

Abstract
Asthma is a common medical condition affecting 300 million people worldwide. Airway inflammation, smooth muscle bronchoconstriction leading to airflow obstruction, and mucous hypersecretion are clinical hallmarks of asthma. The NHLBI Expert Panel Report 3 recommends inhaled corticosteroids (ICS) for patients with moderate to severe persistent asthma. Inhaled corticosteroids (ICS) target gene transcription through their interactions with the glucocorticoid (GC) receptor (GR) at the glucocorticoid response element (GRE). The GC/GR complex enhances anti-inflammatory but inhibits pro-inflammatory mediator production. Classically, asthma has been described as a Th2-associated eosinophil-predominant disease, but recently alternative models have been described including a Th17-mediated neutrophil-predominant phenotype resulting in patients with more severe disease who may be less responsive to steroids. Additional mechanisms of steroid resistance include increased activity of GR phosphorylating kinases which modify the interactions of GR with transcription factors to inhibit the ability of GR to bind with GRE, leading to an increase in pro-inflammatory gene transcription. Oxidative stress also affects the balance between pro-inflammatory and anti-inflammatory gene transcription through the modification of transcription factors and cofactors (such as PI3K) leading to the inhibition of histone deacetylase 2. Continued investigations into the mechanisms behind glucocorticoid resistance will lead to novel treatments that improve control of severe refractory asthma.
Keywords: asthma, glucocorticoids, oxidative stress, refractory, steroid resistance
Asthma is one of the most common chronic medical diseases globally, affecting over 25 million people in the United States and 300 million people worldwide, with that number expected to rise 1. Additionally, it is one of the most costly chronic conditions, in terms of dollars and human lives, with an estimated 15 million daily-adjusted life years (DALYs) lost annually, and it is implicated in one of every 250 deaths worldwide 1.
Asthma is described as a chronic inflammatory condition affecting the airways consisting of a cellular component resulting in airway inflammation, and smooth muscle hyper-responsiveness in response to direct or indirect stimuli leading to bronchoconstriction. Indirect stimuli, such as that of exercise, result in the release of inflammatory mediators from cells present in the airway contributing to airflow obstruction 2. The clinical result of this inflammation and bronchoconstriction is airflow obstruction that is typically reversible with the use of glucocorticoids, particularly for individuals with an eosinophil-, rather than neutrophil-predominant phenotype. It has been shown that decreases in FEV1 correlate with increases in eosinophil and neutrophil counts in the airways of asthmatic patients. Interestingly, increases in airway eosinophilia, but not neutrophilia, appear to correlate with an increased bronchial hyper-responsiveness based on methacholine challenge testing, suggesting the possibility that targeting the neutrophil may be a potential means of controlling chronic, less reversible airflow obstruction 3. Although inflammatory cell infiltration in the airway has been broadly correlated with the degree of bronchial hyper-responsiveness, there has not been any definitive evidence proving causation.
The disease is diagnosed by eliciting a history of symptoms consistent with asthma (cough, dyspnea, and wheeze) in combination with evidence of reversible airflow obstruction as demonstrated by spirometry, or a positive methacholine challenge test. Current therapy for asthma is based on the severity and control of symptoms, with the backbone of treatment consisting of inhaled corticosteroids (ICS) for those with moderate persistent disease and beyond 4. Unfortunately, about 10 percent of asthmatics appear to have refractory disease despite receiving optimal therapy, leading to increased morbidity and cost associated with treatment 5.
Pathophysiology of asthma
Cells and cytokines
The classic model of allergic airway inflammation is characterized by submucosal infiltration of activated Th2 lymphocytes, eosinophils, macrophages, mast cells, and neutrophils 6. This model of asthma is a complex web of cells and cell signaling molecules interacting to elicit an inflammatory response (Fig.1). Allergen/antigen presentation by antigen-presenting cells to Th0 cells results in Th2 cell differentiation. This primary switching may be induced by the production of thymic stromal lymphopoietin (TSLP) by airway epithelial cells in response to antigen stimulation. Thymic stromal lymphopoietin then acts upon TSLP receptors (TSLPR) expressed by dendritic cells (DCs) to promote the transcription of OX-40L. OX-40L, a member of the TNF family of cytokines, induces expression of Th2 cytokines by the activated DCs leading to inflammatory Th2 cell differentiation 7. The Th2 lymphocytes further produce IL-4, IL-5, and IL-13 cytokines that stimulate B cells to synthesize and release IgE. According to the classic paradigm of allergic airway disease, IgE is then bound to the surface of mast cells where inhaled allergens bind to these receptors precipitating the release of histamine, prostaglandins, and leukotrienes during cellular degranulation. These cell signaling molecules induce smooth muscle bronchoconstriction and further propagate the inflammatory response. Th2 lymphocytes also produce IL-9 which stimulates mast cell proliferation in the airway 8, and IL-5, a cytokine associated with eosinophil survival 6. Eosinophils are believed to participate in the inflammatory response through the release of mediators including cysteinyl leukotrienes and reactive oxygen species resulting in bronchoconstriction, mucous secretion, and structural damage to the airways 9–12.
