

Abstract
Introduction
Akt plays a pivotal role in cell survival and proliferation through a number of downstream effectors; unregulated activation of the PI3K/PTEN/Akt pathway is a prominent feature of many human cancers. Akt is considered an attractive target for cancer therapy by the inhibition of Akt alone or in combination with standard cancer chemotherapeutics. Both preclinical animal studies and clinical trials in humans have validated Akt as an important target of cancer drug discovery.
Area covered
A historical perspective of Akt inhibitors, including PI analogs, ATP-competitive and allosteric Akt inhibitors, along with other inhibitory mechanisms are reviewed in this paper with a focus on issued patents, patent applications and a summary of clinical trial updates since the last review in 2007.
Expert opinion
A vast diversity of inhibitors of Akt, both small molecule and biologic, have been developed in the past 5 years, with over a dozen in various phases of clinical development, and several displaying efficacy in humans. While it is not yet clear which mechanism of Akt inhibition will be optimal in humans, or which Akt isoforms to inhibit, or whether a small molecule or biologic agent will be best, data to all of these points will be available in the near future.
Keywords: Akt, allosteric, apoptosis, ATP-competitive, cancer, chemotherapy, clinical trial, inhibitors, kinase, PH domain, PKB, schizophrenia
1. Introduction
Cancer, a complex disease driven by a combination of external (smoking, diet, etc.) and internal factors (hormones, genetics, etc.), remains the leading cause of death in economically developed countries and has quickly earned the second place in developing countries [1,2]. Based on GLOBOCAN 2008 statistics, there were 12.7 million cancer cases and 7.6 million cancer deaths in 2008, with both figures expected to climb annually due to population aging/growth and adoption of smoking and Western-style diets [1]. By 2030, cancer deaths are expected to exceed 11 million [1,2]. Currently, cancer is viewed as a term to encompass hundreds of distinct disorders, and clinicians treat each case on an individual basis based on the molecular signaling mechanisms driving the tumor versus the classical approach of administering general cytotoxic agents. Thus, drug development is aimed at targeted therapies (specific action at tumor cells vs healthy cells) and biased patient populations (personalized medicine) for clinical trials [1,2]. From the search for new mechanisms for targeted therapies, Akt emerged in the early 2000s as an exciting target for drug development, leading to a concentrated effort to develop inhibitors of Akt [3–12]. Akt has a key role in the regulation of cell survival, proliferation and growth. Activation of the Akt pathway is common in cancer cells and efforts to understand the role of Akt in the transformed phenotype has led to a large body of research that has been the subject of numerous basic science reviews [3–12] and a patent review in 2007 in this journal [13]. This review covers small molecule and biological inhibition of Akt from both a historical perspective as well as current status of the patent landscape and clinical trials.
Akt (protein kinase B or PKB) is a serine/threonine kinase that belongs to the AGC family of kinases and shares high homology with protein kinase A (PKA) and PKC. There are three isozymes of Akt (Akt1, 2 and 3, also referred to in the literature as PKB-α,-β and -γ), and each of the three Akt isozymes has distinct functions and expression profiles [3–15]. Moreover, the three isoforms share similar kinetic mechanisms, but differing kinetic constants, that is, the affinity of the three isoforms for ATP (with Akt 1 and Akt3 both comparable Km values, but Akt2 ~ 5-fold weaker Km) and catalytic efficiency [3–15]. Akt1 and 2 are found throughout the body, while Akt3 is predominantly expressed in the brain, kidneys and heart [16]. The functional differences between Akt isozymes have also been studied in knockout mice, where Akt1 was found to be important for overall growth (Akt1 knockout mice are healthy, but have impaired fetal and postnatal growth), Akt2 primarily involved in glucose metabolism (Akt2 knockout mice have normal growth characteristics, but have a mild to severe insulin resistance phenotype) and Akt3 a critical mediator of brain development [17–21]. Recent knock-out data with shRNA in tumor-bearing mice suggest that inhibition of all three Akt isoforms would afford maximum efficacy [22]; however, data with isoform-specific, allosteric small molecule inhibitors in capsase-3 assays suggest that inhibition of Akt1 and Akt2 would be optimal [23,24]. Akt2 is the most commonly observed isozyme overexpressed in tumors while Akt1 and Akt3 overexpression appear to be less common [21]. The identification of isozyme-specific inhibitors provides valuable tools to study Akt activity, although it remains to be determined if isozyme selective inhibitors will have a larger therapeutic window in humans. From a drug design perspective, differences in regional distribution, coupled with differing affinity for ligands at the ATP-binding site, could complicate ATP-competitive Akt inhibitor development.
Structurally, each Akt isozyme contains an N-terminal pleckstrin homology (PH) domain, a 39 amino-acid hinge region, a kinase domain, an ~ 260 amino-acid ATP-binding domain and a 21 amino-acid C-terminal hydrophobic motif [3–15]. The PH domains of the three Akt isoforms contain ~ 110 amino acids and are only about 60% identical. The hinge regions of the three isoforms are only about 25% identical while the kinase domains have 90% homology. As expected, the residues of the ATP-binding site are highly conserved (estimates of 97 – 100% sequence identity) suggesting ATP-competitive ligands will be unable to distinguish the three Akt isozymes [3–16]. Moreover, due to the high homology with PKA, many reported ATP-competitive Akt inhibitors have little selectivity versus PKA (a master regulator of multiple, critical functions in the cell), and engineering selectivity for Akt has represented a required attribute and a formidable challenge [3–16,25].
Akt is a highly flexible protein [26–30]. In the cytoplasm, a dynamic equilibrium of ‘PH-in’ (or closed) and ‘PH-out’ (or open) conformations exist. On growth factor stimulation, PI3K phosphorylates PIP2 to PIP3, which recruits Akt to the plasma membrane. Here, the PH domain of ‘PH-out’ conformation engages PIP3 to enable phosphorylation of T308 (by PDK1) and Ser473 (by mammalian target of rapamycin (mTOR)C2, previously described as a putative PDK2) leading to kinase activation and downstream signaling pathways involved in cell survival, cell proliferation, glucose metabolism, neuroscience and protein synthesis (Figure 1) [3–32]. As such, Akt is a high profile target for both oncology and numerous CNS disorders, with oncology being the most advanced (vide infra).
Figure 1. Akt activation.

Extracellular signaling events such as growth factor-induced receptor dimerization result in receptor autophosphorylation and recruitment of PI3K. Once PI3K is at the membrane, it phosphorylates the 3′-OH of PI(4,5)P2 and converts it to PI(3,4,5)P3. The interfacial concentration of PI(3,4,5)P3 is regulated by specific phosphatases. PTEN is the phosphatase which removes the phosphate from the 3′-postion and if PTEN function is abrogated through mutation, the PI (3,4,5)P3 levels are elevated. Akt and PDK1 bind to PI(3,4,5)P3 and this results in their colocalization and subsequent phosphorylation of a T308 in the activation loop of Akt by PDK1 and Sr473 by mTORC2. Akt exists in an inactive ‘PH-in’ conformation in the cytoplasm. On recruitment of the plasma membrane, Akt opens to an active, ‘PH-out’ conformation where PIP3 binds to the Akt PH domain allowing phosphorylation to occur. Inset. A) model of PIP analog inhibition, where the PIP analog binds to the PH domain; B) model of ATP competitive inhibition, where inhibitor competes for ATP-binding pocket and leaves kinase in ‘PH-out’ conformation; C) model for allosteric inhibition wherein inhibitor binds within both other PH and kinase domain, forcing the kinase to assume the ‘PH-in’ conformation and thereby inhibiting both the activity of Akt as well as the activation/phosphorylation.
mTOR: Mammalian target of rapamycin; PH: Pleckstrin homology; PTEN: Phosphatase and tensin homolog.
Based on the strong rationale for targeting Akt inhibition, many efforts to identify Akt inhibitors with acceptable pharmaceutical properties have been pursued [3–16]. These attempts can be classified by where the compounds bind to Akt and which specific molecular functions are blocked; importantly, Akt has a number of specific binding pockets which can be targeted with small molecules (Figure 1). Binding in these pockets would be expected to block specific aspects of Akt function and signaling through Akt. The most clearly described pockets are the PI(3,4,5)P3 binding pocket on the PH domain, the phosphorylated carboxy terminus binding hydrophobic pocket and the ATP/substrate pocket. In addition to these three, there may be small molecule binding sites which are created in the interface of the interacting PH domain/hinge region and the kinase domain or pockets created by conformational changes created by the interacting domains [3–16]. This review covers the primary, patent and clinical literature for PIP analogs, alkyl phosphocholines and sulfonamide PH domain inhibitors (ligands that have been shown to bind to the PH-domain and block PIP3 binding), ATP-competitive kinase inhibitors (ligands that complete for ATP and shown to bind to the activated state, ‘PH-out’ conformation) and allosteric kinase inhibitors (non-ATP competitive ligands that possess binding sites in both the PH and kinase domains, and stabilize the ‘PH-in’ conformation of the inactive enzyme).
2. Akt and cancer
Activation of the PI3K/phosphatase and tensin homolog (PTEN)/Akt pathway is common in cancer cells and efforts to understand the role of Akt in the transformed phenotype have led to a large body of research which has been the subject of numerous reviews [3–15,33–35]. The Akt pathway is often improperly regulated in cancer cells due to PTEN deletions or mutations, growth factor mutations leading to high levels of PI3K recruitment to the plasma membrane, mutationally-activated PI3K, Akt overexpression/gene amplification or constitutive activity of Akt [33–41]. As would be expected, activation of the pathway upstream of Akt results in elevated levels of Akt phosphorylation, as observed in prostate cancer, breast cancer and colorectal carcinoma. Additional support for the Akt pathway target rationale has come from the analysis of patients treated with tyrosine kinase inhibitors [42–46]. These inhibitors include gleevec (inhibitor of BCR-ABL, PDGFR and c-Kit), iressa (EGFR inhibitor), tarceva (EGFR inhibitor) and herceptin (Her-2/Neu antibody). In general, patients who have the pathway activated are more likely to respond and those patients who respond have reduced phospho-Akt levels. In these studies, it was shown that patients with elevated phospho-Akt levels in tumors had a higher response rate and longer time to progression than patients with low levels of phospho-Akt. In another example, herceptin-resistant tumors were shown to have elevated levels of phospho-Akt and low levels of PTEN. Similar results have been observed with tarceva and gleevec [47,48]. Together, these results suggest that the transformed phenotype in some tumors is driven by activation of the Akt pathway as a result of receptor activation or overexpression. In these cases, inhibition of receptor function may have a therapeutic benefit. Additional evidence for the importance of the Akt pathway in tumorigenesis has been reported in several papers describing the effects of reducing the levels of PDK1 or Akt1 in PTEN+/− mice. Heterozygous PTEN+/− mice have elevated PI(3,4,5)P3 levels, higher phospho-Akt levels and develop several tumor types including those of the prostate, endometrium, thyroid, adrenal medulla and the intestine [49–51]. Mice carrying a hypomorphic mutation in PDK1 express 80 – 90% less PDK1 than normal mice. PDK1 hypomorphic mice were crossed with PTEN+/− mice and the resulting mice had a greatly reduced tumor incidence [52]. In a similar set of experiments, PTEN+/− mice were mated with Akt1−/−mice. The resulting progeny had dramatically reduced levels of endometrial and prostate neoplasia and there were reduced levels of adrenal medulla and thyroid tumors and intestinal polyps [53]. This large and growing body of genetic and inhibitor data strongly suggest that activation of the Akt pathway has an important role in generating and maintaining the transformed phenotype. Finally, recent preclinical data in animals [3–16] and, more importantly, clinical trial data [54] in humans with multiple Akt inhibitors possessing diverse mechanisms of kinase inhibition further substantiate Akt as a target for cancer therapy.
