

Abstract
The number of single genes associated with neurodevelopmental disorders has increased dramatically over the past decade. The identification of causative genes for these disorders is important to clinical outcome as it allows for accurate assessment of prognosis, genetic counseling, delineation of natural history, inclusion in clinical trials, and in some cases determines therapy. Clinicians face the challenge of correctly identifying neurodevelopmental phenotypes, recognizing syndromes, and prioritizing the best candidate genes for testing. However, there is no central repository of definitions for many phenotypes, leading to errors of diagnosis. Additionally, there is no system of levels of evidence linking genes to phenotypes, making it difficult for clinicians to know which genes are most strongly associated with a given condition. We have developed the Developmental Brain Disorders Database (DBDB: https://www.dbdb.urmc.rochester.edu/home), a publicly available, online-curated repository of genes, phenotypes, and syndromes associated with neurodevelopmental disorders. DBDB contains the first referenced ontology of developmental brain phenotypes, and uses a novel system of levels of evidence for gene-phenotype associations. It is intended to assist clinicians in arriving at the correct diagnosis, select the most appropriate genetic test for that phenotype, and improve the care of patients with developmental brain disorders. For researchers interested in the discovery of novel genes for developmental brain disorders, DBDB provides a well-curated source of important genes against which research sequencing results can be compared. Finally, DBDB allows novel observations about the landscape of the neurogenetics knowledge base.
Keywords: developmental brain disorders, database, bioinformatics, levels of evidence
INTRODUCTION
The discovery of single genes associated with neurodevelopmental disorders has increased dramatically over the past decade, with the number of associations with malformations of cortical development (MCD) alone increasing from 27 in 2005 [Barkovich et al., 2005] to more than 100 in the most recent MCD classification [Barkovich et al., 2012]. The identification of numerous genes associated with developmental delay, intellectual disability, autism, and epilepsy brings the number still higher. The maturation of copy number variation studies of the human genome [Itsara et al., 2009; Mefford et al., 2009; Cooper et al., 2011], and the introduction of whole exome sequencing [O’Roak et al., 2011; Chahrour et al., 2012; Iossifov et al., 2012; Sanders et al., 2012] promise a further increase in the number of genes associated with human neurodevelopmental phenotypes.
For the clinician, this rapid expansion of knowledge poses a challenge in arriving at diagnoses in a timely and cost-effective manner. Recognizing that textbooks and print journals quickly become out of date, the genetics community moved many of its clinical resources online to assure more rapid dissemination of new knowledge. The Online Mendelian Inheritance in Man (OMIM) and GeneReviews have become primary sources of information for accurate information regarding genetic syndromes. However, these resources, while encyclopedic, are limited by the state of organization of the knowledge base. A number of problems exist. First, there is currently no system of levels of evidence for gene-phenotype associations. It is often not apparent from OMIM or other sources which of many genes are most highly associated with certain phenotypes. Therefore, it is difficult for clinicians to judge which genes should really be tested and when. Second, there is no common ontology (a shared vocabulary and taxonomy containing the definition of objects and their relationships) of neurodevelopmental phenotypes. This void makes it difficult for many clinicians to recognize key features, particularly when the features themselves are poorly defined. A related issue is that a variety of partial terminologies exist, drawn from Medical Subject Headings (MeSH), the Unified Medical Language System (UMLS), the Human Phenotype Ontology (HPO), and DSM V [American Psychiatric Association and DSM-5 Task Force, 2013]. All of these resources contain terms that remain undefined, are defined in multiple conflicting ways, or the definitions provided are different from how clinicians generally use them.
To address these issues, we curated the genes associated with neurodevelopmental phenotypes, assembled an ontology of these phenotypes from a number of sources, and developed a system of levels of evidence for gene-phenotype associations. The result is the Developmental Brain Disorders Database (DBDB), a publicly available, online-curated repository of genes, phenotypes, and syndromes associated with neurodevelopmental disorders available at: https://www.dbdb.urmc.rochester.edu/home.