Figure 1.

Th2 and Th17 allergen responses in the asthmatic airway. Upon allergen presentation to Th0 cells by antigen-presenting cells (APC), Th cells differentiate into Th2 cells in the presence of IL-4, and Th17 cells in the presence of IL-23. Th2 cells then go on to produce IL-4-, IL-5-, and IL-13-activating B cells to release IgE which attaches to the surface of mast cells. When stimulated by antigen, mast cells release histamine, prostaglandins, and leukotrienes resulting in smooth muscle bronchoconstriction, airway inflammation, and mucous hypersecretion. Eosinophils activated by IL-5 produce cysteinyl leukotrienes and reactive oxygen species (ROS), which act in a similar manner on the airways, and additionally contribute to oxidative stress. Th17 cells producing IL-17 act on airway epithelial cells to stimulate the release of multiple factors. These factors include macrophage chemoattractant protein-1 (MCP-1) which recruits macrophages, IL-5, regulated on activation, normal T cell expressed and secreted (RANTES), and GM-CSF (granulocyte–macrophage colony-stimulating factor) which activate eosinophils, IL-8 which mobilizes neutrophils, stem cell factor (SCF) which works to promote mast cell survival, and IL-25 which induces myeloid cells to release Th2-type cytokines. Neutrophils release matrix metalloproteinase 9 (MMP9), elastase, leukotriene B4, and platelet-activating factor (PAF), which work to enhance the activity of eosinophils. Activated macrophages release IL-1, tumor necrosis factor alpha (TNF-α), and IL-6 which interact with other inflammatory cells and result in a positive feedback loop with airway epithelial cells.
However, current evidence supports the role of respiratory viruses, as opposed simply to exposure to environmental allergens, on the development of asthma exacerbations. Most likely, exacerbations frequently arise from a complex interaction between the two. Respiratory viruses, in particular human rhinovirus (HRV), have been shown to be present during asthma exacerbations 13,14 and been demonstrated to induce asthma exacerbations in susceptible individuals following inoculation 15. The mechanism of action for the viral induction of asthma exacerbations appears to be related to insufficient IFN-γ and IL-10 response and augmented IL-4, IL-5, IL-13 response. This suggests either impaired Th1 or heightened Th2 immunity as a mechanism for virus-induced airway inflammation.
Although asthma is typically associated with Th2 cytokines and eosinophilia, a subset of asthmatics have neutrophil-predominant Th17-associated disease (Fig.1). Patients with mild to moderate asthma typically have disease characterized by Th2 cytokine expression with eosinophilic inflammation and respond well to ICS although a subset of Th2 high eosinophil-predominant asthmatics will have refractory disease despite receiving optimal treatment. These individuals appear to be particularly responsive to treatment with an anti-IL-5 antibody such as mepolizumab 16. Those with more severe, steroid-resistant disease appear to have their cellular milieu defined by Th1/Th2 cytokine expression and neutrophilic airway inflammation with less reversible airflow obstruction, but without increased bronchial hyper-responsiveness based on methacholine challenge testing 3,17,18. Among those with neutrophil-predominant disease, Th17 cells and their associated cytokine IL-17 have been noted to play a significant role in airway inflammation. IL-17 expression has been shown to be increased in the bronchoalveolar lavage (BAL) fluid, sputum, and sera of patients with asthma, where the severity of the disease correlates with an incremental increase in the presence of IL-17. IL-17 expression by Th17 cells has been demonstrated to augment in vitro glucocorticoid beta (GR-β) expression by airway epithelial cells. Glucocorticoid receptor beta (GR-β), an alternative isoform of glucocorticoid receptor alpha (GR-α), functions to suppress GR-α-mediated anti-inflammatory gene transcription through competitive inhibition of transcription at the glucocorticoid response element (GRE) 19. It has also been shown that IL-17 recruits neutrophils by promoting release of IL-8 from airway epithelial cells, and may be the link between T lymphocytes and granulocytes in the asthmatic airway 20,21. In vitro studies have examined GC responsiveness of human airway epithelial cells following preincubation with IL-17A. Cells exposed to IL-17A were less able to inhibit tumor necrosis factor alpha (TNF-α)-induced IL-8 production after GCs were introduced, suggesting that the presence of IL-17-producing cells may render airway epithelial cells less responsive to GCs 22. Additionally, IL-17 has been shown to be a potent activator of endothelial cells, promoting transmigration of neutrophils to sites of inflammation 23. Induced sputum obtained from severe asthmatics demonstrates relatively high levels of neutrophils and appears to correlate with the severity of disease 24. Airway neutrophils produce proteases and lipid mediators, such as matrix metalloproteinase 9, elastase, leukotriene B4, and platelet-activating factor, that further propagate the inflammatory cascade and also appear to be responsible for the recruitment of eosinophils 20.
