

Abstract
Over the past ten years, proteasome inhibition has emerged as an effective therapeutic strategy for treating multiple myeloma (MM) and some lymphomas. In 2003, Bortezomib (BTZ) became the first proteasome inhibitor approved by the U.S. Food and Drug Administration (FDA). BTZ-based therapies have become a staple for the treatment of MM at all stages of the disease. The survival rate of MM patients has improved significantly since clinical introduction of BTZ and other immunomodulatory drugs. However, BTZ has several limitations. Not all patients respond to BTZ-based therapies and relapse occurs in many patients who initially responded. Solid tumors, in particular, are often resistant to BTZ. Furthermore, BTZ can induce dose-limiting peripheral neuropathy (PN). The second generation proteasome inhibitor Carfizomib (CFZ; U.S. FDA approved in August 2012) induces responses in a minority of MM patients relapsed from or refractory to BTZ. There is less PN compared to BTZ. Four other second-generation proteasome inhibitors (Ixazomib, Delanzomib, Oprozomib and Marizomib) with different pharmacologic properties and broader anticancer activities, have also shown some clinical activity in bortezomib-resistant cancers. While the mechanism of resistance to bortezomib in human cancers still remains to be fully understood, targeting the immunoproteasome, ubiquitin E3 ligases, the 19S proteasome and deubiquitinases in pre-clinical studies represents possible directions for future generation inhibitors of ubiquitin-proteasome system in the treatment of MM and other cancers.
Keywords: Bortezomib, carfilzomib, combination therapy, immunoproteasomes, multiple myeloma, proteasome inhibitor, resistance to proteasome inhibitors, ubiquitin-proteasome pathway
INTRODUCTION
The balance of protein synthesis and degradation is tightly regulated in healthy, normal cells. Most intracellular proteins are degraded via the ubiquitin-proteasome system (UPS), in which proteins are tagged by ubiquitin and then recognized by the 26S proteasome complex, which degrades them into small peptides (Fig. 1) [1–3]. Since dysfunction of this system is linked to many human diseases, including neurodegenerative disorders, genetic diseases, autoimmue diseases, and many cancers, much work has been conducted by targeting the UPS as a potential treatment of these conditions [4–8]. In the years since the U.S. FDA’s 2003 approval of the proteasome inhibitor bortezomib (BTZ; Table 1) for the treatment of multiple myeloma (MM) and mantle cell lymphoma (MCL), the drug has provided ample proof that targeting the UPS is a viable route for the treatment of human cancer. The treatment of MM, in particular, has been revolutionized by the advent of BTZ and immunomodulatory drugs [5–11], with the current overall survival in the MM patients increased by 2 to 3-fold [12]. However, some limitations of BTZ treatment have become evident, including baseline resistance in some patients with MM and virtually all patients with solid tumors, the development of acquired BTZ resistance in many initially-responding MM and MCL patients, and the emergence of potentially permanent peripheral neuropathy (PN) in many of BTZ-treated patients [6–13]. To overcome the limitations of BTZ, several second-generation proteasome inhibitors have been developed with the aim of improving anti-tumor efficacy (by increasing the potency of proteasome inhibition) while reducing toxicity (by improving proteasome binding duration and specificity, thereby reducing “off-target” effects) [14–18].
Fig. 1. The ubiquitin-proteasome system (UPS).

The UPS-mediated protein degradation is an ATP-dependent process, involving two distinct steps, ubiquitination and degradation. First, ubiquitin (Ub) is covalently linked to a target protein by a multi-enzymatic system consisting of Ub-activating (E1), Ub-conjugating (E2), and Ub-ligating (E3). E1 activates an Ub monomer and transfers it to E2. E3 facilitates E2 to transfer the Ub to a reactive lysine residue of the target protein. The polyubiquitinated protein is then recognized by the 19S regulatory complex of the 26S proteasome and fed into the 20S catalytic core for degradation into oligopeptides and the ubiquitin molecules recycled.
Table 1.
Bortezomib and second generation proteasome inhibitors.
| Proteasome Inhibitor | Chemical Structure |
Structural Class |
Inhibition Type |
Inhibition Profile |
Development Status |
Route of Administration |
|---|---|---|---|---|---|---|
| Bortezomib (Velcade®, PS-341) | 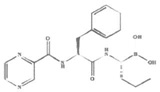 |
Peptide boronic acid | Reversible | β5 > β1 β5i | Approved | IV |
| Carfizomib (Kyprolis®, PR171) | 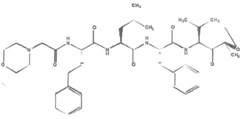 |
Peptide epoxyketone | Irreversible | β5 β5i | Approved | IV |
| Ixazomib (MLN-9708/2238) | 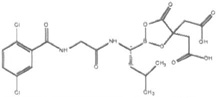 |
Peptide boronic acid | Reversible | β5>β1 | Phase III | Oral, IV |
| Delanzomib (CEP-18770) | 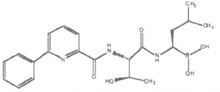 |
Peptide boronic acid | Reversible | β5>β1 | Phase I/II | Oral, IV |
| Oprozomib (ONX-0912, PR-047) | 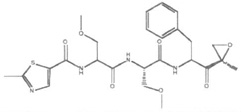 |
Peptide epoxyketone | Irreversible | β5 β5i | Phase I/II | Oral |
| Marizomib (NPI-0052, Salinosporamide A) | 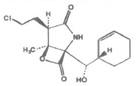 |
β-Lactone-γ-lactam | Irreversible | β5> β2> β1 20Si | Phase Ib | IV |
Herein, we review proteasome function and the status of preclinical and clinical research on first- and second-generation proteasome inhibitors. We will briefly review the development of BTZ, including the clinical efficacy that led to its FDA approval for the treatment of MM and MCL as well as its activity in AL Amyloidosis. We will then review the status of several second-generation proteasome inhibitors, including the recently approved drug carfilzomib (CFZ). We will also describe some important ongoing translational work focused on upstream UPS events, including targeting the immunoproteasome, ubiquitin E3 ligases, the 19S proteasome and deubiquitinases (DUBs) in MM and other cancer types. There are high hopes in the field that elucidating the mechanisms of action and resistance to proteasome inhibition will help advance future cancer management.
CONSTITUTIVE PROTEASOME, IMMUNOPROTEASOME AND CANCER
The UPS is responsible for degrading 80–90% of intracellular proteins [1–3]. Proteins targeted for proteasomal degradation are ubiquitinated by a series of enzymes, including the ubiquitin-activating enzyme E1, ubiquitin-conjugating enzyme E2, and the ubiquitin E3 ligases (Fig. 1), which allow the targeted proteins to be recognized by the 26S proteasome (MW 2,400 kDa), a multi-subunit protease complex composed of the 20S catalytic core (MW 700 kDa) and one or two 19S regulatory particles (MW 700 kDa) (Fig. 2). The 20S core is formed by two sets of identical α rings and two sets of identical β rings stacked in a symmetrical manner with the outside α rings surrounding the inner β rings (αββα). Each α or β ring contains seven different subunits, named α1-α7 or β1-β7, respectively, among which mainly β1, β2, and β5 possess proteolytic activity (Fig. 2). The β1 subunit is responsible for caspase-like (or peptidyl-glutamyl peptide-hydrolyzing-like/ PGPH-like) activity that preferentially cleaves after acidic residues (e.g., aspartate and glutamate), the β2 subunit has trypsin-like activity that preferentially cleaves after basic residues (e.g., arginine and lysine), and the β5 subunit has chymotrypsin- like activity preferentially cleaving after hydrophobic residues (e.g., tyrosine and phenylalanine) (Fig. 2) [1–3, 19]. The 19S regulatory particle is composed of a “lid” and a “base”, and binds to both ends of the 20S core proteasome (Fig. 2). The 19S proteasome lid contains nine or more non-ATPase subunits, which recognize polyubiquitinated proteins, bind them, and remove the polyubiquitin chain from the substrate proteins by a process called deubiquitination. Meanwhile, the 19S base contains six ATPase and four non-ATPase subunits responsible for the unfolding of substrate proteins and promotion of their entry into the 20S proteasome (Fig. 2) [1–3, 19]. The 19S regulatory particle is far less well characterized than the 20S core.
Fig 2. Proteasome structures.

The 26S proteasomes is a multiple-subunit protease complex composed of the 20S catalytic core and two 19S regulatory particles (A). The 20S core proteasome contains four stacked rings in a αββα arrangement, each ring containing 7 subunits (1 to 7). The outer α subunits serve as a gate to regulate protein entry into the inner catalytic site, while the inner β subunits possess proteolytic chymotrypsin-like (β5), trypsin-like (β2), and the PGPH-like (β1) activities (B and insert). Upon cytokine (e.g., interferon-γ or TNFα) stimulation, expression of β1i, β2i, and β5i dramatically increases and immunoproteasome 20Si forms, which is capped on each end by 11S proteasomes (B, insert, and C).
As the UPS is essential for maintenance of cell homeostasis, it is not surprising that dysregulation of protein degradation plays an essential role in the cell growth, development and survival of human cancer [5, 6]. Proteasomal mRNA levels are consistently markedly increased in a variety of malignant human hematopoietic cell lines compared with peripheral lymphocytes and monocytes from healthy adults [20]. Also, increased proteasome activity is associated with malignant disease, including those of the colon [21], prostate [22], and leukemia [20]. Tumorigenic cells are more dependent upon proteasomal activity and thus more sensitive to its blockage. Indeed, researchers found that many types of actively proliferating tumor cells are more sensitive to proteasome inhibitors than non-tumorigenic cells [23, 24]. Various studies have demonstrated that pharmacological inhibition of the proteasome results in the shift in balance of pro- and anti-apoptotic proteins, including the accumulation of tumor suppressor proteins, resulting in cell cycle arrest and apoptosis [22, 25, 26]. In some tumor cell types, proteasome inhibitors exhibit more effective apoptosis-inducing capability compared to standard cytotoxic agents [27, 28]. Proteasome inhibition is found to be an effective strategy for chemosensitization and overcoming resistance [29].
In addition to the above described constitutive 20S proteasome, which contains catalytic subunits β1, β2, and β5, there also exists the immunoproteasome 20Si, which harbors different sets of catalytic subunits designated correspondingly as β1i, β2i, and β5i (Fig. 2) [30, 31]. Upon cytokine stimulation, e.g., interferon-γ and tumor necrosis factor-α, the expression of β1i, β2i, and β5i dramatically increases and cooperatively assembles into the nascent 20S core particles to form immunoproteasome 20Si (Fig. 2). The 20Si core is capped on each end by 11S proteasomes, rather than 19S proteasomes (Fig. 2) [32]. The immunoproteasome has a key function related to antigen processing in the immune response [30, 31]. Compared to the constitutive 20S proteasome, the immunoproteasome 20Si possesses enhanced chymotrypsin- and trypsin-like activities and reduced caspase-like activity. This specialized enzymatic property endows the ability of the immunoproteasome to generate peptides that are suitable for MHC class I-mediated antigen presentation [33, 34]. Increased expression of the immunoproteasome complex has been reported in myeloma [35, 36]. Both BTZ and CFZ inhibit the immunoproteasome, albeit to a lesser degree than to which they inhibit the constitutive proteasome (Table 1).
BORTEZOMIB, THE FIRST FDA APPROVED PROTEASOME INHIBITOR FOR THE TREATMENT OF CANCER
BTZ is a dipeptide boronic acid derivative containing pyrazinoic acid, phenylalanine and leucine with boronic acid in its structure (Table 1). Synthesized in 1995 by Myogenics, it was originally called MG-341, and later PS-341. The compound was later acquired by Millennium Pharmaceuticals in 1999 and renamed bortezomib. The chemical IUPAC name of BTZ is [3-methyl-1-(3-phenyl-2-pyrazin-2-ylcarbonylamino-propanoyl) amino-butyl] boronic acid, with a molecular formula of C19H25BN4O4. BTZ is a reversible inhibitor of the 20S proteasome. The boronic acid group of BTZ binds and forms a complex with the active site of threonine hydroxyl group in the β5-subunit, blocking the chymotrypsin-like activity of the proteasome. It also binds the β1 subunit with lower affinity, inhibiting its PGPH-like activity (Table 1) [5, 6], Proteasome inhibition results in growth suppression and apoptosis induction in tumor cells, mediated through resultant effects on NF-κB signaling under some, but not other experimental conditions [5, 37], or the balance of pro- and anti-apoptotic Bcl-2 proteins [38, 39] and other mechanisms [5, 6, 10, 11]. BTZ triggers p53-induced apoptosis which partially depends on upregulation of Noxa and functional repression of Mcl-1 in lymphoma cells [40, 41]. Cytotoxic effects of BTZ may be abrogated by induction of survival mechanisms involving Akt upregulation [42], the endoplasmic reticulum stress response, the unfolded protein response, histone deacetylase (HDAC)-mediated aggresomal degradation of misfolded proteins and autophagy [43, 44]. Proteasome inhibition in non-tumor cells, including bone marrow stromal cells [45] and osteoblasts and osteoclasts [46], as well as other tissues [47, 48] has potential clinical relevance in the treatment of plasma cell dyscrasias.
The pharmacokinetic properties of BTZ were investigated in two phase I trials in cancer patients who received combination therapy of BTZ and other anticancer agents [49, 50]. After intravenous (IV) injection, BTZ quickly distributes widely into tissues (except adipose and the central nervous system) from the plasma within 10 minutes [51]. The elimination half-life of BTZ is more than 40 hours [52]. BTZ is metabolized primarily through intracellular oxidative deboronation mediated by multiple cytochrome p450 enzymes [53]. Pharmacodynamic studies showed dose-dependent inhibition of proteasome activity by BTZ with intravenous doses ranging from 0.13 to 2.0 mg/m2, with peak inhibition occurring within one hour after dosing [54, 55]. BTZ levels become undetectable about 72 hours after administration, accompanied by recovery of proteasome activity [56]. Pharmacokinetic properties of BTZ after subcutaneous (SC) administration are somewhat different: lower maximal plasma concentration, longer time to maximal plasma concentration, and longer time to maximal inhibition of proteasome function (albeit with similar levels of proteasome inhibition eventually achieved) [57]. It is possible that these differences may account for the reduced neurotoxicity observed with SC administration compared with the IV route. Impaired renal function predictably has little-to-no effect on BTZ PK [58].
Myeloma
On the basis of impressive Phase II data from two trials (CREST [59, 60] and SUMMIT [61]), BTZ received accelerated approval by the U.S. FDA in 2003 for the treatment of patients with previously treated MM, with the brand name Velcade® [5, 6, 53]. The APEX trial was a phase III trial comparing BTZ to dexamethasone (DEX) in 669 patients with relapsed MM. A subgroup analysis showed that response rates were higher for patients receiving BTZ as second-line therapy (45%) as compared to receiving it as a later line of treatment (34%) [61]. These results essentially recapitulated those previously observed in the CREST [59, 60] and SUMMIT [62] trials. The median time to progression for patients randomized to BTZ in the APEX trial was only 6.2 months. The response rate to BTZ + DEX as first line therapy is approximately 80% [63–65]. Frontline BTZ monotherapy results are less impressive, with only 40% of patients achieving a partial response or better after 2 cycles of treatment [63, 64].
There is also extensive published clinical experience with BTZ used in combination with other anti-myeloma therapies. These BTZ-based regimens are a logical extension of pre-clinical work demonstrating synergistic anti-cancer activity when BTZ is combined with several other classes of drugs used in the treatment of multiple myeloma, including alkyating agents, anthracyclines, and immunomodulatory drugs [9, 12, 66]. This work has served as the basis for trials evaluating BTZ-based combination regimens. In general, compared to BTZ or BTZ+DEX, such regimens (Table 2) have increased anti-cancer activity, including PAD/VDD [67–69], VTD [70], VMP [71], RVD [72], CyBorD [73, 74], VMPT [75], and VTD-PACE [76]. The current labeled indications for BTZ in MM treatment include BTZ + melphalan + DEX as first-line therapy in patients who are not candidates for autologous stem cell transplant, and also BTZ + DEX + pegylated doxorubicin for previously treated MM. There is a growing experience using BTZ as maintenance therapy in patients with MM. The available data, summarized in Table 3, suggest that BTZ maintenance is tolerable and prolongs disease control [75–80].
