

Abstract
Fluorescence imaging enables the uniquely sensitive observation of functional‐ and molecular‐recognition events in living cells. However, only a limited range of biological processes have been subjected to imaging because of the lack of a design strategy and difficulties in the synthesis of biosensors. Herein, we report a facile synthesis of emission‐tunable and predictable Seoul‐Fluors, 9‐aryl‐1,2‐dihydrolopyrrolo[3,4‐b]indolizin‐3‐ones, with various R1 and R2 substituents by coinage‐metal‐catalyzed intramolecular 1,3‐dipolar cycloaddition and subsequent palladium‐mediated C—H activation. We also showed that the quantum yields of Seoul‐Fluors are controlled by the electronic nature of the substituents, which influences the extent of photoinduced electron transfer. On the basis of this understanding, we demonstrated our design strategy by the development of a Seoul‐Fluor‐based chemosensor 20 for reactive oxygen species that was not accessible by a previous synthetic route.
Keywords: C—H activation, fluorescence, fluorophore design, quantum yield, switchable sensors
Fluorescence‐based sensors for biomolecules have received much attention because of their excellent selectivity and sensitivity, large linear range of analysis, and high spatial resolution through microscopic imaging.1 Despite the high demand for fluorescent biosensors, the rational design of fluorescent sensors remains challenging because it is difficult to predict changes in the emission wavelength or quantum yield upon a specific recognition event or reaction with analytes. Therefore, an understanding of structure–photophysical‐property relationships in fluorophores is needed for the strategic design of novel fluorescent biosensors. Even though there are dozens of reports on correlations between the emission wavelength and chemical structure of fluorophores, such as BODIPY and cyanine dyes,2 only a few studies have addressed the systematic explanation of fluorescence quantum yields through changes in their substructure.3
We previously reported the discovery of an emission‐tunable indolizine‐based fluorescent core skeleton, namely, Seoul‐Fluor.4a Alteration of the substituents at the C7 and C9 positions of Seoul‐Fluor resulted in dramatic changes in emission wavelength over a wide range of visible‐light wavelengths. Furthermore, we demonstrated that the emission wavelength of Seoul‐Fluors can be predicted by computational analysis of the energy gap between the excited and ground states.4b Because of the ability to predict the emission wavelength, we were able to develop a ratiometric pH sensor,5a a turn‐on lipid‐droplet bioprobe,5b,c and a selective turn‐off probe of the dual‐specific phosphatase DUSP3.5d With these studies, we clearly demonstrated the importance of the substituents at the C7 and C9 positions in perturbing the photophysical properties of Seoul‐Fluors. However, the established synthetic route involves an intramolecular 1,3‐dipolar cycloaddition of an azomethine ylide with a cinnamaldehyde‐derived olefin. The success of the cycloaddition reaction is significantly influenced by the electronic character of the R1 and R2 substituents (Scheme 1 a). In the case of strongly electron rich R2 substituents, various attempts failed to afford the desired cycloadducts. The preparation of cinnamaldehydes with electron‐rich substituents has also proven difficult. More importantly, the introduction of R1 substituents at an early stage of the synthetic route was strategically unfavorable for the systematic diversification of Seoul‐Fluors. For the efficient and facile synthesis of Seoul‐Fluors, we envisioned a new synthetic route based on a silver‐ or gold‐catalyzed intramolecular 1,3‐dipolar cycloaddition to prepare the lactam‐embedded indolizine core and a subsequent palladium‐mediated site‐specific C—H activation for the late‐stage diversification of the Seoul‐Fluors (Scheme 1 b).
Scheme 1.
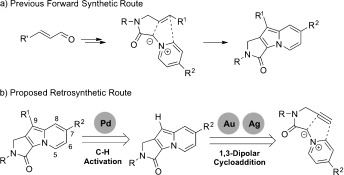
Comparison of a) our previous forward synthetic route and b) our newly proposed retrosynthetic route for the efficient synthesis of Seoul‐Fluors containing diverse R1 and R2 substituents.