Airway epithelial cells are key players in the inflammatory response. They too have been shown to release IL-5 in addition to stem cell factor, a cytokine that supports survival of mast cells within the airway, and macrophage chemoattractant protein-1 (MCP-1). Alveolar macrophages, recruited by MCP-1, may also play an important role in the inflammatory process. It is thought that these macrophages may be a source of IL-1β, TNF-α, and IL-6 which they release following allergen binding to low-affinity IgE receptors. These cytokines might act on epithelial cells to stimulate the release of GM-CSF, IL-8, and regulated on activation, normal T cell expressed and secreted (RANTES). Both RANTES and GM-CSF work to recruit eosinophils to the airway and promote their survival 6,8. Macrophages have also been shown to secrete elastase and metalloproteinases which are capable of degrading elastin in the airway extracellular matrix 25,26.
Airway smooth muscle cells (ASMCs) play a role in the pathogenesis of airway inflammation. It is thought that viral infections may precipitate an asthmatic response in the airway through increased production of interferons and tumor necrosis factor alpha (IFNs/TNF-α) as demonstrated through in vitro exposure of ASMCs to these cytokines. Following exposure, increased levels of pro-inflammatory molecules were produced by ASMCs, GR-β expression was found to be upregulated, and increased contractility was noted through the production of calcium regulatory protein CD38 27. Airway smooth muscle cells from patients with severe asthma were also noted to be corticosteroid unresponsive based on measured levels of cytokine expression following pretreatment with dexamethasone and stimulation with TNF-α, compared to those with nonsevere asthma. This may occur through the actions of TNF-α-induced p38 mitogen-activated protein kinase (MAPK) activity inhibiting anti-inflammatory gene transcription 28.
Additionally, myeloid-derived regulatory cells have also recently been implicated as critical regulators of allergic airway inflammation. Oxidative stress during airway inflammation regulates the expansion, activation, recruitment, and function of these immunoregulatory cells. Differential regulation by nitric oxide- or superoxide-producing subsets of these immature myeloid cells contributes to the balance of immune suppression and exacerbation of airway hyper-responsiveness 29.
Structural changes of the airways
Airway remodeling as a consequence of inflammation is another characteristic of asthma 30–33. Structural changes that occur due to inflammation include thickening of the basement membrane, subepithelial fibrosis, goblet cell metaplasia, neovascularization, and increased airway smooth muscle mass 34. Examination of the relationship between airway remodeling and degree of asthma severity determined that clinical and functional severity scores of asthma were the strongest predictors of increased subepithelial layer thickness, independent of duration of disease, FEV1, or PC20M 31. This suggests that all patients with severe, poorly controlled asthma can experience deleterious effects on the structure and function of their airways, regardless of the duration of disease. While a definitive cause/effect relationship has yet to be conclusively established, airway remodeling has been associated with irreversible decline in FEV1, loss of bronchodilator reversibility, and increased airway hyper-responsiveness 35,36. Additionally, correlation was found between increases in basement membrane thickness in the cartilaginous airways and cases of severe fatal asthma in a retrospective study 37.
IL-33, a member of the IL-1 family, has been implicated in the airway remodeling process. It is produced by airway epithelial cells and smooth muscle cells to stimulate collagen synthesis by fibroblasts, resulting in increased reticular basement membrane thickness. IL-33 is important in switching from Th1 to Th2 responses in vivo 38, producing Th2 cytokines IL-5 and IL-13, as well as inducing cytokine release from mast cells 39 and increasing expression of IL-17 by Th17 cells 40. The action of IL-33 is unaffected by exposure to high-dose steroids, suggesting that this interleukin plays an important role in the pathogenesis of steroid-resistant asthma 41. These various mechanisms lead to structural changes that result in a loss of elastic recoil with increasing lung compliance, most pronounced at the peri-bronchiolar level, resulting in irreversible obstructive small airways disease 42.