METHODS
Curation of Genes Associated With Neurodevelopmental Disorders
The first iteration of DBDB focused on phenotypes of abnormal brain development (Table I). A list of genes associated with these phenotypes was collected and expanded upon through review of the literature, including PubMed search (for autism, developmental delay, mental retardation, intellectual disability, epilepsy, movement disorders, and specific terms for congenital brain abnormalities). Brain disorders with acquired, inflammatory, infectious, and neoplastic etiologies were not included in DBDB. Also not represented were the majority of metabolic diseases and primarily neurodegenerative conditions. An edge group were the hereditary ataxias, some of which present with developmental delay during childhood. New gene publications considered for update are received on a weekly basis using the web service PubCrawler, and reviewed by the authors.
TABLE I.
Ontology of Developmental Brain Disorders Included in DBDB
|
Ontology of Neurodevelopmental Phenotypes
Since no common ontology existed for neurodevelopmental phenotypes, we compiled definitions from a variety of expert sources: (1) MeSH and UMLS (when those definitions were complete), (2) Barkovich and Raybaud’s Pediatric Neuroimaging 5th edition [Barkovich and Raybaud, 2012], (3) Firth and Hurst’s Oxford Desk Reference: Clinical Genetics [Firth et al., 2005], Panayiotopoulos’ A Clinical Guide to Epileptic Syndromes and their Treatment [Panayiotopoulos, 2010], and (5) the Diagnostic and Statistical Manual of Mental Disorders, 5th edition [American Psychiatric Association and DSM-5 Task Force, 2013]. When no standard definition existed or was unsatisfactory, we developed a definition based on our collective long-standing experience with these disorders.
Levels of Evidence for Gene-Phenotype Associations
Using generally accepted principles of evidence-based medicine [Haynes, 2006], we reviewed the literature on recommendations for evaluation of neurodevelopmental disorders specifically [Shevell et al., 2003; Shaffer, 2005; Moeschler, 2008] and other genetic syndromes [Wilson, 2006; Zhang et al., 2011]. We also reviewed the methods of the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) working group [Teutsch et al., 2009]. Using this broad approach, we assembled criteria for three levels of evidence by which to judge the association genes with neurodevelopmental phenotypes (Table II).
TABLE II.
Levels of Evidence for Neurodevelopmental Gene-Phenotype Associations in DBDB
Level 1: Strongest gene-phenotype association
|
Level 2: Moderate gene-phenotype association
|
Level 3: Weak gene-phenotype association
|
Copy number studies in which a gene is deleted/duplicated as part of a multi-gene copy number variant, or genome-wide association studies of single/simple nucleotide polymorphisms with phenotypes, or “burden” of variants/polymorphisms/copy number variations, or where the phenotype is not specific are considered below Level 3 and are not included in DBDB.
Also not included are genes where an animal model demonstrated a gene-phenotype association, but an association in humans has not been reported.
Training Data Set
Each of the authors chose five gene-phenotype associations judged as Level 1 evidence based on their experience, and five gene-phenotype associations with weaker evidence, and presented the evidence to the other authors for discussion. This led to refinement of the levels of evidence further in to the criteria listed in Table II.
Software
DBDB 1.2 was written in Perl and Javascript and implemented with the Catalyst 5.90011 web framework linked to a MySQL database.
Statistical and Data Analysis
Statistical tests were performed for this paper in R v.3.0.1 (http://cran.r-project.org/). Hiveplots, a mapping tool for visualizing networks, were constructed using jhive v.0.1.1 [Krzywinski et al., 2012].
RESULTS
Website
The central page of the DBDB website is the Associations page (https://www.dbdb.urmc.rochester.edu/associations/list). This page displays a table listing genes, modes of inheritance, brain phenotypes, associated syndromes, and levels of evidence with references linked dynamically to PubMed. Where available, GeneReviews chapters are also linked. The user may sort by any of the columns. Links within the table take users to external websites for further gene-specific information such as the University of Chicago Computation Institute’s Lynxbio informatics platform [Sulakhe et al., 2013] and the University of California Santa Cruz (UCSC) Genome Browser, and internal DBDB pages for gene information, phenotype definitions and syndrome associations. Further pages include Phenotypes indexed by DBDB, and Syndromes. A search function allows users to query the entire dataset. DBDB may also be accessed in a RESTful format through our web service (https://www.dbdb.urmc.rochester.edu/webserv). Currently, a simple search is implemented, but a search algorithm allowing for Boolean terms will be included in the next release. Clinical and research tutorials for users are available (https://www.dbdb.urmc.rochester.edu/tutorials).
