

Abstract
Diabetic retinopathy is the leading cause of blindness worldwide. It is caused by the abnormal growth of the retinal blood vessels. Plasminogen activator inhibitor1 (PAI1) is the key growth factor and the inhibition of PAI1 can reduce the angiogenesis. In this study, currently available inhibitors are taken and tested for the toxicity, binding affinity, and bioactivities of the compounds by in silico approach. Five toxic free inhibitors were identified, among which N-acetyl-D-glucosamine shows the significant binding affinity and two of the molecules are having the better bioactivity properties. The molecular optimization of 2-(acetylamino)-2-deoxy-A-D-glucopyranose and alpha-L-fucose can be used for the treatment of diabetic retinopathy.
1. Introduction
Almost half of the diabetes mellitus patients have the high risk of diabetic retinopathy. Worldwide, 17 million people are affected with proliferative diabetic retinopathy [1]. In diabetic patients, the sugar molecules accumulate in retinal blood vessels and damage them; sometimes the molecules block the vessels. Due to this, supply of oxygen and other nutrition to the retina will be reduced; this condition is called ischemia. To overcome this situation, the retina will produce new blood vessels, the process known as neovascularization. But the newly produced vessels are abnormal and fragile; they leak fluid into macula, a part of the retina which is responsible for clear central vision, and cause vision loss [2]. Presently, laser treatment is in use to treat retinopathy, but it leads to peripheral vision loss as it burns the retina. The alternative strategy is to control the expression of the growth factors which induce angiogenesis.
Plasminogen activator inhibitor1 (PAI1) is one of the growth factors responsible for neovascularization in diabetic patients. After ischemia, it is secreted from endothelial cells [3]. It is reported that inhibition of PAI1 will lead to 53% reduction in retinal angiogenesis and prevent tumor invasion and vascularization [4, 5]. In this study, an attempt was made to identify the better therapeutic inhibitor for PAI1.
2. Materials and Methods
2.1. PAI1 Structure Retrieval and Active Site Identification
The 3D structure of PAI1 was retrieved from Protein Data Bank (PDB) [6]. To identify the active site of the protein, the depth and solvent accessible surface area (SASA) were computed and based on that the probability values are assigned to each amino acid using DEPTH server [7]. The residue with high depth and SASA values are likely to form the active site.
2.2. Identification of Inhibitors
The inhibitor compounds (used as ligands in docking studies) so far identified against PAI1 protein were collected from various databases, namely, Human Metabolome Database (HMDB) [8], DrugBank [9], Pharmacogenomic knowledgebase (PharmaGKB) [10], and PDB.
2.3. Toxicity Screening
The collected ligand compounds were screened for toxicity using the online server ToxPredict (http://apps.ideaconsult.net:8080/ToxPredict). It estimates the hazard of chemical structures mainly based on Lipinski's rule and Cramer's rule. The molecules which are having the hydrogen donors ≤ 5, hydrogen bond acceptor ≤ 10, molecular mass ≤ 500 daltons, and logP ≤ 5 are likely to obey Lipinski's rule, and Cramer's rule classifies the chemical compounds into three classes based on the 33 metabolic activities. The compounds belonging to class I are of low order of toxicity, class II are more innocuous than the other two classes, and class III are of significant toxicity.
2.4. Docking
Docking calculations were carried out using interactive molecular graphics programs ArgusLab [11] and PatchDock [12]. Ligand was placed on a search point in the binding site which was calculated by DEPTH server; a set of diverse and energetically favorable rotations was created. In ArgusDock, exhaustive search methods for flexible ligand docking were used to calculate the binding energy. PatchDock algorithm divided the surface representation of the molecules into concave, convex, and flat patches. Then, complementary patches were matched in order to generate candidate transformations and evaluated by scoring functions. The results were visualized by Molegro Molecular Viewer (http://www.molegro.com).
2.5. Bioactivity Prediction
The bioactivities of the biologically significant ligands were predicted by OSIRIS Property Explorer (http://www.organicchemistry.org/prog/peo/). The calculations were originally optimized on training sets of more than 5000 compounds with measured logP values and more than 2000 compounds with measured logS values. The drug score ranges between 0 and 1.