Table 2.
Selected BTZ-containing multi-agent combination regimens for initial treatment of MM.
| Regimen | Agents and Schedule | Notes |
|---|---|---|
| PAD | BTZ 1.3 mg/m^2 IV d 1,4,8,11 DEX 40 mg PO d1–4 (also d8–11,15–18 cycle 1) Doxorubicin 9 mg/m^2 continuous IV d1–4 (21-day cycle) |
95% overall response rate 24% complete remission No difficulties with subsequent ASCT |
| VDD | BTZ 1.3 mg/m^2 IV d 1,4,8,11 DEX 40 mg PO d1,2,4,5,8,9,11,12 (20 mg cycle 2+) Pegylated doxorubicin 30 mg/m^2 d1 (21-day cycle) |
92.5% overall response rate 90% incidence of Grade 1–2 PN |
| VTD | BTZ 1.3 mg/m^2 IV d1,4.8,11 DEX 40 mg PO d1–4,9–12 Thalidomide PO 200 mg/d (100 mg/d d1–14, cycle 1) (three 21d cycles pre-ASCT, two more 35d cycles after) |
93% overall response rate 19% complete response pre-ASCT 49% CR after post-ASCT consolidation 34% PN (10% Grade 3–4) |
| VMP | BTZ 1.3 mg/m^2 d1,8,22,29 IV (+ d4,11,25,32 cycles 1–4) Melphalan PO 9 mg/m^2 d1−4 Prednisone PO 60 mg/m^2 d1–4 (42-day cycle) |
“VISTA” study 71% overall response rate Non-transplantable patients |
| VMPT | BTZ IV 1.3 mg/m^2 d1,8,15,22 Melphalan PO 9 mg/m^2 d1–4 Prednisone PO 60 mg/m^2 d1–4 Thalidomide PO 50 mg/d (35-day cycle) |
Original schedule of V,M, P as per “VISTA” Grade 3–4 PN dropped from 16% to 3% with modification to weekly BTZ |
| CyBorD | BTZ IV 1.5 mg/m^2 d 1,8,15,22 Cyclophosphamide PO 300 mg/m^2 DEX PO 40 mg d1–4,9–12, 17–20 × 2 cycles (then 40 mg/wk) (28-day cycle) |
90% overall response rate RR same as d1,4,8,11 BTZ dosing, with less toxicity |
| RVD | BTZ 1.3 mg/m^2 IV d 1,4,8,11 Lenalidomide 25 mg PO d1–14 DEX 20 mg d1,2,4,5,8,9,11,12 (21-day cycle) |
100% overall response rate 75% 18-month PFS without ASCT 80% PN Antibiotic, antiviral, aspirin prophylaxis |
| VTD-PACE | BTZ 1.0 mg/m^2 SC d 1,4,8,11 Thalidomide 200 mg PO d4–7 DEX 40 mg PO d4–7 Cisplatin 10 mg/m^2/day IV d4–7 Adriamycin 10 mg/m^2/d IV d4–7 Cyclophosphamide 400 mg/m^2/d IV d4–7 Etoposide 40mg/m^2/d d4–7 (up to two 56-day cycles) |
At many centers, requires hospitalization Used as part of “Total Therapy 3” regimen 2 additional post ASCT cycles (lower dosing) |
Table 3.
BTZ-based maintenance after transplant or non-transplant induction therapy.
| Maintenance Agent(s) and Schedule | Notes |
|---|---|
| BTZ 1.3 mg/m^2 IV q 2 weeks Thalidomide PO 50 mg/day (following VMPT induction, maint × 2 years) |
Superior to No Maintenance 56% 3-year PFS |
| BTZ 1.3 mg/m^2 IV q 2 weeks (following PAD induction then ASCT, maint × 2 yrs) |
Only 9% discontinuation rate for toxicity Superior to Thal maintenance 48% 3-year PFS |
| BTZ 1.3 mg/m^2 IV d 1,4,8,11 Thalidomide PO 100 mg/d (following ASCT, BTZ given every 3 months × 3 yrs) |
Marginally superior to Thal alone or Interferon as maintenance |
| BTZ 1.3 mg/m^2 IV d1,4,8,11 Thalidomide PO 50 mg/d OR Prednisone 50 mg every other day (after non-transplant induction, BTZ given q 3 mos) |
Study design made comparison of VT to VP difficult; med PFS approx. 3 yrs |
| BTZ SQ 1.3 mg/m^2 d1,4,8,11 Thalidomide 100 mg/d PO DEX 20 mg/wk PO (Following Total Therapy 3, VTD × 1 yr maint, then TD × 2 yr) |
More recent iteration of this regimen replaces Thal with Lenalidomide 15 mg/day d1–20, 5 mg/d d21–28 q 28 d × 3 yrs |
BTZ has effects on other tissues beyond the malignant clone that are relevant for patients with MM. The drug promotes bone formation by mechanisms which may be independent of its anti-tumor effects [81]. Bone metabolism is shifted from the catabolic state typical of myeloma to an anabolic state by means of the drug’s effects on both osteoblasts and osteoclasts [82–85]. Further, BTZ also has radio-sensitizing effects that ought to be considered during radiotherapy to bone lesions concomitantly with BTZ treatment [86–91]. Interstitial inflammation and progressive renal failure stemming from sustained proteinuria in myeloma (and amyloidosis) is mediated by NF-κB [92, 93]. It has been suggested that the sometimes rapid improvement in renal function seen in BTZ-treated patients [94] may be due to inhibition of NF-κB signaling [95]. Thus, BTZ may not only target the malignant plasma cells, but also help reverse some of the myeloma-associated end-organ damage commonly seen in patients. However, a preclinical study suggests that BTZ induces canonical NF-κB activation in MM cells and therefore, BTZ-induced cytotoxicity cannot be fully attributed to NF-κB inhibition [37]. Further studies are needed to investigate whether NF-κB is the key target of BTZ in MM and other cancers under clinical setting condtions.
The side effect profile of BTZ in myeloma patients is well characterized. Neurotoxicity, fatigue, gastrointestinal effects, and modest cytopenias are common. Peripheral sensory neuropathy (PN), sometimes painful, is the most problematic side effect for many patients. The reported incidence of treatment-emergent PN in trials involving newly diagnosed myeloma patients receiving twice-weekly IV BTZ ranges from 31% with BTZ (+/−DEX) [63, 64] to 80% (including a 32% incidence of neuropathic pain) with the combination of BTZ + lenalidomide + DEX [72]. Risk factors for the development of treatment-emergent PN include pre-existing PN [96] and possibly genetic predisposition [97]. Proposed mechanisms include increased tubulin polymerization [98] and dysregulated transcription, nuclear mRNA processing, and cytoplasmic translation in dorsal root ganglion cells [99]. There may also be non-proteasomal (i.e., “off-target”) neuronal effects specific to BTZ that contribute to the different rates of PN observed in clinical trials involving various proteasome inhibitors [10, 18]. BTZ dose reduction [59], weekly (rather than twice-weekly) administration [75], and SC (rather than IV) administration [100] have all been shown to reduce the incidence of PN during myeloma therapy. BTZ-associated PN often improves and in many cases is completely reversible with treatment interruption [101].
AL Amyloidosis
After the successes achieved with BTZ therapy in MM, exploration of its use in the treatment of a closely related monoclonal plasma cell disorder, AL amyloidosis, soon followed. BTZ monotherapy was shown to be effective and tolerable in an international study evaluating different treatment schedules [102]. The incidence of treatment-emergent PN in this study was only 14% in patients treated with weekly or otherwise reduced dosing, which was perhaps surprising, since over 60% of these patients had a baseline history of neurologic symptoms. BTZ-containing combinations (CyBorD) [103] and melphalan + prednisone + BTZ [104] induce high-quality hematologic responses (as determined by deep reduction in serum levels of amyloidogenic light chains) in ≥80% of newly-diagnosed AL amyloidosis patients. Currently, an ongoing multicenter trial is evaluating BTZ + PEX plus the immunomodulatory drug pomalidomide as initial therapy for AL amyloidosis [105].
Other Hematologic Malignancies
Extensive preclinical work has demonstrated synergistic anti-tumor activity between BTZ and other drugs used in the treatment of non-Hodgkin lymphoma (NHL) [106, 107]. Phase I studies demonstrated the maximal tolerated dose of BTZ in previously-treated NHL patients is similar to that for MM [108, 109]. In 2006, the phase II PINNACLE trial of 155 relapsed or refractory MCL patients treated with BTZ monotherapy showed an overall response rate (ORR) of 32%, prompting the FDA to add MCL to the labeled indications [110, 111]. Phase II trials combining BTZ with rituximab and either purine analogues [112–114] or alkylating agents [115, 116] generally show overall response rates of 50–75%. When BTZ is combined with a standard regimen (Rituxan + HyperCVAD) in newly-diagnosed MCL, the response rate is 90–100% [117, 118]. The response rates reported for patients with relapsed/refractory follicular NHL (F-NHL) seem to be similar to lower than those reported for pretreated MCL [119, 120], though the median time to response appears to be longer for F-NHL than for other subtypes of low-grade NHL - 12 weeks versus 4 weeks [121]. Phase II studies combining BTZ with rituximab and alkylator-based frontline combination regimens are tolerated; it is unclear whether the 83–90% overall response rates observed truly represent an improvement over the same regimens without BTZ added [122,123].
Waldenstrom’s Macroglobulinemia (WM) is another low grade NHL against which BTZ (1.6 mg/m2/wk IV) appears to have significant activity: 85% overall response rate when combined with rituximab as first line therapy [124]. The rate of PN in this 59-patient Phase II study was 46% with 8% of patients discontinuing BTZ due to PN. It is plausible that the dosing schedule, route of administration used, and/or baseline incidence of WM-associated PN contributed to this degree of neurotoxicity.
BTZ-based combination therapy may hold promise in the treatment of more aggressive B-cell NHL, specifically diffuse large B-cell lymphoma (DLBCL). NF-κB signaling is constitutively activated in the “activated B-Cell origin” (ABC) subtype of DLBCL, which provides a strong rationale for evaluating the potential utility of BTZ as therapy. Several studies combining BTZ with R-CHOP as initial therapy have confirmed response rates in excess of 90% [125–127]. In one study in which BTZ was combined with the R-CHOP-like regimen R-EPOCH, high response rates were observed amongst patients with the ABC subtype of DLBCL only (with little activity seen in patients with the “germinal center B-Cell origin” (GCB) type), consistent with the original rationale for incorporating BTZ into DLBCL therapy [128]. Constitutive NF-κB activation has also been described in Reed-Stemberg cells, the malignant cells in Hodgkin’s lymphoma (HL) [129]. Despite this, single agent BTZ in relapsed HL was a complete failure in a Phase II study, with zero out of 30 treated patients achieving even a partial response [130]. BTZ has also been evaluated in the treatment of myeloid malignancies, specifically acute myeloid leukemia. In a small Phase II study, half of the 14 patients treated had some reduction in peripheral blood blasts or improvement in hematologic parameters, albeit with a high incidence of severe PN [131]. The drug has perhaps more promise in combination with standard cytotoxic regimens [132, 133], or with differentiating agents such as all-trans retinoic acid [134].
Solid Tumors
The results of BTZ therapy versus solid tumors have thus far been less impressive. To date, efficacy has not been established versus breast cancer alone [135,136] or combined with chemotherapy [137], hormone-resistant prostate cancer in combination with either prednisone [138] or docetaxel [139], chemotherapy-naïve advanced stage non-small cell lung cancer [140], unresectable/metastatic gastric and gastroesophageal junction adenocarcinoma [141], metastatic neuroendocrine tumors [142], or advanced renal cell carcinoma [143]. Determining whether there are mechanisms of BTZ-resistance specific to any or all of these tumor types will be instrumental in developing effective BTZ-based therapies.
Issues Identified with BTZ as Cancer Therapy
Examining the collective published experience using BTZ to treat cancer, the two main problems encountered are (i) toxicities related to proteasomal inhibition and off-target actions, and (ii) inherent and acquired resistance to the drug, as evidenced by the fact that up to half of patients with “sensitive” tumors such as myeloma may not respond to BTZ monotherapy and responses, when achieved, may be short-lived [5–11].
Cell-based studies have suggested several potential mechanisms for the emergence of BTZ resistance: (i) mutations/overexpression of the BTZ-binding proteasome β5 submit [144, 145]; (ii) overexpression/activation of downstream effectors, e.g., Bcl-2 [146] or NF-κB [147]; (iii) reduced levels of proapoptotic proteins and increased levels of anti-apoptotic proteins (Bcl-2, AKT, MKP-1) [43, 44] after proteasome inhibition; or (iv) altered protein production, processing, and trafficking via high secretion of unfolded protein response (UPR) chaperone protein GRP-78, overexpression of a heat shock proteins, phosphorylation of eIF2α by heme-regulated inhibitor kinase (HRI) [148–151] or induction of autophagy [43, 44]. However, the relative importance of each of these alterations to clinical BTZ resistance in cancer patients remains undefined. It is known, for example, that some patients displaying clinical BTZ resistance do not harbor acquired mutations of proteasome β5 subunit [152].
It has been shown that adhesion of MM cells to extracellular matrix proteins and bone marrow stromal cells plays a role in tumor growth, survival, migration and development of drag resistance [153]. Studies of the signaling cascades mediating these events have identified multiple novel therapeutic targets in MM cells and their bone marrow microenvironment, which may provide a novel strategy to overcome BTZ resistance. Most recently, it has been reported [154] that endoplasmic reticulum to nucleus signaling 1 (ERN1), also called IRE1, is critical for BTZ response in MM cells, and suppression of the IRE-activated downstream transcription factor Xbp1 induces BTZ resistance through de-commitment to plasma cell maturation and immunoglobulin production [154]. Finally, exogenous agents such as antioxidants and green tea catechins may contribute to BTZ resistance through drug-drug interactions [155].
CARFILZOMIB: A SECOND GENERATION PROTEASOME INHIBITOR APPROVED FOR THE TREATMENT OF CHEMOTHERAPY-RESISTANT MULTIPLE MYELOMA PREVIOUSLY TREATED WITH BTZ
With the goal of overcoming the limitations of BTZ, several second-generation proteasome inhibitors (Table 1) have been developed, each with unique chemical structure and biochemical properties, binding affinity, binding reversibility, potency and selectivity. All of these factors could be expected to have an effect on the anti-cancer efficacy as well as the toxicity profiles of second-generation proteasome inhibitors [6, 14–17, 156].
Carfilzomib (CFZ, also called PR-171, marketed as Kyprolis®) is an irreversible peptide epoxyketone class proteasome inhibitor with a molecular formula of C40H57 N5O7 (Table 1) [157]. This class includes the most specific and potent proteasome inhibitors discovered thus far. The α,β-epoxyketone moiety interacts with both the hydroxyl group and the free α-amino group of Thr1 in the catalytic β subunits of the proteasome, leading to the formation of the morpholino adduct in an irreversible fashion [158, 159]. The irreversible binding by epoxyketone-based proteasome inhibitors leads to more sustained as activity compared to BTZ [160], as new subunit synthesis and proteasome assembly are required for restoration of proteasome activity.
CFZ has also been shown to be more specific than BTZ, with little or no off-target activity outside of the proteasome [161]. The free α-amino group required for adduct formation with CFZ is not present in serine and cysteine proteases that can be inhibited by BTZ [18]. In addition to targeting the β5 subunit in the constitutive 20S proteasome, CFZ also targets the correlated β5i subunit of the immunoproteasome 20Si, which appears to be preferentially expressed in MM [35, 162]. The affinity of CFZ for other β-subunits (β1, β2) is lower compared to BTZ, so the trypsin-like and chemotrypsin-like activities of the proteasome are less inhibited.