Each phenotype page in DBDB gives a definition, a list of genes and syndromes associated with that phenotype, the level of evidence (LOE) for the gene-phenotype association, often a representative illustration, and a reference. Each syndrome page displays cardinal features, mode of inheritance, references, and a list of the genes and phenotypes associated with that syndrome. The intention of the individual syndrome pages is not to reproduce information already presented at OMIM or GeneReviews, but rather to list the essential features in a text-light manner and then link the user to those resources. The entire DBDB dataset is updated monthly and is freely available to users in SQL format through GitHub.
DBDB 1.2 at the time of manuscript preparation contained 438 genes, 897 gene-phenotype associations, 72 phenotypes, and 168 syndromes with primary neurodevelopmental features. Curation resulted in 247 Level 1 (strongest) gene-phenotype associations, 330 Level 2, and 320 Level 3 (weakest) associations. In the DBDB ontology, every term is part of a parent–child relationship and includes a definition and a reference. The use of parent–child term identifiers helps when searching for associations, as higher level parent terms will yield broader query results than narrower child-level terms. For example, a search for the parent-level term “epilepsy” will capture all genes associated with all forms of epilepsy. Searching a child term such as “infantile spasms” yields a more restrictive list.
Curation using our levels of evidence restricted the knowledge base presented in DBDB compared to other sources. For example, the Autism Database presented 546 genes associated with autism, but review of these data resulted in only 36 genes associated with autism that met any of our three levels of evidence. This is in part because DBDB does not include genes identified by genome wide association studies, or genes that are solely recognized within the context of chromosomal copy number variations. In these cases, the association of the gene with the phenotype may be speculation. Although DBDB’s methods are more restrictive, the result is a knowledge base of greater practical value to the clinician.
Example of Clinical DBDB Use
A clinician is seeing a 2-year-old child in her office with a complex neurodevelopmental phenotype characterized by severe intellectual disability, hypotonia, and pontocerebellar hypoplasia (PCH) on brain MRI. After a review of the history, brain imaging, prior records, and physical exam she forms a differential diagnosis. Genetic causes seem most likely. She accesses DBDB on her office computer via the Internet. Clicking on the “Associations” link at the top of the page results in the main table of gene-phenotype associations. She surveys the information on this page, and sees that there are 11 gene-phenotype associations for PCH that include 6 Level 2 associations and 5 Level 3 associations. Clicking on “pontocerebellar hypoplasia” as a phenotype for one of the associations takes her to an information page about PCH. The entry can be rapidly perused during a busy clinic. Included on this page is a standardized definition to remove as much ambiguity about terminology as possible. An image is usually given to illustrate the phenotype in some helpful way. A reference for the definition is given. Then at the bottom of the page, DBDB gives the clinician a table with the genes associated with the term “pontocerebellar hypoplasia.” The clinician can sort gene-phenotype associations by the LOE column. This puts the genes most strongly associated with a phenotype at the top of the list. In cases where a clinician may have limited access to genetic testing, this aids the prioritization of testing. The clinician can use the LOE score to evaluate available next-generation sequencing panels, and choose one based on whether all or most of the most strongly associated genes with the highest LOE are included. Clicking on the gene name takes the clinician to a gene-specific page, where additional phenotypes and syndromes associated with that gene may be reviewed quickly. Should more detail be needed, links to PubMed, GeneReviews, Lynx, and UCSC browser are available.
Example of Research DBDB Use
DBDB may also be used by researchers in the analysis of next-generation sequencing data for gene discovery. For example, the list of genes that contain novel nonsynonymous variants from whole exome sequencing can be compared against the list of DBDB genes (https://www.dbdb.urmc.rochester.edu/rest/genes/) to rapidly find “knowns” in that data. To facilitate this, DBDB data are integrated into the publicly available SOLVE-Brain annotation tool. The DBDB genelist can also be broadened using tools such as Lynx to develop a list of candidate interacting genes in the same biological pathways. Further specific research uses of DBDB are in development.