3. Results and Discussion
3.1. PAI1 Structure Retrieval and Identification of Active Site
There are 9 structures with IDs 3LW2, 3Q02, 3R4L, 1C5G, 1DB2, 1DVN, 1DVM, 1LJ5, and 1B3K which are available for PAI1 in PDB, among which the structure IB3K, which consists of 4 chains, was selected as it is in active form and is free from being bound with other molecules. The active site region was identified, represented in Figure 1, and the amino acid composition of the active site is represented in Figure 2.
Figure 1.
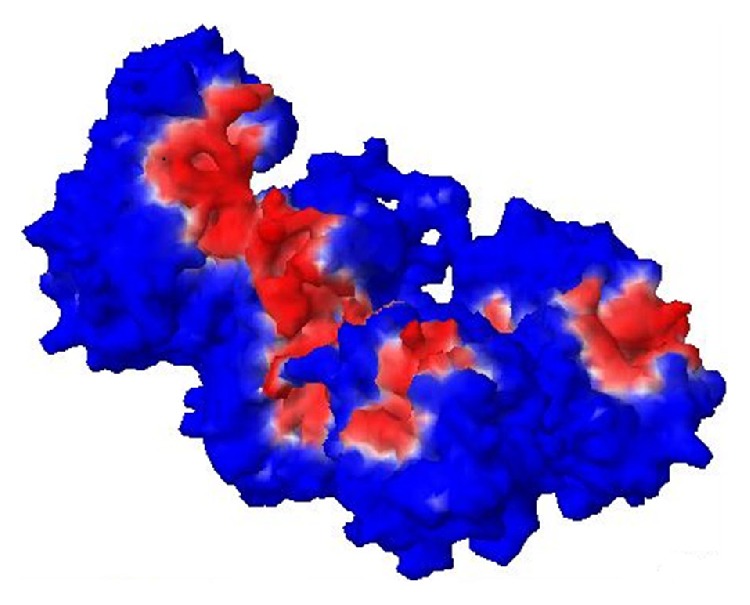
Active site region of the protein is red in colour.
Figure 2.

Amino acid composition of active site of PAI1.
3.2. Identified Ligand Compounds for PAI1
The inhibitors to the protein of our interest are listed in Table 1.
Table 1.
Ligand molecules of PAI1 protein.
| Database | Inhibitor molecules |
|---|---|
| HMDB | Atorvastatin, dimethyl sulfoxide, and simvastatin. |
| DrugBank | Troglitazone. |
| PharmaGKB | Antidepressants (including amitriptyline hydrochloride, amoxapine, clomipramine hydrochloride, and desipramine hydrochloride), citalopram, and fluoxetine. |
| PDB | 2,5-Dihydroxy-3-undecyclohexa-2,5-diene-1,4,-dione; 1,2-ethanediol; beta-D-mannose; alpha-L-fucose; N-acetyl-D-glucosamine; 2-acetylamino-2-deoxy-A-D-glucopyranose; ribose; acetic acid. |
3.3. Toxicity Prediction
The toxicity of the molecules, based on Lipinski's rule and Cramer's rule whether they induce carcinogen or eye irritation, is represented in Table 2.
Table 2.
Toxicity prediction of the inhibitors of PAI1.
| Ligand molecule | Obey Lipinski's rule? | Cramer's rule | Carcinogen | Eye irritation |
|---|---|---|---|---|
| Troglitazone | Yes | Class III | No | No |
| Dimethyl sulfoxide | Yes | Class III | No | No |
| Atorvastatin | No | Class III | No | No |
| Simvastatin | Yes | Class III | No | No |
| Citalopram | Yes | Class III | No | No |
| Fluoxetine | Yes | Class III | No | No |
| 2,5,Dihydroxy-3-undecyciohexa-2,5-diene-1,4,dione | No | Class III | No | No |
| 1,2-Ethanediol | Yes | Class I | No | Yes |
| Beta-D-mannose | Yes | Class I | No | No |
| Alpha-L-fucose | Yes | Class I | No | No |
| N-Acetyl-D-glucosamine | Yes | Class I | No | No |
| 2-(Acetylamino)-2-deoxy-A-D- glucopyranose | Yes | Class I | No | No |
| Ribose | Yes | Class I | No | No |
| Acetic acid | Yes | Class III | No | No |
| Amitriptyline hydroxychloride | No | Class III | No | No |
| Amoxapine | Yes | Class III | No | No |
| Clomipramine hydrochloride | Yes | Class III | No | No |
| Desipramine hydrochloride | Yes | Class III | No | No |
Based on the lower toxicity, the ligands were filtered for further studies; the molecules include 2-acetylamino-2-deoxy-A-D-glucopyranose, alpha-L-fucose, beta-D-mannose, N-acetyl-D-glucosamine, and ribose.
