons (Lein and Higgins, 1989; Lein et al., 1995, 1996, 2002; Guo et al., 1999). The cultures were prepared as previously described from postnatal day 1–2 pups of either sex (He et al., 2005) from timed-pregnant Sprague Dawley rats purchased from Taconic. The neurons were cultured in L-15 medium (Life Technologies, Grand Island, NY) supplemented with D(+)glucose (Sigma-Aldrich, St. Louis, MO), GlutaMax-I (Life Technologies), penicillin/streptomycin (Sigma-Aldrich), fetal bovine serum (FBS; HyClone, Waltham, MA), and nerve growth factor (NGF; Millipore, Darmstadt, Germany). The following day, 5 μM arabinose C (Calbiochem, Darmstadt, Germany) was added to the medium to suppress proliferation of dividing (nonneuronal) cells. At DIV 3, to enhance dendritic differentiation, the medium was replaced with F12/DMEM (Life Technologies) supplemented with bovine serum albumin (BSA; Calbiochem), GlutaMax-I, N2 (Life Technologies), FBS, and NGF. Half of the F12/DMEM-supplemented medium was replenished every 2 d from then on. Neurons were plated on either plastic 35-mm dishes (for protein-level analysis) or dishes with a 1-cm hole covered by a glass coverslip (for morphological analysis; MatTek, Ashland, MA). All dishes were coated with 1 mg/ml PDL (Sigma-Aldrich) at +4°C overnight, washed extensively with water, and then kept in water overnight at +37°C. For morphological studies, neurons were plated directly after dissection and dissociation onto glass-bottomed dishes that had been further coated with Matrigel (for 3 h; BD Biosciences, San Jose, CA) to hasten dendritic development. Under these conditions, first indications of dendritic differentiation occurred at DIV 3 with the appearance in the cultures of slightly thickened processes that we considered to be proto-dendrites, with bona fide dendrites developing by DIV 5–6.
Protein quantification studies by Western blotting were done on cortical primary neurons obtained from E18 rat pups. Primary motor cortex was dissected out into dissection medium (HBSS [Life Technologies], HEPES buffer [Life Technologies], and penicillin-streptomycin [Sigma-Aldrich]) and treated with trypsin (Life Technologies) and briefly with DNAse (Sigma-Aldrich). Cells were then washed in plating medium (Neurobasal [Life Technologies], B27 [Life Technologies], glucose [Sigma-Aldrich], GlutaMax [Life Technologies], NaCl, and FBS) and dissociated using a Pasteur pipette. Each condition consisted of 1 × 106 neurons plated on a poly-l-lysine (PLL)-coated plastic dish (Sigma-Aldrich).
CHO cells were cultured in F-12 medium (Life Technologies) supplemented with FBS, GlutaMax-I, and penicillin-streptomycin and subcultured at 85% confluency. For experiments, cells that had been growing on plastic culture dishes were subcultured onto noncoated glass-bottomed MatTek dishes.
Drug treatments
Monastrol (Calbiochem) dissolved in DMSO (Sigma-Aldrich) was used in neuronal cultures at 100 μM final concentration and fully replenished with fresh medium every 2 d. Previous studies on neurons established that this concentration is effective at inhibiting kinesin-5, while producing no obvious nonspecific or toxic effects (Yoon et al., 2005; Myers and Baas, 2007; Nadar et al., 2008). DMSO alone was used in control experiments at 0.1% final concentration. For morphological studies, monastrol or DMSO control was added to the dishes on DIV 3 and replenished until day 9 (DIV 3–9), or was added to the dishes either on DIV 6 and replenished until day 12 (DIV 6–12), or was added to the dishes either on DIV 14 and replenished until DIV 16 (DIV 14–16). The neurons were either fixed and processed for immunohistochemistry at the end of last day of treatment or transfected with EGFP-EB3 24 h before they were live imaged at the end of the last day of treatment. Laminin (Invitrogen, Grand Island, NY) was used in axon study for 24 h before fixation. Cortical neurons were treated for 24 h between DIV 2 and DIV 4 with 0.1% DMSO as control, roscovitine (Calbiochem), SB203580 (LC Laboratories, Woburn, MA), or SB216763 (Tocris Bioscience, Minneapolis, MN).