Mechanisms of action of corticosteroids
Corticosteroids comprise the backbone of asthma therapy and act to reduce inflammation through both gene activation and suppression 43. Corticosteroids cross the cell membrane and bind to the glucocorticoid receptor (GR) in the cytoplasm (Fig.2). Once bound, conformational changes occur that result in the release of nuclear chaperone proteins such as heat shock protein 90 (hsp-90) exposing two nuclear localization sequences (NL1 and NL2) on GR 44, allowing it to cross the nuclear membrane and interact with the GRE on DNA. The GC/GR complex interacts in a complex and dynamic manner with various transcriptional factors and kinases to alter the transcription of genes 6. One of these interactions involves the transcription of genes such as β-adrenergic receptor, glucocorticoid-induced leucine zipper, and mitogen-activated protein kinase phosphatase-1 (MKP-1). The latter two work as anti-inflammatory proteins with MKP-1 functioning as an inhibitor of MAPK pathways which promote the transcription of pro-inflammatory genes 43,45. Glucocorticoids also act to suppress the transcription of genes associated with inflammation through their interactions with GR. GR attenuates the activity of transcription factors responsible for transcribing genes of pro-inflammatory proteins, such as nuclear factor-kappa B (NF-κB) and AP-1, by inhibiting histone acetylase through the recruitment of histone deacetylase 2 (HDAC2). HDAC2 deacetylates the GR allowing it to form a complex with NF-κB and AP-1, thereby inhibiting their ability to transcribe pro-inflammatory genes 43,46.
Figure 2.
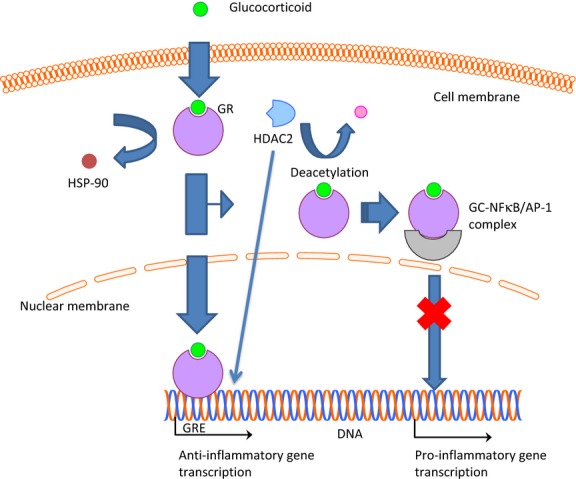
Mechanisms of action of glucocorticoids (GC). GC diffuse across the cell membrane where they bind with GC receptors (GR) in the cytoplasm. Upon binding of the GC, this causes release of inhibitory proteins such as heat shock protein 90 (hsp-90), allowing the GC-bound GR to diffuse across the nuclear membrane where it binds to the glucocorticoid response element (GRE). The GRE is responsible for transcribing anti-inflammatory proteins. Additionally, binding of GC to GR results in recruitment of histone deacetylase 2 (HDAC2), which is responsible for deacetylating GR, permitting its binding to nuclear factor-kappa B (NF-κB) and activating protein-1 (AP-1). Upon binding, these transcription factors are deactivated, thereby inhibiting the transcription of pro-inflammatory proteins. Additionally, HDAC2 deacetylates the histone permitting transcription of anti-inflammatory genes by GR.
Steroid resistance in asthma
Severe/refractory asthma is poorly controlled disease despite optimal therapy and requires that one major criterion and two minor criteria be satisfied in order to meet the clinical definition of disease (see Table1) 5,47,48. Steroid-resistant asthma is a subset of severe asthma defined as a failure to increase FEV1 by 15% following a 7-day course of oral steroid therapy at a dose of 20 mg of prednisolone daily or equivalent 49. Approximately 10 percent of asthmatic patients meet this definition of steroid-resistant/refractory asthma, consuming a disproportionate amount of healthcare dollars due to increased utilization of resources from outpatient clinic visits, emergency room care, and hospitalizations 50,51. Multiple pathways have been implicated in the pathogenesis of refractory asthma. Some of these pathways include immune-mediated dysregulation of cytokines, defects in the ability of GR to bind drug and translocate into the nucleus, increased glucocorticosteroid receptor β (GRβ) expression, excessive activation of activating peptide-1 (AP-1) and NF-κB, and abnormal histone acetylation 43.
Table 1.
American Thoracic Society Criteria for Severe/Refractory Asthma*
| Major criteria |
| Treatment with continuous or near continuous (>50% of the year) oral corticosteroids |
| Need for treatment with high-dose inhaled corticosteroids |
| Minor criteria |
| Need for additional daily treatment with a controller medication (long-acting β-agonist, leukotriene receptor antagonist, theophylline) |
| Asthma symptoms needing short-acting β-agonist use on a daily or near daily basis |
| Persistent airway obstruction (FEV1 < 80% predicted, diurnal peak expiratory flow variability >20% predicted) |
| One of more urgent care visits for asthma per year |
| Three or more oral steroid bursts per year |
| Prompt deterioration with <25% reduction in oral or intravenous corticosteroid use |
| Near fatal asthma event in the past |
One major criterion plus two minor criteria required for diagnosis; other diseases have been excluded, exacerbating factors treated, and patient is generally adherent.