3.4. Docking Study of PAI1 with Selected Ligand Molecules
The graphical representations of the docking of PAI1 protein with 2-acetylamino-2-deoxy-A-D-glucopyranose, alpha-L-fucose, beta-D-mannose, N-acetyl-D-glucosamine, and ribose are in Figures 3, 4, 5, 6, and 7; yellow lines represent hydrogen bonds, pale blue dots represent hydrogen atom acceptors, yellow dots represent hydrogen atom donors, red dots represent positive ions, and dark blue dots represent negative ions. The corresponding binding energy values are presented in Tables 3, 4, 5, 6, and 7 and the overall energy of each inhibitor binding with PAI1 is in Table 8.
Figure 3.
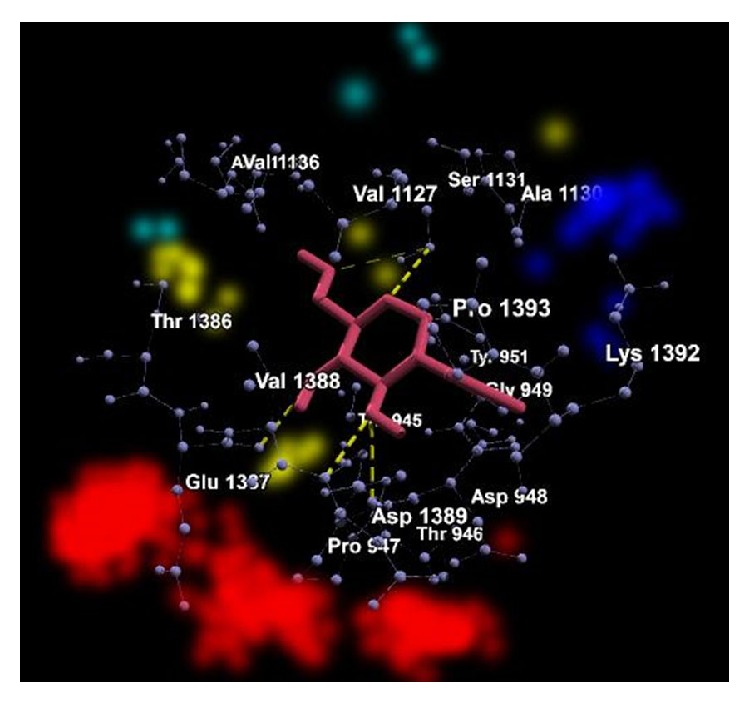
Binding of 2-acetylamino-2-deoxy-A-D-glucopyranose with PAI1.
Figure 4.
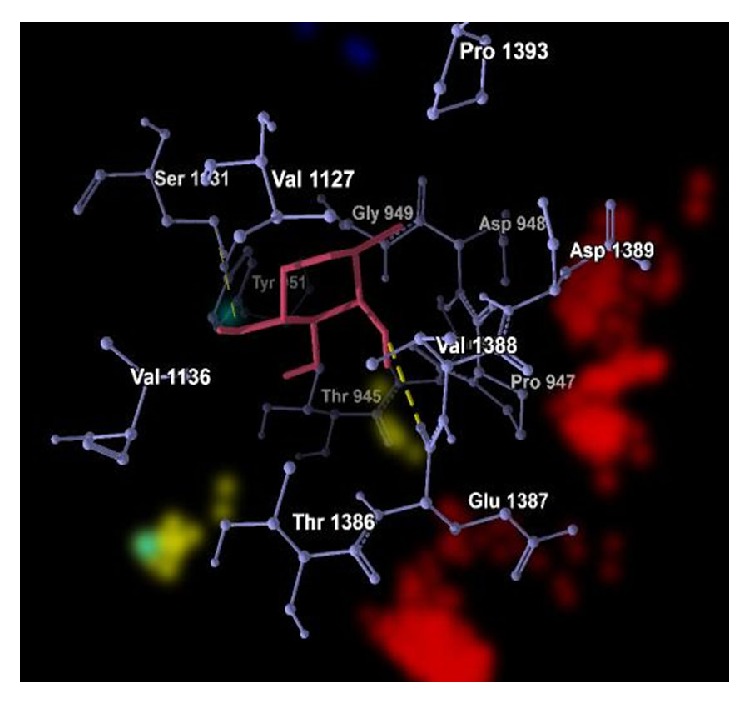
Binding of alpha-L-fucose with PAI1.