Transfection
To deplete proteins at the time of plating and onward, we used siRNA smartpools against kinesin-5 (Sigma-Aldrich), TTL (Sigma-Aldrich), CDK5 (Sigma-Aldrich), GSK3β (Sigma-Aldrich), or control (Ambion, Grand Island, NY) and introduced them into cells with a Nucleofector device (Amaxa, Basel, Switzerland) just before plating, as described previously (He et al., 2005). The same method was used to introduce EGFP-kinesin-5 into CHO cells. For studies on developed adhered cells, EGFP-EB3 in electroporation solution (Mirus, Madison, WI) was introduced into neurons 24 h before imaging using the Cellaxess CX1 device (Cellectricon, Mölndal, Sweden), which produces a series of high-voltage pulses that enable plasmids and siRNA to enter adherent cells (Lin et al., 2012).
Live imaging
Neurons expressing EGFP-EB3 were imaged every 1 s using the multidimensional acquisition module of the AxioVision software for 180 cycles at 100× objective magnification on an Observer Z1 inverted microscope (Carl Zeiss, Jena, Germany) equipped with a heated chamber and CO2 control. Sixty consecutive in-focus frames were analyzed for each dendrite. Retrograde and anterograde comets (corresponding to minus end–distal and plus end–distal microtubules, respectively) were quantified in the proximal region of each dendrite within 10 μm of the cell body. Comets crossing a line drawn perpendicular to the long axis of the dendrite were counted over the 60 frames. This was repeated three times for each dendrite over a 15-μm portion of the proximal dendrite.
Antibodies
Rabbit polyclonal antibody against MAP2 was provided by Itzhak Fischer (Drexel University College of Medicine, Philadelphia, PA; Nunez and Fischer, 1997). Other antibodies were purchased as noted: detyrosinated tubulin rabbit polyclonal antibody (Millipore), acetylated tubulin mouse monoclonal antibody (Sigma-Aldrich), tyrosinated tubulin YL1/2 rat monoclonal antibody (Accurate Scientific, Westbury, NY), β3-tubulin rabbit polyclonal and mouse monoclonal antibody (Covance, Princeton, NJ), β-tubulin fluorescein isothiocyanate–conjugated monoclonal mouse antibody (Sigma-Aldrich), kinesin-5 phosphorylated at Thr-926 rabbit polyclonal antibody (Phospho Solutions, Aurora, CO), total kinesin-5 rabbit polyclonal antibody (Abcam, Cambridge, UK), and mouse monoclonal GAPDH antibody (Abcam). Appropriate fluorescent secondary antibodies were purchased from Jackson Immunoresearch (West Grove, PA).
Plasmids
EGFP-kinesin-5 plasmid was previously generated and used as in earlier studies, with empty EGFP plasmid used as control (Nadar et al., 2008, 2012). EGFP-EB3 was also previously obtained (Lin et al., 2011).
Western blotting
Total protein from cultures of rat SCG neurons was extracted using Cell Lytic (Sigma-Aldrich) and sample buffer from neurons plated on plastic 35-mm dishes coated with 1 mg/ml PDL. The Bio-Rad Mini-PROTEAN Tetra Cell system (Hercules, CA) was used for electrophoretic separation of the protein samples according to the manufacturer's instructions. Protein samples were resolved using 7.5% SDS–PAGE. The gel was transferred onto nitrocellulose membranes (Bio-Rad). The blots were blocked with 5% milk in Tris-buffered saline with Tween 20 (TBST; Bio-Rad), and then probed with primary antibodies at +4°C overnight. After being washed, the blots were incubated with peroxidase-conjugated secondary immunoglobulin G (Jackson Immunoresearch). Membrane-bound peroxidase was visualized on Pierce CL-XPosure Film after treatment with ECL Western Blotting Substrate (Thermo Scientific, Waltham, MA). Levels of protein of interest were normalized with GAPDH for each group and expressed as densitometric ratios against the control lane using ImageJ (Bethesda, MD).
Quantitative fluorescence microscopy
Neurons were fixed with 4% paraformaldehyde and 0.2% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA) simultaneously with extraction (0.2% Triton X-100; Fisher, Waltham, MA) in PHEM buffer for 15 min and then washed with phosphate-buffered saline. Immunofluorescence staining was conducted as previously described (Liu et al., 2010). Primary antibodies were incubated overnight at +4°C, and the secondary antibodies were applied the next day for 1 h at +37°C. All images were taken using an Axiovert 200 inverted microscope (Carl Zeiss), and image analysis was done using the AxioVision 4.6 software. Each cell was imaged with a 40× objective, and images used for quantification did not contain saturated pixels in axons or dendrites. All images within comparable groups were taken with appropriate rigor and under identical settings. Only neurons with clearly definable axons and dendrites that were not obscured by neighboring cells were chosen for analysis. Ratio images were made using the Pascal software of tyrosinated tubulin channel over β3-tubulin channel (Lin et al., 2012). Images were taken with a Zeiss Axiovert microscope LSM 5 Pascal system with an HeNe laser (but with the pinhole wide open, and hence no optical sectioning).
