

Abstract
Angiogenesis represents a rate-limiting step during tumor progression. Targeting angiogenesis is already applied in cancer treatment, yet limits of anti-angiogenic therapies have emerged, notably because tumors adapt and recur after treatment. Therefore, there is a strong need to better understand the molecular and cellular mechanisms underlying tumor angiogenesis. Using the RIP1-Tag2 transgenic murine model, we identified 298 genes that are deregulated during the angiogenic switch, revealing an ingression/expansion of specific stromal cell types including endothelial cells and pericytes, but also macrophages and perivascular mesenchymal cells. Canonical TGF-β signaling is up-regulated during the angiogenic switch, especially in tumor-associated macrophages and fibroblasts. The matrisome, comprising extracellular matrix (ECM) and ECM-associated molecules, is significantly enriched, which allowed us to define the AngioMatrix signature as the 110 matrisomal genes induced during the RIP1-Tag2 angiogenic switch. Several AngioMatrix molecules were validated at expression level. Ablation of tenascin-C, one of the most highly induced ECM molecules during the switch, resulted in reduced angiogenesis confirming its important role. In human glioma and colorectal samples, the AngioMatrix signature correlates with the expression of endothelial cell markers, is increased with tumor progression and finally correlates with poor prognosis demonstrating its diagnostic and therapeutic potential.
Keywords: Tumor angiogenesis, angiogenic switch, extracellular matrix, matrisome
INTRODUCTION
Angiogenesis, a fundamental biological process by which novel blood vessels are formed from pre-existing ones [1], represents a rate-limiting step during tumor progression [2]. Studies from murine models have indicated that angiogenesis occurs relatively early along tumor formation and progression [2]. In particular, the murine RIP1-Tag2 model of pancreatic neuroendocrine tumorigenesis (PNET; ref. [3]) has recurrently allowed to gain novel insights into the molecular and cellular mechanisms governing tumor angiogenesis and progression. In this model of multistep tumorigenesis, pancreatic beta cells of the Langerhans islets over-express the SV40 T antigen oncogene which stochastically drives the sequential transformation of a fraction of normal islets into hyperplastic, angiogenic and macroscopic tumor islets [2]. This in vivo PNET model was key to provide evidences demonstrating that a fraction of islets undergoes an angiogenic switch early during tumor progression [4]. Several molecular and cellular mechanisms were described to promote the RIP1-Tag2 angiogenic switch. These include the crucial role of VEGFA and its signaling [5] and in particular, the matrix metalloprotease MMP-9-mediated regulation of VEGF bioavailability [6]. Neutrophils appear to be a source of MMP-9 hereby promoting the angiogenic switch [7, 8].
The RIP1-Tag2 model is widely used in a pre-clinical setting to evaluate anti-tumor therapeutic strategies, including angiogenesis inhibitors [9–15]. Importantly, several key conceptual advances in our understanding of how tumors adapt and become resistant to anti-angiogenic therapies, a major clinical challenge that has emerged [16, 17], were also obtained using this model [18, 19].
Here we used a genome-wide gene expression profiling strategy to uncover potential novel mechanisms underlying the angiogenic switch during RIP1-Tag2 tumor progression. We show that the angiogenic switch is associated with the deregulation of a limited number of genes, some of which reflect the expansion and ingression of stromal cells and the activation of canonical TGF-β signaling in tumor-associated macrophages and fibroblasts. Furthermore, a significant part of these genes encodes ECM and ECM-associated molecules, together defining the AngioMatrix signature. We show that this signature correlates with endothelial cell (EC) markers and tumor progression in human colorectal cancer (CRC) and glioma. Finally, its high expression correlates with poor prognosis for CRC, low grade glioma and glioblastoma (GBM) patients.
RESULTS
Gene expression profiling of the tumor angiogenic switch in a murine PNET model
We used the RIP1-Tag2 mouse model as a prototypical in vivo model of the tumor angiogenic switch [2, 20] to comprehensively address the underlying mechanisms. We chose an early time point (8 weeks) when a subset of pancreatic islets has undergone the angiogenic switch (Fig. 1A and Supplementary Fig. S1A) but had not yet progressed into macroscopic tumors. Islets were isolated from RIP1-Tag2 mice and classified as angiogenic or non angiogenic based on their appearance. RNA was extracted from the isolated islets to determine genome-wide gene expression levels using microarrays (Fig. 1B). The comparison of the transcriptome of non angiogenic versus angiogenic islets yielded a restricted list of 298 significantly deregulated genes, the “AngioSwitch signature”. We first noted that this signature included several markers of stromal cells (Fig. 1C). Characteristic markers of EC (e.g. Pecam1 and Cdh5 encoding CD31 and VE-cadherin, respectively), perivascular cells (Acta2 encoding alpha-smooth muscle actin or αSMA, Cspg4 encoding NG2, Pdgfrb) and monocytes/macrophages (Emr1 encoding F4/80, Csf1r) were found up-regulated in angiogenic islets, which was confirmed by RT-qPCR (Fig. 1D) and tissue staining (Fig. 1, E-G and Supplementary Fig.S1, B-D).
Figure 1. Transcriptomic profiling of the RIP1-Tag2 angiogenic switch reveals the up-regulation of stromal cell markers.