Figure 5.
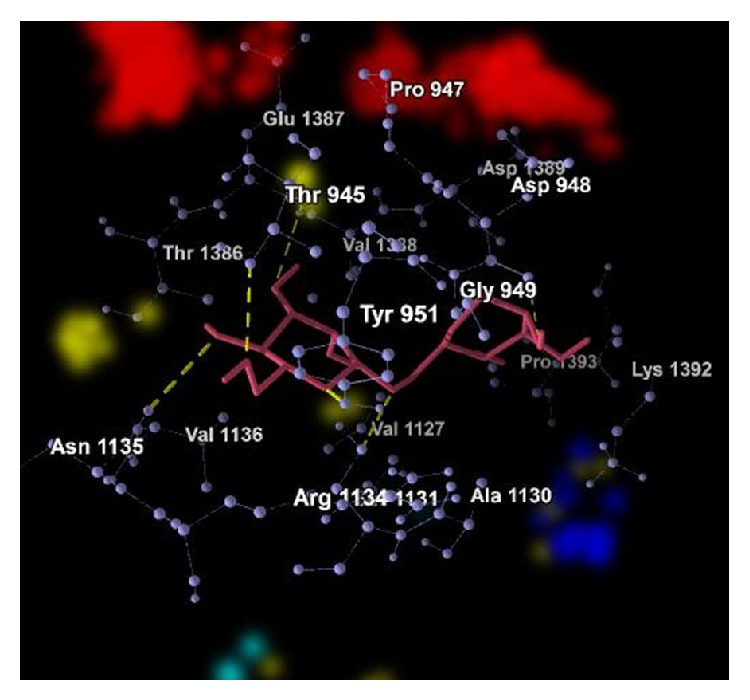
Binding of beta-D-mannose with PAI1.
Figure 6.
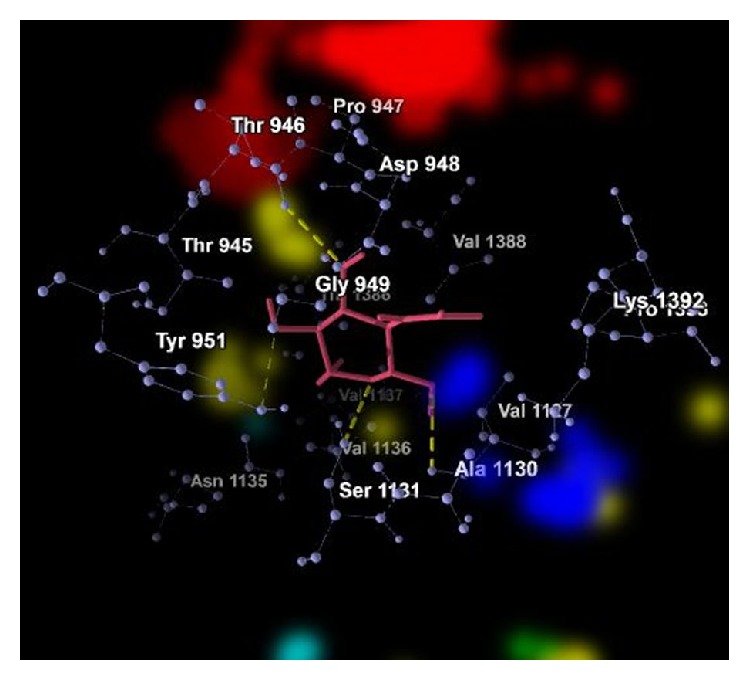
Binding of N-acetyl-D-glucosamine with PAI1.
Figure 7.