Dendrite number, length, and width were measured using MAP-2 immunostained cultures. Dendrite width was measured at the base of the dendrite within 5 μM of the cell body. Dendrite length was measured from that point until the visible end of the dendrite. Average immunofluorescence intensity was measured within a box drawn over the region of interest (ROI) in the dendrite (the brightest part of the dendrite) and a collection of axons. Axon immunofluorescence for neurons growing in different substrates was measured at 24-h postplating. Size of ROIs was kept consistent throughout the experiments. Background intensity was subtracted from all measurements. Measurements were normalized against volume differences between dendrites and axons measured from β3-tubulin–stained neurons.
CHO cells were fixed 48 h after transfection with EGFP-kinesin-5 with 4% paraformaldehyde and glutaraldehyde simultaneously with extraction (as described above) and stained for tyrosinated, detyrosinated, or acetylated tubulin. Images of both channels were obtained at 40× using a Zeiss microscope, transported to 16-bit TIFF, and analyzed in WCIF-ImageJ for colocalization using colocalization analysis within flat regions of the cells. Background was subtracted from the ROI using a factor of 3 within WCIF-ImageJ before colocalization analysis. A Pearson's r correlation mean value was reported.
Statistical analyses
All data analyses, statistical comparisons, and graphs were generated using SPSS 20 (IBM, Armonk, NY) and Excel (Microsoft, Redmond, WA). Data represent mean ± SEM of at least two repeats for each experiment. For statistical analysis, the mean difference was considered to be significant at the 0.05 level (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Specific statistical analyses are indicated in the figure legends.
Presentation of images
Figures were processed with Adobe Photoshop CS2 and Illustrator CS2 (San Jose, CA). In some cases, fluorescence images were subjected to the “invert” function in Adobe Photoshop that converts black to white and white to black and inverts gray levels proportionally, because we found that fine details were more clearly visualized. Heat maps of images were generated in ImageJ.
Acknowledgments
This work was supported by a grant to P.W.B. from the National Institutes of Health. O.I.K. is the recipient of a predoctoral National Research Service Award from the National Institutes of Health. We thank Lotfi Ferhat for helpful advice on the CHO cell experiments.
Abbreviations used:
- BSA
bovine serum albumin
- CDK1/5
cyclin-dependent kinase 1/5
- DMSO
dimethyl sulfoxide
- FBS
fetal bovine serum
- Lambda PP
Lambda protein phosphatase
- PDL
poly-d-lysine
- PLL
poly-l-lysine
- ROI
region of interest
- SCG
superior cervical ganglia
- siRNA
small interfering RNA
- TTL
tubulin tyrosine ligase.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-08-1313) on October 29, 2014.
REFERENCES
- Ahmad FJ, Echeverri CJ, Vallee RB, Baas PW. Cytoplasmic dynein and dynactin are required for the transport of microtubules into the axon. J Cell Biol. 1998;140:391–401. doi: 10.1083/jcb.140.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad FJ, Pienkowski TP, Baas PW. Regional differences in microtubule dynamics in the axon. J Neurosci. 1993;13:856–866. doi: 10.1523/JNEUROSCI.13-02-00856.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ari C, Borysov SI, Wu J, Padmanabhan J, Potter H. Alzheimer amyloid β inhibition of Eg5/kinesin 5 reduces neurotrophin and/or transmitter receptor function. Neurobiol Aging. 2014;35:1839–1849. doi: 10.1016/j.neurobiolaging.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Black MM. Individual microtubules in the axon consist of domains that differ in both composition and stability. J Cell Biol. 1990;111:495–509. doi: 10.1083/jcb.111.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Gonzalez-Billault C. A gathering of neuronal cytoskeletal scientists in South America. Cytoskeleton (Hoboken) 2012;69:407–415. doi: 10.1002/cm.21038. [DOI] [PubMed] [Google Scholar]