Rat pharmacokinetic studies showed that CFZ administered intravenously was rapidly cleared from the plasma compartment with a half-life of < 20 minutes [163, 164], Other preclinical and early clinical work with CFZ confirmed rapid clearance of CFZ from plasma with a half-life of < 30 min with widespread tissue distribution with the exception of the brain [165, 166].
Proteasome activity in blood and PBMCs was inhibited by CFZ in a dose-dependent manner, >75% inhibition at 15 mg/m2 after the first dose, and >90% inhibition after five doses. As predicted by the binding properties of the drug, the observed proteasomal inhibition by CFZ persisted after the drug had been cleared. Proteasome function in PBMC eventually recovered > 2 wks after dosing. Like BTZ, both CFZ and another epoxyketone proteasome inhibitor, oprozomib (discussed, below), inhibit osteoclast function and promote osteoblastogenesis [167].
CFZ monotherapy has been studied in several Phase 1 and II trials involving patients with previously treated MM [168–171], In BTZ-naïve patients, the overall response rate to CFZ monotherapy was 47% with a median progression free survival of 8.2 months in patients treated at a dose of 20 mg/m2 two consecutive days for 3 out of every 4 weeks [169]. Higher dosing (27 mg/m2 after cycle one, same schedule otherwise) led to more responses with longer duration. The question of a dose-response association has been explored further in a Phase I dose escalation trial dosing CFZ at 56 mg/m2/dose in combination with DEX. The response rate was 55%, with dyspnea, fatigue, and cytopenias (but not neuropathy) commonly seen [172]. This escalated dosing schedule will be formally evaluated in patients with previously treated, progressing MM not refractory to prior BTZ therapy in the ENDEAVOR trial [173]. CFZ can also induce responses in patients refractory to BTZ, albeit at a lower rate. In a Phase II study involving 266 patients with MM progressing on last therapy, the overall response rate was 23.7%, with a median duration of response of 7.8 months [168]. For patients progressing on BTZ as their last line of therapy prior to enrollment, the response rate was 18.6%, demonstrating that CFZ can overcome BTZ resistance in at least some cases. An integrated safety analysis of 526 patients treated with single agent CFZ in Phase II trials revealed fatigue (55%), anemia (47%), nausea (45%), cardiac events (22%, including congestive heart failure in 12%), respiratory events 69% (dyspnea 42%), but PN in only 14% (generally grade 1–2) [174]. Essentially no patients required discontinuation or dose adjustments due to neurotoxicity, permitting long-term treatment with CFZ in responding patients [175].
A recent study comparing BTZ and CFZ further pointed out that the neurotoxicity associated with BTZ was related to its off-target inhibition of HtrA2/Omi, a protease known to be involved in neuronal survival [18]. The etiology of the cardiac and pulmonary events noted is unclear, but both are listed as part of a black-box warning on the package insert for CFZ. It remains to be seen whether these or other toxicities will be more pronounced at escalated doses. Little-to-no information is available regarding alternate dosing schedules. Currently, the medication is only administered intravenously. On July 20, 2012, CFZ was granted FDA approval for treatment of patients with MM progressing on or after treatment with BTZ and an immunomodulatory agent. Its use in the U.S. was launched on August 1, 2012 [157].
CFZ is starting to be evaluated as a substitute for BTZ in combination regimens. A handful of trials have treated patients with the combination of CFZ, lenalidomide, and DEX in relapsed and newly diagnosed patients [176–178]. In previously untreated patients, the responses are rapid and the overall response rate ultimately reaches about 100%, which is not surprising given the similarly high response rates seen with BTZ-based triplet regimens [72]. Based on these encouraging results, the pivotal phase III ASPIRE trial, a 700+ patient trial comparing CFZ + lenalidomide + DEX to lenalidomide + DEX was conducted. Enrollment was completed as of early 2013. A large EGOG-ACRIN-led study comparing CFZ/LEN/DEX to BTZ/LEN/DEX has also recently been activated.
As for other potential indications, CFZ has been studied in NHL [165, 171] with early results that appear less impressive than those seen in MM patients. There is an ongoing study of CFZ with or without DEX in patients with AL amyloidosis [179]. A large (79 patient) Phase I/II study evaluating escalated dose CFZ in patients with advanced solid tumors was conducted, with 6 or 48 response evaluable patients demonstrating reduction in tumor size (albeit not sufficiently so to meet RECIST criteria for a partial response) [180]. Side effects in this study were quite manageable, with all deaths the result of progressive disease, only one patient developing congestive heart failure, and no severe respiratory or Grade 3+ PN reported.
OTHER SECOND GENERATION PROTEASOME INHIBITORS IN CLINICAL DEVELOPMENT
Ixazomib (MLN-9708/2238)
Ixazomib (MLN-9708) (Table 1) is an orally bioavailable boronic ester prodrug [10, 11, 113, 158, 161]. Ixazomib (IXZ), a reversible proteasome inhibitor, has the advantage of being the first oral proteasome inhibitor to enter clinical investigation in MM patients. In preclinical studies, the anti-myeloma activity of the drug compared favorably to BTZ [181]. Phase I and II trials suggest that single-agent IXZ has clinical activity in heavily pretreated relapsed and/or refractory MM patients, with infrequent peripheral neuropathy [182, 183]. It is currently being tested in many clinical trials in a wide range of clinical indications, including previously untreated MM, relapsed MM, advanced stage solid tumors, lymphoblastic leukemia, non-Hodgkin’s lymphoma, and AL amyloidosis [184]. There is interest in developing the drug as maintenance therapy in MM.
Delanzomib(CEP-18770)
Delanzomib (DLZ) is another reversibly binding boronate-based, second-generation proteasome inhibitor (Table 1) [10, 11, 158, 161, 184] with both oral and IV bioavailability. DLZ has shown proteasome-inhibitory activity similar to that of BTZ in hematologic and solid tumor cell lines, as well as in primary cells front multiple myeloma patients. Delanzomib is currently in phase I/II clinical investigations for myeloma, lymphoma and solid tumors still using intravenous administration [185]. A recent phase I study of DLZ in patients with advanced solid tumors and MM demonstrated a favorable safety profile with minimal neurotoxicity. The half-life of the drug was rather long, 62 hrs, with linear PK. The degree of proteasome inhibition was lower than that described for BTZ or CFZ (45%), and rash occurred in over half of patients [186]. It remains to be determined whether the drug will have meaningful anti-MM activity in BTZ-resistant patients.
Oprozomib (ONX-0912, PR-047)
Oprozomib (OPZ) is an orally bioavailable peptide epoxyketone-based, irreversible proteasome inhibitor (Table 1) that showed similar potency to CFZ in cytotoxicity assays [14–16]. Orally administered oprozomib showed equivalent antitumor activity to intravenously administered CFZ in human tumor xenograft and mouse syngeneic models [187]. Early studies were hampered by significant gastrointestinal toxicity, prompting the manufacturer to develop a modified formulation. The formulation currently being investigated in Phase I and II trials for patients with hematological cancers (including multiple myeloma) or solid tumors appears more tolerable. The optimal dosing schedule of the drug is still being worked out. A Phase I/II trial combining OPZ with lenalidomide and DEX in newly diagnosed MM patients is being planned.
Marizomib (NPI-0052, Salinosporamide A)
Marizomib is an irreversible proteasome inhibitor with a β-lactone backbone (Table 1) [11, 17, 156, 158]. The carbonyl group of the β-lactone can interact with the hydroxyl group of Thrl in catalytic β subunits, leading to the formation of an acyl-ester adduct. This process is reversible, but the covalent adduct only dissociates at a very slow rate. In addition to the acyl-ester bond, marizomib also forms a cyclic tetrahydrofuran ring with the proteasome, making the reaction irreversible [17, 158, 188]. Compared to other proteasome inhibitors, marizomib produces rapid, broad and prolonged inhibition of all three 20S proteasome catalytic subunits [17]. Marizomib is the only non-peptide-based inhibitor in clinical trials [184]. It is administrated intravenously twice weekly and is being investigated for Phase Ib for recurrent MM, solid tumors, lymphoma and leukemia. Marizomib exhibits an extremely short half-life (< 5 minutes) and wide tissue distribution (including penetration of the blood brain barrier) [189]. Responses to marizomib were found in patients with BTZ-refractory MM. The safety profile of marizomib differs from BTZ, with no significant treatment-emergent PN, myelosuppression or thrombocytopenia reported. The dose limiting toxicities include transient hallucinations, cognitive changes and loss of balance, perhaps reflecting CMS penetration.
NOVEL DRUGS INHIBITING IMMUNOPROTEASOME AND OTHER UPS COMPONENTS WITH TRANSLATIONAL FEASIBILITY
Immunoproteasome Inhibitors
It has been shown that the immunoproteasome complex plays a role in relapsed myeloma and BTZ resistance [35, 36, 162, 190, 191]. Immunoproteasome Specific Inhibitor-001 (IPSI-001; Table 4) preferably inhibits the immunoproteasome 20Si activity (mainly by binding to β 1i subunit) over the constitutive 20S proteasome activity, potently suppressing proliferation and inducing apoptosis in MM cell lines as well as in patient samples [14]. More importantly, it was able to overcome conventional and novel drug resistance, including resistance to BTZ [192].
Table 4.
Inhibitors of immunoproteasome, E3 ligases, 19S proteasome and DUBs.
| Target | Inhibitor | Structural Class | Inhibition Property | Major biological effects | Development Stage |
|---|---|---|---|---|---|
| Immunoproteasome | IPSI-001 | Aldehyde | β1i> β5i> 20S | Overcome BTZ resistance | Preclinical |
| Immunoproteasome | ONX-0914 PR-924 |
Epoxyketone | β5i> β5, β1i | Overcome BTZ resistance | Preclinical |
| Ubiquitin E3 ligase (HDM2) |
Nutlin-3 | cis-Imidazoline | p53 binding pocket on HDM2 |
Accumulation of p53 and increased expression of p53 targets |
Prototype |
| Ubiquitin E3 ligase (HDM2) |
RO5045337 RO5503781 |
cis-Imidazoline | p53 binding pocket on HDM2 |
Accumulation of p53 and increased expression of p53 targets |
Phase I |
| Ubiquitin E3 ligase (IAP) |
LCL161 | Smac peptide mimetic | Binding and inhibiting IAP |
IAP degradation | Phase I-II |
| Ubiquitin E3 ligase (IAP) |
AEG 35156 | IAP antisense | Binding and inhibiting IAP |
IAP degradation | Phase I-II |
| Ubiquitin E3 ligase (Cereblon) |
Lenalidomide Pomalidomide |
Thalidomide analogs | Cereblon | Immunomodulatory and anti- tumorigenic effects |
Approved |
| 19S proteasome (proteasome recognition site) |
Ubistatins | Quinolines | Ub-Ub interface | Blocking the binding of ubiquitinated substrates to the proteasome |
Pre-clinical |
| 19S proteasome- associated DUBs |
b-AP15 | bis-Nitrobenzylidene- piperidinone |
USP14, UCH-L5 | Blocking cleavage of K48-linked ubiquitin chains and accumulating polyubiquitinated proteins |
Pre-clinical |
| DUBs | P5091 P22077 |
Thiophene | USP7 | Deubiquitinating p53 and HDM2; overcoming BTZ resistance |
Pre-clinical |
| DUBs | WP-1130 | Tyrphostin | Multiple DUBs (USP9x, USP5, USP14, UCH-L5) |
Regulating survival protein stability and 26S proteasome function; synergetic effect with BTZ |
Pre-clinical |
ONX-0914 and PR-924 are peptide epoxyketone-based immunoproteasome inhibitors (Table 4), both of which are able to selectively and irreversibly inhibit the β5i subunit ONX-0914 is 20 to 40-fold more selective towards the β5i subunit than both the β5 and β1i subunits [193]. The ability of specific immunoproteasome inhibitors to overcome BTZ resistance in the pre-clinical setting suggests that they may provide an alternative therapeutic option for cancer patients resistant to BTZ.
Inhibitors of Ubiquitin E3 Enzymes
Ubiquitin E3 ligases (Fig. 1) are a large group of ligase enzymes that facilitate the transfer of ubiquitin from E2 ubiquitin-conjugating enzyme to substrate proteins to be targeted for proteasomal degradation. Ubiquitin E3 ligases can be classified into three major groups; N-end rule ubiquitin ligases [194], HECT (Homologous to E6AP C-Terminus) [195] and RING (Really Interesting New Gene) [196, 197]. Ubiquitin E3 ligases regulate almost all cellular processes including the cell cycle, transcription, DNA repair, signal transduction, endocytosis, cellular transport and development, and their inhibition could lead to growth suppression and apoptosis of neoplastic cells. The substrate binding specificity of a given E3 ligase determines what protein(s) will be ubiquitinated. E3 ligases represent a group of potentially “druggable” targets, albeit a rather large group. Therefore, E3 ligase small molecule therapeutics might offer potential advantages over proteasomal blockade as they will target a more defined set of proteins that could further limit off-target effects currently observed with 20S proteasomal inhibitors.
One example is HDM2, the E3 ligase that regulates the degradation of p53 [198]. Inhibition of HDM2 results in accumulation of p53 and increased expression of its target gene p21, eventually leading to cell cycle arrest and apoptosis in cancer cells with wild-type p53 but not in those with mutant p53 [199]. There are at least seven HDM2 inhibitors currently in Phase I clinical trials [200, 201]. These include non-peptide small molecule HDM2 antagonists that competitively bind the deep hydrophobic p53-binding pocket in HDM2. Nutlins (Table 4), a group of cis-imidazoline analogs, are another group of HDM2 antagonists in clinical development that disrupt the interaction between HDM2 and p53 by binding to the p53-binding pocket on HDM2. Two nutlin derivatives, RO5045337 (RG7112) and RO5503781, are currently being evaluated in clinical studies (Table 4).
IAPs (Inhibitors of Apoptosis) are a group of E3 ligases that include XIAP, cIAP1, cIAP2, and ML-IAP. IAPs function as regulators of apoptosis. Smac (Second Mitochondria-Derived Activator of Caspases, also called DIABLO) is an IAP-binding protein that regulates IAP levels and function by triggering auto-ubiquitination and destruction [202]. Several Smac mimetics (e.g., LCL161) and IAP antisense molecules (e.g., AEG 35156) are currently in Phase I/II clinical trials (Table 4) [201].
The E3 ligase Cereblon has been found to be a direct target for immunomodulatory drugs (IMiDs) thalidomide, lenalidomide and pomalidomide (Table 4). Cereblon binding appears to be necessary for the drugs to exert their cytotoxic effects. At least one report has shown a correlation between pre-treatment cereblon expression in myeloma cells and sensitivity to IMiD therapy [203].
Inhibitors to 19S Proteasome and Deubiquitinases
Proteasome Recognition Inhibitors
Ubistatin A and ubistatin B are two small molecules that disrupt substrate recognition by the 19S regulatory particle of the proteasome (Table 4) [204]. They block the binding of ubiquitinated substrates to the proteasome by targeting the ubiquitin-ubiquitin interface of K48-linked chains, the same interface recognized by ubiquitin-chain receptors of the proteasome [204]. This supports the idea that the ubiquitin chain itself could be another potential target for pharmacological intervention in the ubiquitin-proteasome pathway.
Inhibitors of 19S Proteasome-Associated Deubiquitinases
Around 100 human deubiquitinases (DUBs) have been identified, most of which belong to the cysteine protease family [205, 206]. The 19S proteasome is linked to three DUBs, UCHL5 (or UCH37), USP14 and POU1 (Rpn11) [207]. UCHL5 and USP14 can be inhibited by a small molecule called b-AP15 in a reversible manner. The drug b-AP15, which was identified in a screen for compounds that induce the lysosomal apoptosis pathway [208], inhibits UCHL5 and USP14 in a reversible manner (Table 4). Resultant accumulation of polyubiquitinated proteins induces G2/M arrest without causing genotoxicity. Treatment with b-AP15 inhibited tumor progression in four different in vivo solid tumor models and inhibited organ infiltration in an acute myeloid leukemia model [208]. More importantly, although the proteasome is targeted by b-APl5, the cellular response to the drug is distinct from that of BTZ. b-AP15 induced apoptosis in tumor cells with mutant p53 or overexpressed Bcl-2, two characteristics that greatly reduce BTZ sensitivity. Experimental evidence (e.g., differing cytotoxicity profiles versus the NCI-60 panel) supports the notion that b-AP15 may have anti-cancer efficacy against tumors resistant to BTZ [208].