3. Akt and CNS disorders
In addition to oncology, Akt is center stage as a preclinical target for CNS drug discovery as research has shown that dopamine regulates Akt and its downstream substrate GSK-3β [55]. In particular, substantial neuropharmacological and genetic risk factor data have identified the dysregualtion of the Akt signaling pathway in both schizophrenia and bipolar disorder [55,56]. Interestingly, it has been shown that multiple anti-psychotic agents, such as clozapine and olanzapine, activate the Akt pathway, presumably through G protein; thus, for schizophrenia, Akt activators would be required [57]. Recently, Aamodt and co-workers showed that antipsychotic drugs activate the Caenorhabditis elegans Akt pathway via the DAF-2 insulin/IGF-1 receptor [58]. In addition, protein levels of Akt1 are reduced in post-mortem schizophrenic brains, and an Akt1 haloptype is associated with increased risk for the disease [59]. Disrupted in schizophrenia 1, a susceptibility gene for schizophrenia, is intimately linked to Akt and the pathogenesis of schizophrenia [60,61].
Dopamine hyperfunction has guided drug development for schizophrenia for decades, and both dopaminergic and noradrenergic signaling play critical roles in mood, memory, movement, cognition and reward [62]. Recent studies by Galli and co-workers have shown that Akt signaling is a regulator of norepinephrine transporter (NET) trafficking and norepinephrine homeostasis by controlling NET surface availability [63,64]. The related dopamine transporter is subject to Akt-dependent and isoform-specific (Akt2) regulation of cell surface expression and dopamine homeostasis [65]. Intense efforts are now focused on the pharmacological manipulation of Akt in the CNS, and this story will quickly evolve and therapeutic relevance will be evaluated. At issue is the pro-oncogenic potential of Akt activation by small molecules or biologics.
4. Small molecule Akt inhibitors
4.1 Inhibitors targeting the pleckstrin homology domain of Akt
An alternative approach to classical ATP competitive inhibitors would be to identify compounds that block and/or compete with PI(3,4,5)P3 binding to the PH domain [3–16]. This mode of inhibition would prevent Akt translocation to the plasma membrane by trapping Akt in the cytoplasm and thus preventing activation [66,67]. The feasibility of this approach was suggested by the demonstration that D-3-deoxy-myo-inositols inhibited the growth of transformed cells, and it was subsequently found that the inositol derivative DPI (1) had an IC50 of 35 μM against HT-29 colon cancer growth [68,69]. As shown in Figure 2, replacement of the two ester functionalities with ether linkages led to improved cell stability and resulted in DPIEL (2) which showed improved inhibition (IC50 = 2.1 μM) [70].
Figure 2.

Pleckstrin homology domain inhibitors 1 – 10 that inhibit Akt activation.
Recently, a study examined a range of PI analog inhibitors and found that PIA5 (3), in addition to other simple analogs of DPIEL, was active at inhibiting Akt in H1603 cells (IC50 = 4.13 μM) [71]. PIA5 (3) did not affect phosphorylation of PDK-1 but was found to inhibit phosphorylation of proteins downstream of Akt and significantly increase apoptosis in cells with high levels of constitutive Akt activity. It was also shown to inhibit translocation of a fluorescent Akt-PH construct to the cell membrane which is consistent with the expected mechanism of these inhibitors.
A somewhat related class of Akt inhibitors, the alkyl phosphocholines (Figure 2), is the most advanced in the clinic with perifosine (4), a phospholipid derivative of alkyl phosphocholine, in which Phase III trials are underway [54,72–76]. As expected with this class, 4 also disrupts both MAPK and JNK pathways in addition to Akt signaling. Perifosine (4) is manufactured by AEterna Zentaris, and successfully moved through Phase I into Phase II trials, where 4 showed efficacy in hematological malignancies and solid tumors. Phase III trials are ongoing in multiple myeloma and metastatic colorectal cancer. Erucylphopshocholine (also referred to as eruphosphine or ErPC3) (5) is the back-up to 4, which is in preclinical development. A recent report in 2010 highlighted that ErPC3 induces apoptotic cell death in prostate cancer cells and synergizes with short-term ionizing radiation [77]. The patent cooperation treaty (PCT) covering these compounds was published in 2004, and preferred embodiments are captured in markush 6.
Researchers at the University of Arizona and M.D. Anderson set out to discover novel compounds that bind to the PH domain of Akt and thereby inhibit Akt activation [78–80]. By application of proprietary software, 22 scaffolds were identified and surface plasmon resonance was then used to measure binding of the compounds to the PH domain of Akt. From this effort, a series of 4-aliphatic-N-(1,3,4-thiadiazol-2-yl)benzensulfonamides was discovered, with PHT-427 (7) being the optimal compound. PHT-427 inhibited Akt activation and induced apoptosis at low micro-molar concentrations, inhibited Akt and downstream targets in cells and displayed efficacy in pancreatic tumor xenografts. SAR studies have also been reported surveying multiple domains of the PHT-427 core 8 [78–80]. In addition, a patent application appeared in 2009 covering PHT-427 with a very broad genus 9, and this series also resulted in formation of a small company, PHusis Therapeutics, and a second patent application with a far more narrow genus 10 and describing a topical formulation of PHT-427 (Figure 2) [81,82].
While this approach obviates the selectivity issues associated with the ATP-binding domain homology among kinases, there is the potential to interfere with the function of other PH domain containing proteins [78–80]. Over 500 human proteins possess a 100 – 120 amino-acid PH domain, and the PH domain is the 11th most common domain in the proteome [83,84]. While the amino-acid sequence of PH domains is not universally conserved, the tertiary structure is highly conserved. A subset of ~ 40 PH domains display a remarkable selectivity for binding to phosphorylated PtdIns lipids, which are vital for signal transduction pathways involved in a multitude of critical cellular processes [78–80,83,84]. From all reports to date, little selectivity data are reported versus the ~ 40 PH domains of concern (except for PDK1 and PDPK1), and this leaves a question mark on this approach [78–80].
4.2 ATP-competitive inhibitors
Most kinase inhibitors bind in the enzyme active site and display ATP-competitive behavior [85]. Due to the high degree of homology in the ATP-binding pocket among Akt, PKA and PKC, many typical PKA and PKC inhibitors have been identified as inhibitors of Akt. Indeed, most ATP-competitive programs are driven by achieving selectivity versus PKA as a first tier, as inhibition of PKA, a master regulator of key cell functions with multiple feedback loops, is not acceptable in a development candidate [3–12,25]. As a notable example, the prototypical kinase inhibitor staurosporine has an Akt IC50 of 11 nM and bis-indolemaleimide analogs have subsequently been studied as Akt inhibitors [85]. There are numerous patent applications that list inhibitors of either Akt or PKB, which require searching multiple strings in patent databases. In general, these sources lack data on specificity and mechanism of inhibition; therefore, after a review of compounds in the primary literature, the reader will find tables of markush structures from the patent literature.
Based on a crystal structure of an analog (11) of balanol, a non-selective kinase inhibitor binding in PKA, isosteric analogs were rationally designed [86]. In particular, the diamide compound 12 has an IC50 against Akt of 4 nM and against PKA of 2 nM as well as improved plasma stability relative to 11 (Figure 3). Other ester replacements, including less constrained ether and amine analogs, were significantly less active than 11 and 12. Another known PKA inhibitor that has been the subject of optimization for Akt activity is H-89 (13). H-89 is a potent inhibitor of PKA (IC50 = 0.035 μM) with modest activity at Akt1 (IC50 = 2.5 μM) [87]. Libraries around the lipophilic amino-terminus of 13 identified NL-71 – 101 (14) with an Akt1 IC50 of 3.7 μM and a PKA IC50 of 9 μM. Both 13 and 14 induced apoptosis in OVCAR-3 ovarian carcinoma cells at high concentrations (> 25 μM). Structure–activity relationship (SAR) studies of H-89 showed that modification of the isoquinoline function, the sulfonamide moiety or the diamine linker resulted in loss of Akt1 inhibitory activity. A more potent and moderately selective analog of H-89, 15 inhibits Akt with an IC50 of 0.17 μM (PKC IC50 = 1.4 μM, PDK1 IC50 > 30 μM) [88].
Figure 3.

Early ATP-competitive Akt kinase inhibitors.
High-throughput screening (HTS) of Abbott’s sample collection identified trans-3,4′-bispyridinylethylene 16 as a weak inhibitor against Akt1 (IC50 = 5 μM), and this class of compounds is broadly characterized as ATP-competitive, reversible pan-Akt kinase inhibitors (Figure 4) [89–95]. Potency was improved by appending an aromatic group to the ether-linked side chain. In particular, incorporating an indole (17) led to a 350-fold enhancement in Akt1 activity relative to 16. Cyclizing the olefin onto the central pyridine ring was not well tolerated; however, constraining the conformation of the olefin as a bicyclic isoquinoline significantly improved Akt potency to give compound 18 (IC50 = 1.3 nM). Given the high homology of the ATP-binding pocket, compound 18 showed poor selectivity over closely related kinases from the CMGC family and the other members of the AGC family of kinases, and was even more potent for PKA than either of the other two Akt isoforms, Akt2 (IC50 = 6.8 nM) and Akt3 (IC50 = 35 nM).
Figure 4.

Published ATP-competitive Akt inhibitors, part I.
Abbott then replaced the isoquinoline in 18 with alternate azaheterocycles, which led to the identification of indazole 19, the most potent Akt1 inhibitor reported from this class of compounds (IC50 = 0.16 nM) with an improved kinase selectivity profile. However, compound 19 displayed a poor PK profile with a short half-life, high clearance and no oral bioavailability. Tumor cells treated with 19 caused a dose-dependent decrease in cell growth and in Akt-mediated phosphorylation of downstream targets (including GSK3, FOXO3, TSC2 and mTOR) at concentrations comparable to staurosporine. Compound 19 showed antiproliferative activity on MiaPaCa-2 human pancreatic cancer cells with an IC50 = 100 nM and equal potency against all three isoforms of Akt in cells and also inhibited phosphorylation of GSKα/β and TSC2 with an EC50 of 0.3 uM [89–95].