- Baas PW, Lin S. Hooks and comets: the story of microtubule polarity orientation in the neuron. Dev Neurobiol. 2011;71:403–418. doi: 10.1002/dneu.20818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Slaughter T, Brown A, Black MM. Microtubule dynamics in axons and dendrites. J Neurosci Res. 1991;30:134–153. doi: 10.1002/jnr.490300115. [DOI] [PubMed] [Google Scholar]
- Badding MA, Dean DA. Highly acetylated tubulin permits enhanced interactions with and trafficking of plasmids along microtubules. Gene Ther. 2013;20:616–624. doi: 10.1038/gt.2012.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blangy A, Lane HA, d'Herin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–1169. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- Brown A, Li Y, Slaughter T, Black MM. Composite microtubules of the axon: quantitative analysis of tyrosinated and acetylated tubulin along individual axonal microtubules. J Cell Sci. 1993;104:339–352. doi: 10.1242/jcs.104.2.339. [DOI] [PubMed] [Google Scholar]
- Buster DW, Baird DH, Yu W, Solowska JM, Chauviere M, Mazurek A, Kress M, Baas PW. Expression of the mitotic kinesin Kif15 in postmitotic neurons: implications for neuronal migration and development. J Neurocytol. 2003;32:79–96. doi: 10.1023/a:1027332432740. [DOI] [PubMed] [Google Scholar]
- Cahu J, Olichon A, Hentrich C, Schek H, Drinjakovic J, Zhang C, Doherty-Kirby A, Lajoie G, Surrey T. Phosphorylation by Cdk1 increases the binding of Eg5 to microtubules in vitro and in Xenopus egg extract spindles. PLoS One. 2008;3:e3936. doi: 10.1371/journal.pone.0003936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde C, Caceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–332. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- Dunn S, Morrison EE, Liverpool TB, Molina-Paris C, Cross RA, Alonso MC, Peckham M. Differential trafficking of Kif5c on tyrosinated and detyrosinated microtubules in live cells. J Cell Sci. 2008;121:1085–1095. doi: 10.1242/jcs.026492. [DOI] [PubMed] [Google Scholar]
- Erck C, Peris L, Andrieux A, Meissirel C, Gruber AD, Vernet M, Schweitzer A, Saoudi Y, Pointu H, Bosc C, et al. A vital role of tubulin-tyrosine-ligase for neuronal organization. Proc Natl Acad Sci USA. 2005;102:7853–7858. doi: 10.1073/pnas.0409626102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falnikar A, Tole S, Baas PW. Kinesin-5, a mitotic microtubule-associated motor protein, modulates neuronal migration. Mol Biol Cell. 2011;22:1561–1574. doi: 10.1091/mbc.E10-11-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenz NP, Gable A, Wadsworth P. Mitotic functions of kinesin-5. Semin Cell Dev Biol. 2010;21:255–259. doi: 10.1016/j.semcdb.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferhat L, Cook C, Chauviere M, Harper M, Kress M, Lyons GE, Baas PW. Expression of the mitotic motor protein Eg5 in postmitotic neurons: implications for neuronal development. J Neurosci. 1998;18:7822–7835. doi: 10.1523/JNEUROSCI.18-19-07822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumy LF, Chew DJ, Tortosa E, Katrukha EA, Kapitein LC, Tolkovsky AM, Hoogenraad CC, Fawcett JW. The kinesin-2 family member KIF3C regulates microtubule dynamics and is required for axon growth and regeneration. J Neurosci. 2013;33:11329–11345. doi: 10.1523/JNEUROSCI.5221-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen GG, Khawaja S, Bulinski JC. Postpolymerization detyrosination of alpha-tubulin: a mechanism for subcellular differentiation of microtubules. J Cell Biol. 1987;105:251–264. doi: 10.1083/jcb.105.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Chandrasekaran V, Lein P, Kaplan PL, Higgins D. Leukemia inhibitory factor and ciliary neurotrophic factor cause dendritic retraction in cultured rat sympathetic neurons. J Neurosci. 1999;19:2113–2121. doi: 10.1523/JNEUROSCI.19-06-02113.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond JW, Huang CF, Kaech S, Jacobson C, Banker G, Verhey KJ. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol Biol Cell. 2010;21:572–583. doi: 10.1091/mbc.E09-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque SA, Hasaka TP, Brooks AD, Lobanov PV, Baas PW. Monastrol, a prototype anti-cancer drug that inhibits a mitotic kinesin, induces rapid bursts of axonal outgrowth from cultured postmitotic neurons. Cell Motil Cytoskeleton. 2004;58:10–16. doi: 10.1002/cm.10176. [DOI] [PubMed] [Google Scholar]