We first introduced terminal alkynes instead of olefins to overcome the limited substrate scope of pyridine‐based dipoles and facilitated the 1,3‐dipolar cycloaddition of the terminal alkyne with the resulting azomethine ylide by the use of a coinage‐metal catalyst.6 This approach enabled the synthesis of Seoul‐Fluors containing diverse R2 substituents. To optimize the synthesis conditions for the lactam‐embedded indolizine core, we prepared the secondary amine 1 through a substitution reaction of propargyl amine with Boc‐protected 3‐bromopropan‐1‐amine. The resulting amine 1 was acylated with bromoacetyl bromide and treated with pyridine derivatives to generate pyridinium intermediates (Scheme 2). Without purification, the resulting pyridinium ion was converted by treatment with 1,8‐diazabicycloundec‐7‐ene (DBU) into an azomethine ylide, which underwent intramolecular [3+2] cycloaddition with the terminal acetylene. The tricyclic adducts underwent spontaneous aromatization to give lactam‐embedded indolizines 2–7 and 9 without the addition of oxidants, such as 2,3‐dichloro‐5,6‐dicyanobenzoquinone (DDQ), which was required in the previous synthetic method.4 In the case of the electron‐rich dimethylaminopyridine, we failed to synthesize the desired indolizine core by our previous method because of the inherently low reactivity of the electron‐rich dipole. However, we succeeded in synthesizing the cyclized adduct 8 with a dimethylamino group by a new synthetic route in the presence of chloro(triphenylphosphine)gold(I) as a catalyst. The addition of a catalytic amount of silver(I) oxide also improved the yield of the 1,3‐dipolar cycloaddition for pyridines containing weakly electron donating R2 substituents (products 6 and 7) or isoquinoline (product 9). Therefore, this new synthetic route not only expands the range of possible R2 substituents, but also improves the efficiency of the synthesis of Seoul‐Fluors. Under these conditions, we were able to synthesize lactam‐embedded indolizine cores with various R2 substituents.
Scheme 2.
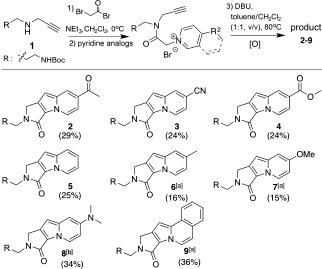
Synthesis of lactam‐embedded indolizine core structures. The yields given are for the isolated product after four steps. [a] Ag2O was used to catalyze the 1,3‐dipolar cycloaddition. [b] [AuPPh3Cl] was used as a catalyst; DDQ was added for the oxidative aromatization. Boc=tert‐butoxycarbonyl.
After the formation of the indolizine core, we aimed to use a transition‐metal‐catalyzed site‐specific C—H activation7 at the C9 position for the late‐stage diversification of the R1 group. Metal‐catalyzed C—H activation methods are powerful, straightforward, and atom‐economical synthetic tools for carbon–carbon and carbon–heteroatom bond formation.7a,b Palladium(II)‐catalyzed C—H activation has emerged as an important catalytic transformation because of its versatility for the installation of many different types of bonds and its exceptional practicality under ambient conditions in the presence of moisture.7c,d Therefore, we envisioned the efficient synthesis of Seoul‐Fluor analogues containing diverse R1 groups that were unobtainable by our previously reported route by the cross‐coupling of the lactam‐embedded indolizine cores with aryl iodides. We selected indolizine 2, which contains an acetyl group at the C7 position, as a model substrate for C—H activation owing to its excellent photophysical properties. By screening various catalysts, ligands, oxidants, solvents, and temperatures (results not shown), we found that the optimal cross‐coupling conditions for the site‐specific C—H activation of indolizine at the C9 position were conventional heating in the presence of palladium(II) acetate and silver acetate.8 Under these optimized conditions, we successfully synthesized a series of Seoul‐Fluor analogues with various R1 substituents, ranging from electron‐rich 4‐diethylaminophenyl to electron‐deficient phthalonitrile (Scheme 3, products 10–19), in moderate to good yield, as well as other R1 substituents, including heterocycles (see Table S1 in the Supporting Information). This palladium‐mediated cross‐coupling reaction also enabled the introduction of R1 substituents onto various indolizine cores (compounds 3–6) containing R2 substituents with a range of electronic characteristics (see Table S2), and even onto indolizine 9 with an electron‐rich isoquinoline moiety (see Table S3). Notably, the yield of the palladium‐catalyzed cross‐coupling reaction increased as the electron‐donating ability of the R1 substituent decreased.
Scheme 3.