Another DUB, USP7 (HAUSP), has been a focus of research due to its role in deubiquitinating and stabilizing p53 and its E3 ligase HDM2 [206]. P5091 (Table 4) actively inhibits USP7 at concentrations as low as 5–10 µM in the cellular environment, resulting in HDM2 polyubiquitination and accelerated degradation by the proteasome, and consequently increased steady-state protein levels of p53 and p21. P5091 induced apoptosis in BTZ-resistant MM cells, supporting the concept that alternate (upstream) targeting of the UPS has potential utility in cancer therapy. In mice, P5091 was well tolerated, inhibited MM tumor growth and angiogenesis, triggered MM cell apoptosis and prolonged survival. Furthermore, combined treatment of P5091 and lenalidomide, dexamethasone, or the HDAC inhibitor vorinostat (SAHA) induced synergistic anti-multiple myeloma activity in vitro [209].
P22077 (Table 4) is an analog of P5091 which, in addition to decreasing levels of HDM2 and increasing levels of p53 and p21, also caused reduced levels of DNA damage proteins DDB1, RBX1, DCAF7 and DCAF11, all of which are subunits of E3 ligases. This suggests a previously unknown functional link between USP7 and enzymes involved in DNA repair [210].
HBX19818 is a recently identified selective USP7 inhibitor with an IC50 of 28.1 µM (211). In tumor cells, HBX19818 treatment caused accumulation of functionally active p53 in p53 wild-type cells with the expected G1 arrest. Importantly, however, cell viability was similarly reduced even in cells missing p53, implying alternate mechanisms mediating the cellular response [211].
Other DUB inhibitors are also in development. For example, WP1130 (degrasyn) (Table 4), an inhibitor of multiple DUBs (e.g., USP5, USP9X, USP14 and UCHL5) may induce apoptosis by regulating MCL-1 and p53 [206]. Further, combination of WP1130 and BTZ showed a synergistic anti-tumor effect versus mantle cell lymphoma cells [212].
CONCLUSIONS (SEE BOX 1)
Box 1. Key points.
BTZ, the first U.S. FDA approved proteasome inhibitor for treating MM and MCL, is an established component of induction therapy for these patients but has several limitations, including emergence of resistance in MM patients, neuto-toxicities, and little efficacy in solid cancers.
Rationally designed BTZ-based combination therapies, including BTZ + cyclophosphamide, or BTZ + lenalidomide, are associated with improved outcomes, but are still non-curative.
CFZ, the second U. S. FDA approved proteasome inhibitor for treating relapsed MM, has increased proteasome-targeting selectivity and potency, with an excellent safety profile.
Ixazomib, Delanzomib, Oprozomib and Marizomib are second-generation proteasome inhibitors with different pharmacologic characteristics compared to BTZ; they are currently being tested in clinical trials.
Targeting the immunoproteasome, ubiquitin E3 ligases, 19S proteasome and DUBs may hold promise in the future as proteasome/UPS-targeted cancer therapies.
In the last decade, the proteasome has been validated as a novel, valuable molecular target in the treatment of MM and other hematologic cancers. BTZ, the first FDA-approved proteasome inhibitor, is an important treatment of MM and MCL. However, challenges remain, such as resistance, dose limiting toxicities, and low potency against solid tumors.
The successes and limitations of BTZ, the first proteasome inhibitor anticancer drug, have taught researchers useful lessons and encouraged the development of next generation proteasome inhibitors with reduced toxicities, broader anticancer activities, and - in some cases - more convenient oral dosing. Alternate chemical structures have had effects on pharmacokinetic properties, binding affinity and selectivity, breadth and potency of proteasome inhibition, efficacy spectrum and anticancer activities, etc. CFZ, the second FDA approved proteasome inhibitor, has increased binding specificity for the proteasome and reduced off-target effects, resulting in activity versus some BTZ-resistant MM with decreased neurologic toxicities. Other second generation proteasome inhibitors with improved therapeutic windows are in development as treatment for bortezomib-resistant MM, as well. The potential reversal strategies for bortezomib resistance also include identifying new targets in the UPS, such as the immunoproteasome, E3 ligases, 19S proteasome, and DUBs in bortezomib-resistant cancer cells and developing novel combinational therapies. Such novel inhibitors have demonstrated preclinical activities in cancer cells, including BTZ-resistant MM cells in some cases, which needs a confirmation under clinical settings.
ACKNOWLEDGEMENTS
We thank Jean Guerin for her assistance. This work was partially supported by the National Cancer Institute grants (1R01CA20009, 5R01CA127258-05 and 1R21CA184788-01 to QPD) and NIH center grant P30 CA022453 (to Karmanos Cancer Center).
LIST OF ABBREVIATIONS
- ABC
Ativated B-Cell origin
- Akt
protein kinase B
- AL
Amyloid Light-chain amyloidosis
- ATP
Adenosine triphosphate
- BTZ
Bortezomib, PS-341, Velcade
- CFZ
Carfizomib PR171
- CNS
Central Nervous System
- DEX
Dexamethasone
- CyBorD
Cyclophosphamide+BTZ+DEX
- DLBCL
Dffuse large B-cell lymphoma
- DLZ
Delanzomib
- DUBs
Deubiquitinases
- ECOG
Eastern Cooperative Oncology Group
- ERN1
Edoplasmic reticulum to nucleus signaling 1, or IRE1
- FDA
Food and Drug Administration
- F-NHL
Fllicular NHL
- GBC
Grminal center B-Cell origin
- HAUSP
a ubiquitin specific protease also call USP7
- HBX
Hepatitis B Virus X protein
- HDAC
Histone deacetylase
- HDM2
Human Double Minute 2
- HECT
Homologous to E6AP C-Terminus
- HL
Hodgkin’s lymphoma
- HRI
Heme-regulated inhibitor kinase
- IAPs
Inhibitors of Apoptosis
- IC50
Inhibitory Concentration 50
- IMiDs
immunomodulatory drugs
- IPSI
Immunoproteasome Specific Inhibitor
- IV
Intravenous
- IXZ
Ixazomib, MLN-9708
- kDa
Kilodaltons
- LEN
Lenalidomide
- MCL
Myeloid Cell Lymphoma
- MG
Myogenics
- MLN-9708
Ixazomib
- MM
multiple myeloma
- MRNA
messenger Ribonucleic acid
- MW
Molecular weight
- NCI
National Cancer Institute
- NF-κB
Nuclear Factor-KappaB
- NHL
non-Hodgkin lymphoma
- OPZ
Oprozomib, ONX-0912, PR-047
- PAD
VDD, BTZ+DEX+Doxorubicin
- PGPH
Peptidyl-glutamyl peptide-hydrolyzing
- PK
Pharmacokinetics
- PN
peripheral neuropathy
- POU1
Rpn1
- RECIST
Response Evaluation Criteria In Solid Tumors
- RING
Really Interesting New Gene
- RVD
BTZ+Lenalidomide+DEX
- SAHA
suberoylanilide hydroxamic acid
- SC
subcutaneous
- Smac
Second Mitochondria-Derived Activator of Caspases
- UCHL5
Ubiquitin carboxyl-terminal hydrolase L5, or UCH37
- UPS
ubiquitin-proteasome system
- USP7
a ubiquitin specific protease also called HAUSP
- VDD
PAD, BTZ+DEX+Doxorubicin
- VMP
BTZ+ Melphalan+ Prednisone
- VMPT
BTZ+Melphalan+Prednisone+Thalidomide
- VTD
BTZ+DEX+Thalidomide
- VTD-PACE
BTZ+DEX+Thalidomide+ Cisplatin+ Adriamycin+ Cyclophosphamide+ Etoposide
- WM
Waldenstrom’s Macroglobulinemia
- WP1130
degrasyn
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 2.Hochstrasser M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell. Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg AL, Akopian TR, Kisselev AF, Lee DH, Rohrwild M. New insights into the mechanisms and importance of the proteasome in intracellular protein degradation. Biol. Chem. 1997;378:131–140. [PubMed] [Google Scholar]
- 4.Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov. 2006;5(7):596–613. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]
- 5.Adams J. The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer. 2004;4:349–360. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]
- 6.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr. Cancer Drug Targets. 2011;11(3):239–253. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta. doi: 10.1016/j.bbamcr.2013.08.012. doi: pii: 2013 Aug 27 S0167-4889(13)00310-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skrott Z, Cvek B. Linking the activity of bortezomib in multiple myeloma and autoimmune diseases. Crit. Rev. Oncol. Hematol. 2014 May 10; doi: 10.1016/j.critrevonc.2014.05.003. pii: S1040-8428(14)00082-1. [Epub ahead of print] Review. [DOI] [PubMed] [Google Scholar]
- 9.Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A, Harousseau JL. Proteasome inhibitors in multiple myeloma: 10 years later. Blood. 2012;120(5):947–959. doi: 10.1182/blood-2012-04-403733. (2012 Aug). Epub May 29 Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010;15(5–6):243–249. doi: 10.1016/j.drudis.2010.01.008. 2010, Epub Jan 29 Review. [DOI] [PubMed] [Google Scholar]
- 11.Buac D, Shen M, Schmitt S, Kona FR, Deshmukh R, Zhang Z, Neslund-Dudas C, Mitra B, Dou QP. From Bortezomib to other Inhibitors of the proteasome and beyond. Curr. Pharm. Des. 2013;19(22):4025–4038. doi: 10.2174/1381612811319220012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson KC. Therapeutic advances in relapsed or refractory multiple myeloma. J. Natl. Compr. Canc. Netw. 2013 May;11(5 Suppl):676–679. doi: 10.6004/jnccn.2013.0199. [DOI] [PubMed] [Google Scholar]
- 13.Argyriou AA, Iconomou G, Kalofonos HP. Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood. 2008 Sep 1;112(5):1593–1599. doi: 10.1182/blood-2008-04-149385. 2008, Epub Jun 23. Review. [DOI] [PubMed] [Google Scholar]
- 14.Kuhn DJ, Orlowski RZ, Bjorklund CC. Second generation proteasome inhibitors: carfilzomib and immunoproteasome-specific inhibitors (IPSIs) Curr. Cancer Drug Targets. 2011;11(3):285–295. doi: 10.2174/156800911794519725. [DOI] [PubMed] [Google Scholar]
- 15.Nooka A, Gleason C, Casbourne D, Lonial S. Relapsed and refractory lymphoid neoplasms and multiple myeloma with a focus on carfilzomib. Biologics. 2013;7:13–32. doi: 10.2147/BTT.S24580. 2013, Epub Jan 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson JL. Carfilzomib: A second-generation proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. Ann Pharmacother. 2013 Jan;47(1):56–62. doi: 10.1345/aph.1R561. Epub Jan 8. Review (2013). [DOI] [PubMed] [Google Scholar]
- 17.Potts BC, Albitar MX, Anderson KC, Baritaki S, Berkers C, Bonavida B, Chandra J, Chauhan D, Cusack JC, Jr, Fenical W, Ghobrial IM, Groll M, Jensen PR, Lam KS, Lloyd GK, McBride W, McConkey DJ, Miller CP, Neuteboom ST, Oki Y, Ovaa H, Pajonk F, Richardson PG, Roccaro AM, Sloss CM, Spear MA, Valashi E, Younes A, Palladino MA. A proteasome inhibitor for ail seasons; preclinical profile and a framework for clinical trials. Curr. Cancer Drug Targets. 2011;11(3):254–284. doi: 10.2174/156800911794519716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arastu-Kapur S, Anderl JL, Kraus M, Pariati F, Shenk KD, Lee SJ, Muchamuel T, Bennett MK, Driessen C, Ball AJ, Kirk CJ. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin. Cancer Res. 2011 May 1;17(9):2734–2743. doi: 10.1158/1078-0432.CCR-10-1950. 2011 Epub Mar 1. [DOI] [PubMed] [Google Scholar]
- 19.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 20.Kumatori A, Tanaka A, Inamura N, Sone S, Ogura T, Matsumoto T, Tachikawa T, Shin S, Ichihara A. Abnormally high expression of proteasomes in human leukemic cells. Proc. Natl. Acad. Sci USA. 1990;87:7071–7075. doi: 10.1073/pnas.87.18.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loda M, Cukor B, Tam SW, Lavin P, Fiorentinc M, Draetta GF, Jessup JM, Pagano M. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat. Med. 1997;3:231–234. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- 22.Li B, Dou QP. Bax degradation by the ubiquitin/proseasome-dependent pathway: involvement in tumor survival and progression. Proc. Natl. Acad. Sci. U S A. 2000;97:3850–3855. doi: 10.1073/pnas.070047997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dou QP, Li B. Proteasome inhibitors as potential novel anticancer agents. Drug Resist. Updat. 1999;2:215–223. doi: 10.1054/drup.1999.0095. [DOI] [PubMed] [Google Scholar]
- 24.Almond JB, Cohen GM. The proteasome: a novel target for cancer chemotherapy. Leukemia. 2002;16:433–443. doi: 10.1038/sj.leu.2402417. [DOI] [PubMed] [Google Scholar]
- 25.Sun J, Nam S, Lee CS, Li B, Coppola D, Hamilton AD, Dou QP, Sebti SM. CEP1612, a dipeptidyl proteasome inhibitor, induces p21WAF1 and p27KIP1 expression and apoptosis and inhibits the growth of the human lung adenocarcinoma A-549 in nude mice. Cancer Res. 2001;61:1280–1284. [PubMed] [Google Scholar]