- He Y, Francis F, Myers KA, Yu W, Black MM, Baas PW. Role of cytoplasmic dynein in the axonal transport of microtubules and neurofilaments. J Cell Biol. 2005;168:697–703. doi: 10.1083/jcb.200407191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, Peterman EJ, Kwok BH, Kim JH, Kapoor TM, Schmidt CF. The bipolar mitotic kinesin Eg5 moves on both microtubules that it crosslinks. Nature. 2005;435:114–118. doi: 10.1038/nature03503. [DOI] [PubMed] [Google Scholar]
- Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol. 2000;150:975–988. doi: 10.1083/jcb.150.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashina AS, Baskin RJ, Cole DG, Wedaman KP, Saxton WM, Scholey JM. A bipolar kinesin. Nature. 1996;379:270–272. doi: 10.1038/379270a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollins KM, Bell RL, Butts M, Withers GS. Dendrites differ from axons in patterns of microtubule stability and polymerization during development. Neural Dev. 2009;4:26. doi: 10.1186/1749-8104-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Setou M. Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat Neurosci. 2009;12:559–567. doi: 10.1038/nn.2314. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Finch EA. MAP2 and tau segregate into dendritic and axonal domains after the elaboration of morphologically distinct neurites: an immunocytochemical study of cultured rat cerebrum. J Neurosci. 1987;7:3142–3153. doi: 10.1523/JNEUROSCI.07-10-03142.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer G, Liao G, Gundersen GG. Detyrosination of tubulin regulates the interaction of intermediate filaments with microtubules in vivo via a kinesin-dependent mechanism. Mol Biol Cell. 1999;10:1105–1118. doi: 10.1091/mbc.10.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein PJ, Beck HN, Chandrasekaran V, Gallagher PJ, Chen HL, Lin Y, Guo X, Kaplan PL, Tiedge H, Higgins D. Glia induce dendritic growth in cultured sympathetic neurons by modulating the balance between bone morphogenetic proteins (BMPs) and BMP antagonists. J Neurosci. 2002;22:10377–10387. doi: 10.1523/JNEUROSCI.22-23-10377.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein P, Guo X, Hedges AM, Rueger D, Johnson M, Higgins D. The effects of extracellular matrix and osteogenic protein-1 on the morphological differentiation of rat sympathetic neurons. Int J Dev Neurosci. 1996;14:203–215. doi: 10.1016/0736-5748(96)00008-1. [DOI] [PubMed] [Google Scholar]
- Lein PJ, Higgins D. Laminin and a basement membrane extract have different effects on axonal and dendritic outgrowth from embryonic rat sympathetic neurons in vitro. Dev Biol. 1989;136:330–345. doi: 10.1016/0012-1606(89)90260-1. [DOI] [PubMed] [Google Scholar]
- Lein P, Johnson M, Guo X, Rueger D, Higgins D. Osteogenic protein-1 induces dendritic growth in rat sympathetic neurons. Neuron. 1995;15:597–605. doi: 10.1016/0896-6273(95)90148-5. [DOI] [PubMed] [Google Scholar]
- Lin S, Liu M, Mozgova OI, Yu W, Baas PW. Mitotic motors coregulate microtubule patterns in axons and dendrites. J Neurosci. 2012;32:14033–14049. doi: 10.1523/JNEUROSCI.3070-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Liu M, Son YJ, Timothy Himes B, Snow DM, Yu W, Baas PW. Inhibition of kinesin-5, a microtubule-based motor protein, as a strategy for enhancing regeneration of adult axons. Traffic. 2011;12:269–286. doi: 10.1111/j.1600-0854.2010.01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Nadar VC, Kozielski F, Kozlowska M, Yu W, Baas PW. Kinesin-12, a mitotic microtubule-associated motor protein, impacts axonal growth, navigation, and branching. J Neurosci. 2010;30:14896–14906. doi: 10.1523/JNEUROSCI.3739-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruta H, Greer K, Rosenbaum JL. The acetylation of alpha-tubulin and its relationship to the assembly and disassembly of microtubules. J Cell Biol. 1986;103:571–579. doi: 10.1083/jcb.103.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- McAllister AK. Cellular and molecular mechanisms of dendrite growth. Cereb Cortex. 2000;10:963–973. doi: 10.1093/cercor/10.10.963. [DOI] [PubMed] [Google Scholar]