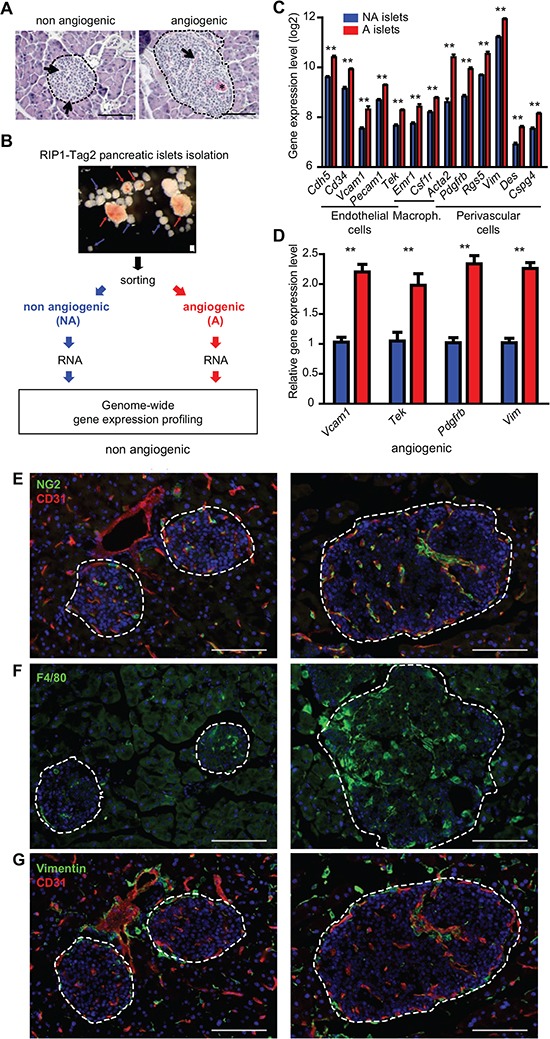
(A) patterns of non-angiogenic (left) and angiogenic (right) islets in H&E stained tissue sections from RIP1-Tag2 pancreata. Examples of normal capillaries in a non angiogenic islet (arrows) and of a dilated vessel (arrow) and micro-hemorrhaging (asterisk) in the angiogenic islet. (B) strategy used to compare angiogenic and non angiogenic pancreatic tumor islets by gene expression profiling upon their differential isolation, sorting and RNA extraction. (C) up-regulation of specific stromal cell markers in the transcriptome of RIP1-Tag2 angiogenic (red) compared to non angiogenic (blue) islets: markers for EC (Cdh5, Cd34, Vcam1, Pecam1, Tek or Tie2, also expressed by macrophages), macrophages/monocytes (Emr1, Tek and Csf1r) perivascular and smooth muscle cells (Acta2, Pdgfrb, Rgs5, Vim, Des and Cspg4 encoding NG2). Measures represent the mean expression level from two independent profiling experiments, error bars the SEM. ** p < 5×10−3. (D) RT-qPCR confirmation of the up-regulation of stromal cell markers (Vcam1, EC; Tek: ECs and macrophages/monocytes; Pdgfrb: pericytes; Vim: perivascular SMC) in angiogenic (red) compared to non angiogenic (blue) islets. Measures represent the mean expression level from two independent experiments, error bars the SEM. ** p < 5×10−3. (E-G) expression (immunofluorescence) of stromal cell markers in non angiogenic and angiogenic islets, CD31 and NG2 (E), F4/80 (F) and Vimentin and CD31 (G). Nuclei are stained with Dapi (blue). In A and E-G dashed lines encircle islets. Scale bars, 100 μm.
Stromal-specific activation of canonical TGF-β signaling during the RIP1-Tag2 angiogenic switch
Several TGF-β pathway members and target genes were found up-regulated, including ligands and extracellular regulators, Cd105 (encoding endoglin, a TGF-β co-receptor) and known target genes (Fig. 2A). The up-regulation of genes encoding TGF-β ligands (Tgfb1 and Tgfb3) and prototypical SMAD2/3 target genes (Tgfbi, Serpine1 and Plat encoding PAI-1 and t-PA, respectively) was confirmed by RT-qPCR (Fig. 2B). We hypothesized that TGF-β signaling may occur preferentially within stromal cells, as a previous study revealed the presence of ALK5 (Tgf-β receptor 1)-positive cells of presumably stromal origin within RIP1-Tag2 angiogenic islets [21], which suggested that these unidentified stromal cell type(s) could undergo canonical TGF-β signaling. We used Gene Set Enrichment Analysis (GSEA) to determine the enrichment of stromal cell-specific TGF-β response signatures (TBRS; ref [22]) and found that the fibroblast- and the macrophage-specific TBRS were significantly enriched in angiogenic islets (Fig. 2C), suggesting that these stromal cell types may undergo canonical TGF-β signaling. To test this hypothesis, we analyzed the expression and sub-cellular localization of SMAD3 phosphorylated on S423/S425 (pSMAD3), as readout for TGF-β signaling activation, in RIP1-Tag2 tissue sections co-stained with stromal markers. While in non angiogenic islets an exclusively cytoplasmic staining was observed in some cells, within angiogenic islets pSMAD3 expression and nuclear localization was recurrently detected in some tumor cells but also more strikingly in both αSMA-positive fibroblasts and F4/80-positive macrophages (Fig. 2, D and E), demonstrating that these stromal cells undergo canonical TGF-β signaling in the angiogenic islets.
Figure 2. TGF-β signaling activation during the RIP1-Tag2 angiogenic switch.
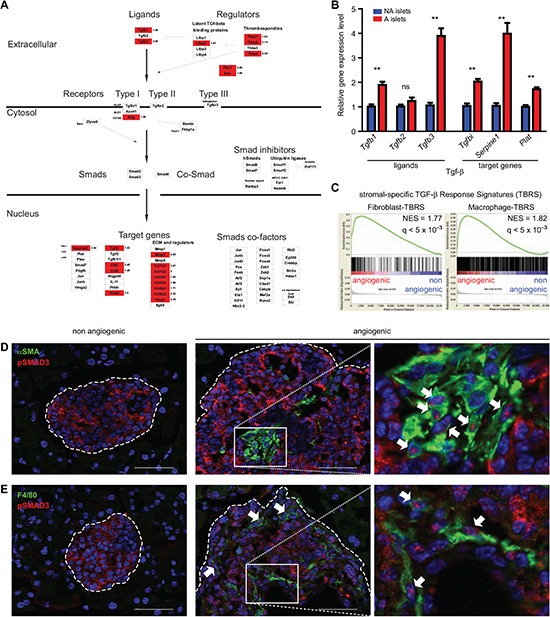
(A) schematic depiction of gene expression profiling data on a TGF-β signaling GenMapp. Genes (represented by boxes) in red indicate genes significantly up-regulated in angiogenic islets. No gene in this pathway was down-regulated. Several TGF-β ligands, extracellular regulators, the Endoglin co-receptor (Eng, encoding CD105), together with target genes are up-regulated in angiogenic islets. (B) RT-qPCR analysis of TGF-β ligands and target gene expression in non angiogenic (blue) and angiogenic (red) islets. The Tgfb1 and Tgfb3 genes are significantly up-regulated in angiogenic islets together with the prototypical Smad2/3 target genes Tgfbi, Serpine1 and Plat. Measures represent the mean of two independent experiments, error bars the SEM, ** p < 5×10−3, ns not significant. (C) GSEA demonstrates significant enrichment of fibroblast- and macrophage- specific TGF-β response signatures in the transcriptome of angiogenic islets. The Normalized Enrichment Score (NES) and the FDR q-value assessing the significance of enrichment are indicated. (D-E) co-staining of phosphorylated SMAD3 (pSMAD3) with αSMA (D) or F4/80 (E) in RIP1-Tag2 islets. Nuclear localization of pSMAD3 is observed in angiogenic islets, predominantly in tumor-associated αSMA+ fibroblasts (D) and F4/80+ macrophages (E) (arrows). Nuclei are stained in blue (DAPI). Dashed lines encircle islets; non angiogenic: left column, angiogenic: middle column and higher magnification pictures corresponding to the boxed areas within angiogenic islets are presented (right column). Scale bars, 50 μm.
Up-regulation of ECM and ECM-associated genes during the RIP1-Tag2 angiogenic switch: identification of the AngioMatrix signature
We then addressed whether groups of functionally-related genes are over-represented in the AngioSwitch signature using GeneOntologies (GO). This revealed significant enrichment of angiogenesis-related GO categories, supporting the biological relevance of the profiling data, but also of several GO categories related to ECM and secreted molecules (Fig. 3A). By RT-qPCR we validated the up-regulation of 12 of these genes (Fig. 3B), leading to a total of 25 validated genes with a significant correlation between array and RT-qPCR data (Fig. 3C).