Consistent with the in vitro studies, indazole 19 inhibited Akt activity in vivo in a dose-dependent manner and significantly slowed tumor growth as monotherapy in two mouse xenograft models, MiaPaCa-2 and 3T3-Akt1. Although Akt activity was not fully inhibited in tumors, concentrations of 19 above the cellular EC50 for 12 h were sufficient to effectively induce apoptosis in tumors. In addition to being effective as a monotherapy, 19 sensitized H460 human lung carcinoma cells to apoptosis when combined with several chemotherapeutic agents including doxorubicin, camptothecin and paclitaxel. Dosing of this class of Akt inhibitors was limited in both magnitude and duration by severe toxicities including lethargy and weight loss. The authors reported a narrow therapeutic window for 19 with maximum tolerated doses only twofold lower than the efficacious doses. Other observations from treated animals include an increase in the total amount of phosphorylated Akt and an increase in plasma insulin levels, a general concern with inhibition of Akt2 noted in Akt2-knockout animals. In addition, all of these analogs suffered from CV toxicity (QT prolongation and hypotension) [89–95]. The addition of a nitrogen atom to the 6-position of the indazole ring provided 20, a compound of comparable Akt inhibition (Akt1 IC50 = 0.6 nM), oral bioavailability (F = 25%) and exposure (AUC = 1.7 μM h); however, no statistically significant hypotension was observed when dosed orally at 150 mg/kg in conscious mice [94].
Chiron recently disclosed another series of ATP-competitive kinase inhibitors that show activity against Akt. Optimization of a weak Akt3 inhibitor (21) led to the identification of compound 22, a potent inhibitor of Akt1 and Akt3 (IC50 = 6 and 2.6 nM, respectively) and a moderately potent inhibitor of Akt2 (IC50 = 23 nM) [96]. Compound 22 showed a poor selectivity profile with closely related kinases PKCα and Kit, and was 30- to 230-fold more potent on PKA than the three Akt isozymes. X-ray of a complex between 22 and PKA showed similar binding interactions as the Abbott compounds (16 – 20) [89–95]. Compound 22 inhibited cell proliferation in DOV13 ovarian carcinoma cells (EC50 = 1 uM) as well as phosphorylation of GSK3 (EC50 = 0.3 uM); however, the superior PKA activity over Akt was also observed in cells. No in vivo activity was reported for this series of kinase inhibitors [96]. GSK reported on a related series derived from HTS hit 23 (Akt1 IC50 = 0.6 nM) in 2009 [97]. SAR studies, aided by a co-crystal of 23 in Akt2, led to the identification of a potent Akt inhibitor 24 (Akt1 IC50 = 0.03 nM): a structure nearly identical to Chiron’s 22. Moreover, a dose-dependent reduction in pGSK3β levels was observed when 24 was dosed in BT474 tumor bearing xenograft mice [97].
In 2008, GSK described the development of a novel series of Akt inhibitors [98]. Once again, using modeling and co-crystallization approaches, optimized preclinical candidate Akt inhibitor 25 (GSK690693) was developed (Figure 4). 25 was a potent Akt inhibitor (Akt1 IC50 = 2 nM, Akt2 IC50 = 13 nM, Akt3 IC50 = 9 nM) which also inhibited the phosphorylation of the downstream target GSK3β in cells. Intraperitoneal administration of 25 in uncompromised mice resulted in the inhibition of GSK3β and repeated dosing of 25 in BT474 breast carcinoma xenograft mice produced a significant delay in tumor growth [98–100]. GSK690693 (25) entered Phase I clinical development as an intravenous formulation, but the trial was halted due to interim results suggesting transient drug-related hyperglycemia [54,98–100]. This, and additional preclinical data, point to GSK690693 causing peripheral insulin resistance. A second Akt inhibitor, GSK2141795, entered Phase I in January 2011 as an oral agent in 70 patients with solid tumors [54,101–103]. Very little is known about GSK2141795 and the structure in unavailable. Thus, it is unclear if GSK2141795 is the same chemical series as 25 or a novel series [54,98–103].
Pfizer disclosed a novel series of Akt inhibitors (Figure 5) based on HTS pyrrolopyrimidine lead 26 (Akt1 IC50 = 210 nM) [104]. Utilizing an X-ray co-crystal structure of several inhibitors in this series with Akt1, the team was able to identify key interactions and launch a structure-based design effort which culminated in 27, with an ~ 100-fold increase in potency (Akt1 IC50 = 2.4 nM). In a cell-based Akt1 assay, 27 displayed an IC50 of 50 nM; however, ancillary kinase activity for 27 and this series was a key issue. None the less, mice xenografted with a Rat-1a tumor line, activated with human (Myr-Akt1), were dosed at 50 mg/kg orally for 10 days, which resulted in tumor growth inhibition of 68% versus vehicle control [104]. In late 2010, Pfizer disclosed 28, a more potent analog with an improved kinase selectivity profile (> 900-fold vs PKA) [105].
Figure 5.


Published ATP-competitive Akt inhibitors, part II.
In 2006, Ko et al. described a unique series of ATP-competitive inhibitors based on a 1H-indazole-4,7-dione scaffold 29 [106]. Though weak (Akt1 IC50 values in the 4.9 – 20 μM range), prototypical inhibitor 29 (Akt1 IC50 = 4.9 μM) afforded a 27% reduction in tumor growth in a PC-3 tumor xenograft model when dosed 5 mg/kg/day for 16 days. Moreover, studies in their lab show that 29 and related congeners have dual inhibitory effects on both activity and phosphorylation of Akt in PC-3 cells (Figure 5) [106].
In 2010, Array Biopharma reported on a pyrrolopyrimidine class of Akt inhibitors represented by 30 (Figure 5) [107]. This series shows potent activity at all three Akt isoforms, modest selectivity versus PKA, robust knockdown of phosphor-PRAS40 in LNCaP cells and U87 tumor xenografts at 20 mg/kg. GSK has also described a third class of tetra-substituted pyridines as ATP-competitive Akt inhibitors exemplified by 31 [108]. Compound 31 displayed dose-dependent inhibition of the phosphorylation of GSK3β and robust efficacy in a BT474 tumor xenograft model in mice. Amgen has described a novel series of thiazole Akt inhibitors, such as 32, which displayed good pharmacokinetic and efficacy in U87 tumor xenograft models [109,110]. In 2011, Array BioPharma and Genetech reported on a new chemotype of Akt inhibitor based on a spirochromane core [111,112]. A HTS campaign identified 33 with modest selectivity versus PKA and mid-micromolar potency against Akt1. Resolution of the hydroxyl diastereomers (they were unable to resolve the spiro core) indicated that the Akt inhibitory activity resided preferentially in the d1 diastereomer 34 (d1Akt1 IC50 = 2.1 μM vs d2Akt1 IC50 = 40 μM). Further optimization and replacement of the isoxazole ring with a 2,6-dimethyl phenyl moiety provided 35, a 260 nM Akt1 inhibitor with > 385-fold selectivity versus PKA. More extensive changes led to the discovery of a 2,7-diazas-piro[4.5]decane core with an amino pyrimidine head group 36 that possessed single digit nanomolar potency (Akt1 IC50 = 8 nM) and > 1000-fold selectivity versus PKA (Figure 5) [111,112].
Figures 6 and 7 depict the markush structures 37 – 68 of granted US patents and patent applications claiming Akt (or PKB) inhibitors that are competitive with ATP [113–140]. Here, Eli Lilly, GSK, Pfizer, Abbott, Amgen, Vertex, Astex, Aventis and Exelixis have numerous patent applications and several issued US patents. A diversity of chemotypes are claimed, and the majority of these patents reflect series for which primary publications have described SAR and activity in detail (vide supra). Related to 30, Array and Genentech have multiple patent applications claiming hydroxylated pyr-imidyl cyclopentanes, 54 – 59 [127–133], with an advanced pan-Akt inhibitor, GDC-0068 (structure not disclosed, though probably closely related to 59 based on narrowed patent claims), in Phase I clinical trials [54,133]. Similarly, Exe-lixis advanced XL-418, a dual Akt–p70S6K inhibitor, into Phase I trials in patients with solid tumors [134–136,141]. Due to low exposure, the trial was halted, and the structure of XL-418 has not been disclosed; however, based on the patent literature it is most likely within the 62 genus [136]. As mentioned above, potential issues with insulin regulation, due to Akt2 inhibition, persist for pan-Akt inhibitors, as well as the traditional issues with kinome selectivity, and PKA in particular, as it has been used as a basis for Akt ligand design in numerous instances [3–13,54].
Figure 6. Markush structures of issued patents and patent applications for ATP-competitive Akt (PKB) inhibitors, part I.


PKB: Protein kinase B.
Figure 7. Markush structures of issued patents and patent applications for ATP-competitive Akt (PKB) inhibitors, part II.


PKB: Protein kinase B.
4.3 Allosteric Akt kinase inhibitors
The study of Akt as a potential therapeutic target for cancer therapy has been hampered by a lack of small molecule kinase inhibitors specific for Akt as well as isoform selective inhibitors that would enable one to test the role of the three isozymes in tumor formation and maintenance. The development of Akt specific and isozyme selective inhibitors that bind to the catalytic kinase domain was predicted to be difficult due to high sequence identity [3–13,142–148]. Therefore, efforts to identify allosteric inhibitors, and thus potential isoform selective inhibitors, were undertaken primarily by Merck [3–13,23,24]. A moderate screening effort directed at identifying compounds capable of inhibiting activated full-length Akt1 produced compounds 60 and 61 (Figure 8) that were subsequently shown to exhibit isozyme selectivity [149]. Compound 60 was selective for Akt 1 (Akt1 IC50 = 4.6 μM, Akt2 IC50 > 250 μM) while compound 61 proved to be a dual Akt1–Akt2 inhibitor (Akt1 IC50 = 2.1 μM, Akt2 IC50 = 21 μM). A detailed kinetic analysis indicated that these inhibitors were not competitive with ATP or peptide substrate. Another unexpected finding was that neither compound inhibited Akt3, PH domain deleted mutants or any of the closely related AGC kinases (PKA, PKC, SGK) at concentrations up to 250 μM. Similar selectivity profiles were also observed in cell-based assays. The dependence on the PH domain for inhibition coupled with a kinetic analysis indicating that these inhibitors were noncompetitive with respect to ATP and peptide substrate supported an allosteric mode of inhibition for these compounds [149]. Further work demonstrated that these inhibitors can prevent phosphorylation of T308 by PDK1 in cells. It was also reported that antibodies bound to either the PH domain or hinge region did not affect enzymatic activity but did prevent inhibition. Based on these findings, a model has been proposed to describe this novel mode of inhibition wherein inhibitors bind to a site that exists only in the presence of the PH domain. This site could be at the interface of the two domains or could be formed as the result of conformational changes induced by domain interactions. In either case, inhibitor binding is proposed to stabilize Akt in a ‘PH-in’ conformation that is not capable of being activated by PDK1 (Figure 1) [149]. A definitive explanation of the mechanism of inhibition will be detailed later in the text as improved allosteric inhibitors became available.
Figure 8.

Early allosteric, isoform selective Akt inhibitors from Merck that established roles for the individual isoforms and provided mechanistic understanding for inhibition.