Cells and signaling pathways contributing to steroid resistance
Th17 lymphocytes appear to play a key role in immune-mediated diseases including steroid-resistant asthma. While its causative role remains uncertain, accumulating evidence suggests a correlation between high levels of IL-17 production by Th17 cells and steroid-resistant disease 52. CD4+ lymphocytes differentiate into Th17 cells in the presence of IL-1β and IL-23, produced by antigen-presenting cells. IL-23 in particular drives the regulation, proliferation, and cytokine production by Th17 cells 19. As discussed, Th17 cells produce IL-17 which is a pro-inflammatory cytokine and appears to drive neutrophil-predominant steroid-resistant asthma 53,54. Neutrophils have been shown to release TNF-α which induces bronchial hyper-responsiveness in a murine model. Additionally, neutrophils have been found to produce reactive oxygen species leading to an increased transcription of IL-8 by airway epithelial cells, further propagating the chemotactic neutrophil response 24. High concentrations of neutrophils have been found in the airways of severe asthmatic patients hospitalized for life-threatening attacks, suggesting a role for this cell type in the pathogenesis of acute allergic airway inflammation. Although steroids are known to increase neutrophil numbers by prolonging survival through decreased apoptosis 55, airway samples taken over time from this cohort showed an initial increase in neutrophil count that, interestingly, continued to increase throughout resolution of the exacerbation 56. This has led some to speculate that the neutrophil may not only be part of the problem, but may also help play a role in the recovery process.
Although not much is known regarding the functional relationship of immunoregulatory myeloid cells which are regulators of airway inflammation and steroid resistance in asthma, glucocorticoids induce the in vitro expansion of these cells 57. Eosinophils, mast cells, and airway epithelial cells have been shown to produce IL-25 in the lungs of asthmatics 58. IL-25, a member of the IL-17 family, regulates multiple aspects of mucosal immunity and has been shown to induce a population of pulmonary myeloid cells capable of producing Th2-associated cytokines IL-4 and IL-13. Recent evidence also indicates that blocking the glucocorticoid signal in a murine trauma model using the antagonist of the glucocorticoid receptor RU486 blocked the in vivo expansion of these cells 59. Further investigations are therefore necessary to provide mechanistic insight for the regulation of steroid resistance by these cellular phenotypes and identifying these regulatory cells as new therapeutic targets for asthma therapy.
In addition to driving Th2 responses in the airways, IL-25 has also been implicated in airway remodeling. Using a murine model, investigators blocked IL-25 in allergen-exposed mice and noted subsequent reduction in eosinophil count and Th2 cytokine levels. Reduction in airway smooth muscle hyperplasia, neovascularization, collagen deposition, and bronchial hyper-responsiveness were also noted in those animals undergoing IL-25 neutralization. Additionally, production of IL-33 and TSLP was also decreased when IL-25 was blocked 60.
Glucocorticoid receptor inhibition by kinases
Glucocorticoids exert their effects by binding to GR in the cytoplasm which then translocates into the nucleus where it interacts with DNA to enhance or inhibit the transcription of genes affecting the inflammatory response. It has been demonstrated that GR function is reduced through phosphorylation by several kinases, most notably p38MAPK. Stress-induced phosphorylation of serine 134 on the GR in a p38MAPK-dependent manner 61 leads to steroid resistance by impeding nuclear translocation, protein stabilization, and binding to DNA, thus decreasing the transcription of anti-inflammatory genes 43,62. p38MAPK activity is enhanced by IL-2, IL-4, and IL-13 and inhibited by p38MAPK inhibitors, where inhibition of its action correlates with decreased phosphorylation of the GR, resulting in increased nuclear translocation and ability to bind GRE 63,64. Along with MAPKs, extracellular signal-regulated kinase (ERK) can phosphorylate the GC receptor when stimulated by superantigens, preventing nuclear translocation. Upon treatment with an ERK inhibitor, GC response is restored 65.
Glucocorticoid receptor and transcription factors
Nuclear factor-kappa B is a transcription factor playing an important role in the pro-inflammatory response. It exists in the cytoplasm bound to an inhibitory protein, IκB. Upon binding of cell signaling molecules such as IL-1β, IL-2, GM-CSF, and TNF-α, a kinase cascade ensues leading to phosphorylation and ubiquitination of IκB, liberating NF-κB to translocate to the nucleus where it then transcribes pro-inflammatory cytokines 66. AP-1, a heterodimer of Fos and Jun, is a member of the leucine zipper transcription family which functions to enhance or inhibit transcription by dimerizing with other transcription factors. AP-1 has been demonstrated to physically interact with the GR to prevent binding to GREs 6,67.