- 26.Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–4765. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- 27.Wojcik C. Proteasomes in apoptosis: villains or guardians? Cell Mol. life Sci. 1999;56:908–917. doi: 10.1007/s000180050483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin. Cancer Res. 2008;14:1649–1657. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 29.Orlowski RZ. The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Differ. 1999;6:303–313. doi: 10.1038/sj.cdd.4400505. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka K. Role of proteasomes modified by interferon-gamma in antigen processing. J. Leukoc. Biol. 1994;56(5):571–575. doi: 10.1002/jlb.56.5.571. [DOI] [PubMed] [Google Scholar]
- 31.Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011 Nov 11;11(12):823–836. doi: 10.1038/nri3084. Review. [DOI] [PubMed] [Google Scholar]
- 32.Hill CP, Masters EI, Whitby FG. The 11S regulators of 20S proteasome activity. Curr. Top. Microbial. Immunol. 2002;268:73–89. doi: 10.1007/978-3-642-59414-4_4. [DOI] [PubMed] [Google Scholar]
- 33.Strehl B, Seifert U, Krüger E, Heink S, Kuckelkorn U, Kloetzel PM. Interferon-gamma, the functional plasticity of the ubiquitin-proteasome system, and MHC class I antigen processing. Immunol. Rev. 2005;207:19–30. doi: 10.1111/j.0105-2896.2005.00308.x. [DOI] [PubMed] [Google Scholar]
- 34.Angeles A, Fung G, Luo H. Immune and non-immune functions of the immunoproteasome. Front. Biosci. 2012;17:1904–1916. doi: 10.2741/4027. [DOI] [PubMed] [Google Scholar]
- 35.Altun M, Galardy PJ, Shringarpure R, Hideshima T, LeBlanc R, Anderson KC, Ploegh HL, Kessler BM. Effects of PS-341 on the activity and composition of proteasomes in multiple myeloma cells. Cancer Res. 2005;65(17):7896–7901. doi: 10.1158/0008-5472.CAN-05-0506. [DOI] [PubMed] [Google Scholar]
- 36.Kuhn DJ, Orlowski RZ. The immunoproteasome as a target in hematologic malignancies. Semin. Hematol. 2012 Jul;49(3):258–262. doi: 10.1053/j.seminhematol.2012.04.003. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hideshima T, Ikeda H, Chauhan D, Okawa Y, Raje N, Podar K, Mitsiades C, Munshi NC, Richardson PG, Carrasco RD, Anderson KC. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood. 2009 Jul 30;114(5):1046–1052. doi: 10.1182/blood-2009-01-199604. 2009, Epub May 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, Bennett F, Pollock PM, Trent JM, Hendrix MJ, Rizzo P, Miele L, Nickoloff BJ. Proteasome inhibitors trigger NOXA-mecliated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65:6282–6293. doi: 10.1158/0008-5472.CAN-05-0676. [DOI] [PubMed] [Google Scholar]
- 39.Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N. Noxa, a BH3-only member of the Be1-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 40.Nasr R, El-Sabban ME, Karam JA, Dbaibo G, Kfoury Y, Arnulf B, Lepelletier Y, Bex F, de Thé H, Hermine O, Bazarbachi A. Efficacy and mechanism of action of the proteasome inhibitor PS-341 in T-cell lymphomas and HTLV-I associated adult T-cell leukemia/lymphoma. Oncogene. 2005;24(3):419–430. doi: 10.1038/sj.onc.1208212. [DOI] [PubMed] [Google Scholar]
- 41.Ri M, Iida S, Ishida T, Ito A, Yano H, Inagaki A, Ding J, Kusumoto S, Komatsu H, Utsunomiya A, Ueda R. Bortezomib-induced apoptosis in mature T-cell lymphoma cells partially depends on upregulation of Noxa and functional repression of Mcl-1. Cancer Sci. 2009 Feb;100(2):341–348. doi: 10.1111/j.1349-7006.2008.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ikeda H, Hideshima T, Fulciniti M, Perrone G, Miura N, Yasui H, Okawa Y, Kiziltepe T, Santo L, Vallet S, Cristea D, Calabrese E, Gorgun G, Raje NS, Richardson P, Munshi NC, Lannutti BJ, Puri KD, Giese NA, Anderson KC. PI3K/p110 {delta} is a novel therapeutic target in multiple myeloma. Blood. 2010 Sep 2;116(9):1460–1468. doi: 10.1182/blood-2009-06-222943. 2010, Epub May 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller CP, Singh MM, River Del-Valle N, Manton CA, Chandra J. Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J. Biomed. Biotechnol. 2011;2011:514261. doi: 10.1155/2011/514261. 2011, Epub Jun 28. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist. Updat. 2008 Aug-Oct;11(4–5):164–179. doi: 10.1016/j.drup.2008.08.002. 2008 Epub Sep 24. Review. [DOI] [PubMed] [Google Scholar]
- 45.Mitsiades CS, Mitsiades NS, Munshi NC, Richardson PO, Anderson KC. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma; interplay of growth factors, their receptors and stromal interactions. Eur. J. Cancer. 2006 Jul;42(11):1564–1573. doi: 10.1016/j.ejca.2005.12.025. 2006, Epub Jun 9. Review. [DOI] [PubMed] [Google Scholar]
- 46.Zangari M, Terpos E, Zhan F, Tricot G. Impact of bortezomib on bone health in myeloma: a review of current evidence. Cancer Treat Rev. 2012 Dec;38(8):968–980. doi: 10.1016/j.ctrv.2011.12.007. 2012 Epub Jan 9. Review. [DOI] [PubMed] [Google Scholar]
- 47.Palladini G, Merlini G. Current treatment of AL amyloidosis. Haematologica. 2009;94(8):1044–1048. doi: 10.3324/haematol.2009.008912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dimopoulos MA, Terpos E, Chanan-Khan A, Leung N, Ludwig H, Jagannath S, Niesvizky R, Giralt S, Fermand JP, Blade J, Comenzo RL, Sezer O, Palumbo A, Harousseau JL, Richardson PG, Barlogie B, Anderson KC, Sonneveld P, Tosi P, Cavo M, Rajkumar SV, Durie BGM, Miguel JS. Renal impairment in patients with multiple myeloma: a consensus statement on behalf of the International Myeloma Working Group. J. Clin. Oncol. 2010;28(33):4976–4984. doi: 10.1200/JCO.2010.30.8791. [DOI] [PubMed] [Google Scholar]
- 49.Supko JG, E J, Lynch TJ. Pharmacokinetics of irinotecan and the proteasome inhibitor bortezomib in adult patients with solid malignancies (abstract/poster 544) Proc. Am. Soc. Clin. Oncol. 2003:136. [Google Scholar]
- 50.Appleman LJRD, Clark JW. Phase I dose escalation study of bortezomib and gemcitabine safety and tolerability in patients with advanced solid tumors (abstract 839) Proc. Am. Soc. Clin. Oncol. 2003:209. [Google Scholar]
- 51.Nix D, P R, Wehrman T. Tissue distribution and mass balance of bortezomib (Velcade) in non-human primates (abstract M1336). Annual Meeting of the American Association of Pharmaceutical Scientists; Salt Lake City. 2003. [Google Scholar]
- 52.Nix D, M M, Pligavko C. Pharmacokinetics of the proteasome inhibitor bortezomib (Velcade) in male cynomolgus monkeys (abstract C245). American Association for Cancer Research-National Cancer Institute-European Organization for Research and Treatment of Cancer Molecular Targets and Cancer Therapeutics Discovery, Biology, and Clinical Applications meeting; Boston. 2003. [Google Scholar]
- 53.Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003;8:508–513. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]
- 54.Aghajanian C, Soignet S, Dizon DS, Pien CS, Adams J, Elliott PJ, Sabbatini P, Miller V, Hensley ML, Pezzulli S, Canales C, Daud A, Spriggs DR. A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin. Cancer Res. 2002;8:2505–2511. [PubMed] [Google Scholar]
- 55.Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, Kim J, Adams J, Elliott P, Esseltine D, Petrusich A, Dieringer P, Perez C, Logothetis CJ. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J. Clin. Oncol. 2004;22:2108–2121. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 56.Schwartz R, Davidson T. Pharmacology, pharmacokinetics, and practical applications of bortezomib. Oncology (Williston Park) 2004;18:14–21. [PubMed] [Google Scholar]
- 57.Moreau P, Karamanesht II, Domnikova N, Kysetyova MY, Vilchevska KV, Doronin VA, Schmidt A, Hulin C, Leleu X, Esseltine DL, Venkatakrishnan K, Skee D, Feng H, Girgis S, Cakana A, van de Velde H, Deraedt W, Facon T. Pharmacokinetic, pharmacodynamic and covariate analysis of subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma. Clin. Pharmacokinet. 2012 Dec;51(12):823–829. doi: 10.1007/s40262-012-0010-0. [DOI] [PubMed] [Google Scholar]
- 58.Leal TB, Remick SC, Takimoto CH, Ramanathan RK, Davies A, Egorin MJ, Hamilton A, LoRusso PA, Shibata S, Lenz HJ, Mier J, Sarantopoulos J, Mani S, Wright JJ, Ivy SP, Neuwirth R, von Moltke L, Venkatakrishnan K, Mulkerin D. Dose-escalating and pharmacological study of bortezomib in adult cancer patients with impaired renal function: a National Cancer Institute Organ Dysfunction Working Group Study. Cancer Chemother. Pharmacol. 2011;68(6):1439–1447. doi: 10.1007/s00280-011-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jagannath S, Barlogie B, Berenson J, Siegel D, Irwin D, Richardson PG, Niesvizky R, Alexanian R, Limentani SA, Alsina M, Adams J, Kauffman M, Esseltine DL, Schenkein DP, Anderson KC. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br. J. Haematol. 2004;127(2):165–172. doi: 10.1111/j.1365-2141.2004.05188.x. [DOI] [PubMed] [Google Scholar]
- 60.Jagannath S, Barlogie B, Berenson JR, Siegel DS, Irwin D, Richardson PG, Niesvizky E, Alexanian R, Limentani SA, Alsina M, Esseltine DL, Anderson KC. Updated survival analyses after prolonged follow-up of the phase 2, multicenter CREST study of bortezomib in relapsed or refractory multiple myeloma. Br. J. Haematol. 2008;43(4):537–540. doi: 10.1111/j.1365-2141.2008.07359.x. [DOI] [PubMed] [Google Scholar]
- 61.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Bladé J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators. Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005;352(24):2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 62.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkcin DP, Anderson KC. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003;348(26):2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 63.Jagannath S, Durie BG, Wolf J, Camacho E, Irwin D, Lutzky J, McKinley M, Gabayan E, Mazumder A, Schenkein D, Crowley J. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br. J. Haematol. 2005;129(6):776–783. doi: 10.1111/j.1365-2141.2005.05540.x. [DOI] [PubMed] [Google Scholar]
- 64.Jagannath S, Durie BG, Wolf XL, Camacho ES, Irwin D, Lutzky J, McKinley M, Polts P, Gabayan AE, Mazumder A, Crowley J, Vescio R. Extended follow-up of a phase 2 trial of bortezomib alone and in combination with dexamethasone for the frontline treatment of multiple myeloma. Br. J. Haematol. 2009;146(6):619–626. doi: 10.1111/j.1365-2141.2009.07803.x. [DOI] [PubMed] [Google Scholar]
- 65.Harousseau JL, Attal M, Avet-Loiseau H, Marit G, Caillot D, Mohty M, Lenain P, Hulin C, Facon T, Casassus P, Michallet M, Maisonneuve H, Benboubker L, Maloisel F, Petillon MO, Webb I, Mathiot C, Moreau P. Bortezomib plus dexamethasone is superior to vincristine plus doxorubicin plus dexamethasone as induction treatment prior to autologous stem-cell transplantation in newly diagnosed multiple myeloma: results of the IFM 2005-01 phase III trial. J. Clin. Oncol. 2010 Oct 20;28(30):4621–4629. doi: 10.1200/JCO.2009.27.9158. 2010, Epab Sep 7. [DOI] [PubMed] [Google Scholar]
- 66.Kapoor P, Ramakrishnan V, Rajkumar SV. Bortezomib combination therapy in multiple myeloma. Semin. Hematol. 2012;49(3):228–242. doi: 10.1053/j.seminhematol.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oakervee HE, Popat R, Curry N, Smith P, Morris C, Drake M, Agrawal S, Stec J, Schenkein D, Esseltine DL, Cavenagh JD. PAD combination therapy (PS-341/bortezomib, doxorubicin and dexamethasone) for previously untreated patients with multiple myeloma. Br. J. Haematol. 2005;129(6):755–762. doi: 10.1111/j.1365-2141.2005.05519.x. [DOI] [PubMed] [Google Scholar]
- 68.Popat R, Oakervee HE, Hallam S, Curry N, Odeh L, Foot N, Esseltine DL, Drake M, Morris C, Cavenagh JD. Bortezomib, doxorubicin and dexamethasone (PAD) front-line treatment of multiple myeloma: updated results after long-term follow-up. Br. J. Haematol. 2008;141(4):512–516. doi: 10.1111/j.1365-2141.2008.06997.x. [DOI] [PubMed] [Google Scholar]
- 69.Jakubowiak AJ, Kendall T, Al-Zoubi A, Khaled Y, Mineishi S, Ahmed A, Campagnaro E, Brozo C, Braun T, Talpaz M, Kaminski MS. Phase II trial of combination therapy with bortezomib, pegylated liposomal doxorubicin, and dexamethasone in patients with newly diagnosed myeloma. J. Clin. Oncol. 2009;27(30):5015–5022. doi: 10.1200/JCO.2008.19.5370. [DOI] [PubMed] [Google Scholar]
- 70.Cavo M, Tacchetti P, Patriarca F, Petrucci MT, Pantani L, Galli M, Di Raimondo F, Crippa C, Zamagni E, Palumbo A, Offidani M, Corradini P, Narni F, Spadano A, Pescosta N, Deliliers GL, Ledda A, Cellini C, Caravita T, Tosi P, Baccarani M. GIMEMA Italian Myeloma Network. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: a randomised phase 3 study. Lancet. 2010;376(9758):2075–2085. doi: 10.1016/S0140-6736(10)61424-9. [DOI] [PubMed] [Google Scholar]