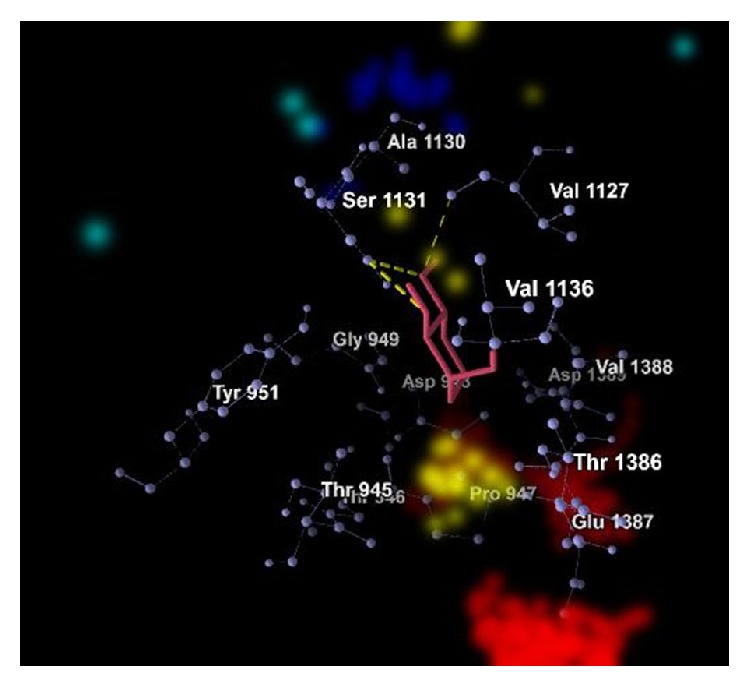
Binding of ribose with PAI1.
Table 3.
Binding energy of 2-acetylamino-2-deoxy-A-D-glucopyranose with PAI1.
| Bond | Energy | Residues involved | |
|---|---|---|---|
| Hydrogen | −2.5 (kcal/mol) | 2.64532 Å (length) | Glu 1387 |
| Hydrogen | −2.5 (kcal/mol) | 2.99992 Å (length) | Val 1388 |
| Hydrogen | −2.5 (kcal/mol) | 2.69638 Å (length) | Pro 947 |
| Hydrogen | −0.835562 (kcal/mol) | 3.43289 Å (length) | Ser 1131 |
| Hydrogen | −2.5 (kcal/mol) | 2.72334 Å (length) | Ser 1131 |
|
| |||
| Hydrogen (nondirectional) | −13.551 (kcal/mol) | — | |
|
| |||
| Steric |
−71.96 (kcal/mol) (by PLP) −24.70 (kcal/mol) (by LJ16-6) |
Asp 948, Gly 949, Thr 945, Thr 946, Thr 951, Ala 1130, Asn 1135, Asp 1389, Lys 1392, Pro 1393, Thr 1386, Val 1127, Val 1136 | |
Table 4.
Binding energy of alpha-L-fucose with PAI1.
| Bond | Energy | Residues involved | |
|---|---|---|---|
| Hydrogen | −2.50 (kcal/mol) | 2.84262 Å (length) | Val 1388 |
| Hydrogen | −0.91 (kcal/mol) | 3.41776 Å (length) | Ser 1131 |
|
| |||
| Hydrogen (nondirectional) | −4.04 (kcal/mol) | — | |
|
| |||
| Steric |
−52.65 (kcal/mol) (by PLP) −17.65 (kcal/mol) (by LJ16-6) |
Asp 948, Gly 949, Pro 947, Thr 945, Thr 946, Tyr 951, Ala 1130, Asp 1389, Glu 1387, Pro 1393, Thr 1386, Val 1127, Val 1136 | |
Table 5.
Binding energy of beta-D-mannose with PAI1.
| Bond | Energy | Residues involved | |
|---|---|---|---|
| Hydrogen | −1.34 (kcal/mol) | 3.05 Å (length) | Glu 1387 |
| Hydrogen | −0.23 (kcal/mol) | 3.51 Å (length) | Glu 1387 |
| Hydrogen | −2.45 (kcal/mol) | 3.11 Å (length) | Val 1136 |
| Hydrogen | −1.78 (kcal/mol) | 3.24 Å (length) | Thr 945 |
| Hydrogen | −2.50 (kcal/mol) | 2.86 Å (length) | Tyr 951 |
|
| |||
| Hydrogen (nondirectional) | −15.91 (kcal/mol) | — | |
|
| |||
| Steric |
−35.10 (kcal/mol) (by PLP) −445.37 (kcal/mol) (by LJ16-6) |
Asp 948, Gly 949, His 950, Thr 946, Ala 1130, Arg 1134, Asn 1135, Asp 1389, Gln 1126, Lys 1392, Pro 1393, Thr 1386, Val 1127, Val 1388 | |
Table 6.