The Landscape of the Neurogenetics Knowledgebase
DBDB provides an opportunity to draw together data that were previously scattered and survey the landscape of the neurogenetics knowledge base. Figure 1 shows the distribution of genetic associations per phenotype. Most of phenotypes (74%) have 10 or fewer genetic associations and only 4 phenotypes included in DBDB have no genetic associations documented. The top three phenotypes with the most known genetic associations are, perhaps not surprisingly, intellectual disability, microcephaly, and epilepsy. Using a quantitative approach, the “specificity” of a genetic cause of a phenotype can be measured by the ratio of Level 1 (strongest) gene associations for that phenotype divided by the total number of gene associations for that phenotype. A ratio of one indicated the highest specificity for a genetic cause. Only six phenotypes (central hypoventilation, craniosynostosis, subcortical band heterotopia, subependymal nodules, tubers, and early myoclonic epilepsy) met this level of specificity, where all of the genetic associations were Level 1 associations (Fig. 2A). When Levels 1 and 2 associations were combined in the calculation of specificity, the number of phenotypes rose to 24 (Fig. 2B). Only four phenotypes (absent anterior commissure, band-like calcifications, benign epilepsy of childhood with centrotemporal spikes, and hemimegalencephaly) had Level 3 genetic associations only.
FIG. 1.

The distribution of genetic associations per phenotype, showing that most of phenotypes (74%) have 10 or fewer genetic associations, and four phenotypes have no known genetic associations.
FIG. 2.

Only six phenotypes (central hypoventilation, craniosynostosis, subcortical band heterotopia, subependymal nodules, tubers, and early myoclonic epilepsy) had a specificity of 1, where all of the genetic associations were Level 1 associations (A). When Levels 1 and 2 associations were combined in the calculation of specificity, the number of phenotypes rose to 24 (B). The distribution of phenotypes with level of evidence 1 genetic associations proceeded in a step-wise fashion, with 25 phenotypes having no Level 1 associations, with the number rising gradually toward the top four phenotypes (molar tooth malformation, retinal dystrophy, microcephaly, and intellectual disability) with the most level of evidence 1 genetic associations (C). When all genetic associations across all levels of evidence were compared, most phenotypes clustered under 20 genes/phenotype, with a mean of 10.6 genetic associations/phenotype (D). The specificity of genetic associations for the top four phenotypes with the most level of evidence 1 genetic associations (E). All four phenotypes were associated with significantly fewer genes linked to other phenotypes, compared to all other phenotypes with Level 1 genetic associations (*P < 2.2 × 10−16; **P = 0.00023; ***P = 0.0001; ****P = 0.0098). RDYST, retinal dystrophy; MTM, molar tooth malformation; MIC, microcephaly; ID, intellectual disability.
The phenotypes with LOE 1 genetic associations were distributed in a step-wise fashion, with 25 phenotypes having no Level 1 associations, and the number rose gradually toward the top four phenotypes (molar tooth malformation, retinal dystrophy, microcephaly, and intellectual disability) with the most LOE 1 genetic associations (Fig. 2C). These were molar tooth malformation (9 LOE 1 associations), retinal dystrophy (17), microcephaly (24), and intellectual disability (30). When all genetic associations across all levels of evidence were compared, most phenotypes clustered under 20 genes/phenotype, with a mean of 10.6 genetic associations/phenotype (Fig. 2D).
Next, we examined the specificity of the genetic associations for the top four phenotypes with the most LOE 1 genetic associations—intellectual disability, microcephaly, molar tooth malformation, and retinal dystrophy. We found that all four phenotypes were associated with significantly fewer genes linked to other phenotypes, compared to all other phenotypes with Level 1 genetic associations (Fig. 2E). Genes associated with retinal dystrophy were the most specific, and were extremely unlikely to be associated with other phenoytpes.
Although genes with Level 1 associations with intellectual disability, microcephaly, molar tooth malformation, or retinal dystrophy were less likely to also be associated with other phenotypes, there was variability among these “highly specific” phenotypes. This can be illustrated using hiveplots, showing the increasingly specific nature of genetic associations from intellectual disability with relatively more additional phenotypic associations which also included autism, various brain malformations, and epilepsy subtypes (Fig. 3A), through microcephaly (Fig. 3B), molar tooth malformation (Fig. 3C), to retinal dystrophy (Fig. 3D), with only one gene with one other phenotypic association.
FIG. 3.