- 71.San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samoilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Mateos MV, Anderson KC, Esseltine DL, Liu K, Cakana A, van de Velde H, Richardson PG. VISTA Trial Investigators. VISTA Trial Investigators. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N. Engl. J. Med. 2008;359(9):906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 72.Richardson PG, Weller E, Lonial S, Jakubowiak AJ, Jagannath S, Raje NS, Avigan DE, Xie W, Ghobrial IM, Schlossman RL, Mazumder A, Munshi NC, Vesole DH, Joyce R, Kaufman JL, Doss D, Warren DL, Lunde LE, kaster S, Delaney C, Hideshima T, Mitsiades CS, Knight R, Esseltine DL, Anderson KC. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood. 2010;116(5):679–686. doi: 10.1182/blood-2010-02-268862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reeder CB, Reece DE, Kukreti V, Chen C, Trudel S, Hentz J, Noble B, Pirooz NA, Spong JE, Piza JG, Zepeda VH, Mikhael JR, Leis JF, Bergsagel PL, Fonseca R, Stewart AK. Cyclophosphamide, bortezomib and dexamethasone induction for newly diagnosed multiple myeloma: high response rates in a phase II clinical trial. Leukemia. 2009;23(7):1337–1341. doi: 10.1038/leu.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reeder CB, Reece DE, Kukreti V, Chen C, Trudel S, Laumann K, Hentz J, Pirooz NA, Piza JG, Tiedemann R, Mikhael JR, Bergsagel PL, Leis JF, Fonseca R, Stewart AK. Once-versus twice-weekly bortezomib induction therapy with CyBorD in newly diagnosed multiple myeloma. Blood. 2010;115(16):3416–3417. doi: 10.1182/blood-2010-02-271676. [DOI] [PubMed] [Google Scholar]
- 75.Palumbo A, Bringhen S, Rossi D, Cavalli M, Larocca A, Ria R, Offidani M, Patriarca F, Nozzoli C, Guglielmelli T, Benevolo G, Callea V, Baldini L, Morabito F, Grasso M, Leonardi G, Rizzo M, Falcone AP, Gottardi D, Montefusco V, Musto P, Petrucci MT, Ciccone G, Boccadoro M. Bortezomib-melphalan-prednisone-thalidomide followed by maintenance with bortezomib-thalidomide compared with bortezomib-melphalan-prednisone for initial treatment of multiple myeloma: a randomized controlled trial. J. Clin. Oncol. 2010;28(34):5101–5109. doi: 10.1200/JCO.2010.29.8216. [DOI] [PubMed] [Google Scholar]
- 76.Barlogie B, Anaissie E, van Rhee F, Haessle RJ, Hollmig K, Pineda-Roman M, Cottler-Fox M, Mohiuddin A, Alsayed Y, Tricot G, Bolejack V, Zangari M, Epstein J, Petty N, Steward D, Jenkins B, Gurley J, Sullivan E, Crowley J, Shaughnessy JD., Jr Incorporating bortezomib into upfront treatment for multiple myeloma: early results of total therapy 3. Br. J. Haematol. 2007;138(2):176–185. doi: 10.1111/j.1365-2141.2007.06639.x. [DOI] [PubMed] [Google Scholar]
- 77.Nair B, van Rhee F, Shaughnessy JD, Jr, Anaissie E, Szymonifka J, Hoering A, Alsayed Y, Waheed S, Crowley J, Barlogie B. Superior results of Total Therapy 3 (2003–33) in gene expression profiling-defined low-risk multiple myeloma confirmed in subsequent trial 2006–66 with VRD maintenance. Blood. 2010;115(21):4168–4173. doi: 10.1182/blood-2009-11-255620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mateos MV, Oriel A, Martínez-López J, Gutiérrez N, Teruel AI, López de la Guía A, López J, Bengoechea E, Pérez M, Polo M, Palomera L, de Arriba F, González Y, Hernández JM, Granell M, Bello JL, Bargay J, Peñalver FJ, Ribera JM, Martín-Mateos ML, Garcia-Sanz R, Lahuerta JJ, Bladé J, San-Miguel JF. Maintenance therapy with bortezomib plus thalidomide or bortezomib plus prednisone in elderly multiple myeloma patients included in the GEM2005MAS65 trial. Blood. 2012;120(13):2581–2588. doi: 10.1182/blood-2012-05-427815. 2012, Epub Aug 13. [DOI] [PubMed] [Google Scholar]
- 79.Sooneveld P, Schmidt-Wolf IG, van der Holt B, El Jarari L, Bertsch U, Salwender H, Zweegman S, Vellenga E, Broyl A, Blau IW, Weisel KC, Wittebol S, Bos GM, Stevens-Kroef M, Scheid C, Pfreundschuh M, Hose D, Jauch A, van der Velde H, Raymakers R, Schaafsma MR, Kersten MJ, van Marwijk-Kooy M, Duehrsen U, Lindemann W, Wijermans PW, Lokhorst HM, Goldschmidt HM. Bortezomib induction and maintenance treatment in patients with newly diagnosed multiple myeloma: results of the randomized phase III HOVON-65/ GMMG-HD4 trial. J. Clin. Oncol. 2012;30(24):2946–2955. doi: 10.1200/JCO.2011.39.6820. [DOI] [PubMed] [Google Scholar]
- 80.Rosifiol L. Maintenance Therapy After Stem-Cell Transplantation for Multiple Myeloma with Bortezomib/Thalidomide Vs. Thalidomide Vs. alfa2b-Interferon: Final Results of a Phase III Pethema/GEM Randomized. ASH Annual Mtg Abstracts. 2012;120:334. [Google Scholar]
- 81.Heider U, Kaiser M, Müller C, Jakob C, Zavrski I, Schulz CO, Fleissner C, Hecht M, Sezer O. Bortezomib Increases osteoblast activity in myeloma patients irrespective of response to treatment. Eur. J. Haematol. 2006;77(3):233–238. doi: 10.1111/j.1600-0609.2006.00692.x. [DOI] [PubMed] [Google Scholar]
- 82.Boissy P, Andersen TL, Lund T, Kupisiewicz K, Plesner T, Delaissé JM. Pulse treatment with the proteasome inhibitor bortezomib inhibits osteoclast resorptive activity in clinically relevant conditions. Leuk. Res. 2008;32(11):1661–1668. doi: 10.1016/j.leukres.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 83.Munemasa SA, Kuroda Y, Okikawa Y, Katayama Y, Asaoku H, Kubo T, Shimose S, Kimura A. Osteoprogenitor differentiation is not affected by immunomodulatory thalidomide analogs but is promoted by low bortezomib concentration, while both agents suppress osteoclast differentiation. Int. J. Oncol. 2008;33(1):129–136. [PubMed] [Google Scholar]
- 84.Kaiser MF, Heider U, Mieth M, Zang C, von Metzler I, Sezer O. The proteasome inhibitor bortezomib stimulates osteoblastic differentiation of human osteoblast precursors via upregulation of vitamin D receptor signalling. Eur. J. Haematol. 2013;90(4):263–272. doi: 10.1111/ejh.12069. [DOI] [PubMed] [Google Scholar]
- 85.Pennisi A, Li X, Ling W, Khan S, Zangari M, Yaccoby S. The proteasome inhibitor, bortezomib suppresses primary myeloma and stimulates bone formation in myelomatous and nonmyelomatous bones in vivo. Am. J. Hematol. 2009;84(1):6–14. doi: 10.1002/ajh.21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Edelman MJ. The potential role of bortezomib in combination with chemotherapy and radiation in non-small-cell lung cancer. Clin. Lung Cancer. 2005;(7 Suppl 2):S64–S66. doi: 10.3816/clc.2005.s.011. [DOI] [PubMed] [Google Scholar]
- 87.Russo SM, Tepper JE, Baldwin AS, Jr, Liu R, Adams J, Elliott P, Cusack JC., Jr Enhancement of radiosensitivity by proteasome inhibition: implications for a role of NF-kappaB. Int. J. Radiat. Oncol. Biol. Phys. 2001;50:183–193. doi: 10.1016/s0360-3016(01)01446-8. [DOI] [PubMed] [Google Scholar]
- 88.Messersmith WA, Baker SD, Lassiter L, Sullivan RA, Dinh K, Almuete VI, Wright JJ, Donehower RC, Carducci MA, Armstrong DK. Phase I trial of bortezomib in combination with docetaxel in patients with advanced solid tumors. Clin. Cancer Res. 2006;12:1270–1275. doi: 10.1158/1078-0432.CCR-05-1942. [DOI] [PubMed] [Google Scholar]
- 89.Orlowski RZ, Voorhees PM, Garcia RA, Hall MD, Kudrik FJ, Allred T, Johri AR, Jones PE, Ivanova A, Van Deventer HW, Gabriel DA, Shea TC, Mitchell BS, Adams J, Esseltine DL, Trehu EG, Green M, Lehman MJ, Natoli S, Collins JM, Lindley CM, Dees EC. Phase 1 trial of the proteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients with advanced hematologic malignancies. Blood. 2005;105:3058–3065. doi: 10.1182/blood-2004-07-2911. [DOI] [PubMed] [Google Scholar]
- 90.Perrone G, Hideshima T, Ikeda H, Okawa Y, Calabrese E, Gorgun G, Santo L, Cirstea D, Raje N, Chauhan D, Baccarani M, Cavo M, Anderson KC. Ascorbic acid inhibits antitumor activity of bortezomib in vivo. Leukemia. 2009;23:1679–1686. doi: 10.1038/leu.2009.83. [DOI] [PubMed] [Google Scholar]
- 91.Ryan DP, O'Neil BH, Supko JG, Rocha Lima CM, Dees BC, Appleman LJ, Clark J, Fidias P, Orlowski RZ, Kashala O, Eder JP, Cusack JC., Jr A Phase I study of bortezomib plus irinotecan in patients with advanced solid tumors. Cancer. 2006;107:2688–2697. doi: 10.1002/cncr.22280. [DOI] [PubMed] [Google Scholar]
- 92.Takase O, Hirahashi J, Takayanagi A, Chikaraishi A, Marumo T, Ozawa Y, Hayashi M, Shimizu N, Saruta T. Gene transfer of truncated IkappaBalpha prevents tabulointerstitial injury. Kidney Int. 2003;63(2):501–513. doi: 10.1046/j.1523-1755.2003.00781.x. [DOI] [PubMed] [Google Scholar]
- 93.Rangan GK, Wang Y, Tay YC, Harris DC. Inhibition of nuclear factor-kappaB activation reduces cortical tubulointerstitial injury in proteinuric rats. Kidney Int. 1999;56(1):118–134. doi: 10.1046/j.1523-1755.1999.00529.x. [DOI] [PubMed] [Google Scholar]
- 94.Ludwig H, Drach J, Graf H, Lang A, Meran JG. Reversal of acute renal failure by bortezomib-based chemotherapy in patients with multiple myeloma. Haematologica. 2007;92(10):1411–1414. doi: 10.3324/haematol.11463. [DOI] [PubMed] [Google Scholar]
- 95.Sitia R, Palladini G, Merlini G. Bortezomib in the treatment of AL amyioidosis: targeted therapy? Haematologica. 2007;92(10):1302–1307. doi: 10.3324/haematol.12136. [DOI] [PubMed] [Google Scholar]
- 96.Dimopoulos MA, Mateos MV, Richardson PG, Schlag R, Khuageva NK, Shpilberg O, Kropff M, Spicka I, Palumbo A, Wu KL, Esseltine DL, Liu K, Deraedt W, Cakana A, Van De Velde H, San Miguel JF. Risk factors for, and reversibility of, peripheral neuropathy associated with bortezomib-melphalan-prednisone in newly diagnosed patients with multiple myeloma: subanalysis of the phase 3 VISTA study. Eur. J. Haematol. 2011;86(1):23–31. doi: 10.1111/j.1600-0609.2010.01533.x. [DOI] [PubMed] [Google Scholar]
- 97.Favis R, Sun Y, van de Velde H, Broderick E, Levey L, Meyers M, Mulligan G, Harousseau JL, Richardson PG, Ricci DS. Genetic variation associated with bortezomib-induced peripheral neuropathy. Pharmacogenet Genomics. 2011;21(3):121–129. doi: 10.1097/FPC.0b013e3283436b45. [DOI] [PubMed] [Google Scholar]
- 98.Staff NP, Podratz JL, Grassner L, Bader M, Paz J, Knight AM, Loprinzi CL, Trushina E, Windebank AJ. Bortezomib alters microtobule polymerization and axonal transport in rat dorsal root ganglion neurons. Neurotoxicology. 2013 Sep 12;39C:124–131. doi: 10.1016/j.neuro.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Casafont I, Berciano MT, Lafarga M. Bortezomib induces the formation of nuclear poly(A) RNA granules enriched in Sam68 and PABPN1 in sensory ganglia neurons. Neurotox. Res. 2010;17(2):167–178. doi: 10.1007/s12640-009-9086-1. [DOI] [PubMed] [Google Scholar]
- 100.Moreau P, Pylypenko H, Grosicki S, Karamanesht I, Leleu X, Grishunina M, Rekhtman C, Masliak Z, Robak T, Shubina A, Arnulf B, Kropff M, Cavet J, Esselline DL, Feng H, Girgis S, van de Velde H, Deraedt W, Harousseau JL. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol. 2011;12(5):431–440. doi: 10.1016/S1470-2045(11)70081-X. [DOI] [PubMed] [Google Scholar]
- 101.Richardson PG, Sonneveld P, Schuster MW, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, Bladé J, Boccadoro M, Cavenagh JD, Boral AL, Esseltine DL, Wen PY, Amato AA, Anderson KC, San Miguel J. Reversibility of symptomatic peripheral neuropathy with bortezomib in the phase III APEX trial in relapsed multiple myeloma: impact of a dose-modification guideline. Br. J. Haematol. 2009;144(6):895–903. doi: 10.1111/j.1365-2141.2008.07573.x. [DOI] [PubMed] [Google Scholar]
- 102.Reece DE, Hegenbart U, Sanchorawala V, Merlini G, Palladini G, Bladé J, Fermand JP, Hassoun H, Heffner L, Vescio RA, Liu K, Enny C, Esseltine DL, van de Velde H, Cakana A, Comenzo RL. Efficacy and safety of once-weekly and twice-weekly bortezomib in patients with relapsed systemic AL amyloidosis: results of a phase 1/2 study. Blood. 2011;118(4):865–873. doi: 10.1182/blood-2011-02-334227. [DOI] [PubMed] [Google Scholar]
- 103.Mikhael JR, Mikhael JR, Schuster SR, Jimenez-Zepeda VH, Bello N, Spong J, Reeder CB, Stewart AK, Bergsagel PL, Fonseca R. Cyclophosphamide-bortezomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyioidosis. Blood. 2012;119(19):4391–4394. doi: 10.1182/blood-2011-11-390930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zonder JA, Saochorawala V, Snyder RM, Matous F, Terebelo H, Janakiraman N, Mapara MY, Lalo S, Tageja N, Webb C, Monsma D, Sellers C, Abrams J, Gasparetto C. Melphalan and dexamethasone plus BTZ induces hematologic and organ responses in AL-amyloidosis with tolerable neurotxicity ASH. 2009 meeting, abstract 746. [Google Scholar]
- 105.Zonder JA. Pomalidomide, Bortezomib, and Dexamethasone as First-Line Treatment in Treating Patients With Amyloid Light-Chain Amyloidosis or Light Chain Deposition Disease. ClinicalTrials.gov Identifier; NCT01728259 2012, ClinicalTrials.Gov National Institute of Health first received: November 13, 2012 Last verified: May 2014.