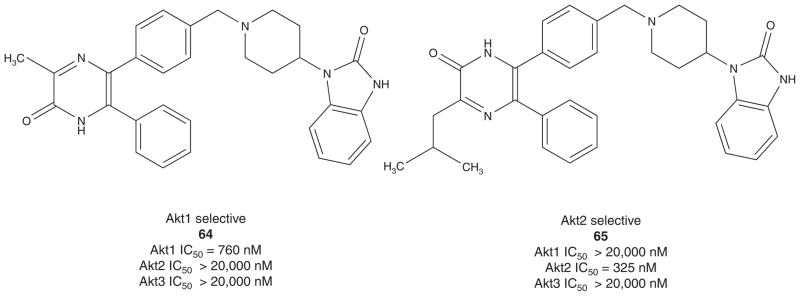
In 2005, Merck disclosed the synthesis and SAR of allosteric Akt inhibitors (Figure 8) derived from the screening lead 61 [23,24]. A library approach generated the more potent dual Akt1–2 inhibitor 62 that still displayed PH domain-dependent inhibition. While a notable advance, 62 displayed poor solubility. Further rounds of library synthesis delivered 63, a potent dual Akt1–2 allosteric inhibitor with improved physical properties and a potential proof of concept compound. Concurrently, monocyclic templates were being synthesized, and this effort surprisingly afforded Akt1- and Akt2-selective inhibitors, 64 and 65, respectively. These isozyme selective tools demonstrated in caspase-3 assays that a maximal apoptotic response required inhibition of both Akt1 and Akt2 in multiple tumor cell lines. Based on these data, compound 63 was examined in a mouse pharmacodynamic assay to determine if it could inhibit Akt and inhibit Akt phosphorylation in vivo. By IP Western, both basal-and IGF-stimulated Akt1 and Akt2 were inhibited in mouse lung (plasma concentrations of 1.5 – 2 μM), with no effect on Akt3 phosphorylation. Similar selectivity profiles were also observed in cell-based C33a IPKA assays, and all of these compounds maintained the same allosteric mode of inhibition observed with 61 [23,24].
Recently, Calleja et al. reported on a mechanistic study on the mode of Akt inhibition by allosteric Akt1–2 dual inhibitor 63, where they identified a critical residue in the PH-domain, Trp 80, which was consistent with the proposed model of PH-induced cavity formation formed in the kinase domain of Akt [150]. This cavity was found in Akt1, but not in Akt3, and hence the high selectivity of 63 for Akt1 versus Akt3. Further in vivo FRET data showed that the presence of 63 locked Akt into its ‘PH-in’ conformation, prohibiting the phosphorylation of Thr 308 and Ser 473 [150]. In late 2010, Brandhuber and co-workers [151] reported on the co-crystal structure of Akt1 with 63 bound, confirming the earlier postulates of Barnett et al. and Calleja et al. [149,150]. Here, they showed a two-site binding mode: the benzimidazolone moiety binds in the PH domain and the imidazoquinoxaline binds in the kinase domain, locking Akt in the ‘PH-in’ or ‘closed’ cytoplasmic conformation thus inhibiting the kinase as well as preventing activation (phosphorylation) of the kinase [151].
One further point of differentiation, Okuzumi et al. demonstrated that ATP-competitive Akt inhibitors such as 19, induce hyperphosphorylation on binding at the ATP site of Akt and, therefore, impart regulatory phosphorylation of their target [152,153]. In contrast, allosteric Akt inhibitors, such as 63, were found to inhibit both the physiological activation of Akt and the drug-induced Akt hyperphosphorylation. These data further highlight the distinct mechanistic differences between ATP-competitive and allosteric Akt inhibitors; however, the ramifications of these differences have yet to be determined in the clinic.
Due to compound liabilities, 63 could not be advanced further, and efforts focused on improving physical properties by the incorporation of a more basic nitrogen into the core ring system, that is, converting the quinoxaline core to a quinoline core (Figure 9) [154]. Both quinoline isomers, 66 and 67, were prepared wherein either the N4 or N1 nitrogen atoms of the quinoxline core of 62 were excised, respectively. Quinoline 66, with the N1 nitrogen intact, was equipotent to 62, whereas quinoline 67 displayed moderate inhibition of Akt1 and almost no inhibition of Akt2. Indeed, the more basic 66 displayed improved physical properties; however, molecular mass was still an issue. To further increase basicity and reduce molecular mass, the quinoline core was truncated to provide a pyridine template. Subsequent rounds of library synthesis afforded pyridine 68, a potent dual Akt1 – 2 allosteric inhibitor that displayed excellent aqueous solubility at pH 4.5. Of note, inhibitors in the pyridine series uniformly displayed greater inhibition of Akt2 than Akt1 in contrast to the bicyclic templates [154].
Figure 9.


Advance iterations of allosteric, isoform selective Akt inhibitors from Merck culminating in MK-2206 in Phase I.
Following these initial disclosures, Merck published seven manuscripts during 2008 – 2009 describing additional refinements to their allosteric Akt inhibitors [155–161], culminating in a successful oral Phase I trial with MK-2206 74. The first manuscript described 69, a congener of 62 with improved aqueous solubility and balanced biochemical and cell-based activity [155]. Then, reports of naphthyridine 70 and naphthyridonones 71 appeared in which 71 was efficacious in inhibiting Akt1 and Akt2 in lung tissue (80 and 75%, respectively) and in tumors (95 and 54%, respectively) in an A2780 tumor xenograft model [156–159]. A series of pyridopyrimidines soon followed, such as 72, and SAR efforts addressed hERG liabilities [160]. The most recent primary literature report describes a series of [1,2,4]triazolo[3,4-f][1,6]naphthyridines, exemplified by 73, with potent and balanced Akt inhibition properties and devoid of hERG liabilities [161]. Further optimization and truncation led to MK-2206 74 (Akt1 IC50 = 8 nM, Akt2 IC50 = 12 nM, Akt3 IC50 = 65 nM). Merck’s MK-2206 is non-competitive with ATP and, like its progenitors, induces a conformational change in the Akt protein to the ‘PH-in’ cytoplasmic conformation which inhibits both the activity and activation of the kinase and blocks signal transduction through the PTEN pathway [12,54]. An oral Phase I study just completed with MK-2206 in patients with solid tumors was found to be generally well tolerated at doses up to 60 mg QOD at concentrations that align with activity in preclinical models [162–164]. Currently, MK-2206 is being studied in combination with various cytotoxic therapies in HER-2 overexpressing breast cancer [54]. In addition, Merck and AstraZeneca recently announced a collaboration around Merck’s MK-2206 and AstraZeneca’s AZD6244 (a highly selective ATP-uncompetitive MEK 1/2 inhibitor) as a novel combination therapy to treat cancer [165,166]. Recent reports detail how the combination of MK-2006 and AZD6244 is more effective than either drug alone in human NSCLC both in vitro and in vivo. Moreover, initial clinic reports on the combination, as well as MK-2206 alone, are quite promising.
Figure 10 depicts the markush structures 75 – 89 of granted US patents claiming allosteric Akt (or PKB) inhibitors based on HTS lead 61 [167–181]. A smaller group of US patents, markush structures 90 – 92, claim analogs based on HTS hit 60 (Figure 11) [182–184]. Virtually all of these patents reflect series for which primary publications have described SAR and activity in detail (vide supra), and are exclusively developed by Merck with one definitive exception. A PCT has recently published from Bayer Schering describing an analog 98 [185] closely related to the Merck series 75 – 89 and 93 – 98 [186–190], but with a different core heterocycle (Figure 12). For all of these, there are multiple use patents describing co-treatment with other cytotoxic agents and chemosensitizers. Finally, in 2005, Imclone described a series of 2,3-dithienylquinoxalines 99 – 101 as Akt inhibitors with Akt1 IC50 values ranging from 66 μM to 300 nM [191]. Though no mechanism of action studies have ever been reported, based on the structural similarity to the Merck allosteric inhibitors, it is presumed that these are also allosteric; however, this presumption needs to be verified [191].
Figure 10. Markush structures of issued US patents for allosteric Akt (PKB) inhibitors.



PKB: Protein kinase B.
Figure 11. Markush structures of issued US patents for allosteric Akt (PKB) inhibitors.

PKB: Protein kinase B.
Figure 12. Markush structures of published patent applications for confirmed and presumed allosteric Akt (PKB) inhibitors.

PKB: Protein kinase B.
4.4 ‘Other’ inhibitors of Akt activation
Triciribine (102), a DNA synthesis inhibitor that entered and failed clinical trials > 20 years ago as the phosphate salt 103, has regained new interest after the discovery in an NCI screen that triciribine inhibited Akt activation (Figure 13) [192–199]. Championed, and patented, by Cheng at the University of South Florida [200–202], 102 was shown to block phosphorylation of all three Akt isoforms at both Ser473 and Thr308; interestingly, 102 did not directly inhibit Akt and was highly selective versus the AGC family of kinases as well as other kinases [199]. 102 also inhibited cell growth, induced apoptosis, decreased phosphorylation levels of downstream substrates and was efficacious in multiple tumor xenograft models. Since 2006, triciribine (102/103, VQD-002 or TCN-PM) has been in two clinical trials (one in adult subjects with metastatic cancer and one in adult patients with advanced hematologic malignancies) conducted by the H. Lee Moffitt Cancer Center in collaboration with VioQuest Pharmaceuticals [202,203]. No SAR or mechanism of action studies have been published. However, a recent study found that in prostate cancer cell lines with constitutively active Akt, such as PC-3 and LNCaP, 102 was sensitized to death receptor-induced apoptosis with TRAIL [199]. Thus, trial design is focusing on patients with elevated Akt to avoid the toxicity seen with 102/103 in early high dose clinical trials as a DNA synthesis inhibitor [203,204].
Figure 13. Triciribine and the analogous phosphate congener.

DNA synthesis inhibitors that have been shown to inhibit Akt activation through a yet undefined mechanism and currently in clinical trials.
5. Biologics
There have been a number of patents filed describing antibodies specific for individual Akt ioszymes as well as general Akt antibodies. Nuclea Biotechnologies has patented antibodies that immunospecifically bind to phospho-Akt and certain p-Akt substrates [205]. Nastech has disclosures on meroduplex ribonucleic acid molecules (mdRNA) capable of decreasing or silencing AKT gene expression [206], and Isis has been granted US patents for antisense modulation, via antisense oligonucleotides, of the expression of all three Akt isoforms [207–209]. Of the biologic approaches, antisense oligonucleotides are the most advanced, with clinical trials underway. Rexhan is advancing RX-0201, a 20-mer antisense oligonucleotide to the RNA encoding Akt1 through the clinic. RX-0201 has completed Phase I trials and is now in Phase II trials for pancreatic cancer in combination with gemcitabine [210–213].