- Myers KA, Baas PW. Kinesin-5 regulates the growth of the axon by acting as a brake on its microtubule array. J Cell Biol. 2007;178:1081–1091. doi: 10.1083/jcb.200702074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadar VC, Ketschek A, Myers KA, Gallo G, Baas PW. Kinesin-5 is essential for growth-cone turning. Curr Biol. 2008;18:1972–1977. doi: 10.1016/j.cub.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadar VC, Lin S, Baas PW. Microtubule redistribution in growth cones elicited by focal inactivation of kinesin-5. J Neurosci. 2012;32:5783–5794. doi: 10.1523/JNEUROSCI.0144-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez J, Fischer I. Microtubule-associated proteins (MAPs) in the peripheral nervous system during development and regeneration. J Mol Neurosci. 1997;8:207–222. doi: 10.1007/BF02736834. [DOI] [PubMed] [Google Scholar]
- Palazzo A, Ackerman B, Gundersen GG. Cell biology: tubulin acetylation and cell motility. Nature. 2003;421:230. doi: 10.1038/421230a. [DOI] [PubMed] [Google Scholar]
- Papasozomenos SC, Binder LI. Phosphorylation determines two distinct species of tau in the central nervous system. Cell Motil Cytoskeleton. 1987;8:210–226. doi: 10.1002/cm.970080303. [DOI] [PubMed] [Google Scholar]
- Peris L, Wagenbach M, Lafanechere L, Brocard J, Moore AT, Kozielski F, Job D, Wordeman L, Andrieux A. Motor-dependent microtubule disassembly driven by tubulin tyrosination. J Cell Biol. 2009;185:1159–1166. doi: 10.1083/jcb.200902142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, Powers J, Strome S, Saxton WM. Kinesin-5 acts as a brake in anaphase spindle elongation. Curr Biol. 2007;17:R453–R454. doi: 10.1016/j.cub.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin KE, Mitchison TJ. Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc Natl Acad Sci USA. 1995;92:4289–4293. doi: 10.1073/pnas.92.10.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp DJ, Rogers GC, Scholey JM. Microtubule motors in mitosis. Nature. 2000;407:41–47. doi: 10.1038/35024000. [DOI] [PubMed] [Google Scholar]
- Sharp DJ, Yu W, Ferhat L, Kuriyama R, Rueger DC, Baas PW. Identification of a microtubule-associated motor protein essential for dendritic differentiation. J Cell Biol. 1997;138:833–843. doi: 10.1083/jcb.138.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirajuddin M, Rice LM, Vale RD. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat Cell Biol. 2014;16:335–344. doi: 10.1038/ncb2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23:2655–2664. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronel S, Belnoue L, Grosjean N, Revest JM, Piazza PV, Koehl M, Abrous DN. Adult-born neurons are necessary for extended contextual discrimination. Hippocampus. 2012;22:292–298. doi: 10.1002/hipo.20895. [DOI] [PubMed] [Google Scholar]
- Tronel S, Fabre A, Charrier V, Oliet SH, Gage FH, Abrous DN. Spatial learning sculpts the dendritic arbor of adult-born hippocampal neurons. Proc Natl Acad Sci USA. 2010;107:7963–7968. doi: 10.1073/pnas.0914613107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Wildenberg SM, Tao L, Kapitein LC, Schmidt CF, Scholey JM, Peterman EJ. The homotetrameric kinesin-5 KLP61F preferentially crosslinks microtubules into antiparallel orientations. Curr Biol. 2008;18:1860–1864. doi: 10.1016/j.cub.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waitzman JS, Rice SE. Mechanism and regulation of kinesin-5, an essential motor for the mitotic spindle. Biol Cell. 2014;106:1–12. doi: 10.1111/boc.201300054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Zou DJ, Rajan I, Cline H. Dendritic dynamics in vivo change during neuronal maturation. J Neurosci. 1999;19:4472–4483. doi: 10.1523/JNEUROSCI.19-11-04472.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SY, Choi JE, Huh JW, Hwang O, Lee HS, Hong HN, Kim D. Monastrol, a selective inhibitor of the mitotic kinesin Eg5, induces a distinctive growth profile of dendrites and axons in primary cortical neuron cultures. Cell Motil Cytoskeleton. 2005;60:181–190. doi: 10.1002/cm.20057. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Wildonger J, Ye B, Zhang Y, Kita A, Younger SH, Zimmerman S, Jan LY, Jan YN. Dynein is required for polarized dendritic transport and uniform microtubule orientation in axons. Nat Cell Biol. 2008;10:1172–1180. doi: 10.1038/ncb1777. [DOI] [PMC free article] [PubMed] [Google Scholar]