- 106.Leonard JP, Furman RR, Coleman M. Proteasome inhibition with bortezomib: a new therapeutic strategy for non-Hodgkin’s lymphoma. Int. J. Cancer. 2006;119(5):971–979. doi: 10.1002/ijc.21805. [DOI] [PubMed] [Google Scholar]
- 107.Mato AR, Feldman T, Goy A. Proteasome inhibition and combination therapy for non-Hodgkin’s lymphoma: from bench to bedside. Oncologist. 2012;17(5):694–707. doi: 10.1634/theoncologist.2011-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, Adams J, Esseltine DL, Elliott PJ, Pien CS, Guerciolini R, Anderson JK, Depcik-Smith ND, Bhagat R, Lehman MJ, Novick SC, O’Connor OA, Soignet SL. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J. Clin. Oncol. 2002;20(22):4420–4427. doi: 10.1200/JCO.2002.01.133. [DOI] [PubMed] [Google Scholar]
- 109.O’Connor OA, Wright J, Moskowitz C, Muzzy J, MacGregor-Cortelli B, Stubblefield M, Straus D, Portlock C, Hamlin P, Choi E, Dumetrescu O, Esseltine D, Trehu E, Adams J, Schenkein D, Zelentz AD. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J. Clin. Oncol. 2005;23(4):676–684. doi: 10.1200/JCO.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 110.Fisher RIRI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Stadtmauer EA, O’Conner OA, Shi H, Boral AL, Goy A. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2006;24(30):4867–4874. doi: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 111.Goy A, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Nasta S, O’Connor OA, Shi H, Boral AL, Fisher RI. Bortezomib in patients with relapsed or refractory mantle cell lymphoma: updated time-to-event analyses of the multicenter phase 2 PINNACLE study. Annals of Oncology. 2009;20(3):520–525. doi: 10.1093/annonc/mdn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weigert O, Weidmann E, Mueck R, Bentz M, von Schilling C, Rohrberg R, Jentsch-Ullrich K, Hiddemann W, Dreyling M. A novel regimen combining high dose cytarabine and bortezomib has activity in multiply relapsed and refractory mantle cell lymphoma - long-term results of a multicenter observation study. Leuk. Lymphoma. 2009;50(5):716–722. doi: 10.1080/10428190902856790. [DOI] [PubMed] [Google Scholar]
- 113.Kouroukis CT, Fernandez LA, Crump M, Gascoyne RD, Chua NS, Buckstein R, Turner R, Assouline S, Klasa RJ, Walsh W, Powers J, Eisenhauer E. A phase II study of bortezomib and gemcitabine to relapsed mantle cell lymphoma from the National Cancer Institute of Canada Clinical Trials Group (IND 172) Leuk Lymphoma. 2011;52(3):394–399. doi: 10.3109/10428194.2010.546015. [DOI] [PubMed] [Google Scholar]
- 114.Orciuolo E, Buda G, Pelosini M, Petrini M. Fludarabine, Bortezomib, Myocet and rituximab chemotherapy in relapsed and refractory mantle cell lymphoma. Br. J. Haematol. 2010;148(5):810–812. doi: 10.1111/j.1365-2141.2009.07998.x. [DOI] [PubMed] [Google Scholar]
- 115.Musto PM, Guariglia R, Pietrantuono G, Vilani O, Martorelli MC, D’Auria F, Zonno A, Zupa A, Bianchino G, Improta G, Grieco V, Digiovannantonio L, Feudale E, Lerose R, Pinto A, Ferrara F. A pilot study with rituximab, bortezomib and hyper-fractionated cyclophosphamide (rbc regimen) for the treatment of advanced mantle cell lymphoma in elderly patients. Haematologica. 2008;93(suppl 1) abstr 0273. [Google Scholar]
- 116.Friedberg JW, Vose JM, Kelly JL, Young F, Bernstein SH, Peterson D, Rich L, Blumel S, Proia NK, Liesveld J, Fisher RI, Armitage JO, Grant S, Leonard JP. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood. 2011;117(10):2807–2812. doi: 10.1182/blood-2010-11-314708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Romaguera JE, Fayad LE, McLaughlin P, Pro B, Rodriguez A, Wang M, Weaver P, Hartig K, Kwak LW, Feldman T, Smith J, Ford P, Goldberg S, Pecora A, Goy A. Phase I trial of bortezomib in combination with rituximab-HyperCVAD alternating with rituximab, methotrexate and cytarabine for untreated aggressive mantle cell lymphoma. Br. J. Haematol. 2010;151(1):47–53. doi: 10.1111/j.1365-2141.2010.08315.x. [DOI] [PubMed] [Google Scholar]
- 118.Kahl B, Chang J, Eickhoff J, Gilbert L, Rogers E, Werndli J, Huie M, McFarland T, Volk M, Blank JH, Callander N, Longo W, Peterson C. VcR-CVAD Produces a High Complete Response Rate in Untreated Mantle Cell Lymphoma: A Phase II Study from the Wisconsin Oncology Network. Blood. 2008;112 abstr 265. [Google Scholar]
- 119.Ribrag V, Ribrag V, Herve T, Casasnovas O, Bosly A, Bouabdallah R, Delarue R, Boue F, Bron D, Geugier P, Haioun C, Offner FC, Coiffier B. Final Results of a Randomized Phase 2 Multicenter Study of Two Bortezomib Schedules In Patients with Recurrent or Refractory Follicular Lymphoma. Groups d’Etude Des Lymphomes De l’Adulte (GELA) Study FL-05. Blood. 2010;116 abstr 768. [Google Scholar]
- 120.Di Bella N, Taetle R, Kolibaba K, Boyd T, Raju R, Barrera D, Cochran EW, Jr, Dien PY, Lyons R, Schlegel PJ, Vukelja SJ, Boston J, Boehm KA, Wang Y, Asmar L. Results of a phase 2 study of bortezomib in patients with relapsed or refractory indolent lymphoma. Blood. 2010;115(3):475–480. doi: 10.1182/blood-2009-08-233155. [DOI] [PubMed] [Google Scholar]
- 121.O’Connor OA, Portlock C, Moskowitz C, Hamlin P, Straus D, Gerecitano J, Gonen M, Dumitrescu O, Sarasohn D, Butos J, Neylon E, Mac-Gregor Cortelli B, Blumel S, Evens AM, Zelenetz AD, Wright I, Cooper B, Winter I, Vose J. Time to treatment response in patients with follicular lymphoma treated with bortezomib is longer compared with other histologic subtypes. Clin. Cancer Res. 2010;16(2):719–726. doi: 10.1158/1078-0432.CCR-08-2647. [DOI] [PubMed] [Google Scholar]
- 122.Sehn LH, MacDonald D, Rubin S, Cantin G, Rubinger M, Lemieux B, Basi S, Imrie K, Gascoyne RD, Sussman J, Chen BE, Djurfeldt M, Shepherd L, Couban S, Crump M. Bortezomib ADDED to R-CVP is safe and effective for previously untreated advanced-stage follicular lymphoma: a phase II study by the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2011;29(25):3396–3401. doi: 10.1200/JCO.2010.33.6594. [DOI] [PubMed] [Google Scholar]
- 123.Nabhan C, Dalal N, Tolzien K, Starr A. Bortezomib (VELCADE), rituximab, cyclophosphamide, dexamethasone (VRCD) combination therapy in front-line low-grade non-hodgkin lymphoma (LG-NHL) is active in elderly patient population. Blood. 2010;116 abstr 1769. [Google Scholar]
- 124.Dimopoulos MA, Garcia-Sanz R, Gavriatopoulou M, Morel P, Kyrtsonis MC, Michalis E, Kartasis Z, Leleu X, Palladini G, Tedeschi A, Gika D, Merlini G, Kastritis E, Sonneveld P. Primary therapy of Waldenstrom’s macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone and rituximab (BDR); long term results of a phase II study of the European Myeloma Network (EMN) Blood. 2013 Sep 4; doi: 10.1182/blood-2013-05-503862. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 125.Ruan J, Martin P, Furman RR, Lee SM, Cheung K, Vose JM, Lacasce A, Morrison J, Elstrom R, Ely S, Chadburn A, Cesarman E, Coleman M, Leonard JP. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J. Clin. Oncol. 2011;29(6):690–697. doi: 10.1200/JCO.2010.31.1142. [DOI] [PubMed] [Google Scholar]
- 126.Kim JE, Yoon DH, Jang G, Lee DH, Kim S, Park CS, Huh J, Kim WS, Park J, Lee JH, Lee SI, Suh C. A phase I/II study of bortezomib plus CHOP every 2 weeks (CHOP-14) in patients with advanced-stage diffuse large B-cell lymphomas. Korean J. Hemalol. 2012;47(1):53–59. doi: 10.5045/kjh.2012.47.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Furman RR, Martin P, Ruan J, Cheung YK, Vose JM, LaCasce AS, Elstrom R, Coleman M, Leonard JP. Phase 1 trial of bortezomib plus R-CHOP in previously untreated patients with aggressive non-Hodgkin lymphoma. Cancer. 2010;116(23):5432–5439. doi: 10.1002/cncr.25509. [DOI] [PubMed] [Google Scholar]
- 128.Dunleavy K, Pittaluga S, Czuczman MS, Dave SS, Wright G, Grant N, Shovlin M, Jaffe ES, Janik JE, Staudt LM, Wilson WH. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood. 2009;113(24):6069–6076. doi: 10.1182/blood-2009-01-199679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Krappmann D, Emmerich F, Kordes U, Scharschmidt E, Dörken B, Scheidereit C. Molecular mechanisms of constitutive NF-kappaB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene. 1999;18(4):943–953. doi: 10.1038/sj.onc.1202351. [DOI] [PubMed] [Google Scholar]
- 130.Blum KA, Johnson JL, Niedzwiecki D, Canellos GP, Cheson BD, Battled NL. Single agent bortezomib in the treatment of relapsed and refractory Hodgkin lymphoma: cancer and leukemia Group B protocol 50206. Leuk. Lymphoma. 2007;48(7):1313–1319. doi: 10.1080/10428190701411458. [DOI] [PubMed] [Google Scholar]
- 131.Sarlo C, Buccisano F, Manrillo L, Cefalo M, Di Caprio L, Cicconi L, Ditto C, Ottaviani L, Di Veroli A, Del Principe MI, Grasso MA, Nasso D, De Santis G, Amadori S, Venditti A. Phase II study of bortezomib as a single agent in patients with previously untreated or relapsed/refractory acute myeloid leukemia ineligible for intensive therapy. Leuk. Res. Treatment. 2013;2013:705–714. doi: 10.1155/2013/705714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Attar BC, Johnson JL, Amrein PC, Lozanski G, Wadleigh M, DeAngelo DJ, Kolitz JE, Powell BL, Voorhees P, Wang ES, Blum W, Stone RM, Marcucci G, Bloomfield CD, Moser B, Larson RA. Bortezomib added to daunorubicin and cytarabine during induction therapy and to intermediate-dose cytarabine for consolidation in patients with previously untreated acute myeloid leukemia age 60 to 75 years: CALGB (Alliance) study 10502. J. Clin. Oncol. 2013;31(7):923–929. doi: 10.1200/JCO.2012.45.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Howard DS, Liesveld J, Phillips GL, 2nd, Hayslip J, Weiss H, Jordan CT, Guzman ML. A phase I study using bortezomib with weekly idarubicin for treatment of elderly patients with acute myeloid leukemia. Leuk. Res. 2013 Sep 8; doi: 10.1016/j.leukres.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ying M, Zhou X, Zhong L, Lin N, Jing H, Luo P, Yang X, Song H, Yang B, He Q. Bortezomib sensitizes human acute myeloid leukemia cells to all-trans-retinoic acid-induced differentiation by modifying the RARα/STATl axis. Mol. Cancer Ther. 2013;12(2):195–206. doi: 10.1158/1535-7163.MCT-12-0433. [DOI] [PubMed] [Google Scholar]
- 135.Engel RH, Brown JA, Von Roenn JH, O’Regan RM, Bergan R, Badve S, Rademaker A, Gradishar WJ. A phase II study of single agent bortezomib in patients with metastatic breast cancer: a single institution experience. Cancer Invest. 2007;25:733–737. doi: 10.1080/07357900701506573. [DOI] [PubMed] [Google Scholar]
- 136.Yang CH, Gonzalez-Angulo AM, Reuben JM, Booser DJ, Pusztai L, Krishnamurthy S, Esseltine D, Stec J, Broglio KR, Islam R, Hortobagyi GN, Cristofanilli M. Bortezomib (VELCADE) in metastatic breast cancer: pharmacodynamics, biological effects, and prediction of clinical benefits. Ann. Oncol. 2006;77:813–817. doi: 10.1093/annonc/mdj131. [DOI] [PubMed] [Google Scholar]
- 137.Irvin WJ, Orlowski RZ, Chiu WK, Carey LA, Collichio FA, Bernard PS, Stijleman IJ, Perou C, Ivanova A, Dees EC. Phase II study of bortezomib and pegylated liposomal doxorubicin in the treatment of metastatic breast cancer. Clin. Breast Cancer. 2011;10(6):465–470. doi: 10.3816/CBC.2010.n.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Morris MJ, Kelly WK, Slovin S, Ryan C, Eicher C, Heller G, Scher HI. A phase II trial of bortezomib and prednisone for castration resistant metastatic prostate cancer. J. Urol. 2007;178(6):2378–2383. doi: 10.1016/j.juro.2007.08.015. discussion 83-4. [DOI] [PubMed] [Google Scholar]
- 139.Hainsworth JD, Meluch AA, Spigel DR, Barton J, Jr, Simons L, Meng C, Gould B, Greco FA. Weekly docetaxel and bortezomib as first-line treatment for patients with hormone-refractory prostate cancer: A minnie pearl cancer research network phase II trial. Clin. Genitourinary Cancer. 2007;5(4):278–283. doi: 10.3816/CGC.2007.n.004. [DOI] [PubMed] [Google Scholar]
- 140.Li TH, Ho L, Piperdi B, Elrafei T, Camacho FJ, Rigas JR, Perez-Soler R, Gucalp R. Phase II study of the proteasome inhibitor bortezomib (PS-341, Velcade) in chemotherapy-naive patients with advanced stage non-small cell lung cancer (NSCLC) Lung Cancer. 2010;68(1):89–93. doi: 10.1016/j.lungcan.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 141.Shah MA, Power DG, Kindler HL, Holen KD, Kemeny MM, Ilson DH, Tang L, Capanu M, Wright JJ, Kelsen DP. A multicenter, phase II study of Bortezomib (PS-341) in patients with unresectable or metastatic gastric and gastroesophageal junction adenocarcinoma. Investigational New Drugs. 29(6):1475–1481. doi: 10.1007/s10637-010-9474-7. [DOI] [PubMed] [Google Scholar]
- 142.Shah MH, Young D, Kindler HL, Webb I, Kleiber B, Wright J, Grever M. Phase II study of the proteasome inhibitor bortezomib (PS-341) in patients with metastatic neuroendocrine tumors. Clin. Cancer Res. 2004;10:6111–6118. doi: 10.1158/1078-0432.CCR-04-0422. [DOI] [PubMed] [Google Scholar]
- 143.Kondagunta GV, Drucker B, Schwartz L, Bacik J, Marion S, Russo P, Mazumdar M, Motzer RJ. Phase II trial of bortezomib for patients with advanced renal cell carcinoma. J. Clin. Oncol. 2004;22:3720–3725. doi: 10.1200/JCO.2004.10.155. [DOI] [PubMed] [Google Scholar]
- 144.Ocrlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BA, Scheper RJ, Jansen G. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112(6):2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 145.Lü S, Yang J, Song X, Gong S, Zhou H, Guo L, Song N, Bao X, Chen P, Wang J. Point mutation of the proteasome beta5 subunit gene is an important mechanism of bortezomib resistance in bortezomib-selected variants of Jurkat T cell lymphoblastic lymphoma/leukemia line. J. Pharmacol. Exp. Ther. 2008;326(2):423–431. doi: 10.1124/jpet.108.138131. (2008). 2008, Epub May 23. [DOI] [PubMed] [Google Scholar]
- 146.Smith AJ, Dai H, Correia C, Takahashi R, Lee SH, Schmitz I, Kaufmann SH. Noxa/Bc1-2 protein interactions contribute to bortezomib resistance in human lymphoid cells. J. Biol. Chem. 2011;286(20):17682–17692. doi: 10.1074/jbc.M110.189092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang DT, Young KH, Kahl BS, Markovina S, Miyamoto S. Prevalence of bortezomib-resistant constitutive NF-kappaB activity in mantle cell lymphoma. Mol Cancer. 2008;7:40. doi: 10.1186/1476-4598-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kern J, Untergasser G, Zenzmaier C, Sarg B, Gastl G, Gunsilius E, Steurer M. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood. 2009;114(18):3960–3967. doi: 10.1182/blood-2009-03-209668. [DOI] [PubMed] [Google Scholar]
- 149.Hirai M, Kadowaki N, Kitawaki T, Fujita H, Takaori-Kondo A, Fukui R, Miyake K, Maeda T, Kamihira S, Miyachi Y, Uchiyama T. Bortezomib suppresses function and survival of plasmacytoid dendritic cells by targeting intracellular trafficking of Toll-like receptors and endoplasmic reticulum homeostasis. Blood. 2010 Jan 13;117(2):500–509. doi: 10.1182/blood-2010-05-284737. Epub Oct 18. [DOI] [PubMed] [Google Scholar]
- 150.De Wilt LH. Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochem Pharmacol. 2012 Jan 15;83(2):207–217. doi: 10.1016/j.bcp.2011.10.009. 2011, Epub Oct 18. [DOI] [PubMed] [Google Scholar]
- 151.Fournier MJ, Gareau C, Mazroui R. The chemotherapeutic agent bortezomib induces the formation of stress granules. Cancer Cell Int. 2010;12(10) doi: 10.1186/1475-2867-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang L, Kumar S, Fridley BL, Kalari KR, Moon I, Pelleymounter LL, Hildebrandt MA, Batzler A, Eckloff BW, Wieben ED, Greipp PR. Proteasome beta subunit pharma-cogenomics: gene resequencing and functional genomics. Clin. Cancer Res. 2008;14(11):3503–3513. doi: 10.1158/1078-0432.CCR-07-5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hideshima T, Chauhan D, Richardson P, Anderson KC. Identification and validation of novel therapeutic targets for multiple mycloma. J. Clin. Oncol. 2005;23(26):6345–6350. doi: 10.1200/JCO.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 154.Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, Reece DE, Chung KC, Tiedetmann RE. Xbpls-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer Cell. 2013;24(3):289–304. doi: 10.1016/j.ccr.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Golden EB, Lam PY, Kardosh A, Gaffney KJ, Cadenas E, Louie SG, Petasis NA, Chen TC, Schönthal AH. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood. 2009;113(23):5927–5937. doi: 10.1182/blood-2008-07-171389. [DOI] [PubMed] [Google Scholar]