6. Expert opinion
Recent progress in the field of Akt kinase inhibitors and the patent landscape has been reviewed. Since the first disclosures in 2003, our understanding of Akt (PKB) biology, mechanisms of Akt inhibition and the design of small molecule Akt inhibitors has grown tremendously. While the reports of Akt inhibition by ATP-competitive inhibitors and PH domain inhibitors have increased, key issues regarding Akt specificity (vs kinome, and PKA especially, as well as PH domain-containing proteins) and Akt isozyme selectivity remain. With the recent discovery of allosteric Akt kinase inhibitors, these issues have begun to be addressed. Several series of allosteric and PH domain-dependent inhibitors have demonstrated specificity for Akt versus other kinases. Data obtained with these inhibitors indicate that a development candidate should inhibit both Akt1 and Akt2, while shRNA suggests all three. It remains to be determined if a compound that only inhibits Akt1 or Akt2 will have an improved therapeutic window when compared to an inhibitor of all three isozymes, and there may be tumor types in which Akt3 activation is important. The distinct mechanistic underpinnings of the ATP-competitive versus allosteric approaches will probably not be definitively addressed until molecules have been studied in patients beyond the acute setting. Despite the issues and divergent approaches, there is tremendous interest in the target with Amgen, Abbott, Pfizer, GSK, Chiron, Bayer Schering, Eli Lilly, Vertex, Array, Genetech and Merck all publishing and possessing diverse patent portfolios of small molecule Akt inhibitors. Based on the extensive number of granted US patents, Merck is firmly in the lead advancing allosteric inhibitors. All of the other major efforts are focused on ATP-competitive inhibitors, and each with formidable, yet less extensive, patent suite. However, despite a large number of published PCT applications in the time period of 2005 – 2008, few have converted to US patents, suggesting the organizations halted development. Triciribine, a molecule that inhibits Akt activation by an as yet unknown mechanism, is progressing through clinical trials as well. Multiple biologic approaches (antibodies, mdRNA, antisense oligonucleotides) are also underway, with significant IP and clinical success. At the time of this writing, there are > 10 ongoing clinical trials testing the hypothesis that inhibition of Akt will have therapeutic benefit in cancer patients, representing numerous mechanisms (small molecule and biologic) for Akt inhibition and drugs (perifosine, MK-2206, RX-0201, PBI-05204, GSK2141795, GDC-0068, triciribine and ErPC). In addition, many more are staging Phase I trials. Efficacy data from late stage Phase III clinical trials, and a new therapeutic agent for the treatment of cancer, are eagerly awaited. Within the next 3 years, many of the basic science questions will be answered from the human studies: this is a very exciting time for Akt.
Article highlights.
Significant advancements have been made in the design and synthesis of both small molecule and biological inhibitors of Akt activity through multiple, distinct mechanisms. The benefits and concerns of each approach are discussed in detail.
A review of the primary literature on the chemistry, structure–activity relationship and pharmacology of pleckstrin homology domain inhibitors of Akt, ATP-competitive inhibitors of Akt and allosteric inhibitors of Akt is described.
Proof of concept has been achieved for Akt inhibition in multiple preclinical models as well as in humans in early Phase I and Phase II trials, both alone and in combination with other chemotherapeutics.
The current status of preclinical and clinical biologic approaches (antibodies, mdRNA, antisense oligonucleotides) is discussed.
Both a historical and an up-to-date review of the patent literature for inhibitors of Akt is presented for both small molecules and biologics. Companies have aligned themselves as either pursuing ATP-competitive or allosteric ligand design efforts.
The present status of Akt inhibitors in various stages of clinical development is discussed.
This box summarizes key points contained in the article.
Footnotes
Declaration of interest
Lindsley is a professor of phramacology at Vanderbilt University Medical Center. Mattmann is a postdoctoral fellow in the Lindsley laboratory. Sydney Stoops is a graduate student in the Lindsley laboratory. None of the authors have any financial stake or interest in Akt or any of the corporations discussed in this review.
Bibliography
- 1.Jemal A, Bray F, Center MM, et al. Global Cancer Statistics. CA Cancer J Clin. 2011;21:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2•.American Cancer Society; Available from: http//www.cancer.org. This website contains all the latest data on cancer statistics and trends. [Google Scholar]
- 3.Kumar C, Madison V. AKT crystal structure and AKT-specific inhibitors. Oncogene. 2005;24:7493–501. doi: 10.1038/sj.onc.1209087. [DOI] [PubMed] [Google Scholar]
- 4•.Cheng JQ, Lindsley CW, Cheng GZ, et al. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene. 2005;24(50):7482–92. doi: 10.1038/sj.onc.1209088. This is one of the most highly cited reviews in the Akt field covering biology, chemistry and therapeiutic potential. [DOI] [PubMed] [Google Scholar]
- 5.Kim D, Cheng GZ, Lindsley CW, et al. Targeting phosphatidylinositol-3 kinase/Akt pathway for cancer therapy. Curr Opin In Invest Drugs. 2005;6:1250–8. [PubMed] [Google Scholar]
- 6.Lu Y, Wang H, Mills GB. Targeting PI3K-AKT pathway for cancer therapy. Rev Clin Exp Hematol. 2003;7:205–28. [PubMed] [Google Scholar]
- 7.Zdychova J, Komers R. Emerging role of Akt kinase/protein kinase B signaling in pathophysiology of diabetes and its complications. Physiol Res. 2005;54(1):1–16. doi: 10.33549/physiolres.930582. [DOI] [PubMed] [Google Scholar]
- 8.Mitsiades CS, Mitsiades N, Koutsilieris M, et al. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets. 2004;4:235–56. doi: 10.2174/1568009043333032. [DOI] [PubMed] [Google Scholar]
- 9.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–62. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 10•.Barnett SF, Bilodeau MT, Lindsley CW. The Akt/PKB family of protein kinases: a review of small molecule inhibitors and progress towards target validation. Curr Topics Med Chem. 2005;5(2):109–25. doi: 10.2174/1568026053507714. This is one of the first comprehensive reviews on the medicnal chemistry of Akt inhibitors. [DOI] [PubMed] [Google Scholar]
- 11.Layton ME, Barnett SF, Bilodeau MT, et al. Recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr Topics Med Chem. 2007;7:1349–63. doi: 10.2174/156802607781696864. [DOI] [PubMed] [Google Scholar]
- 12.Lindsley CW. The Akt/PKB family of protein kinases: a review of small molecule inhibitors and progress towards target validation: a 2009 update. Curr Topics Med Chem. 2010;10:458–77. doi: 10.2174/156802610790980602. [DOI] [PubMed] [Google Scholar]
- 13• •.Li Q. Recent progress in the discovery of Akt inhibitors as anticancer agents. Exp Opin Ther Patents. 2007;17:1077–130. An excellent and comprehensive review of the patent literature on Akt inhibitors up to 2007. [Google Scholar]
- 14.Laine J, Kunstle G, Obata T, et al. Differential regulation of Akt kinase isoforms by the members of TCL1 oncogene family. J Biol Chem. 2002;277:3743–51. doi: 10.1074/jbc.M107069200. [DOI] [PubMed] [Google Scholar]
- 15.Datta SR, Brunet A, Greenberg ME. Cell survival: a play in three Akts. Genes Dev. 1999;13:2905–27. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 16.Masure S, Haefner B, Wesselink JJ, et al. Molecular cloning, expression and characterization of the human serine/threonine kinase Akt-3. Euro J Biochem. 1999;265:353–60. doi: 10.1046/j.1432-1327.1999.00774.x. [DOI] [PubMed] [Google Scholar]
- 17.Cho H, Thorvaldsen JL, Chu QW, et al. Akt1/PKB alpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–52. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 18.Garofalo RS, Orena SJ, Rafidi K, et al. Severe diabetes, age dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho H, Mu J, Kim JK, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–31. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 20.Easton RM, Cho H, Roovers K, et al. Role for Akt3/Protein Kinase Bgamma in Attainment of normal brain size. Mol Cell Biol. 2005;25(5):1869–78. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21•.Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochem Soc Trans. 2007;35:231–5. doi: 10.1042/BST0350231. This reviews summarizes all of the relevant data regarding Akt knockouts and functions of the three Akt isoforms. [DOI] [PubMed] [Google Scholar]
- 22.Degtyarev M, De Maziere A, Orr C, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–16. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23• •.Lindsley CW, Zhao Z, Duggan ME, et al. Allosteric Akt (PKB) kinase inhibitors. Discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15:761–4. doi: 10.1016/j.bmcl.2004.11.011. The first report of allosteric Akt inhibitors and the develeoment of the first isoform selective Akt inhibitors. [DOI] [PubMed] [Google Scholar]
- 24• •.Defeo-Jones D, Barnett SF, Fu S, et al. Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol Cancer Ther. 2005;4(2):271–9. The first report of the role of indivdual Akt isoforms in preclinical cancer assays using small molecules as opposed to genetic manipulations. [PubMed] [Google Scholar]
- 25.Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal. 2008;1:1–6. doi: 10.1126/scisignal.122re4. [DOI] [PubMed] [Google Scholar]
- 26.Milburn CC, Deak M, Kelly SM, et al. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J. 2003;375:531–8. doi: 10.1042/BJ20031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Cron P, Good VM, et al. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol. 2002;9:940–4. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- 28.Yang J, Cron P, Thompson V, et al. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell. 2002;9:1227–40. doi: 10.1016/s1097-2765(02)00550-6. [DOI] [PubMed] [Google Scholar]