Figure 3. Genes encoding the extracellular matrix and regulators, or matrisome, are up-regulated during the RIP1-Tag2 angiogenic switch.
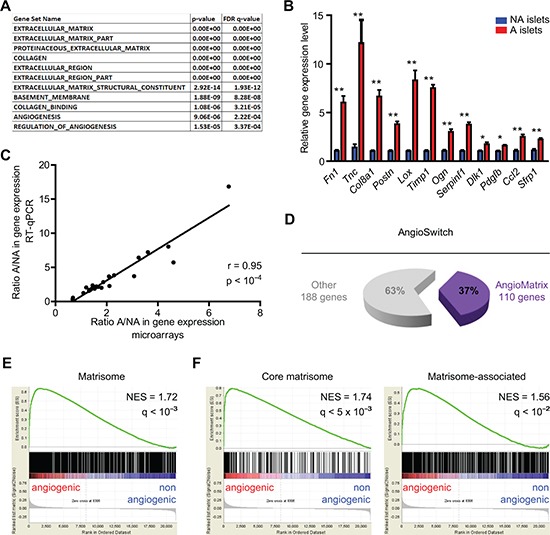
(A) significantly enriched GO categories in the AngioSwitch signature. The p- and FDR q-values indicate the significance of enrichment. (B) RT-qPCR validation of increased expression for 12 candidate genes up-regulated in angiogenic islets. Data (blue, non-angiogenic; red, angiogenic islets) represent mean and error bars the SEM from two independent experiments. **, p < 5×10−3; *, p < 10−2. (C) comparison of the gene expression ratio determined by array profiling and RT-qPCR for 25 validated genes (22 up-regulated, 1 unchanged and 2 down-regulated). The Pearson correlation coefficient and the p-value are indicated. (D) overlap between the AngioSwitch signature and the matrisome: 37% of genes induced during the angiogenic switch belong to the matrisome, defining the AngioMatrix signature (110 genes). (E-F) GSEA demonstrate significant enrichment of the matrisome (E) and its divisions (F) in the transcriptome of angiogenic islets. The Normalized Enrichment Score (NES) and the FDR q-value assessing the significance of enrichment are indicated.
As GO analysis revealed enrichment of several ECM-related categories, we examined the overlap of the AngioSwitch signature with the matrisome [23, 24], a comprehensive list of genes coding for ECM molecules and regulators. Of note, 37% of the genes composing the AngioSwitch signature encode matrisomal proteins (Fig. 3D), and core matrisomal genes are particularly over-represented (Supplementary Fig. S2). Moreover, GSEA revealed significant enrichment of the whole matrisome and its subclasses the core matrisome and matrisome-associated divisions (Fig. 3, E and F). We further defined the AngioMatrix signature as the 110 matrisomal genes induced during the RIP1-Tag2 angiogenic switch (Table 1). The expression of several AngioMatrix molecules, including vascular basement membrane components (collagen IV, laminin α4) and ECM glycoproteins (fibronectin, periostin, tenascin-C and sparc), was confirmed by tissue staining, which revealed their strong and stromal perivascular expression in angiogenic islets (Fig. 4 and Supplementary Fig. S3). Furthermore, we generated RIP1-Tag2 mice knocked-out for tenascin-C (TNC; ref. [25]), an ECM glycoprotein that was among the most highly up-regulated AngioMatrix genes (Fig. 3B and Table 1). We compared the number of angiogenic islets and the relative proportion of non-angiogenic and angiogenic islets from control and TNC-depleted RIP1-Tag2 mice on tissue sections, which revealed a significant decrease in the absence of TNC (Fig. 4, F and G).
Table 1. Composition of the AngioMatrix signature.
Gene symbol, expression ratio during the RIP1-Tag2 angiogenic switch and matrisome classification are indicated. Genes are grouped by matrisome division and categories, and ranked in descending order of expression ratio.
| Matrisome | ||||
|---|---|---|---|---|
| Gene symbol | Ratio A / NA | p-value | Division | Category |
| Col8a1 | 3.25 | 1.1869E-07 | Core matrisome | Collagens |
| Col10a1 | 3.11 | 9.3207E-04 | ||
| Col1a1 | 2.95 | 1.5235E-08 | ||
| Col1a2 | 2.77 | 1.0141E-09 | ||
| Col6a3 | 2.26 | 5.9958E-08 | ||
| Col12a1 | 2.22 | 4.7032E-08 | ||
| Col14a1 | 2.01 | 5.4409E-06 | ||
| Col3a1 | 1.91 | 1.4790E-07 | ||
| Col15a1 | 1.87 | 6.7506E-08 | ||
| Col4a2 | 1.74 | 4.5137E-06 | ||
| Col6a1 | 1.71 | 8.8071E-07 | ||
| Col5a2 | 1.68 | 6.1916E-07 | ||
| Col6a2 | 1.67 | 3.2904E-06 | ||
| Col4a1 | 1.57 | 5.2897E-07 | ||
| Col18a1 | 1.40 | 1.9590E-03 | ||
| Thbs4 | 6.77 | 3.9919E-09 | ECM Glycoproteins | |
| Fn1 | 4.61 | 6.5803E-08 | ||
| Tnc | 3.71 | 1.2366E-08 | ||
| Postn | 3.07 | 7.7478E-08 | ||
| Mfap5 | 2.87 | 3.2983E-09 | ||
| Fbn1 | 2.74 | 7.9111E-08 | ||
| Ctgf | 2.34 | 7.4667E-07 | ||
| Srpx2 | 2.28 | 7.0478E-07 | ||