Binding energy of N-acetyl-D-glucosamine with PAI1.
| Bond | Energy | Residues involved | |
|---|---|---|---|
| Hydrogen | −2.50 (kcal/mol) | 2.85 Å (length) | Val 1127 |
| Hydrogen | −0.21 (kcal/mol) | 3.55 Å (length) | Ser 1131 |
| Hydrogen | −2.50 (kcal/mol) | 3.02 Å (length) | Ser 1131 |
| Hydrogen | −0.90 (kcal/mol) | 3.42 Å (length) | Tyr 951 |
| Hydrogen | −2.50 (kcal/mol) | 2.84 Å (length) | Pro 947 |
|
| |||
| Hydrogen (nondirectional) | −14.54 (kcal/mol) | — | |
|
| |||
| Steric |
−44.44 (kcal/mol) (by PLP) 13.40 (kcal/mol) (by LJ16-6) |
Asp 948, Gly 949, His 950, Thr 945, Thr 946, Ala 1130, Asn 1135, Asp 1389, Gln 1126, Glu 1387, Lys 1392, Pro 1393, Thr 1386, Val 1136, Val 1388 | |
Table 7.
Binding energy of ribose with PAI1.
| Bond | Energy | Residues involved | |
|---|---|---|---|
| Hydrogen | −1.02 (kcal/mol) | 3.39 Å (length) | Val 1127 |
| Hydrogen | −2.30 (kcal/mol) | 2.57 Å (length) | Ser 1131 |
| Hydrogen | −2.49 (kcal/mol) | 2.59 Å (length) | Ser 1131 |
|
| |||
| Hydrogen (nondirectional) | −5.82 (kcal/mol) | — | |
|
| |||
| Steric |
−46.01 (kcal/mol) (by PLP) −17.31 (kcal/mol) (by LJ16-6) |
Asp 948, Gly 949, Pro 947, Thr 945, Thr 946, Tyr 951, Ala 1130, Asp 1389, Glu 1387, Thr 1386, Val 1136, Val 1388 | |
Table 8.
The overall binding affinity of ligands with PAI1.
| Ligand molecule | Binding energy (kcal/mol) | Area covered (Å) |
|---|---|---|
| N-Acetyl-D-glucosamine | −7.83 | 386.10 |
| 2-Acetylamino-2-deoxy-A-D-glucopyranose | −6.03 | 402.30 |
| Beta-D-mannose | −6.00 | 508.40 |
| Alpha-L-fucose | −5.43 | 283.50 |
| Ribose | −5.13 | 251.00 |
3.5. Bioactivity Properties
The bioactivity of the ligands which are toxic free and having biologically significant binding affinity is represented in Table 9.
Table 9.
Bioactivity of the selected ligands of PAI1.
| Ligand | clogP | Solubility | Molecular weight | Druglikeness | Drug score |
|---|---|---|---|---|---|
| 2-Acetylamino-2-deoxy-A-D-glucopyranose | −1.63 | −0.15 | 191.01 | −0.50 | 0.67 |
| Alpha-L-fucose | −1.63 | −0.15 | 101.00 | −0.56 | 0.67 |
| Beta-D-mannose | −3.5 | 0.42 | 312.00 | −5.08 | 0.38 |
| N-Acetyl-D-glucosamine | −2.3 | −0.02 | 221.00 | −3.05 | 0.5 |
| Ribose | −1.3 | −0.06 | 134.00 | −5.68 | 0.4 |
Among the presently identified inhibitors against PAI1, only five of them, namely, 1,2-ethanediol, beta-D-mannose, alpha-L-fucose, N-acetyl-D-glucosamine, and 2-(acetylamino)-2-deoxy-A-D-glucopyranose, come under class I of Cramer's rule. As 1,2-ethanediol causes the irritation in eye, it is excluded from the study. The remaining ligand compounds are used for further study.
The docking results showed that N-acetyl-D-glucosamine is highly biologically significant followed by 2-(acetylamino)-2-deoxy-A-D-glucopyranose, beta-D-mannose, alpha-L-fucose, and ribose in decreasing order. The above five molecules are toxic free and can bind with PAI1, but the bioactivities of the compounds revealed that 2-(acetylamino)-2-deoxy-A-D-glucopyranose and alpha-L-fucose are having the property of druglikeness at moderate level; the rest cannot be used for the purpose of drug.