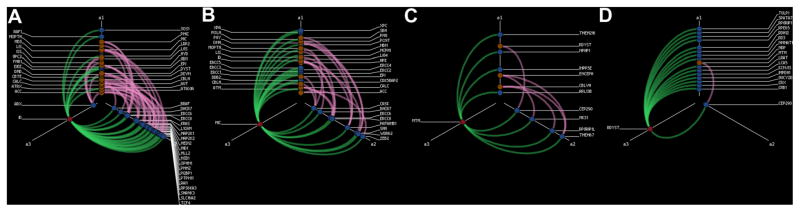
Hiveplots showing additional phenotypic relationships of genes associated by level of evidence 1 with intellectual disability (A), microcephaly (B), molar tooth malformation (C), and retinal dystrophy (D). Red node in all panels indicates primary phenotype. Blue nodes indicate associated genes. Copper nodes represent secondary associated phenotypes. Green edges represent Level 1 associations between genes and the primary phenotype. Pink edges represent other associations (of any level of evidence) between those genes and the secondary phenotypes. Genes for intellectual disability and microcephaly are more likely to have secondary phenotypic associations, and genes for retinal dysplasia are less likely to have secondary phenotypic associations.
DISCUSSION
The practice of medical genetics and child neurology has changed dramatically over the past 10 years, with advances in brain imaging and genetic testing leading to an explosion of newly recognized phenotypes, syndromes, and genetic associations. Families continue to expect diagnoses for reasons of prognosis, family planning, inclusion in natural history studies and, increasingly, treatment [Moeschler, 2008; Makela et al., 2009]. Clinicians must be able to efficiently apply the growing neurogenetics knowledge regarding an increasing number of syndromes and genes to their patient population. DBDB is a publicly available web-based tool that allows clinicians to “cut to the chase” and rapidly evaluate the most current data on genetic associations with developmental brain disorders. DBDB is meant to augment existing web-based resources such as OMIM and GeneReviews by introducing levels of evidence for gene-phenotype associations and a structured ontology of neurodevelopmental disorders. When used with other resources, DBDB should streamline the genetic workup of children with neurodevelopmental disorders.
Levels of evidence are an evolving concept in diagnostic medicine and are essential to move neurogenetics diagnosis from an “expert-driven craft” to a field increasingly based on biological data. This is illustrated by the evolution in our understanding of classically recognized conditions. Twenty years ago, finding a molar tooth malformation by MRI ended with the diagnosis of “Joubert syndrome.” Now, it leads to the analysis of the 17 known (so far) associated genes. To date, mutations in 12 genes have been associated with lissencephaly. Rare are both the trainee and professor who can recite all of these genes from memory. An accurate reference that is easily updated is clearly needed. Which of these genes has the best evidence for association with a phenotype, are therefore more likely causes, and should be tested first? Which next-generation sequencing panel offered best captures the likely diagnosis in a patient? The online implementation of an evidence system for neurodevelopmental disorders that is transparent, dynamically linked to the literature, and curated by experts represents one solution. As new literature emerges, DBDB is easily updated, and references remain current because they are served directly via weblinks.
DBDB also addresses the need for an ontology of definitions to encourage consistency in the terms used for phenotypes. This is as important for trainees as for seasoned clinicians and for families and relatives who go online to learn about prognosis. Further, it is difficult to remain current with evolving understandings of phenotypic interrelationships. As our knowledge of underlying biology matures, these connections inevitably change. One illustration of this is the difficulty separating cobblestone malformation (still known to some as “Type II lissencephaly”) from lissencephaly and polymicrogyria in both the literature and general practice—an important task as all three malformations are distinct with regard to their underlying genetic etiologies, mechanism, and prognosis. The same can be said for Dandy-Walker malformation and its related phenotypic cousins cerebellar vermis hypoplasia and mega cisterna magna. DBDB provides a central repository of terms and definitions for all of these phenotypes, with references. These data are not present in OMIM or in GeneReviews, making DBDB a unique contribution to the evaluation of children with neurodevelopmental disorders.
DBDB allowed general observations about the landscape of the neurogenetics knowledge base. The number of neurodevelopmental phenotypes with no genetic associations is few, and most phenotypes have less than 10 genes associated with them. At the other end of the spectrum, very few phenotypes have exclusively LOE 1 genetic associations. However, when Levels 1 and 2 were grouped, this number rose to 24 phenotypes where some genetic causation is understood.