Once bound to glucocorticoid, GR translocates to the nucleus where it inhibits NF-κB and AP-1 pro-inflammatory cytokine transcription through the recruitment of histone deacetylase 2 (HDAC2) which reverses histone acetylation, thereby inhibiting gene transcription 68. However, in the presence of MAPK phosphorylation, GR is unable to translocate to the nucleus and has reduced ability to induce histone acetylation, precluding its ability to inhibit this NF-κB and AP-1-associated pro-inflammatory transcription process 6,69. Additionally, HDAC2 activity is reduced by phosphoinositide 3-kinase (PI3K) δ phosphorylation 70. Oxidative stress increases the activity of PI3Kδ and thus is responsible for a pro-inflammatory state. Generation of oxidants occurs in the airway of asthmatic patients due to the presence of increased nitric oxide levels resulting in the formation of peroxynitrite, tyrosine nitration, and lipid peroxidation 71. Tyrosine nitration of HDAC2 is purported to inhibit its functional ability, and this appears to be an important mechanism of steroid resistance in asthmatic patients who smoke 72.
Novel therapeutic options
Asthma is typically treated with ICS, along with the addition of long-acting β-agonists (LABA) and/or long-acting muscarinic agonist (LAMA) 73 if ICS alone do not control disease. For patients who remain poorly controlled on this regimen, escalating doses of ICS are provided, along with alternative therapies including the addition of anti-IgE therapy in qualifying patients 74. When all conventional asthma treatments fail, new targets for therapy offer hope. These new drugs target novel pathways in the asthmatic response to gain better control over the disease.
Targeting cytokines
One potential option for treatment is the modulation of chemokines and cytokines to inhibit inflammatory cell migration into airways (Table2). Inhibition of Th2-associated cytokines IL-4, IL-5, IL-9, and IL-13 offer promise as potential treatment targets.
Table 2.
Novel targets currently under investigation for the treatment of steroid-resistant asthma
| Target | Treatment | Stage of development for use in asthma (ref) |
|---|---|---|
| IL-4 | Pitrakinra/dupilumab | Phase II clinical trials 17,75,76/phase II clinical trials 77 |
| IL-5 | Mepolizumab | Phase III clinical trials 16,78,79 |
| IL-9 | MEDI-528 | Phase II clinical trials 81,82 |
| IL-13 | Lebrikizumab/tralokinumab | Phase III clinical trials 82–85/phase II clinical trials 86 |
| IL-17 | IL-17 antibody | Animal model of allergic airway disease only 87–89 |
| IL-23 | IL-23 antibody | Animal model of allergic airway disease only 89 |
| IL-33 | IL-33 antibody | Animal model of allergic airway disease only 90 |
| CCR3 | CCR3 receptor antagonists | Phase II clinical trial 91,92 |
| Kinase inhibitors | Imatinib | Phase II clinical trials 95 |
| Upregulation of HDAC2 | Theophylline | Currently available 96–98 |
| Airway smooth muscle | Bronchial thermoplasty | Currently available 99,100 |
IL-4 monoclonal antibody appears effective for blocking the allergic airway response. The development of recombinant human IL-4, pitrakinra, was shown to block both IL-4 and IL-13 from binding to IL-4Rα/IL-13Rα1, resulting in improved FEV1 following allergen challenge and reduction in the need for rescue medications in humans subjects 17,75,76. Dupilumab, another IL-4Rα monoclonal antibody, was recently shown to reduce asthma exacerbations in subjects on high-dose ICS/LABA with sputum eosinophilia. Individuals were treated with either dupilumab or placebo for 4 weeks prior to withdrawal of LABAs, and then steroids were tapered over weeks 6–9. Those receiving drug experienced an exacerbation rate of 6% relative to those receiving placebo of 44% 77.
A human anti-IL-5 antibody, mepolizumab, was developed based on its ability to decrease eosinophil recruitment into the airways following allergen challenge in animals pretreated with antibody 78. In a recent meta-analysis, mepolizumab was shown to reduce eosinophil counts in the sputum and blood of subjects, but had no effect on other end points such as FEV1, peak flow, or histamine PC20 79. However, the DREAM study (mepolizumab for severe eosinophilic asthma), a multicenter, randomized, placebo-controlled trial, did demonstrate a reduction in the rate of exacerbations in the treatment arm 16, suggesting that this may provide improvement in quality of life.
Humanized anti-IL-9 monoclonal antibody MEDI-528 was shown to have an acceptable safety profile in clinical trials with a trend toward less frequent exacerbations in the treatment group 80. While further study remains, MEDI-528 was able to reduce Th2-associated cytokines IL-4, IL-5, and IL-13 along with eosinophil and lymphocyte counts in BAL fluid from a mouse model. Additionally, MEDI-528 inhibited airway hyper-reactivity in response to a methacholine challenge in the same animal model of allergic airway disease 81.
An anti-IL-13 antibody, lebrikizumab, was developed with the ability to reduce Th2-associated cytokine and IgE production along with eosinophil recruitment. Trials in humans have demonstrated safety 82,83 and have shown significant improvement in FEV1 in severe asthmatics compared to those treated with placebo. This effect was most pronounced in those with higher serum levels of periostin, a marker of high Th2 eosinophil-predominant disease, released from airway epithelial cells in response to increased circulating levels of IL-13 84,85. An alternative IL-13 monoclonal antibody, tralokinumab, was recently shown to produce a statistically significant improvement in FEV1 and decreased use of rescue medications in those on drug relative to placebo in atopic patients with moderate to severe asthma 86.