| Cilp | 2.22 | 3.0748E-06 | ||
| Svep1 | 2.21 | 1.0975E-07 | ||
| Mgp | 2.21 | 3.7883E-05 | ||
| Thbs2 | 2.12 | 8.1086E-07 | ||
| Spon1 | 2.04 | 5.5033E-06 | ||
| Nid1 | 1.98 | 1.4637E-07 | ||
| Ltbp2 | 1.95 | 1.9311E-05 | ||
| Pcolce | 1.92 | 1.9741E-08 | ||
| Mfap4 | 1.91 | 2.4071E-07 | ||
| Thbs1 | 1.90 | 3.1110E-04 | ||
| Lama4 | 1.86 | 4.4791E-07 | ||
| Sparc | 1.83 | 1.0330E-07 | ||
| Aebp1 | 1.71 | 1.4840E-05 | ||
| Lama2 | 1.71 | 8.5452E-06 | ||
| Dpt | 1.70 | 1.5983E-03 | ||
| Gas6 | 1.69 | 1.7581E-04 | ||
| Sparcl1 | 1.64 | 1.3306E-04 | ||
| Slit2 | 1.63 | 2.5898E-07 | ||
| Lamc1 | 1.58 | 9.9984E-08 | ||
| Eln | 1.58 | 8.1302E-05 | ||
| Igfbp4 | 1.57 | 5.5755E-04 | ||
| Fbln5 | 1.56 | 1.5923E-04 | ||
| Fbln2 | 1.49 | 1.3614E-06 | ||
| Tgfbi | 1.48 | 2.0248E-05 | ||
| Mmrn2 | 1.48 | 3.4566E-05 | ||
| Wisp1 | 1.46 | 1.5947E-03 | ||
| Igfbp5 | 1.44 | 3.2103E-03 | ||
| Nid2 | 1.44 | 1.3026E-05 | ||
| Slit3 | 1.43 | 4.6296E-05 | ||
| Pxdn | 1.41 | 8.0373E-06 | ||
| Lum | 2.60 | 2.9885E-05 | Proteoglycans | |
| Fmod | 2.53 | 3.8582E-05 | ||
| Bgn | 2.36 | 5.6766E-08 | ||
| Aspn | 1.91 | 1.0662E-06 | ||
| Ogn | 1.87 | 1.0220E-02 | ||
| Prelp | 1.77 | 2.5035E-04 | ||
| Dcn | 1.66 | 1.9182E-02 | ||
| Vcan | 1.62 | 7.8882E-05 | ||
| Hspg2 | 1.45 | 1.0067E-04 | ||
| Lox | 4.41 | 1.0907E-07 | Matrisome-associated | ECM Regulators |
| Timp1 | 3.61 | 1.0843E-06 | ||
| Mmp2 | 2.70 | 6.4945E-05 | ||
| Serpine1 | 2.29 | 8.7933E-07 | ||
| Adamts2 | 2.17 | 9.2591E-07 | ||
| Serpinf1 | 2.10 | 1.9858E-06 | ||
| Serpine2 | 2.03 | 2.6508E-05 | ||
| Loxl1 | 1.97 | 3.9437E-07 | ||
| Mmp14 | 1.94 | 9.5446E-07 | ||
| Ctsc | 1.70 | 1.5301E-05 | ||
| Ctsh | 1.69 | 3.0313E-02 | ||
| Serpinh1 | 1.65 | 1.5995E-05 | ||
| Adamts12 | 1.61 | 2.0488E-06 | ||
| Adamts5 | 1.59 | 1.0358E-05 | ||
| Adam12 | 1.57 | 4.4749E-07 | ||
| Adamts1 | 1.52 | 5.9751E-06 | ||
| Adamtsl3 | 1.49 | 3.8709E-05 | ||
| Mmp13 | 1.48 | 4.2445E-04 | ||
| Cd109 | 1.47 | 1.1918E-04 | ||
| Serpina3n | 1.47 | 6.5008E-03 | ||
| Sulf2 | 1.43 | 1.1640E-04 | ||
| Loxl3 | 1.40 | 3.8243E-05 | ||
| Adamts9 | 1.40 | 1.9618E-04 | ||
| Anxa1 | 2.20 | 1.9866E-05 | ECM-affiliated Proteins | |
| Clec4n | 2.11 | 7.5384E-04 | ||
| Plxdc2 | 1.81 | 5.2764E-08 | ||
| Frem1 | 1.78 | 4.3323E-06 | ||
| Anxa3 | 1.77 | 4.4351E-03 | ||
| Lgals1 | 1.73 | 1.0923E-06 | ||
| Plxnd1 | 1.73 | 1.2998E-04 | ||
| Anxa2 | 1.72 | 2.6193E-05 | ||
| Colec12 | 1.57 | 6.4004E-04 | ||
| Cspg4 | 1.48 | 3.5828E-05 | ||
| Clec7a | 1.45 | 9.1353E-05 | ||
| C1qb | 1.42 | 2.8805E-04 | ||
| Sema6a | 1.41 | 3.1453E-06 | ||
| Gpc6 | 1.41 | 9.0537E-07 | ||
| Fstl1 | 2.92 | 1.0280E-05 | Secreted Factors | |
| Tgfb3 | 2.09 | 5.2714E-07 | ||
| Igf1 | 1.93 | 5.8883E-07 | ||
| Sfrp1 | 1.80 | 4.0155E-03 | ||
| Cxcl9 | 1.63 | 8.7549E-03 | ||
| Ccl3 | 1.57 | 2.1757E-03 | ||
| S100a6 | 1.54 | 1.7308E-04 | ||
| Angptl2 | 1.45 | 1.2141E-04 | ||
| Ccl2 | 1.45 | 1.7686E-04 | ||
| Pdgfc | 1.42 | 9.5455E-04 | ||
| Tgfb1 | 1.42 | 1.8128E-03 | ||
Figure 4. Analysis of AngioMatrix protein expression and functional validation of tenascin-C role in the RIP1-Tag2 angiogenic switch.
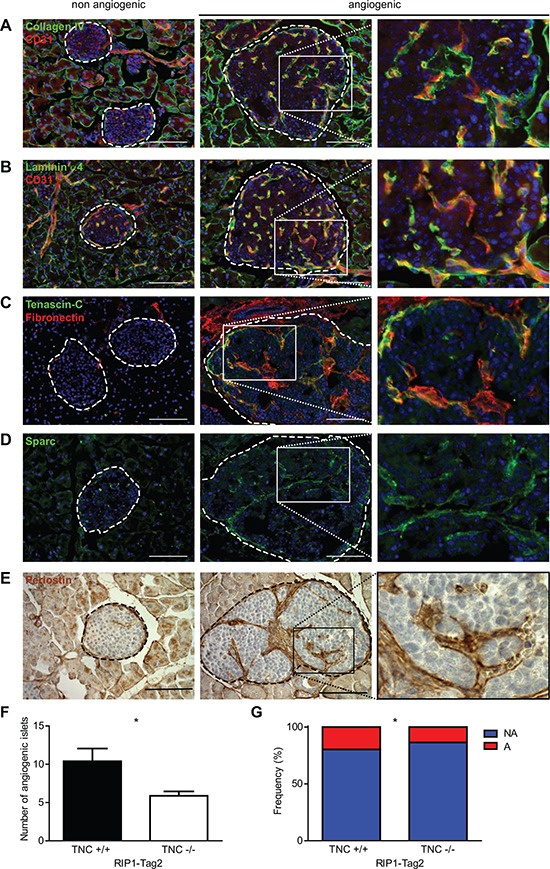
(A-E) expression pattern of the vascular basement membrane components collagen IV (A) and laminin α4 (B), and of the ECM glycoproteins fibronectin and tenascin-C (C), sparc (D) and periostin (E). Dashed lines encircle islets; non angiogenic: left column, angiogenic: middle column and higher magnification pictures corresponding to the boxed areas within angiogenic islets are presented (right column). Scale bars, 100 μm. (F-G) functional validation of tenascin-C contribution to the angiogenic switch. The number (F) and relative proportion (G) of angiogenic islets are significantly decreased in RIP1-Tag2 TNC −/− mice (n = 8 mice) as compared to TNC +/+ controls (n = 5 mice). A: angiogenic islets, NA: non angiogenic islets. * p < 5×10−2.