- 156.Huber EM, Groll M. Inhibitors for the immuno- and constitutive proteasome: current and future trends in drug development. Angew. Chem. Int. Ed. Engl. 2012 Aug 27;51(35):8708–8720. doi: 10.1002/anie.201201616. (2012)Epub Jun 18. Review. [DOI] [PubMed] [Google Scholar]
- 157.Herndon TM, Deisseroth A, Kaminskas E, Kane RC, Koti KM, Rothmann MD, Habtemariam B, Bullock J, Bray JD, Hawes J, Palmby TR, Jee J, Adams W, Mahayni H, Brown J, Dorantes A, Sridhara R, Farrell AT, Pazdur R. Food and drug administration approval: carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 2013;19(17):4559–4563. doi: 10.1158/1078-0432.CCR-13-0755. 2013, Epub Jun 17. [DOI] [PubMed] [Google Scholar]
- 158.Kisselev AF, Van Der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem. Biol. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem. Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 160.Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007 Nov 1;110(9):3281–3290. doi: 10.1182/blood-2007-01-065888. 2007 Epub Jun 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ruschak AM, Slassi M, Kay LE, Schimmer AD. Novel proteasome inhibitors to overcome bortezomib resistance. J. Natl. Cancer Inst. 2011;103:1007–1017. doi: 10.1093/jnci/djr160. [DOI] [PubMed] [Google Scholar]
- 162.Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007;110:3281–3290. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yang J, Wang Z, Fang Y, Jiang J, Zhao F, Wong H, Bennett MK, Molineaux CJ, Kirk CJ. Pharmacokinetics, pharmacodynamics, metabolism, distribution, and excretion of carfilzomib in rats. Drug Metab. Dispos. 2011;39(10):1873–1882. doi: 10.1124/dmd.111.039164. [DOI] [PubMed] [Google Scholar]
- 164.Papadopoulos K, Mendelson DS, Tolcher AW, Patnaik A, Burris HA, Rasco DW, Bendell JC, Gordon MS, Kato G, Wong H, Bomba D, Lee S, Gillenwater HH, Woo T, Infante JR. A Phase 1, open-label, dose-escalation study of the novel oral proteasome inhibitor (PI) ONX 0912 in patients with advanced refractory or recurrent solid tumors. J. Clin. Oncol. 2011;(Suppl 29) Abstr.3075. [Google Scholar]
- 165.O’Connor OA, Stewart AK, Vallone M, Molineaux CJ, Kunkel LA, Gerecitano JF, Orlowski RZ. A Phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR-171) in patients with hematologic malignancies. Clin. Cancer Res. 2009;15(22):7085–7091. doi: 10.1158/1078-0432.CCR-09-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk KD, Smyth MS, Sun CM, Vallone MK, Woo TM, Molineaux CJ, Bennett MK. Antitumora of PR-171 a novel irreversible inhibitor of the proteasome. Cancer Res. 2007;67(13):6383–6391. doi: 10.1158/0008-5472.CAN-06-4086. [DOI] [PubMed] [Google Scholar]
- 167.Hurchla MA, Garcia-Gomez A, Hornick MC, Ocio EM, Li A, Blanco JF, Collins L, Kirk CJ, Piwnica-Worms D, Vij R, Tomasson MH, Pandiella A, San Miguel JF, Garayoa M, Weilbaecher KN. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia. 2013;27(2):430–440. doi: 10.1038/leu.2012.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.diCapua Siegel DS, Martin T, Wang M, vij R, Jakubowiak AJ, Jagannath S, Lonial S, Kukreti V, Bahlis N, Alsina M, chanan-Kahn A, Somolo G, Buadi F, Reu F, Zonder JA, Song K, Stadtmauer E, Wong AF, Vallone M, Chang YL, Kauffman M, Orlowski RZ, Stewart K, singhal SB the MMRC. Results of PX-171-003-A1, an open-label, single-arm, phase 2 (Ph 2) study of carfilzomib (CFZ) in patients (pts) with relapsed and refractory multiple myeloma (MM) Blood. 2010;116(21):433. [Google Scholar]
- 169.Vij R, Kaufman JL, Jakubow A, Wang M, Jagannath S, Kukreti V, McDonagh KT, Alsina M, Bahlis NJ, Belch A, Reu FJ, Gabrail NY, Matous J, Vesole DH, Orlowski RZ, Wear S, Kunkel L, Wong A, Lee P, Stewart AK. Final results from the bortezomib-naive group of PX-171-004, a phase 2 study of single agent carfilzomib in patients with relapsed and/or refractory MM. Blood. 2011;118(21):369–370. [Google Scholar]
- 170.Stewart K, Stewart K, Siegel D, Wang M, Kaufman J, Jakubowiak A, Jagnnath S, Kukreti V, McDonagh K, Alsina M, Bahlis N, Belch A, Gabrail N, Le M, Bennett M, Vij R. Results of Px-171-004, an ongoing open-label, phase Ii study of carfilzomib in patients with relapsed and/or refractory multiple mycloma (R/R Mm) with or without prior bortezomib exposure. Haematologica-the Hematology Journal. 2010;95:452. [Google Scholar]
- 171.Alsina M, Trudel S, Furman RR, Rosen PJ, O’Connor OA, Comenzo RL, Wong A, Kunkel LA, Molineaux CJ, Goy A. A phase I single-agent study of twice-weekly consecutive-day dosing of the proteasome inhibitor carfilzomib in patients with relapsed or refractory multiple myeloma or lymphoma. Clin. Cancer Res. 2012;18(17):4830–4840. doi: 10.1158/1078-0432.CCR-11-3007. [DOI] [PubMed] [Google Scholar]
- 172.Badros AZ, Papadopoulos KP, Zojwalla N, Lee JR, Siegel DS. A Phase 1b study of 30-minute infusion carfilzomib 20/45 and 20/56 Mg/m2 plus 40 Mg weekly dexamethasone in patients with relapsed and/or refractory (R/R) multiple myeloma. ASH. 2012 abstract 4036. [Google Scholar]
- 173.A Randomized, Open-label, Phase 3 Study of Carfilzomib Plus Dexamethasone vs. Bortezomib Plus Dexamethasone in Patients With Relapsed Multiple Myeloma. ClinicalTrials.gov. 2012 Identifier: NCT01568866; First received: March 28, 2012: Last verified: June 2014.
- 174.Siege ID, Martin T, Nooka A, Harvey RD, Vij R, Niesvizky R, Badros AZ, Jagannath S, McCulloch L, Rajangam K, Lonial S. Integrated safety profile of single-agent carfilzomib: experience from 526 patients enrolled in 4 phase 2 clinical studies. Haematologica. 2013 Aug 9; doi: 10.3324/haematol.2013.089334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Vij R, Vij R, Wang L, Orlowski RZ, Stewart AK, Jagannath S, Lonial S, Trudel S, Jakubowiak AJ, Belch A, Alsina M, Bahlis NJ, Le MH, Cruickshank S, Bennett MK, Molineaux S, Kauffman M, Siegel D. The Multiple Myeloma Research Consortium (MMRC) Carfilzomib (CFZ), a novel proteasome inhibitor for relapsed or refractory multiple myeloma, is associated with minimal peripheral neuropathic effects. Blood. 2009;114(22):178–179. [Google Scholar]
- 176.Jakubowiak AJ, Akubowiak AJ, Dytfeld D, Griffith KA, lebovic D, Vesole DH, Jagannath S, Al-Zoubi A, Anderson T, Nordgren B, Detweiler-Short K, Stockerl-Goldstein K, Ahmed A, Jobkar R, Durecki DE, McDonnell K, Mietzel M, Couriel D, Kaminski M, Vig R. Final results of a frontline phase 1/2 Study of carfilzomib, lenalidomide, and low-dose dexamethasone (CRd) in multiple myeloma (MM) Blood. 2011;118(21):288–289. doi: 10.1182/blood-2012-04-422683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang M, Bensinger W, Martin T, Alsina M. Interim results from PX-171-006, a phase (Ph) II multicenter dose-expansion study of carfilzomib (CFZ), lenalidomide (LEN), and low-dose dexamethasone (IoDex) in relapsed and/or refractory multiple myeloma (R/R MM) J. clin. Oncol. 2011;(suppl) abstr 8025. [Google Scholar]
- 178.Niesvizky R, Martin TG, 3rd, Bensinger WI, Alsina M, Siegel DS, Kunkel LA, Wong AF, Lee S, Orlowski RZ, Wang M. Phase Ib dose-escalation study (PX-171-006) of carfilzomib, lenalidomide, and low-dose dexamethasone in relapsed or progressive multiple myeloma. Clin. Cancer Res. 2013;19(8):2248–2256. doi: 10.1158/1078-0432.CCR-12-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.A Phase I Dose Escalation Study of Carfilzomib in Patients With Previously-Treated Systemic Light-Chain (AL) Amyloidosis ClinicalTrials.gov. 2013 Identifier: NCT01789242; First received: February 6, 2013; Last verified: May 2014. [Google Scholar]
- 180.Papadopoulos KP, Burris HA, 3rd, Gordon M, Lee P, Sausville EA, Rosen PJ, Patnaik A, Cutler RE, Jr, Wang Z, Lee S, Jones SF, Infante JR. A phase I/II study of carfilzomib 2-10-min infusion in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013;72(4):861–868. doi: 10.1007/s00280-013-2267-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lawasut P, Chauhan D, Laubach J, Hayes C, Fabre C, Maglio M, Mitsiades C, Hideshima T, Anderson KC, Richardson PG. New proteasome inhibitors in myeloma. Curr Hematol Malig Rep. 2012 Dec 7;(4):258–266. doi: 10.1007/s11899-012-0141-2. Review. [DOI] [PubMed] [Google Scholar]
- 182.Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, Richardson P, Anderson KC. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Cancer Res. 2011;17(16):5311–5321. doi: 10.1158/1078-0432.CCR-11-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Richardson PG, Baz R, Wang L, Jakubowiak AJ, Berg D, Liu G, Gupta N, Di Bacco A, Hui AM, Lonial S. Investigational agent MLN9708, an oral proteasome inhibitor, in patients (Pts) with relapsed and/or refractory multiple myeloma (MM): Results from the expansion cohorts of a phase 1 dose-escalation study. Blood. 2011;118(21):140. [Google Scholar]
- 184.Richardson PG, Berdeja JG, Niesvizky R, Lonial S, Roy V, Hari P, Berg D, Liu G, Gupta N, Di Bacco A, Hui AM, Kumar S. Oral weekly MLN9708, an investigational proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients (pts) with previously untreated multiple myeloma (MM): A phase I/II stydy. J Clin. Oncol. 2012;30(suppl) abstr 8033. [Google Scholar]
- 185.US National Institute of Health. [cited 2012 July 31]; ClincalTrial.gov. 2012 Available from: http://clinicaltrials.gov/.
- 186.Gallerani E, Zucchetti M, Brunelli D, Marangon E, Noberasco C, Hess D, Delmonte A, Martinelli G, Böhm S, Driessen C, De Braud F, Marsoni S, Cereda R, Sala F, D’Incalci M, Sessa C. A first in human phase I study of the proteasome inhibitor CEP-18770 in patients with advanced solid tumours and multiple myeloma. Eur. J. Cancer. 2013;49(2):290–296. doi: 10.1016/j.ejca.2012.09.009. 2012, Epub Oct 8. Review. [DOI] [PubMed] [Google Scholar]
- 187.Chauhan D, Singh AV, Aujay M, Kirk CJ, Bandi M, Ciccarell IB, Raje N, Richardson P, Anderson KC. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood. 2010;116:4906–4915. doi: 10.1182/blood-2010-04-276626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tsukamoto S, Yokosawa H. Inhibition of the ubiquitin-proteasome system by natural products for cancer therapy. Planta Med. 2010;76:1064–1074. doi: 10.1055/s-0029-1240901. [DOI] [PubMed] [Google Scholar]
- 189.Richardson PG, Spencer A, Cannell P, Harrison SJ, Catley L, Underhill C, Zimmerman TM, Hofmeister CC, Jakubowiak AJ, Laubach JP, Palladino MA, Longenecker AM, Lay A, Wear S, Lloyd GK, Hannah AL, Reich S, Spear MA, Anderson KC. Phase 1 clinical evaluation of twice-weekly marizomib (NPI-0052), a novel proteasome inhibitor, in patients with relapsed/refractory multiple myeloma (MM) Ash. 2011;118(21):140–141. [Google Scholar]
- 190.Anderson KC. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol. 2012 Feb 1;30(4):445–452. doi: 10.1200/JCO.2011.37.8919. 2012, Epub Jan 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood. 2009 May 7;113(19):4667–4676. doi: 10.1182/blood-2008-07-171637. 2008, Epub Dec 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ. Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood. 2009;113:4667–4676. doi: 10.1182/blood-2008-07-171637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Muchamuel T, Baster M, Aujay MA, Suzuki E, Kalim KW, Lauer C, Sylvain C, Ring ER, Shields J, Jiang J, Shwonek P, Parlati F, Demo SD, Bennett MK, Kirk CJ, Groettrup M. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009;15:781–787. doi: 10.1038/nm.1978. [DOI] [PubMed] [Google Scholar]
- 194.Varshavsky A. The N-end rule and regulation of apoptosis. Nat. Cell Biol. 2009;3(5):373–376. doi: 10.1038/ncb0503-373. [DOI] [PubMed] [Google Scholar]
- 195.Schwarz SE. Characterization of human hect domain family members and their interaction with UbcH5 and UbcH7. J. Biol. Chem. 1998;273(20):12148–12154. doi: 10.1074/jbc.273.20.12148. [DOI] [PubMed] [Google Scholar]
- 196.Saurin AJ, Borden KL, Boddy MN, Freemont PS. Does this have a familiar RING? Trends Biochem. Sci. 1996;21(6):208–214. [PubMed] [Google Scholar]
- 197.Lorick KL, Jensen JP, Fangd S, Ong AM, Hatakeyama S, Weissman AM. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc. Natl. Acad. Sci. U. S. A. 96(20):11364–11369. doi: 10.1073/pnas.96.20.11364. 199, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 199.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–888. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 200.Suzuki K, Matsubara H. Recent advances in p53 research and cancer treatment. J. Biomed. Biotechnol. 2011;7 doi: 10.1155/2011/978312. 2011, Epub Jun 16 Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Goldenberg SJ, Marblestone JG, Mattern MR, Nicholson B. Strategies for the identification of ubiquitin ligase inhibitors. Biochem Soc Trans. 2010 Feb;38(Pt 1):132–136. doi: 10.1042/BST0380132. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011 Jun 23;12(7):439–452. doi: 10.1038/nrm3143. 2010, Review. [DOI] [PubMed] [Google Scholar]
- 203.Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, Karasawa S, Carmel G, Jackson P, Abbasian M, Mahmoudi A, Cathers B, Rychak E, Gaidarova S, Chen R, Schafer PH, Handa H, Daniel TO, Evans JF, Chopra R. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012 Nov;26(11):2326–2335. doi: 10.1038/leu.2012.119. 2012 Epub May 3,. Erratum in: Leukemia. 2012, Nov; 26(11): 2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Verma R, Peters NR, D’Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW. Ubistatins inhibit proteaosme-dependent degradation by binding the ubiquitin chain. Science. 2004;306:117–120. doi: 10.1126/science.1100946. [DOI] [PubMed] [Google Scholar]
- 205.Fraile JM, Quesada V, Rodriguez D, Freije JM, Lόpez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012 May 10;31(19):2373–2388. doi: 10.1038/onc.2011.443. 2011 Epub Sep 26. Review. [DOI] [PubMed] [Google Scholar]
- 206.Lim KH, Baek KH. Deubiquitinating enzymes as therapeutic targets in cancer. Curr. Pharm. Des. 2013;19(22):4039–4052. doi: 10.2174/1381612811319220013. [DOI] [PubMed] [Google Scholar]
- 207.D’Arcy P, Linder S. Proteasome deubiquitinases as novel targets for cancer therapy. Int. J. Biochem. Cell. Biol. 2012 Nov;44(11):1729–1738. doi: 10.1016/j.biocel.2012.07.011. 2012 Epub Jul. [DOI] [PubMed] [Google Scholar]
- 208.D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, Linder S. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011;17:1636–1640. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- 209.Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov MP, Weinstock J, Kingsbury WD, Hideshima T, Shah PK, Minvielle S, Altuo M, Kessler BM, Orlowski R, Richardson P, Munshi N, Anderson KC. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22:345–358. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Altun M, Kramer HB, Willems Li, McDermott JL, Leach CA, Goldenberg SJ, Kumar KG, Konietzny R, Fischer R, Kogan E, Mackeen MM, McGouran J, Khoronenkova SV, Parsons JL, Dianov GL, Nicholson B, Kessler BM. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 2011;18:1401–1412. doi: 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 211.Reverdy C, Conrath S, Lopez R, Planquette C, Atmanene C, Collura V, Harpon J, Battaglia V, Vivat V, Sippl W, Colland F. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol. 2012;19:467–477. doi: 10.1016/j.chembiol.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 212.Pham LV, Tamayo AT, Li C, Bommann W, Priebe W, Ford RJ. Degrasyn potentiates the antitumor effects of bortezomib in mantle cell lymphoma cells in vitro and in vivo: therapeutic implications. Mol. Cancer Ther. 2010;9(7):2026–2036. doi: 10.1158/1535-7163.MCT-10-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]