Th17-associated cytokine IL-17 is another target of interest. The use of an anti-IL-17 antibody was shown to block the effects of IL-17 following allergen challenge in a murine model where decreased levels of eosinophils, lymphocytes, and neutrophils were detected in BAL fluid from the treated animals 87. Treatment with anti-IL-17 antibody reduced levels of IL-4, IL-5, and IL-13 88. Upstream blocking of IL-17 production by Th17 cells using an anti-IL-23 antibody was shown to be effective in reducing recruitment of neutrophils, eosinophils, and lymphocytes into the airways. Use of both anti-IL-17 and anti-IL-23 antibodies in humans is currently underway in clinical trials for other immune-mediated diseases such as Crohn's and rheumatoid arthritis and is likely to represent a future target in the treatment of asthma 89.
IL-33 can enhance Th2 responses through an increased expression of IL-5 and IL-13, leading to eosinophil influx and IgE production. Treatment with an IL-33 monoclonal antibody in a murine model of allergic airway disease attenuates these features; however, its role in human allergic airway disease remains to be determined 90.
Targeting chemokine receptors
Targeting the receptors responsible for mediating the traffic of inflammatory cells is an alternate pathway under investigation for the treatment of asthma. Chemokine receptors CCR1, CCR2, CCR3, CCR4, CCR5, and CCR8 have been implicated in allergic airway disease through their interactions with chemokine ligands expressed by mast cells, eosinophils, and T-helper lymphocytes among other cells involved in the inflammatory response 91. The most promising of these, CCR3, expressed largely on eosinophils, is the only chemokine receptor for which an antagonist has been developed and tested in clinical trials. Unfortunately, despite demonstrating safety, it did not show efficacy in a phase III clinical trial for the treatment of allergic rhinitis 92. Development of these small molecules to target chemokine receptors for the treatment of asthma may prove to be problematic due to their functional redundancy and binding specificity 93.
Additional targets for treatment
As discussed, certain kinases can phosphorylate glucocorticoid receptors to diminish their glucocorticoid response. p38MAPK inhibitors currently under development can interfere with the phosphorylation of GR to decrease the downstream release of inflammatory mediators by increasing GR nuclear translocation and have been shown to be safe in animal models 94. In vitro studies using peripheral blood mononuclear cells (PBMCs) have examined the effects of p38 MAPK inhibition on steroid responsiveness. Peripheral blood mononuclear cells from severe asthmatics exposed to lipopolysaccharide in addition to a p38 MAPK inhibitor expressed less IL-8 after the addition of dexamethasone than cells not pretreated with p38 MAPK inhibitor. This finding suggests that use of these inhibitors may attenuate inflammatory responses in the presence of steroid 64. The use of tyrosine kinase inhibitor imatinib mesylate in severe asthma represents a promising new therapy 95. Administration of imatinib was shown to attenuate airway hyper-reactivity, eosinophil accumulation, and Th2-associated cytokines IL-4 and IL-13 in a murine model of allergic airway disease. Its proposed mechanism of action is through the inhibition of tyrosine kinase C-kit activity, resulting in decreased production of stem cell factor by airway epithelial cells leading to reduced mast cell activity in the airways. This drug is currently in phase II clinical trials for use in asthma.
The use of drugs to potentiate the production of HDAC2 when levels are decreased as a consequence of oxidative stress is another option for treating refractory asthma. One such drug, theophylline, is speculated to restore steroid responsiveness in previously resistant individuals. The mechanism through which this is thought to occur is increased activity of HDAC2 secondary to NF-κB suppression due to direct inhibition of oxidant activated PI3Kδ by theophylline 96,97. In a study conducted in severe asthmatics controlled on high-dose ICS and other therapies including theophylline, withdrawal of theophylline was shown to worsen control of disease, while re-treatment restored asthma control, suggesting utility in using this drug to augment treatment 98. Another target in the treatment of severe asthma is the airway itself. To reduce bronchoconstriction, a new procedure termed bronchial thermoplasty has been developed to reduce airway smooth muscle mass. For this procedure, a catheter is used to deliver 65°C of thermal energy to the airway in order to reduce smooth muscle mass through uncoupling of contractile tissue. This procedure typically takes place in three sessions scheduled approximately 3 weeks apart. The Asthma Intervention Research 2 Trial for bronchial thermoplasty (AIR2 trial) is a double-blind, randomized, sham-controlled clinical trial examining the efficacy and safety of bronchial thermoplasty in severe asthmatics. Initial data showed that treated patients had a statistically significant improvement in their Asthma Quality of Life Questionnaire (AQLQ) and less missed days of work/school from exacerbations. However, this study also notes that the sham treatment conferred benefit as well, and those receiving the intervention had an increased rate of hospitalization following treatment 99. Recently published data on the AIR2 trial at 5 years of follow-up show that treated patients continued to experience a reduction in their rate of exacerbations between years 2–5, despite remaining on a lower average dose of steroids relative to the 12 months prior to treatment. Additionally, no structural airway abnormalities were observed on CT scan 100.