- 29.Garcia Echeverria C, Traxler P, Evans DB. ATP site-directed competitive and irreversible inhibitors of protein kinases. Medici Res Rev. 2000;20:28–57. doi: 10.1002/(sici)1098-1128(200001)20:1<28::aid-med2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 30.Noble MEM, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–5. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 31.Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B alpha. Cur Biol. 1997;7:261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 32.Stokoe D, Stephens LR, Copeland T, et al. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–70. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 33.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18(1):77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 34.Wetzker R, Rommel C. Phosphoinositide 3-kinases as targets for therapeutic intervention. Curr Pharm Des. 2004;10(16):1915–22. doi: 10.2174/1381612043384402. [DOI] [PubMed] [Google Scholar]
- 35•.Chen KQ, Iribarren P, Gong WH, et al. The Essential role of Phosphoinositide 3-kinases (PI3Ks) Mol Immunol. 2005;2(4):241–52. This reviews summarizes the essential role othe PI3K/PTEN pathway. [PubMed] [Google Scholar]
- 36.Simpson L, Parsons R. PTEN: life as a tumor suppressor. Experi Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- 37.Chow L, Lionel ML, Baker SJ. PTEN function in normal and neoplastic growth. Cancer Lett. 2006;241(2):184–96. doi: 10.1016/j.canlet.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 38.Hede K. PTEN takes center stage in cancer stem cell research, works as tumor suppressor. J Natl Cancer Inst. 2006;98(12):808–9. doi: 10.1093/jnci/djj270. [DOI] [PubMed] [Google Scholar]
- 39.Cully M, You H, Levine AJ, et al. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6(3):184–92. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 40.Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478–83. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 41.Bayascas JR, Leslie NR, Parsons R, et al. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN+/−mice. Curr Biol. 2005;15:1839–46. doi: 10.1016/j.cub.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 42.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105(7):2640–53. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 43.Steinert DM, McAuliffe JC, Trent JC. Imatinib mesylate in the treatment of gastrointestinal stromal tumour. Expert Opin Pharmacother. 2005;6(1):105–13. doi: 10.1517/14656566.6.1.105. [DOI] [PubMed] [Google Scholar]
- 44.Cappuzzo F, Finocchiaro G, Metro G, et al. Clinical experience with gefitinib:an update. Crit Rev Oncol Hematol. 2006;58:31–45. doi: 10.1016/j.critrevonc.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Tang PA, Tsao M-S, Moore MJ. A review of erlotinib and its clinical use. Expert Opin Pharmacother. 2006;7(2):177–93. doi: 10.1517/14656566.7.2.177. [DOI] [PubMed] [Google Scholar]
- 46.Plosker GL, Keam SJ. Trastuzumab: a review of its use in the management of Her2-positive metastatic and early-stage breast cancer. Drugs. 2006;66(4):449–75. doi: 10.2165/00003495-200666040-00005. [DOI] [PubMed] [Google Scholar]
- 47.Cappuzzo F, Marrini E, Ceresoli GL, et al. Akt phosphorylation and gefitinib efficacy in patients with advanced non-small cell lung cancer. J Natl Cancer Inst. 2004;96:1133–41. doi: 10.1093/jnci/djh217. [DOI] [PubMed] [Google Scholar]
- 48.Cappuzzo F, Hirsch FR, Rossi E, et al. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non–small-cell lung cancer. J Nat Cancer Inst. 2005;97(9):643–55. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- 49.Suzuki A, de la Pompa JL, Stambolic V, et al. High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol. 1998;8:1169–78. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 50.Di Cristofano A, Pesce B, Cordon-Cardo C, et al. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 51.Podsypanina K, Ellenson LH, Nemes A, et al. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci USA. 1999;96:1563–8. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bayascas J, Leslie J, Parsons N, et al. Hypomorphic mutation of PDK1 suppresses tumorigenesis in PTEN mice. Curr Biol. 2005;15(20):1839–46. doi: 10.1016/j.cub.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 53.Chen M-L, Xu P-Z, Peng X, et al. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/−mice. Genes Dev. 2006;20:1569–74. doi: 10.1101/gad.1395006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54•.Pal KS, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs. 2010;19:1355–66. doi: 10.1517/13543784.2010.520701. This reviews summarizes all of the relevant Akt clincial trial data through 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.La Cour CM, Salles M-J, Pasteau V, et al. Signaling pathways leading to phosphorylation of Akt and GSK-3beta by activation of cloned human and rat cerebral D2 and D3 receptors. Mol Pharm. 2011;79:91–105. doi: 10.1124/mol.110.065409. [DOI] [PubMed] [Google Scholar]
- 56.Dwyer DS, Weeks KR, Aamodt EJ. Drug discovery based on genetic and metabolic findings in schizophrenia. Expert Rev Clin Pharmacol. 2008;1:773–89. doi: 10.1586/17512433.1.6.773. [DOI] [PubMed] [Google Scholar]
- 57.Lu XH, Dweyer DS. Second generation antipsychotic drugs olanzapine, quetiapine and clozapine enhance neurite outgrowth in PC12 cells via the PI3K/Akt, ERK and pertussis toxin-sensitive pathways. J Mol Neurosci. 2005;27:43–64. doi: 10.1385/jmn:27:1:043. [DOI] [PubMed] [Google Scholar]
- 58.Weeks KR, Dwyer DS, Aamodt EJ. Antipsychotic drugs activate the C. elegans Akt pathway via the DAE-2 insulin/IGF-1 receptor. ACS Chem Neurosci. 2010;1:463–73. doi: 10.1021/cn100010p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59• •.Emamian E, Hall D, Birnbaum M, et al. Convergent evidence for impaired AKT-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–7. doi: 10.1038/ng1296. A key paper linking Akt dysfucntion to schizophrenia. [DOI] [PubMed] [Google Scholar]
- 60.Chubb JE, Bradshaw NJ, Soares DC, et al. The DISC1 locus in psychiatric illness. Mol Psychiatry. 2008;13:36–64. doi: 10.1038/sj.mp.4002106. [DOI] [PubMed] [Google Scholar]
- 61.Kim JY, Duan X, Liu C, et al. DISC1 regulates new neuronal development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron. 2009;63:761–73. doi: 10.1016/j.neuron.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lindsley CW, Shipe WD, Wolkenberg SE, et al. Progress towards validating the NMDA receptor hypofunction hypothesis of schizophrenia. Curr Topics Med Chem. 2006;8:771–84. doi: 10.2174/156802606777057599. [DOI] [PubMed] [Google Scholar]
- 63.Siuta MA, Robertson SD, Kocalis H, et al. Dysregulation of the norepinephrine transporter sustains cortical hypodopaminergia and schizophrenia-like behaviors in neuronal rictor null mice’. PLoS Biol. 2010;8:e1000393–401. doi: 10.1371/journal.pbio.1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robertson SD, Owens AW, Matthies HJG, et al. Insulin reveals Akt signaling as a novel regulator of norepinephrine transporter trafficking and norepinephrine homeostasis. J Neurosci. 2010;30:11305–16. doi: 10.1523/JNEUROSCI.0126-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Speed NR, Matthies HJG, Kennedy JP, et al. Akt dependent and isoform specific regulation of the dopamine transporter cell surface expression. ACS Chem Neurosci. 2010;1:476–481. doi: 10.1021/cn100031t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gills JJ, Dennis PA. The development of phosphatidylinositol ether lipid analogues as inhibitors of the serine/threonine kinase, Akt. Expert Opin Invest Drugs. 2004;13:787–97. doi: 10.1517/13543784.13.7.787. [DOI] [PubMed] [Google Scholar]
- 67.Meuillet EJ, Mahadevan D, Vankayalapati H, et al. Specific inhibition of the Akt1 pleckstrin homology domain by D-3-deoxyphosphatidyl-myo-inositol analogues. Mol Cancer Ther. 2003;2:389–99. [PubMed] [Google Scholar]
- 68.Powis G, Aksoy IA, Melder DC, et al. D-3-deoxy-3-substituted myo-inositol analogues as inhibitors of cell growth. Cancer Chemother Pharmacol. 1991;29:95–104. doi: 10.1007/BF00687317. [DOI] [PubMed] [Google Scholar]
- 69.Kozikowski AP, Kiddle JJ, Frew T, et al. Synthesis and biology of 1D-3-deoxyphosphatidylinositol: a putative antimetabolite of phosphatidylinositol-3-phosphate and an inhibitor of cancer cell colony formation. J Med Chem. 1995;38:1053–6. doi: 10.1021/jm00007a001. [DOI] [PubMed] [Google Scholar]
- 70.Qiao L, Nan F, Kunkel M, et al. 3-Deoxy-D-myo-inositol 1-phosphate, 1-phosphonate, and ether lipid analogues as inhibitors of phosphatidylinositol-3-kinase signaling and cancer cell growth. J Med Chem. 1998;41:3303–6. doi: 10.1021/jm980254j. [DOI] [PubMed] [Google Scholar]
- 71.Castillo SS, Brognard J, Petukhov PA, et al. Preferential inhibition of Akt and killing of Akt-dependent cancer cells by rationally designed phosphatidylinositol ether lipid analogues. Cancer Res. 2004;64:2782–92. doi: 10.1158/0008-5472.can-03-1530. [DOI] [PubMed] [Google Scholar]
- 72.Gills JJ, Dennis PA. Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep. 2009;11:102–10. doi: 10.1007/s11912-009-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73•.Hideshima T, Catley L, Yasui H, et al. Perifosine, an oral bioactive novel alkylphospholipid, inhibits Akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood. 2006;107:4053–62. doi: 10.1182/blood-2005-08-3434. The first report of perifosine showing efficacy in preclincial models by inhibiton of Akt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vink SR, Schellens JHM, Beijnen JH, et al. Phase I and pharmacokinetic study of combined treatment with perifosine and radiation in patients with advanced solid tumours. Radiother Oncol. 2006;80:207–13. doi: 10.1016/j.radonc.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 75.Ghobrial IM, Leleu X, Rubin N, et al. Phase II trial of the novel oral Akt inhibitor perifosine in relapsed and/or refractory Waldenstrom macroglobulinemia (WM). Meeting Abstracts. J Clin Oncol. 2008;26(15 Suppl):8546. [Google Scholar]