Expression of the AngioMatrix signature correlates with angiogenesis markers, tumor progression and poor prognosis for CRC, low grade glioma and GBM patients
To address the potential relevance of the AngioMatrix signature for cancer patients, we analyzed transcriptomic datasets, as this strategy enables investigating large and independent patient cohorts. Since insulinoma is rare and mostly benign (and no dataset could be retrieved), we focused on colorectal cancer and glioma, as their incidence is higher, angiogenesis is known to drive their progression and several independent datasets could be retrieved for CRC [26–30] and glioma [31–33].
We addressed whether expression of the AngioMatrix signature correlates with surrogate markers of blood vessels and angiogenesis in CRC. We determined for each sample the AngioMatrix signature expression level by averaging the expression levels of the 110 genes forming the signature, thereafter referred to as “AngioMatrix expression”, and observed significant correlations with the expression of the EC markers PECAM1 (Fig. 5A) and CDH5 (Fig. 5B). We next analyzed the pattern of AngioMatrix expression along CRC formation and progression. This revealed higher expression in normal tissue compared to adenoma and up-regulation during the adenoma-carcinoma transition (Fig. 5C), which was confirmed in an independent cohort (Supplementary Fig. S4A). We observed significantly higher AngioMatrix expression in primary CRC classified as Duke B or Duke C (versus A; Supplementary Fig. S4B), and higher expression in advanced primary CRC in an independent cohort (stage 3 or 4 versus 0, TNM; Supplementary Fig. S4C). We next asked if AngioMatrix expression could vary according to CRC molecular subtypes [29, 34] and found a significantly higher AngioMatrix expression in the Inflammatory subtype (compared to the Goblet-like or the Transit-amplifying subtypes) and in the Stem-like subtype compared to any other subtype (Fig. 5D). Furthermore, AngioMatrix expression was significantly lower in the C3 and the C1 subtypes and higher in the C4 subtype (Fig. 5E). We then wondered if AngioMatrix expression may vary during the ultimate steps of CRC progression. We found slightly increased AngioMatrix expression in metastatic (compared to non-metastatic) primary CRC (Supplementary Fig. S4D). In CRC metastasis, while no difference is observed in the lung (Supplementary Fig. S4E), AngioMatrix expression is significantly up-regulated in liver metastasis compared to normal tissue (Fig. 5F). The recurrent link between increased AngioMatrix expression and CRC progression prompted us to test a potential correlation with CRC patient survival. We used datasets from two independent cohorts of patients [28, 29], which were stratified using cut-off values into AngioMatrix low or high groups and survival analysis was performed to compare the outcome of these groups. A high expression of the AngioMatrix signature significantly correlated with a shorter relapse-free survival in the two CRC cohorts (Fig. 5, G and H, and Supplementary Figure S4, F and G).
Figure 5. Correlation between AngioMatrix signature expression and EC markers, tumor progression and poor prognosis in human CRC.
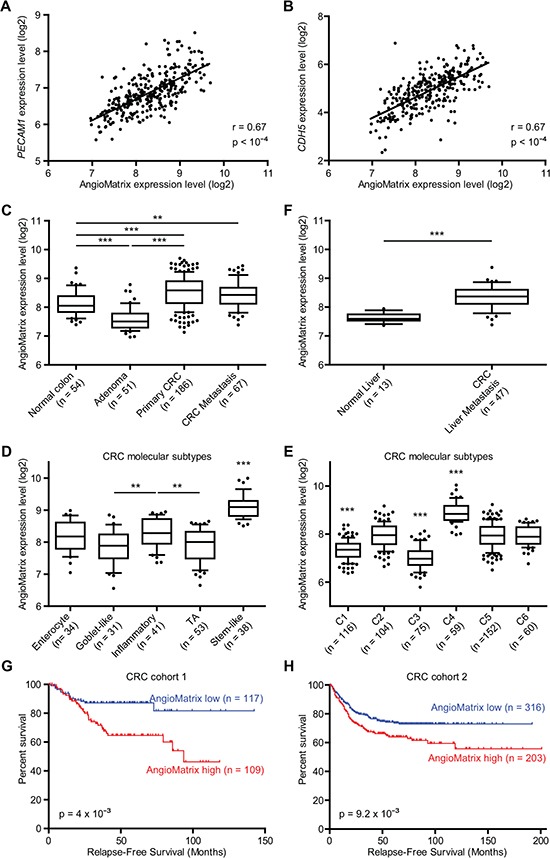
(A-B) correlation between AngioMatrix expression level and PECAM1 (A) or CDH5 (B) expression in normal, adenoma and primary CRC samples. The Pearson correlation coefficient and the p-value are indicated. (C, F) analysis of AngioMatrix expression during CRC progression. Comparison of normal colon, adenoma, primary CRC and CRC metastasis (C) and CRC metastasis versus normal liver (F). (D-E) analysis of AngioMatrix expression in the different primary CRC molecular subytpes. Note the significant higher levels of AngioMatrix in the Stem-like (D) and C4 (E) subtypes. In C-F, ***and ** indicate p-values < 10−3 and 10−2, respectively. (G-H) Kaplan-Meier survival analysis of CRC patients. Patients were stratified according to the average expression of the AngioMatrix signature as AngioMatrix high or low using a cutoff value. In each cohort, high AngioMatrix expression significantly correlates with poor prognosis for patients. P-values indicate the significance of survival difference between the groups of individuals. In C-H, n indicates the number of samples per group.
We analyzed AngioMatrix expression in independent glioma datasets and observed again a significant correlation between AngioMatrix expression and the EC markers PECAM1 (Fig. 6A) and CDH5 (Supplementary Fig. S5A). Comparing glioma histological subtypes revealed higher AngioMatrix expression in GBM compared to astrocytoma or oligodendroglioma (Fig. 6B), which was confirmed in an independent cohort (Supplementary Fig. S5B). Also, AngioMatrix expression increased with grade (Fig. 6C). Differences in AngioMatrix expression were observed between the GBM molecular subtypes [35], of which the highest expression in the mesenchymal subtype was the most significant (Fig. 6D). The recurrent correlation between AngioMatrix expression and glioma progression led us to test the potential use of the signature to stratify glioma patients and analyze their survival. High AngioMatrix expression significantly correlated with poor prognosis for all glioma patients (Supplementary Fig. S5C) and for subgroups of low-grade glioma: astrocytoma, oligodendroglioma, grade II, grade III or combined grade II and III glioma (Supplementary Fig. S5D-H). Finally, we analyzed GBM from two independent cohorts and found that high AngioMatrix expression significantly correlated with shortened patient survival (Fig. 6, E and F, and Supplementary Figure S5, I and J).
Figure 6. Correlation between AngioMatrix signature expression and EC marker, tumor progression and poor prognosis in human glioma.