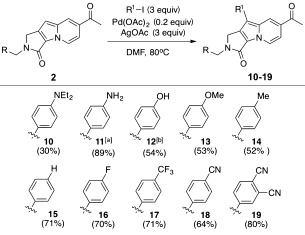
Late‐stage modification of Seoul‐Fluors with diverse R1 substituents by a cross‐coupling reaction involving palladium‐mediated C—H activation. The yields given are for the isolated product. [a] Compound 11 was obtained by the reduction of the nitrophenyl compound. [b] Compound 12 was obtained by deprotection of the tert‐butyldimethylsilyl‐protected hydroxyphenyl compound. DMF=N,N‐dimethylformamide.
Owing to the expanded range of possible substituents at the R1 and R2 positions, we were able to investigate the correlation between the chemical structure of Seoul‐Fluors and their photophysical properties. We measured the absorption maxima (λabs), emission maxima (λem), and absolute quantum yields (ΦF) of ten different Seoul‐Fluors (compounds 10–19) in methanol with systematic variation of the electronic richness of the R1 substituent. To quantify the electronic character of the R1 substituents, we used the Hammett substituent constant σp, as described previously.4, 9 By using density functional theory (DFT), we also calculated the energy level of the highest occupied molecular orbital (HOMO) in the R1 group as a barometer of the electron density of the phenyl moiety. We previously used the energy gap between the ground and excited states of Seoul‐Fluors to predict their emission wavelength, owing to similar patterns of nonradiative energy loss in Seoul‐Fluors.4b However, this energy gap is less well correlated with changes in the quantum yield, ΦF. On the other hand, we observed interesting structure–photophysical‐property relationships that depended on the electronic character of the R′ substituent: Whereas the emission wavelength showed an inverse correlation with the Hammett constant, the quantum yield was positively correlated with the Hammett constant (Table 1). In this study, we focused on the quantum‐yield variation in Seoul‐Fluors, since a systematic understanding of this property is important for the design of on/off‐switchable fluorescent sensors. In fact, Nagano and co‐workers reported a mechanistic explanation of fluorescence off/on switching in fluorescein analogues on the basis of phenyl‐substituent changes.3a Their understanding of quantum yields enabled them to develop fluorescent probes of UDP‐glucuronosyltransferase 1A1.10
Table 1. Electronic effect of R′ substituents on the relationship between the structure and photophysical properties of Seoul‐Fluors.[a]
 .
.
| Compound | R′ | σp[b] | Eh[c] | λabs[d] | λem[e] | ΦF[f] |
|---|---|---|---|---|---|---|
| 10 | 4‐NEt2 | −0.72 | −0.160 | 441 | – | 0.00 |
| 11 | 4‐NH2 | −0.66 | −0.169 | 419 | 546 | 0.03 |
| 12 | 4‐OH | −0.37 | −0.200 | 413 | 572 | 0.12 |
| 13 | 4‐OMe | −0.27 | −0.195 | 412 | 555 | 0.20 |
| 14 | 4‐Me | −0.17 | −0.215 | 404 | 534 | 0.38 |
| 15 | 4‐H | 0 | −0.227 | 403 | 523 | 0.50 |
| 16 | 4‐F | 0.06 | −0.225 | 402 | 525 | 0.47 |
| 17 | 4‐CF3 | 0.54 | −0.249 | 405 | 514 | 0.64 |
| 18 | 4‐CN | 0.66 | −0.247 | 398 | 512 | 0.63 |
| 19 | 3,4‐(CN)2 | – | −0.262 | 402 | 500 | 0.72 |
[a] Measurements were made in methanol. [b] σp is the Hammett constant of the R′ substituent at the para position.9 [c] Calculated energy in hartree of the HOMO level of the phenyl moiety. [d] Longest absorption wavelength maximum. [e] The emission wavelength for excitation at the longest absorption wavelength maximum. [f] Absolute fluorescence quantum yield.