4. Conclusion
For the known inhibitors of PAI1, toxicity, binding affinity, and bioactivity were predicted computationally. There were five molecules identified; moreover they have the feasible binding affinity with PAI1 as well. As the molecular weight of N-acetyl-D-glucosamine and beta-D-mannose is higher and cLog P value is higher for ribose, there is a least priority to these compounds to be used as drug. The molecules 2-(acetylamino)-2-deoxy-A-D-glucopyranose and alpha-L-fucose were identified as better therapeutic inhibitors to PAI1 than other molecules used in this study. Due to the toxic free nature and significant binding energy, this study can be extended at clinical level. For the efficient and quick treatment level, they should be structurally optimized as the drug score of the identified two molecules was moderate.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Yau J. W. Y., Rogers S. L., Kawasaki R., Lamoureux E. L., Kowalski J. W., Bek T., Chen S.-J., Dekker J. M., Fletcher A., Grauslund J., Haffner S., Hamman R. F., Ikram M. K., Kayama T., Klein B. E. K., Klein R., Krishnaiah S., Mayurasakorn K., O'Hare J. P., Orchard T. J., Porta M., Rema M., Roy M. S., Sharma T., Shaw J., Taylor H., Tielsch J. M., Varma R., Wang J. J., Wang N., West S., Zu L., Yasuda M., Zhang X., Mitchell P., Wong T. Y. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35(3):556–564. doi: 10.2337/dc11-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sampath kumar K. P., Bhowmik D., Harish G., Duraivel S., Pragathi kumar B. Diabetic retinopathy-symptoms, causes, risk factors and treatment. The Forma Innovation. 2012;1(8):7–13. [Google Scholar]
- 3.Sakamoto T., Yasue H., Ogawa H., Misumi I., Masuda T. Association of patency of the infarct-related coronary artery with plasma levels of plasminogen activator inhibitor activity in acute myocardial infarction. The American Journal of Cardiology. 1992;70(3):271–276. doi: 10.1016/0002-9149(92)90603-V. [DOI] [PubMed] [Google Scholar]
- 4.Bajou K., Noël A., Gerard R. D., Masson V., Brunner N., Holst-Hansen C., Skobe M., Fusenig N. E., Carmeliet P., Collen D., Foidart J. M. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nature Medicine. 1998;4(8):923–928. doi: 10.1038/nm0898-923. [DOI] [PubMed] [Google Scholar]
- 5.Basu A., Menicucci G., Maestas J., Das A., McGuire P. Plasminogen activator inhibitor-1 (PAI-1) facilitates retinal angiogenesis in a model of oxygen-induced retinopathy. Investigative Ophthalmology & Visual Science. 2009;50(10):4974–4981. doi: 10.1167/iovs.09-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. The protein data bank. Nucleic Acids Research. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan K. P., Varadarajan R., Madhusudhan M. S. DEPTH: a web server to compute depth and predict small-molecule binding cavities in proteins. Nucleic Acids Research. 2011;39(2):W242–W248. doi: 10.1093/nar/gkr356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wishart D. S., Tzur D., Knox C., Eisner R., Guo A. C., Young N., Cheng D., Jewell K., Arndt D., Sawhney S., Fung C., Nikolai L., Lewis M., Coutouly M.-A., Forsythe I., Tang P., Shrivastava S., Jeroncic K., Stothard P., Amegbey G., Block D., Hau D. D., Wagner J., Miniaci J., Clements M., Gebremedhin M., Guo N., Zhang Y., Duggan G. E., MacInnis G. D., Weljie A. M., Dowlatabadi R., Bamforth F., Clive D., Greiner R., Li L., Marrie T., Sykes B. D., Vogel H. J., Querengesser L. HMDB: the human metabolome database. Nucleic Acids Research. 2007;35(1):521–526. doi: 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wishart D. S., Knox C., Guo A. C., Shrivastava S., Hassanali M., Stothard P., Chang Z., Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Research. 2006;34(1):668–672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whirl-Carrillo M., McDonagh E. M., Hebert J. M., et al. Pharmacogenomics knowledge for personalized medicine. Clinical Pharmacology & Therapeutics. 2012;92(4):414–417. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chikhi A., Benseguen A. Docking efficiency comparison of Surflex, a commercial package and Arguslab, a licensable freeware. Journal of Computer Science & Systems Biology. 2008;1:81–86. [Google Scholar]
- 12.Schneidman-Duhovny D., Inbar Y., Nussinov R., Wolfson H. J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Research. 2005;33(2):W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]