Among the phenotypes at the higher end of the Level 1 association spectrum, there was significant specificity that a gene was associated with that single phenotype, and no other. Therefore, genes associated with intellectual disability, microcephaly, molar tooth malformation, and especially retinal dystrophy were unlikely to be also associated with other phenotypes. This may be for historical reasons, however, not related to biology. The search for intellectual disability genes is not a new undertaking, and the sheer number of Level 1 associations may be due to the length of time of scientific study of that phenotype, with the result that the evidence for their association has matured. When there are other phenotypic associations, DBDB illustrates that these may occur in patterns of associations that are recurrent and recognizable. For example, intellectual disability genes cluster in their additional associations with specific forebrain malformations (i.e., agenesis of the corpus callosum), hindbrain malformations (i.e., cerebellar tonsillar ectopia), and numerous epilepsy phenotypes. Microcephaly genetic associations cluster recurrently with intellectual disability and specific forebrain malformations (lissencephaly, pachygyria, polymicrogyria). Genes associated with retinal dystrophy proved to be the most specific of all—genes for retinal dystrophy were less likely to also associate with other phenotypes—with the exception of limited overlap with molartooth malformation. These observations allow clinicians to make connections more easily when managing patients with complex developmental brain disorders.
Research laboratory applications for DBDB represent an area of future growth. Already the gene list that DBDB provides may be used to augment the annotation of research whole exome sequencing results, streamlining the identification of variants in genes known to cause developmental brain disorders. As our data are freely available and regularly updated, they may be incorporated easily into bioinformatics workflows. Additionally, the availability of a well-curated list of known developmental brain disorder genes supplements the annotation of the human genome, and our data have already been incorporated into online platforms such as Lynx and tools such as SOLVE-Brain. Finally, there is interest in including “gene pathway” information in future iterations of DBDB, as these data may allow the easier identification of new genetic causes of developmental brain disorders from whole exome data. There are now examples from congenital microcephaly [Shen et al., 2010], megalencephaly [Mirzaa et al., 2013], and PCH [Wan et al., 2012] among others suggesting this is a valuable tool for gene discovery. However, our observations about the current incomplete state of pathway annotations leads us to conclude that this would be a considerable curation task.
Acknowledgments
The authors wish to thank Mark Peterson of the Academic IT group at the University of Rochester Medical Center for assistance with web hosting. Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health under award numbers R01NS046616 (to W.B.D.), R01NS050375 and R01NS058721 (to W.B.D. and K.J.M.), and K08NS078054 (to A.R.P.).
Footnotes
Conflict of interest: none.
URLS CITED
Autism Database: http://autism.mindspec.org/autdb/Welcome.do
GeneReviews: http://www.genetests.org/by-genereview/
GitHub: https://github.com/Paciorkowski-Lab/DBDB
HivePlots: http://www.hiveplot.net/
Human Phenotype Ontology: http://www.human-phenotype-ontology.org/index.php/hpo_home.html
Lynx: http://lynx.ci.uchicago.edu/
Medical Subject Headings: http://www.ncbi.nlm.nih.gov/mesh
OMIM: http://www.ncbi.nlm.nih.gov/omim
PubCrawler: http://pubcrawler.gen.tcd.ie/
SOLVE-Brain: https://paciorkowski-lab.urmc.rochester.edu/solve_brain
UCSC Browser: http://www.genome.ucsc.edu/
Unified Medical Language System: http://www.nlm.nih.gov/research/umls/
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- American Psychiatric Association and DSM-5 Task Force. Diagnostic and statistical manual of mental disorders: DSM-5. Arlington, VA: American Psychiatric Association; 2013. [Google Scholar]
- Barkovich AJ, Raybaud C. Pediatric neuroimaging. Philadelphia, PA: Wolters Kluwer, Lippincott Williams & Wilkins; 2012. [Google Scholar]
- Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65:1873–1887. doi: 10.1212/01.wnl.0000183747.05269.2d. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: Update 2012. Brain. 2012;135:1348–1369. doi: 10.1093/brain/aws019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, Hill RS, Stevens CR, Schubert CR, Greenberg ME, Gabriel SB, Walsh CA ARRA Autism Sequencing Collaboration. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8:e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, Abdel-Hamid H, Bader P, McCracken E, Niyazov D, Leppig K, Thiese H, Hummel M, Alexander N, Gorski J, Kussmann J, Shashi V, Johnson K, Rehder C, Ballif BC, Shaffer LG, Eichler EE. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, Hurst JA, Hall JG. Oxford desk reference: Clinical genetics. Oxford: Oxford University Press; 2005. [Google Scholar]
- Haynes RB. Of studies, syntheses, synopses, summaries, and systems: The “5S” evolution of information services for evidence-based healthcare decisions. Evid Based Med. 2006;11:162–164. doi: 10.1136/ebm.11.6.162-a. [DOI] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y-H, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI, Mefford H, Ying P, Nickerson DA, Eichler EE. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Birol I, Jones SJM, Marra MA. Hive plots-rational approach to visualizing networks. Brief Bioinformatics. 2012;13:627–644. doi: 10.1093/bib/bbr069. [DOI] [PubMed] [Google Scholar]
- Makela NL, Birch PH, Friedman JM, Marra CA. Parental perceived value of a diagnosis for intellectual disability (ID): A qualitative comparison of families with and without a diagnosis for their child’s ID. Am J Med Genet Part A. 2009;149A:2393–2402. doi: 10.1002/ajmg.a.33050. [DOI] [PubMed] [Google Scholar]
- Mefford HC, Cooper GM, Zerr T, Smith JD, Baker C, Shafer N, Thorland EC, Skinner C, Schwartz CE, Nickerson DA, Eichler EE. A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome Res. 2009;19:1579–1585. doi: 10.1101/gr.094987.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaa GM, Rivière J-B, Dobyns WB. Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am J Med Genet Part C. 2013;163C:122–130. doi: 10.1002/ajmg.c.31361. [DOI] [PubMed] [Google Scholar]
- Moeschler JB. Genetic evaluation of intellectual disabilities. Semin Pediatr Neurol. 2008;15:2–9. doi: 10.1016/j.spen.2008.01.002. [DOI] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayiotopoulos CP. A clinical guide to epileptic syndromes and their treatment. London: Springer-Verlag; 2010. [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Günel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer LG. American College of Medical Genetics guideline on the cytogenetic evaluation of the individual with developmental delay or mental retardation. Genet Med. 2005;7:650–654. doi: 10.1097/01.gim.0000186545.83160.1e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Gilmore EC, Marshall CA, Haddadin M, Reynolds JJ, Eyaid W, Bodell A, Barry B, Gleason D, Allen K, Ganesh VS, Chang BS, Grix A, Hill RS, Topcu M, Caldecott KW, Barkovich AJ, Walsh CA. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet. 2010;42:245–249. doi: 10.1038/ng.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, Majnemer A, Noetzel M, Sheth RD. Practice parameter: Evaluation of the child with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2003;60:367–380. doi: 10.1212/01.wnl.0000031431.81555.16. [DOI] [PubMed] [Google Scholar]
- Sulakhe D, Balasubramanian S, Xie B, Feng B, Taylor A, Wang S, Berrocal E, Dave U, Xu J, Börnigen D, Gilliam TC, Maltsev N. Lynx: a database and knowledge extraction engine for integrative medicine. Nucleic Acids Res. 2014;42:D1007–D1012. doi: 10.1093/nar/gkt1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teutsch SM, Bradley LA, Palomaki GE, Haddow JE, Piper M, Calonge N, Dotson WD, Douglas MP, Berg AO. The Evaluation of Genomic Applications in Practice and Prevention (EGAPP) initiative: Methods of the EGAPP Working Group. Genet Med. 2009;11:3–14. doi: 10.1097/GIM.0b013e318184137c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Yourshaw M, Mamsa H, Rudnik-Schöneborn S, Menezes MP, Hong JE, Leong DW, Senderek J, Salman MS, Chitayat D, Seeman P, von Moers A, Graul-Neumann L, Kornberg AJ, Castro-Gago M, Sobrido MJ, Sanefuji M, Shieh PB, Salamon N, Kim RC, Vinters HV, Chen Z, Zerres K, Ryan MM, Nelson SF, Jen JC. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet. 2012;44:704–708. doi: 10.1038/ng.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BJ. The challenge of developing evidence-based genetics health care in practice. Fam Cancer. 2006;5:55–59. doi: 10.1007/s10689-005-2576-2. [DOI] [PubMed] [Google Scholar]
- Zhang B, Beeghly-Fadiel A, Long J, Zheng W. Genetic variants associated with breast-cancer risk: Comprehensive research synopsis, meta-analysis, and epidemiological evidence. Lancet Oncol. 2011;12:477–488. doi: 10.1016/S1470-2045(11)70076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]