In the case of Seoul‐Fluors, we observed a systematic decrease in the quantum yield from 0.72 (for 19) to 0.00 (for 10) as the electron density of the phenyl group was altered by changing the R1 substituent from a dicyano‐substituted to a diethylamino‐substituted phenyl moiety (Figure 1 a). Similar fluorescence‐quenching behavior, correlated with the electron richness of the R1 substituent, was also observed in three different solvents: methanol, acetonitrile, and dichloromethane (see Figure S1 in the Supporting Information). This dramatic change in the quantum yield can be rationalized by the occurrence of photoinduced electron transfer (PeT), one of the major mechanisms of fluorescence quenching, through electron transfer from a PeT donor to its acceptor in the fluorescent molecule.3, 11 Therefore, we considered the phenyl part of Seoul‐Fluors as a PeT donor and their indolizine core as a PeT acceptor (Figure 1 b). The quantum yields of the Seoul‐Fluors were correlated with two major parameters to enable quantification of the electron‐donating characteristics of the R′ substituents: the Hammett substituent constant (Figure 1 c) and the HOMO energy level (Eh, Figure 1 d) of the phenyl moiety. To confirm the PeT process in Seoul‐Fluor, we tried to detect the existence of radical species transiently generated upon electron transfer from the PeT donor to the acceptor. By using electron paramagnetic resonance (EPR) spectroscopy, we directly observed radical signals in the nonfluorescent Seoul‐Fluor 10 under irradiation at 365 nm as a signature event of the PeT process (Figure 1 e). However, we did not observe any EPR signal from the fluorescent Seoul‐Fluor 15. In fact, the spectrum of 10 observed at 133 K is composed of two signals due to a radical cation (g=1.9742) and a radical anion (g=1.9723), whereas the g values of [N,N‐diethylaniline].+ and 2.− were 1.9744 and 1.9721, respectively. This observation confirmed that the electron‐richness of the R1 moiety can induce the PeT process, which affects the quantum yield of the Seoul‐Fluor.
Figure 1.
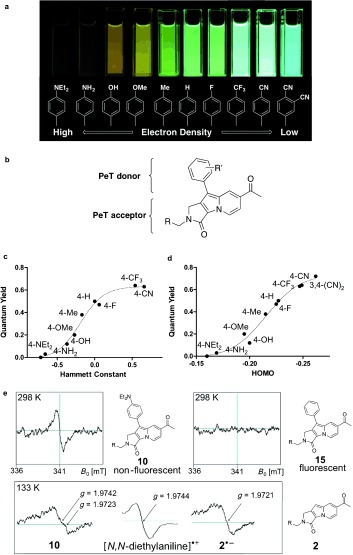
Correlation between the structure of Seoul‐Fluors and their quantum yield, as rationalized by photoinduced electron transfer (PeT). a) Change in the fluorescence brightness of Seoul‐Fluors with various R1 substituents. b) Analysis of the Seoul‐Fluor structure in terms of a PeT donor and acceptor. c) Relationship between the Hammett substituent constant and the quantum yield. d) Relationship between the HOMO energy level of the phenyl moiety and the quantum yield. e) Chemical structures of 2, 10, and 15 and their EPR spectra (1.0 mm) measured in acetonitrile at room temperature and 133 K under irradiation at 365 nm with a high‐intensity xenon lamp.
On the basis of our understanding of the structure–quantum‐yield relationship of Seoul‐Fluors, we were able to rationally explain the fluorescence of on/off‐switchable sensors. For example, we previously reported the discovery of a fluorescence turn‐off bioprobe, SfBP, for the dual‐specific phosphatase DUSP3. Upon the selective dephosphorylation of SfBP by DUSP3, the quantum yield of Seoul‐Fluor drastically decreased from 7.5 to 0.1 %. This 75‐fold quantum‐yield reduction in SfBP can be rationalized by differences in the electron richness of the phosphate group (σp=0.0) and the phenoxide group (σp=−0.81) upon a specific chemical transformation of SfBP by DUSP3 (see Figure S2). The rational predictability of quantum‐yield changes in Seoul‐Fluors enables the design and development of novel fluorescent on/off‐switchable sensors. Therefore, we substantiated our rational designing ability by the development of a Seoul‐Fluor‐based fluorescent H2O2 sensor 20 containing a phenylboronic acid pinacol ester substituent.12 The boronate ester in biosensor 20 for reactive oxygen species (ROS) was transformed into a phenol group upon treatment with H2O2 in a aqueous system to give 12 (Figure 2 a,b). The drastic decrease in the quantum yield from 10.5 to 1.9 % was able to be predicted on the basis of the change in both the HOMO energy (from −0.224 to −0.200 hartree) and the estimated Hammett constant (from 0.3 to 0.12). We also demonstrated that this turn‐off sensor 20 can report real‐time changes in ROS levels of HeLa human cervical cancer cells (Figure 2 c–e). The development of a Seoul‐Fluor‐based cellular H2O2 sensor implies that our study of the structure–quantum‐yield relationship has provided a rational design strategy for the construction of novel fluorescence on/off‐switchable sensors.