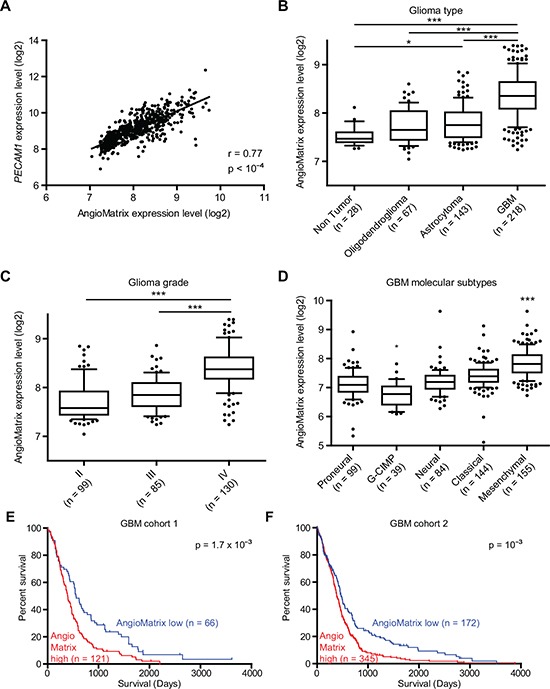
(A) correlation between AngioMatrix expression and PECAM1 in glioma samples. The Pearson correlation coefficient (r) and the p-value are indicated. (B) comparison of AngioMatrix expression between non tumor brain samples and glioma histological subtypes. Note the higher levels of AngioMatrix expression in GBM compared to normal brain tissue, oligodendroglioma or astrocytoma. (C-D) analysis of AngioMatrix expression according to glioma grade (C) and the different GBM molecular subtypes (D). Note the significantly higher levels in grade IV glioma (C) and in the GBM mesenchymal subtype (D). In B-D, * and *** indicate p-values < 5×10−2 and 10−3, respectively. (E-F) Kaplan-Meier survival analysis of GBM patients. Patients were stratified according to the average expression of the AngioMatrix signature as AngioMatrix high or low using a cutoff value. In each cohort, high AngioMatrix expression significantly correlates with poor prognosis for patients. P-values indicate the significance of survival difference between the groups of individuals. In B-F, n indicates the number of samples per group.
DISCUSSION
We have used a strategy based on gene expression profiling to comprehensively describe the angiogenic switch in a prototypical murine cancer model [2, 4]. Our microarray analysis first revealed the up-regulation of cell type specific markers in the angiogenic islets, suggesting an expansion of stromal cells. This was confirmed at tissue level using specific markers for endothelial cells, pericytes and macrophages. Of note, no neutrophil marker was retrieved, although neutrophils have been functionally implicated in the RIP1-Tag2 angiogenic switch [7, 8]. Since we have extracted RNA from whole islets for gene expression profiling, we may have missed the low abundant neutrophils (0.4% of RIP1-Tag2 islet cells; ref. [7]). At the molecular level, we noted a recurrent overlap between the AngioSwitch signature and several cellular signaling pathways that have been functionally implicated in RIP1-Tag2 tumor progression, including the PDGF receptor β and its ligand PDGF-BB [36] or endoglin [37]. We also observed and confirmed up-regulation of several genes encoding canonical TGF-β signaling pathway members, suggesting that TGF-β signaling is activated during the RIP1-Tag2 angiogenic switch. This is in line with a previous report showing the up-regulation of Tgfb1 and the presence of ALK5-positive cells, (expressing TGF-β receptor 1 and therefore susceptible of undergoing canonical TGF-β signaling in the presence of ligand) within RIP1-Tag2 angiogenic islets, and presumably representing stromal cells [21]. We found significant enrichment of fibroblast- and macrophage-specific TBRS, which suggested that these tumor-associated stromal cells may undergo signaling. Accordingly, we demonstrated their presence and that they undergo canonical TGF-β signaling as revealed by the nuclear localization of phosphorylated SMAD3 within these stromal cells in angiogenic but not in non angiogenic RIP1-Tag2 islets. Altogether, these observations strongly support the notion that the AngioSwitch signature is biologically and functionally meaningful, and that the activation of canonical TGF-β signaling within stromal cells may represent a key event driving this transition. Mechanistically it remains to be determined which specific signals trigger TGF-β signaling during the RIP1-Tag2 angiogenic switch and how specific AngioMatrix molecules are implicated. MMP-9 and MMP-2 represent candidate drivers of the transition since both are induced during the RIP1-Tag2 angiogenic switch and MMP9 in particular exerts a crucial role [6] and both MMPs are able to activate latent TGF-β [38]. Furthermore we uncovered that the RIP1-Tag2 angiogenic switch is associated with the up-regulation of genes encoding ECM and ECM-associated molecules. This is in line with our and others findings, as canonical TGF-β signaling regulates the production of ECM and regulators in the microenvironment of tissue under various physio-pathological conditions including cancer [39, 40]. Although beyond the current scope, it will be important to evaluate in the future whether blocking TGF-β signaling potentially impinges on the angiogenic switch affecting the expression of AngioMatrix molecules.
Using an elegant approach combining in silico and proteomic analysis, Naba and co-workers defined the matrisome, a comprehensive list of ECM and ECM-associated molecules [23, 24]. Using this resource we assessed the overlap with the AngioSwitch signature to define the AngioMatrix signature and validated the induction of expression for several AngioMatrix proteins during the angiogenic switch, including the ECM glycoproteins fibronectin, tenascin-C, sparc and periostin. Functionally, we demonstrated that TNC ablation impairs the RIP1-Tag2 angiogenic switch, in line with our macroscopical characterization of the two islet classes [25]. These data again support the notion that components of the AngioMatrix signature promote the RIP1-Tag2 angiogenic switch.
To evaluate the potential translational relevance of the AngioMatrix signature for cancer patients, we showed that AngioMatrix expression significantly correlated with EC markers in human CRC and glioma, supporting the notion that this signature also correlates with the angiogenesis status within human tumors. During CRC progression, AngioMatrix expression is increased at the adenoma-carcinoma transition, in partial agreement with previous studies showing that the angiogenic switch occurs early along the adenoma-carcinoma sequence [41, 42]. In glioma, AngioMatrix expression is significantly up-regulated in GBM compared to lower grade glioma. This may reflect vascular co-option in low-grade glioma in contrast to angiogenesis that is more important for GBM vascularization [20]. AngioMatrix expression varies according to primary CRC molecular subtypes [29, 34]. Although these studies have followed different approaches to define CRC subtypes, we found significantly higher levels of AngioMatrix expression in the stem-like [34] and the C4 [29] subtypes, the latter being also enriched in stem cell-like signatures [29]. We speculate that higher AngioMatrix expression in stem-like CRC reflects a potential role of some AngioMatrix molecules not only in angiogenesis but also in the regulation of cell fate within (cancer) stem cell niches. Moreover, tenascin-C and periostin are both expressed in the hair follicle stem cell niche in murine skin and are crucial for metastatic breast cancer stem cells colonizing the lung [43–45]. It will be interesting to determine if other AngioMatrix molecules represent normal and cancer stem cell niches components. We found lower AngioMatrix expression levels in the C1 and C3 subtypes, and higher level in the C4 subtype, which correlates with the respective enrichment of the GO sprouting angiogenesis category within these subtypes [29], reinforcing the notion that this signature correlates with angiogenesis in human CRC. Also, higher AngioMatrix expression levels are found in the GBM mesenchymal subtype, described as enriched in EC and angiogenesis markers [35]. Finally, the AngioMatrix signature allows to identify CRC, low-grade glioma and GBM patients with a poorer prognosis. It will be important to determine whether this can be extended to other tumor types and if specific AngioMatrix subsets may improve stratification of patients at higher risk of tumor relapse.