- 76.Bethesda, MD: US National Institutes of Health; 2010. [Last accessed 1 September 2010]. NCT01097018: a Phase III randomized study to assess the efficacy and safety of perifosine plus capecitabine versus placebo plus capecitabine in patients with refractory advanced colorectal cancer. Clinical trials.gov. Available from: http://clinicaltrials.gov/ct2/show/NCT01097018?term=NCT01097018&rank=1. [Google Scholar]
- 77.Zentaris GMBH. 2004012744. WO. 2004
- 78.Moses SA, Ali MA, Zuohe S, et al. In vitro and in vivo activity of novel small-molecule inhibitors targeting the pleckstrin homology domain of protein kinase B/Akt. Cancer Res. 2009;69:5073–81. doi: 10.1158/0008-5472.CAN-08-3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meuillet EJ, Zuohe S, Lemos R, et al. Molecular pharmacology and antitumor activity of PHT-427, a novel Akt/phosphotidylinostitide-dependent protein kinase 1 pleckstrin homology domain inhibitor. Mol Cancer Ther. 2010;9:706–17. doi: 10.1158/1535-7163.MCT-09-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ahad AM, Zuohe S, Du-Cuny L, et al. Devleopment of sulfonamide Akt PH domain inhibitors. Bioorg Med Chem Lett. 2011;19:2046–54. doi: 10.1016/j.bmc.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.University of Texas; University of Arizona. 2009129267. WO. 2009
- 82.PHusis Therapeutics. 2011032169. WO. 2011
- 83.Rebecchi MJ, Scarlata S. PLeckstrin homology domains: a common fold with diverse functions. Annu Rev Biophys Biomol Struct. 1998;27:503–28. doi: 10.1146/annurev.biophys.27.1.503. [DOI] [PubMed] [Google Scholar]
- 84.Workman P, Clark PA, Guillard S, et al. Druggin the PI3 kinome. Nat Biotechnol. 2006;24:794–6. doi: 10.1038/nbt0706-794. [DOI] [PubMed] [Google Scholar]
- 85.Li Q, Zhu GD. Targeting serine/threonine protein kinase B/Akt and cell-cycle checkpoint kinases for treating cancer. Curr Top Med Chem. 2002;2:939–71. doi: 10.2174/1568026023393318. [DOI] [PubMed] [Google Scholar]
- 86.Breitenlechner CB, Wegge T, Berillon L, et al. Structure-based optimization of novel azepane derivatives as PKB inhibitors. J Med Chem. 2004;47:1375–90. doi: 10.1021/jm0310479. [DOI] [PubMed] [Google Scholar]
- 87.Chijiwa T, Mishima A, Hagiwara M, et al. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–72. [PubMed] [Google Scholar]
- 88.Reuveni H, Livnah N, Geiger T, et al. Toward a PKB inhibitor: modification of a selective PKA inhibitor by rational design. Biochemistry. 2002;41:10304–14. doi: 10.1021/bi0202530. [DOI] [PubMed] [Google Scholar]
- 89.Li Q, Li T, Zhu GD, et al. Discovery of trans-3,4′′-bispyridinylethylenes as potent and novel inhibitors of protein kinase B (PKB/Akt) for the treatment of cancer: synthesis and biological evalutions. Bioorg Med Chem Lett. 2006;16:1679–85. doi: 10.1016/j.bmcl.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 90.Li Q, Woods KW, Thomas S, et al. Synthesis and strucutre-activity relationship of trans-3,4′′-bispyridinylethylenes: discovery of a potent 3-isoquinolinylpyridine inhibitor of protein kinase B (PKB/Akt) for the treatment of cancer. Bioorg Med Chem Lett. 2006;16:2000–7. doi: 10.1016/j.bmcl.2005.12.065. [DOI] [PubMed] [Google Scholar]
- 91.Thomas SA, Li T, Woods KW, et al. Identification of a novel 3,5-disubstituted pyridine as a potent, selective, and orally active inhibitor of Akt1 kinase. Bioorg Med Chem Lett. 2006;16:3740–4. doi: 10.1016/j.bmcl.2006.04.046. [DOI] [PubMed] [Google Scholar]
- 92.Zhu G-D, Gandhi VB, Gong J, et al. Discovery and SAR of oxindole-pyridine-based protein kinase B/Akt inhibitors for treating cancers. Bioorg Med Chem Lett. 2006;16:3424–9. doi: 10.1016/j.bmcl.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 93.Woods KW, Fischer JP, Claiborne A, et al. Synthesis and SAR of indazole-pyridine based protein kinase B/Akt inhibitors. Bioorg Med Chem Lett. 2006;14:6832–46. doi: 10.1016/j.bmc.2006.06.047. [DOI] [PubMed] [Google Scholar]
- 94.Luo Y, Shoemaker AR, Liu X, et al. Potent and selective inhibitors of Akt kinases slow the progress of tumors in vivo. Mol Cancer Ther. 2005;4:977–86. doi: 10.1158/1535-7163.MCT-05-0005. [DOI] [PubMed] [Google Scholar]
- 95.Zhou G-D, Gandhi VB, Gong J, et al. Synthesis of potent, selective and orally bioavailable indazole-pyridine series of protein kinase B/Akt inhibitors with reduced hypertension. J Med Chem. 2007;50:2990–3003. doi: 10.1021/jm0701019. [DOI] [PubMed] [Google Scholar]
- 96.Lin X, Murray JM, Rico AC, et al. Discovery of 2-pyrimidyl-5-amidothiophenes as potent inhibitors for AKT: synthesis and SAR studies. Bioorg Med Chem Lett. 2006;16:4163–8. doi: 10.1016/j.bmcl.2006.05.092. [DOI] [PubMed] [Google Scholar]
- 97.Seefeld MA, Rouse MB, McNulty KC, et al. Discovery of 5-pyrrolpyridinyl-2-thiopenecarboxamides as potent Akt inhibitors’. Bioorg Med Chem Lett. 2009;19:2244–8. doi: 10.1016/j.bmcl.2009.02.094. [DOI] [PubMed] [Google Scholar]
- 98.Heerding DA, Rhodes N, Leber JD, et al. Identification of 4-(2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{(3S)-3-piperidinylmethyl]oxy}-1H-imidazo4,5-c] pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a Novel Inhibitor of AKT Kinase’. J Med Chem. 2008;51:5663–79. doi: 10.1021/jm8004527. [DOI] [PubMed] [Google Scholar]
- 99.Rouse MB, Seefeld MA, Leber JD, et al. Aminofurazans as potent inhibitors of AKT kinase. Bioorg Med Chem Lett. 2009;19:1508–11. doi: 10.1016/j.bmcl.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 100.Altomare DA, Zhang L, Deng J, et al. GSK690693 delays tumor onset and progression in genetically defined mouse models expressing activated Akt. Clin Cancer Res. 2010;16:486–96. doi: 10.1158/1078-0432.CCR-09-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bethesda, MD: US National Institutes of Health; 2007. NCT00493818: open-label study to investigate the safety, tolerability, PK, and pharmacodynamics of the AKT inhibitor GSK690693. Clinical trials.gov. Available from: http://clinicaltrials.gov/ct2/show/NCT00493818. [Google Scholar]
- 102.LoRusso P, Hurwitz H, Chiorean E, et al. AKT inhibitor GSK690693: preliminary results from the first time in human study. 1 Annual Meeting. AACR Meeting Abstracts; 2008; p. LB-68. [Google Scholar]
- 103.Bethesda, MD: US National Institutes of Health; 2009. NCT00920257: a phase I, open-label, two-stage study to investigate the safety, tolerability, pharmacokinetics and pharmacodynamics of the oral AKT inhibitor GSK2141795 in subjects with solid tumors or lymphomas. Clinical trials.gov. Available from: http://clinicaltrials.gov/ct2/show/NCT00920257?term=NCT00920257. [Google Scholar]
- 104.Lippa B, Pan G, Corbett M, et al. Synthesis and structure based optimization of novel Akt inhibitors. Bioorg Med Chem Lett. 2008;18:3359–63. doi: 10.1016/j.bmcl.2008.04.034. [DOI] [PubMed] [Google Scholar]
- 105.Freeman-Cook KD, Autyr C, Borzillo G, et al. Design of selective ATP-competitive inhibitors of Akt. J Med Chem. 2010;53:4615–22. doi: 10.1021/jm1003842. [DOI] [PubMed] [Google Scholar]
- 106.Ko JH, Yeon SW, Ryu JS, et al. Synthesis and biological evaluation of 5-arylamino-6-chloro-1H-indazole-4,7-diones as inhibitors of protein kinase B/Akt. Bioorg Med Chem Lett. 2006;16:6001–5. doi: 10.1016/j.bmcl.2006.08.120. [DOI] [PubMed] [Google Scholar]
- 107.Blake JF, Kallan NC, Xiao D, et al. Discovery of pyrrolopyrimidine inhibitors of Akt. Bioorg Med Chem Lett. 2010;20:5607–12. doi: 10.1016/j.bmcl.2010.08.053. [DOI] [PubMed] [Google Scholar]
- 108.Lin H, Yamashita DS, Xie R, et al. Tetrasubstituted pyridines as potent and selective Akt inhibitors: reduced CYP450 and hERG inhibition of aminopyridines. Bioorg Med Chem Lett. 2010;20:648–88. doi: 10.1016/j.bmcl.2009.11.061. [DOI] [PubMed] [Google Scholar]
- 109.Zeng Q, Allen JG, Bourbeau MP, et al. Azole-based inhibitors of Akt/PKB for the treatment of cancer. Bioorg Med Chem Lett. 2010;20:1559–64. doi: 10.1016/j.bmcl.2010.01.067. [DOI] [PubMed] [Google Scholar]
- 110.Zeng Q, Bourbeau MP, Wohlheiter GE, et al. 2-Aminothiadiazole inhibitors of Akt1 as potential cancer therapeutics. Bioorg Med Chem Lett. 2010;20:1652–6. doi: 10.1016/j.bmcl.2010.01.046. [DOI] [PubMed] [Google Scholar]
- 111.Xu R, Banka A, Blake F, et al. Discovery of spirocyclic sulfonamides as potent Akt inhibitors with exquisite selectivity against PKA. Bioorg Med Chem Lett. 2011;21:2335–40. doi: 10.1016/j.bmcl.2011.02.098. [DOI] [PubMed] [Google Scholar]
- 112.Kallan NC, Spencer KL, Blake JF, et al. Discovery and SAR of spirochromane Akt inhibitors. Bioorg Med Chem Lett. 2011;21:2410–14. doi: 10.1016/j.bmcl.2011.02.073. [DOI] [PubMed] [Google Scholar]
- 113.Eli Lilly and Co. 7449477. US. 2008
- 114.Eli Lilly and Co. 7414063. US. 2008
- 115.Amgen, Inc. 20090298836. WO. 2009
- 116.Amgen, Inc. 20090275592. WO. 2009
- 117.Astra Zeneca AB. 20090163524. WO. 2009
- 118.SmithKline Beecham Corp. 20100137338. WO. 2010
- 119.SmithKline Beecham Corp. 20100267759. WO. 2010
- 120.Imperial College Innovations, Ltd. 20080039527. WO. 2008
- 121.Eli Lilly & Co. 20090221633. WO. 2009
- 122.SmithKline Beecham Corp. 20100056523. WO. 2010
- 123.SmithKline Beecham Corp. 20090209607. WO. 2009
- 124.St. Jude’s Children Research Hospital. 20060241108. WO. 2006
- 125.SmithKline Beecham Corp. 20080318947. WO. 2008
- 126.St. Jude’s Children Research Hospital. 2006094207. WO. 2006
- 127.Array Biopharma. 2008006025. WO. 2008
- 128.Array Biopharma. 2008006032. WO. 2008
- 129.Array Biopharma. 2009006569. WO. 2009
- 130.Array Biopharma. 2008006039. WO. 2008
- 131.Array Biopharma. 2008006040. WO. 2008
- 132.Array Biopharma. 2009006567. WO. 2009
- 133.Array Biopharma. 2009089453. WO. 2009
- 134.Exelixis 2005117909. WO. 2005
- 135.Exelixis 2006071819. WO. 2005
- 136.Vertex 2005030776. WO. 2005
- 137.Vertex 7041687. US. 2006
- 138.Vertex 2005039564. WO. 2005
- 139.Astex 2005061463. WO. 2005; 2006136823. WO. 2006; 2006136830. WO. 2006; 2427406. GB. 2006
- 140.Aventis Pharma. 0248884. US. 2004; 2006010641. WO. 2006; 2006010642. WO. 2006; 2006010643. WO. 2006
- 141. NCT00460278: a phase 1 dose-escalation study of the safety and pharmacokinetics of XL418 administered orally daily to subjects with solid tumors. Available from: http://www.clinicaltrials.gov.