Figure 2.
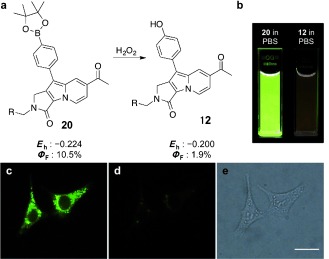
Structural change from 20 to 12 and change in the quantum yield upon the addition of H2O2, and cellular fluorescence images of 20‐treated HeLa cells before and after treatment of H2O2. a) Correlation of the quantum yield with the HOMO energy level of 20 and its counterpart 12. b) Photographic images of 20 and 12 in phosphate‐buffered saline (PBS). c) Fluorescence image of HeLa cells upon treatment with 20. d) Fluorescence image of the same 20‐treated HeLa cells after the addition of 1 % H2O2. e) Bright‐field image of (d). Scale bar is 20 μm.
In this study, we developed an efficient method for the facile synthesis of Seoul‐Fluors by the silver‐ and gold‐catalyzed intramolecular 1,3‐dipolar cyclization of terminal acetylenes with substituted azomethine ylides and subsequent palladium‐mediated C—H activation for late‐stage diversification of the Seoul‐Fluors. By using this new synthetic route, we were able to synthesize diverse Seoul‐Fluor analogues containing a wide variety of R1 and R2 substituents that were unobtainable by our previous method. On the basis of the construction of a series of Seoul‐Fluors, we discovered that their quantum yield can be perturbed by systematic changes in the electronic character of the R′ substituents, as quantified by Hammett constants and the HOMO energy level of the R1 group. Furthermore, we demonstrated that the occurrence of a photoinduced electron‐transfer process might cause a reduction in the quantum yield of Seoul‐Fluors with increasing electron richness of the R1 substituent. By using our fundamental understanding of the structure–photophysical‐property relationship, we designed and developed a Seoul‐Fluor‐based fluorescent turn‐off chemosensor for cellular hydrogen peroxide; this compound was not accessible by our previous synthetic route. Therefore, we envision the development of novel fluorescent on/off‐switchable sensors by rational design with respect to PeT‐based changes in quantum yield.
Supplementary Material
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
This research was supported by the Bio & Medical Technology Development Program (2012M3A9C4048780), the Basic Research Laboratory (2010‐0019766), and a Global Frontier Project Grant (2013M3A6A4044245) funded by the National Research Foundation of Korea. E.J.C., Y.L., and A.J. are grateful for their predoctoral fellowships awarded by the BK21 Program.
References
- 1.
- 1a.Lakowicz J. R., Principles of Fluorescence Spectroscopy, Springer, New York, 2006; [Google Scholar]
- 1b.Rettig W., Strehmel B., Schrader S., Seifert H., Applied Fluorescence in Chemistry, Biology, and Medicine, Springer, New York, 1999; [Google Scholar]
- 1c.Haugland R. P., Handbook of Fluorescence Probes and Research Products, Molecular Probes, Eugene, OR, 2002; [Google Scholar]
- 1d.Lavigne J. J., Anslyn E. V., Angew. Chem. 2011, 123, 3174–3175; [Google Scholar]; Angew. Chem. Int. Ed. 2011, 50, 3118–3130; [Google Scholar]
- 1e.Zhang J., Campbell R. E., Ting A. Y., Tsien R. Y., Nat. Rev. Mol. Cell Biol. 2002, 3, 906–918; [DOI] [PubMed] [Google Scholar]
- 1f.Domaille D. W., Que E. L., Chang C. J., Nat. Chem. Biol. 2008, 4, 168–175. [DOI] [PubMed] [Google Scholar]
- 2.Demchenko A. P., Advanced Fluorescence Reporters in Chemistry and Biology I, Springer, Berlin, 2010. [Google Scholar]
- 3.