ECM molecules and regulators exert key functions during vascular remodeling in tumors and play instrumental roles in promoting tumor progression by multiple mechanisms as e.g. providing pro-angiogenic niches and favoring tumor cell survival and dissemination. Importantly, ECM molecules represent potential therapeutic targets as functional studies have underlined their importance in the process of blood vessel regrowth after anti-angiogenic therapy [12]. Whether AngioMatrix molecules are potentially relevant in tumor vessel regrowth is unknown and important to be addressed in the future. It is interesting to note that the ECM glycoproteins fibronectin, tenascin-C and periostin, that were found here among the most highly up-regulated genes during the angiogenic switch, have also been identified as crucial for metastatic colonization in other cancer models in vivo [44–46]. Further studies are warranted to assess if additional AngioMatrix molecules also contribute to the generation of metastatic niches. Finally, AngioMatrix expression is significantly higher in hepatic metastases, the most common metastatic site for CRC. It will be interesting to determine if some AngioMatrix molecules represent metastasis-specific components as these could represent novel opportunities to develop targeted therapies.
In summary, we have shown that the angiogenic switch, a rate-limiting and early step during PNET progression in a murine model, is associated with a specific transcriptome, which allowed us to define the AngioMatrix signature and show that it correlates with tumor progression and poor prognosis for CRC, low-grade glioma and GBM patients. Our study paves the way for the identification of novel molecular and cellular mechanisms that are key to tumor angiogenesis and might unravel novel opportunities for diagnosis and therapeutic targeting.
METHODS
A detailed description is available from the Supplementary methods.
RIP1-Tag2 mice
Experiments involving RIP1-Tag2 animals [3] were done at 8 weeks and in accordance with the guidelines from INSERM (National Institute for Health and Medical Research), as described [25].
Genome-wide gene expression profiling and data mining
Pools of angiogenic and non angiogenic pancreatic islets were sorted as described [25] and RNA was extracted for labeling and hybridization (Affymetrix arrays). Data are deposited in the Gene Expression Omnibus (NCBI, GSE51637). Significantly deregulated genes were selected using the BRB-ArrayTools software (NCI, USA). The matrisome [23, 24] was used to compare the overlap with the AngioSwitch signature and define the AngioMatrix signature. Enrichments of TBRS from specific stromal cell types [22], the matrisome and its divisions [23] in the profiling dataset of RIP1-Tag2 angiogenic and non angiogenic islets we generated (GSE51637) were analyzed using GSEA [47]. Correlations between AngioMatrix expression and various parameters were analyzed in independent cohorts of CRC [26–30] and glioma [31–33]. Molecular subtypes of CRC [29, 34] and GBM [35] were previously defined. Kaplan-Meier survival analysis was performed by analyzing transcriptomic datasets from independent cohorts of human CRC [28, 29], glioma and subtypes [31] and glioblastoma [31, 33].
Statistical analysis and graphical representation
Analysis was performed using GraphPad Prism (GraphPad Software, Inc. USA), Epi Info (Centers for Disease Control and Prevention, USA), BRB-ArrayTools (NCI, USA) and GSEA [47]. Histograms represent data expressed as mean +/− SEM. When comparing two groups, data were analyzed using two-tailed Mann Whitney U or unpaired Student t tests. When comparing three groups or more, data were analyzed using one-way ANOVA (with Tukey's post-test) or Kruskal-Wallis (with Dunn's post-test) tests. In box plots, whiskers represent the 10th and 90th centiles, and data points outside this interval are represented. The false discovery rate (FDR) q-value and the log-rank test were used to assess the significance of GSEA enrichments and of survival differences, respectively. P-values and q-values < 0.05 were considered as significant.
SUPPLEMENTARY METHODS, FIGURES AND TABLE
Acknowledgments
We are grateful to Gerhard Christofori, Joerg Huelsken, Lydia Sorokin and Patricia Simon-Assman and the MN3T team for sharing material and comments on the manuscript. We acknowledge animal caretakers and the IGBMC Microarray Facility (Christelle Thibault).
AUTHORs' CONTRIBUTIONS
Conception and design: G. Orend, T. Hussenet
Development of methodology: B. Langlois, F. Saupe, T. Rupp, C. Arnold, M. van der Heyden, G. Orend, T. Hussenet
Acquisition of data: B. Langlois, F. Saupe, T. Rupp, C. Arnold, M. van der Heyden, T. Hussenet
Analysis and interpretation of data: B. Langlois, F. Saupe, G. Orend, T. Hussenet
Writing, review, and/or revision of the manuscript: B. Langlois, F. Saupe, G. Orend, T. Hussenet
Administrative, technical, or material support: B. Langlois, F. Saupe, T. Rupp, C. Arnold, M. van der Heyden, G. Orend, T. Hussenet
Study supervision: G. Orend, T. Hussenet
GRANT SUPPORT
This work was supported by grants to G.O. from the Institut National du Cancer (INCa), Association pour la Recherche sur le Cancer (ARC), Oncosuisse, the Agence Nationale de la Recherche (ANR) and Ligue Régionale Grand-Est Contre le Cancer. F.S. was supported by the Fondation des Treilles.
Conflicts of interest
The authors disclose no potential conflicts of interest.