- 142.Breitenlechner CB, Wegge T, Berillon L, et al. Structure-based optimization of novel azepane derivatives as PKB inhibitors. J Med Chem. 2004;47:1375–90. doi: 10.1021/jm0310479. [DOI] [PubMed] [Google Scholar]
- 143.Zinda MJ, Johnson MA, Paul JD, et al. AKT-1,−2, and-3 are expressed in both normal and tumor tissues of the lung, breast, prostate, and colon. Clin Cancer Res. 2001;7(8):2475–9. [PubMed] [Google Scholar]
- 144.Hsu JH, Shi Y, Hu L, et al. Role of the AKT kinase in expansion of multiple myeloma clones: effects on cytokine-dependent proliferative and survival responses. Oncogene. 2002;21:1391–400. doi: 10.1038/sj.onc.1205194. [DOI] [PubMed] [Google Scholar]
- 145.Brognard J, Clark AS, Ni Y, et al. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001;61:3986–97. [PubMed] [Google Scholar]
- 146.Kozikowski AP, Sun H, Brognard J, et al. Novel PI analogues selectively block activation of the pro-survival serine/threonine kinase Akt. J Am Chem Soc. 2003;125:1144–5. doi: 10.1021/ja0285159. [DOI] [PubMed] [Google Scholar]
- 147.Graff JR. Emerging targets in the AKT pathway for treatment of androgen-independent prostatic adenocarcinoma. Expert Opin Ther Targets. 2002;6:103–13. doi: 10.1517/14728222.6.1.103. [DOI] [PubMed] [Google Scholar]
- 148.Yang ZZ, Tschopp O, Baudry A, et al. Physiological functions of protein kinase B/Akt. Biochem Soc Trans. 2004;32:350–4. doi: 10.1042/bst0320350. [DOI] [PubMed] [Google Scholar]
- 149.Barnett SF, Defeo-Jones D, Fu S, et al. Identification and characterization of pleckstrin homology domain dependent and isozyme specific Akt inhibitors. Biochem J. 2004;385:399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Calleja V, Laguerre M, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: Structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:189–200. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151• •.Wu WI, Voegtil WC, Sturgis HL, et al. Crystal structure of humanAKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One. 2010;5:e12913. doi: 10.1371/journal.pone.0012913. The first crystal strucutre of full length Akt with an allosteric inhibitor bound. This paper confirms the two-site binding pose postulated earlier by Barnnet using biochemical and genetic approaches. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Okuzumi T, Fielder D, Zhang C, et al. Inhibitor hijacking of Akt activation. Nat Chem Bio. 2009;5:484–93. doi: 10.1038/nchembio.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ananthanarayanan B, Fosbrink M, Radhar M, et al. Live cell molecular analysis of Akt reveals roles for activation loop phosphorylation. J Biol Chem. 2007;282:36634–41. doi: 10.1074/jbc.M706227200. [DOI] [PubMed] [Google Scholar]
- 154.Zhao Z, Duggan ME, Barnett SF, et al. Discovery of 2,3,5-trisubstituted pyridine derivatives as potent Akt1 and Akt2 dual inhibitors. Bioorg Med Chem Lett. 2005;15:905–9. doi: 10.1016/j.bmcl.2004.12.062. [DOI] [PubMed] [Google Scholar]
- 155.Zhao Z, Robinson RG, Barnett SF, et al. Development of potent, allosteric Akt1 and Akt2 dual inhibitors with improved physical properties and cell activity. Bioorg Med Chem Lett. 2008;18:49–53. doi: 10.1016/j.bmcl.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 156.Bilodeau MT, Balitza AE, Hoffman JM, et al. Allosteric inhibitors of Akt1 and Akt2: a naphthyridinone with efficacy in an A2780 tumor xenograft model. Bioorg Med Chem Lett. 2008;18:3178–82. doi: 10.1016/j.bmcl.2008.04.074. [DOI] [PubMed] [Google Scholar]
- 157.Wu Z, Robinson RG, Fu S, et al. Rapid assembly of diverse and potent allosteric Akt inhibitors. Bioorg Med Chem Lett. 2008;18:2211–14. doi: 10.1016/j.bmcl.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 158.Wu Z, Hartnett JC, Neilson LA, et al. Development of pyridopyrimidines as potent Akt1/2 inhibitors. Bioorg Med Chem Lett. 2008;18:1274–9. doi: 10.1016/j.bmcl.2008.01.054. [DOI] [PubMed] [Google Scholar]
- 159.Sui T, Liang J, Arruda J, et al. Discovery of potent and cell-active allosteric dual Akt1 and 2 inhibitors’. Bioorg Med Chem Lett. 2008;18:4186–90. doi: 10.1016/j.bmcl.2008.05.085. [DOI] [PubMed] [Google Scholar]
- 160.Sui T, Li Y, Nagasawa J, et al. The design and synthesis of potent and cell-active allosteric dual Akt1 and 2 inhibitors devoid of hERG activity. Bioorg Med Chem Lett. 2008;18:4191–4. doi: 10.1016/j.bmcl.2008.05.084. [DOI] [PubMed] [Google Scholar]
- 161.Li Y, Liang J, Sui T, et al. Allosteric inhibitors of Akt 1 and Akt2: discovery of 1,2,4]triazolo3,4-f]1,6]naphthyridines with potent and balanced activity. Bioorg Med Chem Lett. 2009;19:834–6. doi: 10.1016/j.bmcl.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 162.Hirai H, Sootome H, Nakatsuru Y, et al. An allosteric Akt inhibitor, MK-2206 enhanced anti-tumor efficacy by standard of care agents or molecular targeted drugs in vitro and in vivo. AACR Meeting Abstracts; 2009. p. 3707. [DOI] [PubMed] [Google Scholar]
- 163.Yap TA, Patnaik A, Fearen I, et al. First-in-class Phase I trial of a selective Akt inhibitor, MK2206 (MK), evaluating alternate day (QOD) and once weekly (QW) doses in advanced cancer patients (pts) with evidence of target modulation and antitumor activity. Meeting Abstracts. J Clin Oncol. 2010;28(15 Suppl):3009. [Google Scholar]
- 164.Hirai H, Sootome H, Nakatsuru Y, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–67. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 165.Lu W, Defeo-Jones D, Davis LJ, et al. In vitro and in vivo antitumor activities of MK-2206, a new allosteric Akt inhibitor. AACR Meeting Abstracts; 2009. p. 3714. [Google Scholar]
- 166.Johnson LA. AstraZeneca, Merck collaborate on cancer treatment. AP Business Wire. 2009 Jun 1; [Google Scholar]
- 167.Merck 7034026. US. 2006
- 168.Merck 7273869. US. 2007
- 169.Merck 7223738. US. 2007
- 170.Merck 7638530. US. 2009
- 171.Merck 7579355. US. 2009
- 172.Merck 7576209. US. 2009
- 173.Merck 7544677. US. 2009
- 174.Merck 7524850. US. 2009
- 175.Merck 7399764. US. 2008
- 176.Merck 7396832. US. 2008
- 177.Merck 7414055. US. 2008
- 178.Merck 7304063. US. 2008
- 179.Merck 7750151. US. 2010
- 180.Merck 7655649. US. 2010
- 181.Merck 7589068. US. 2009
- 182.Merck 6960584. US. 2005
- 183.Merck 7098208. US. 2006
- 184.Merck 6958334. US. 2005
- 185.Bayer Schering Pharma. 20090091824. WO. 2009
- 186.Merck 20100222321. WO. 2010
- 187.Merck 20100075970. WO. 2010
- 188.Merck 20090062327. WO. 2009
- 189.Merck 20090253734. WO. 2009
- 190.Merck 20100022573. WO. 2010
- 191.Imclone. 2005007099. WO. 2005
- 192.Schramm KH, Townsend LB. The synthesis of 6-amino-4-methyl-8-(beta-D-ribofuranosyl)-(4H,8H)-pyrrolo-4,3,2-de] pyrimido4,5-c]pyridazine, a new tricyclic nucleotide. Tetrahedron Lett. 1971;49:4757–60. [Google Scholar]
- 193.Yang L, Dan HC, Sun M, et al. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumour activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–9. doi: 10.1158/0008-5472.CAN-04-0343. [DOI] [PubMed] [Google Scholar]
- 194.Schweinsberg PD, Smith RG, Loo TL. Identification of the metabolites of an antitumour tricyclic nucleoside (NSC-154020) Biochem Pharmacol. 1981;30:2521–6. doi: 10.1016/0006-2952(81)90577-3. [DOI] [PubMed] [Google Scholar]
- 195.Mittelman A, Casper ES, Godwin TA, et al. Phase I study of tricyclic nucleoside phosphate. Cancer Treat Rep. 1983;67:159–62. [PubMed] [Google Scholar]
- 196.Feun LG, Savaraj N, Bodey GP, et al. Phase I study of tricyclic nucleoside phosphate using a five-day continuous infusion schedule. Cancer Res. 1984;44:3608–12. [PubMed] [Google Scholar]
- 197.Schilcher RB, Haas CD, Samson MK, et al. Phase I evaluation and clinical pharmacology of tricyclic nucleoside 5′-phosphate using a weekly intravenous regimen. Cancer Res. 1986;46:3147–51. [PubMed] [Google Scholar]
- 198.Hoffman K, Holmes FA, Fraschini G, et al. Phase I – II study: triciribine (tricyclic nucleoside phosphate) for metastatic breast cancer. Cancer Chemother Pharmacol. 1996;37:254–8. doi: 10.1007/BF00688325. [DOI] [PubMed] [Google Scholar]
- 199.Dieterle A, Orth R, Daubrawa M, et al. The Akt inhibitor triciribine sensitizesd prostate carcinoma cells to TRAIL-induced apopotsis. Int J Cancer. 2009;125:932–41. doi: 10.1002/ijc.24374. [DOI] [PubMed] [Google Scholar]
- 200.University of South Florida. 0028339. US. 2010
- 201.University of South Florida. 0008327. US. 2011
- 202.University of South Florida. 0113381. US. 2010
- 203. NCT00363454: Phase I Study of Triciribine Phosphae Monohydrate (TCN-PM, VD-002) in Adult Subjects with Metastatic Cancer. 2011 Clinical trials.gov. Available from: http://clinicaltrials.gov/ct2/show/NCT00363454.
- 204. NCT00642031: Triciribine Phosphae Monohydrate (TCN-PM, VD-002) in Adult Patients with Advanced Hematologic Malignancies. 2011 Clinicaltrials.gov. Available from: http://clinicaltrials.gov/ct2/show/NCT00642031.
- 205.Nuclea Biotechnologies LLC. 20100092473. WO. 2010
- 206.Nastech Pharmaceuticals. 20080293136. WO. 2008
- 207.Isis Pharmaceuticals. 5958773. US. 1999
- 208.Isis Pharmaceuticals. 6043090. US. 2000
- 209.Isis Pharmaceuticals. 6187586. US. 2001
- 210.Yoon H, Kim DJ, Ahn EH, et al. Antitumor activity of a novel antisense oligonucleotide against Akt1. J Cell Biochem. 2009;108:832–8. doi: 10.1002/jcb.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.US Patent Granted for Rexahn Pharmaceuticals Leading Cancer Candidate. Rockville, MD: rexahn pharmaceuticals press release; Nov 6, 2006. Available from: http://www.rexahn.com/cms/index.php/2006/11/us-patent-grantedfor-rexahn-pharmaceuticals-leadingcancer-candidate-2. [Google Scholar]
- 212.Marshall J, Posey J, Hwang J, et al. A Phase I trial of RX-0201 (AKT anti-sense) in patients with an advanced cancer. Meeting Abstracts. J Clin Oncol. 2007;25(18 Suppl):3564. [Google Scholar]
- 213.Bethesda, MD: US National Institutes of Health; 2009. NCT01028495: a safety and efficacy study of RX-0201 plus gemcitabine in metastatic pancreatic cancer. Clinicaltrials.gov. Available from: http://clinicaltrials.gov/ct2/show/NCT01028495. [Google Scholar]