- 3a.Miura T., Urano Y., Tanaka K., Nagano T., Ohkubo K., Fukuzumi S., J. Am. Chem. Soc. 2003, 125, 8666–8671; [DOI] [PubMed] [Google Scholar]
- 3b.Ueno T., Urano Y., Setsukinai K., Takakusa H., Kojima H., Kikuchi K., Ohkubo K., Fukuzumi S., Nagano T., J. Am. Chem. Soc. 2004, 126, 14079–14085; [DOI] [PubMed] [Google Scholar]
- 3c.Urano Y., Kamiya M., Kanda K., Ueno T., Hirose K., Nagano T., J. Am. Chem. Soc. 2005, 127, 4888–4894; [DOI] [PubMed] [Google Scholar]
- 3d.Ueno T., Urano Y., Kojima H., Nagano T., J. Am. Chem. Soc. 2006, 128, 10640–10641. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a.Kim E., Koh M., Ryu J., Park S. B., J. Am. Chem. Soc. 2008, 130, 12206–12207; [DOI] [PubMed] [Google Scholar]
- 4b.Kim E., Koh M., Lim B. J., Park S. B., J. Am. Chem. Soc. 2011, 133, 6642–6649. [DOI] [PubMed] [Google Scholar]
- 5a.Kim E., Lee S., Park S. B., Chem. Commun. 2012, 48, 2331–2333; [DOI] [PubMed] [Google Scholar]
- 5b.Kim E., Lee S., Park S. B., Chem. Commun. 2012, 48, 2331–2333; [DOI] [PubMed] [Google Scholar]
- 5c.Lee Y., Na S., Lee S., Jeon N. L., Park S. B., Mol. BioSyst. 2013, 9, 952–956; [DOI] [PubMed] [Google Scholar]
- 5d.Jeong M. S., Kim E., Kang H. J., Choi E. J., Cho A. R., Chung S. J., Park S. B., Chem. Commun. 2012, 48, 6553–6555. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a.Hashmi A. S. K., Chem. Rev. 2007, 107, 3180–3211; [DOI] [PubMed] [Google Scholar]
- 6b.Su S., J. A. Porco, Jr., J. Am. Chem. Soc. 2007, 129, 7744–7745. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a.Chen X., Engle K. M., Wang D., Yu J., Angew. Chem. 2009, 121, 5196–5217; [Google Scholar]; Angew. Chem. Int. Ed. 2009, 48, 5094–5115; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b.Dupont J., Consorti C. S., Spencer J., Chem. Rev. 2005, 105, 2527–2572; [DOI] [PubMed] [Google Scholar]
- 7c.Cho S. H., Kim J. Y., Kwak J., Chang S., Chem. Soc. Rev. 2011, 40, 5068–5083; [DOI] [PubMed] [Google Scholar]
- 7d.Lyons T. W., Sanford M. S., Chem. Rev. 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a.Liang Z., Zhao J., Zhang Y., J. Org. Chem. 2010, 75, 170–177; [DOI] [PubMed] [Google Scholar]
- 8b.Stuart D. R., Villemure E., Fagnou K., J. Am. Chem. Soc. 2007, 129, 12072–12073; [DOI] [PubMed] [Google Scholar]
- 8c.Miyasaka M., Hirano K., Satoh T., Miura M., J. Org. Chem. 2010, 75, 5421–5424. [DOI] [PubMed] [Google Scholar]
- 9.Hansch C., Leo A., Taft R. W., Chem. Rev. 1991, 91, 165–195. [Google Scholar]
- 10.Terai T., Tomiyasu R., Ota T., Ueno T., Komatsu T., Hanaoka K., Urano Y., Nagano T., Chem. Commun. 2013, 49, 3101–3103. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a.de Silva A. P., Gunaratne H. Q. N., Gunnlaugsson T., Huxley A. J. M., McCoy C. P., Rademacher J. T., Rice T. E., Chem. Rev. 1997, 97, 1515–1566; [DOI] [PubMed] [Google Scholar]
- 11b.Kollmannsberger M., Rurack K., Resch‐Genger U., Daub J., J. Phys. Chem. A 1998, 102, 10211–10220. [Google Scholar]
- 12.
- 12a.Chang M. C. Y., Pralle A., Isacoff E. Y., Chang C. J., J. Am. Chem. Soc. 2004, 126, 15392–15393; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b.Dickinson B. C., Chang C. J., J. Am. Chem. Soc. 2008, 130, 9638–9639; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c.Albers A. E., Okreglak V. S., Chang C. J., J. Am. Chem. Soc. 2006, 128, 9640–9641; [DOI] [PubMed] [Google Scholar]
- 12d.Abo M., Urano Y., Hanaoka K., Terai T., Komatsu T., Nagano T., J. Am. Chem. Soc. 2011, 133, 10629–10637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information