REFERENCES
- 1.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115–122. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339:58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 5.Inoue M, Hager JH, Ferrara N, Gerber H-P, Hanahan D. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell. 2002;1:193–202. doi: 10.1016/s1535-6108(02)00031-4. [DOI] [PubMed] [Google Scholar]
- 6.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103:12493–12498. doi: 10.1073/pnas.0601807103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shojaei F, Singh M, Thompson JD, Ferrara N. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc Natl Acad Sci U S A. 2008;105:2640–2645. doi: 10.1073/pnas.0712185105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parangi S, O'Reilly M, Christofori G, Holmgren L, Grosfeld J, Folkman J, Hanahan D. Antiangiogenic therapy of transgenic mice impairs de novo tumor growth. Proc Natl Acad Sci U S A. 1996;93:2002–2007. doi: 10.1073/pnas.93.5.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science. 1999;284:808–812. doi: 10.1126/science.284.5415.808. [DOI] [PubMed] [Google Scholar]
- 11.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mancuso MR, Davis R, Norberg SM, O'Brien S, Sennino B, Nakahara T, Yao VJ, Inai T, Brooks P, Freimark B, Shalinsky DR, Hu-Lowe DD, McDonald DM. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest. 2006;116:2610–2621. doi: 10.1172/JCI24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie L, Duncan MB, Pahler J, Sugimoto H, Martino M, Lively J, Mundel T, Soubasakos M, Rubin K, Takeda T, Inoue M, Lawler J, Hynes RO, Hanahan D, Kalluri R. Counterbalancing angiogenic regulatory factors control the rate of cancer progression and survival in a stage-specific manner. Proc Natl Acad Sci U S A. 2011;108:9939–9944. doi: 10.1073/pnas.1105041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.You W-K, Sennino B, Williamson CW, Falcón B, Hashizume H, Yao L-C, Aftab DT, McDonald DM. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011;71:4758–4768. doi: 10.1158/0008-5472.CAN-10-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, Tabruyn SP, You W-K, Chapman HA, Christensen JG, Aftab DT, McDonald DM. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012;2:270–287. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- 17.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8:299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 19.Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 21.Cunha SI, Pardali E, Thorikay M, Anderberg C, Hawinkels L, Goumans M-J, Seehra J, Heldin C-H, ten Dijke P, Pietras K. Genetic and pharmacological targeting of activin receptor-like kinase 1 impairs tumor growth and angiogenesis. J Exp Med. 2010;207:85–100. doi: 10.1084/jem.20091309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Calon A, Espinet E, Palomo-Ponce S, Tauriello DVF, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung P, Zhang XH-F, Byrom D, Riera A, Rossell D, Mangues R, Massagué J, Sancho E, Batlle E. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012;11:M111.014647. doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hynes RO, Naba A. Overview of the matrisome–an inventory of extracellular matrix constituents and functions. Cold Spring Harb Perspect Biol. 2012;4:a004903. doi: 10.1101/cshperspect.a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saupe F, Schwenzer A, Jia Y, Gasser I, Spenlé C, Langlois B, Kammerer M, Lefebvre O, Hlushchuk R, Rupp T, Marko M, van der Heyden M, Cremel G, Arnold C, Klein A, Simon-Assmann P, Djonov V, Neuville-Méchine A, Esposito I, Slotta-Huspenina J, Janssen K-P, de Wever O, Christofori G, Hussenet T, Orend G. Tenascin-C downregulates wnt inhibitor dickkopf-1, promoting tumorigenesis in a neuroendocrine tumor model. Cell Rep. 2013;5:482–492. doi: 10.1016/j.celrep.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 26.Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA, Domany E. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc Natl Acad Sci U S A. 2009;106:7131–7136. doi: 10.1073/pnas.0902232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galamb O, Wichmann B, Sipos F, Spisák S, Krenács T, Tóth K, Leiszter K, Kalmár A, Tulassay Z, Molnár B. Dysplasia-carcinoma transition specific transcripts in colonic biopsy samples. PloS One. 2012;7:e48547. doi: 10.1371/journal.pone.0048547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jorissen RN, Gibbs P, Christie M, Prakash S, Lipton L, Desai J, Kerr D, Aaltonen LA, Arango D, Kruhøffer M, Orntoft TF, Andersen CL, Gruidl M, Kamath VP, Eschrich S, Yeatman TJ, Sieber OM. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15:7642–7651. doi: 10.1158/1078-0432.CCR-09-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi M-C, Schiappa R, Guenot D, Ayadi M, Kirzin S, Chazal M, Fléjou J-F, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe T, Kobunai T, Yamamoto Y, Matsuda K, Ishihara S, Nozawa K, Iinuma H, Konishi T, Horie H, Ikeuchi H, Eshima K, Muto T. Gene expression signature and response to the use of leucovorin, fluorouracil and oxaliplatin in colorectal cancer patients. Clin Transl Oncol Off Publ Fed Span Oncol Soc Natl Cancer Inst Mex. 2011;13:419–425. doi: 10.1007/s12094-011-0676-z. [DOI] [PubMed] [Google Scholar]
- 31.Madhavan S, Zenklusen J-C, Kotliarov Y, Sahni H, Fine HA, Buetow K. Rembrandt: helping personalized medicine become a reality through integrative translational research. Mol Cancer Res. 2009;7:157–167. doi: 10.1158/1541-7786.MCR-08-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bredel M, Bredel C, Juric D, Harsh GR, Vogel H, Recht LD, Sikic BI. Functional network analysis reveals extended gliomagenesis pathway maps and three novel MYC-interacting genes in human gliomas. Cancer Res. 2005;65:8679–8689. doi: 10.1158/0008-5472.CAN-05-1204. [DOI] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Research Network: Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LCG, Lannon WA, Grotzinger C, Del Rio M, Lhermitte B, Olshen AB, Wiedenmann B, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu C-J, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joyce JA, Laakkonen P, Bernasconi M, Bergers G, Ruoslahti E, Hanahan D. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2003;4:393–403. doi: 10.1016/s1535-6108(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 37.Anderberg C, Cunha SI, Zhai Z, Cortez E, Pardali E, Johnson JR, Franco M, Páez-Ribes M, Cordiner R, Fuxe J, Johansson BR, Goumans M-J, Casanovas, et al. Deficiency for endoglin in tumor vasculature weakens the endothelial barrier to metastatic dissemination. J Exp Med. 2013;210:563–579. doi: 10.1084/jem.20120662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts AB, Heine UI, Flanders KC, Sporn MB. Transforming growth factor-beta. Major role in regulation of extracellular matrix. Ann N Y Acad Sci. 1990;580:225–232. doi: 10.1111/j.1749-6632.1990.tb17931.x. [DOI] [PubMed] [Google Scholar]
- 40.Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118:211–215. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi Y, Ellis LM, Mai M. The angiogenic switch of human colon cancer occurs simultaneous to initiation of invasion. Oncol Rep. 2003;10:9–13. [PubMed] [Google Scholar]
- 42.Staton CA, Chetwood ASA, Cameron IC, Cross SS, Brown NJ, Reed MWR. The angiogenic switch occurs at the adenoma stage of the adenoma carcinoma sequence in colorectal cancer. Gut. 2007;56:1426–1432. doi: 10.1136/gut.2007.125286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tucker RP, Ferralli J, Schittny JC, Chiquet-Ehrismann R. Tenascin-C and tenascin-W in whisker follicle stem cell niches: possible roles in regulating stem cell proliferation and migration. J Cell Sci. 2013;126:5111–5115. doi: 10.1242/jcs.134650. [DOI] [PubMed] [Google Scholar]
- 44.Oskarsson T, Acharyya S, Zhang XH-F, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massagué J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17:867–874. doi: 10.1038/nm.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr H-A, Delaloye J-F, Huelsken J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012;481:85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 46.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.