Conclusions
Asthma is a complex disease affecting millions of individuals worldwide with 10 percent of asthmatics being refractory to conventional treatment. Novel therapies are currently being explored to circumvent steroid refractory disease. The European Network for Understanding Mechanisms of Severe Asthma (ENFUMOSA), Severe Asthma Research Project (SARP), and Unbiased BIOmarkers in the PREDiction of respiratory disease outcomes (U-BIOPRED) are currently working to phenotype and subphenotype asthmatic patients. Microarray studies comparing PBMCs from corticosteroid-sensitive and insensitive asthmatic patients have identified 11 discrepant genes which could be used to profile individuals at risk for steroid-resistant asthma 101. Future trends may include phenotyping and genotyping individuals to determine the best course of treatment for each patient.
Acknowledgments
The authors wish to thank Dr. Mark Dransfield and Dr. Victor J. Thannickal for critical review of this manuscript. This work was supported by Young Clinical Scientist Faculty Award from the Flight Attendants Medical Research Institute and Parker B. Francis fellowship awarded to Dr. Jessy Deshane.
Conflicts of interest
The authors have no conflicts of interest to disclose.
References
- Masoli M, Fabian D, Holt S, Beasley R Global Initiative for Asthma (GINA) Program. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59:469–478. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- Brannan JD, Lougheed MD. Airway hyperresponsiveness in asthma: mechanisms, clinical significance, and treatment. Front Physiol. 2012;3:460. doi: 10.3389/fphys.2012.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff PG, Khashayar R, Lazarus SC, Janson S, Avila P, Boushey HA, et al. Relationship between airway inflammation, hyperresponsiveness, and obstruction in asthma. J Allergy Clin Immunol. 2001;108:753–758. doi: 10.1067/mai.2001.119411. [DOI] [PubMed] [Google Scholar]
- National Asthma Education and Prevention Program. Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma-Summary Report 2007. J Allergy Clin Immunol. 2007;120(Suppl 5):S94–S138. doi: 10.1016/j.jaci.2007.09.043. [DOI] [PubMed] [Google Scholar]
- Proceedings of the ATS workshop on refractory asthma: current understanding, recommendations, and unanswered questions. American Thoracic Society. Am J Respir Crit Care Med. 2000;162:2341–2351. doi: 10.1164/ajrccm.162.6.ats9-00. [DOI] [PubMed] [Google Scholar]
- Adcock IM, Caramori G. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol Cell Biol. 2001;79:376–384. doi: 10.1046/j.1440-1711.2001.01025.x. [DOI] [PubMed] [Google Scholar]
- Liu YJ. Thymic stromal lymphopoietin and OX40 ligand pathway in the initiation of dendritic cell-mediated allergic inflammation. J Allergy Clin Immunol. 2007;120:238–244. doi: 10.1016/j.jaci.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118:3546–3556. doi: 10.1172/JCI36130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleich GJ. Mechanisms of eosinophil-associated inflammation. J Allergy Clin Immunol. 2000;105:651–663. doi: 10.1067/mai.2000.105712. [DOI] [PubMed] [Google Scholar]
- Porsbjerg CM, Gibson PG, Pretto JJ, Salome CM, Brown NJ, Berend N, et al. Relationship between airway pathophysiology and airway inflammation in older asthmatics. Respirology. 2013;18:1128–1134. doi: 10.1111/resp.12142. [DOI] [PubMed] [Google Scholar]
- Niimi A. Cough, asthma, and cysteinyl-leukotrienes. Pulm Pharmacol Ther. 2013;26:514–519. doi: 10.1016/j.pupt.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Annema JT, Sparrow D, O'Connor GT, Rijcken B, Koeter GH, Postma DS, et al. Chronic respiratory symptoms and airway responsiveness to methacholine are associated with eosinophilia in older men: the Normative Aging Study. Eur Respir J. 1995;8:62–69. doi: 10.1183/09031936.95.08010062. [DOI] [PubMed] [Google Scholar]
- Wark PA, Johnston SL, Moric I, Simpson JL, Hensley MJ, Gibson PG. Neutrophil degranulation and cell lysis is associated with clinical severity in virus-induced asthma. Eur Respir J. 2002;19:68–75. doi: 10.1183/09031936.02.00226302. [DOI] [PubMed] [Google Scholar]
- Grissell TV, Powell H, Shafren DR, Boyle MJ, Hensley MJ, Jones PD, et al. Interleukin-10 gene expression in acute virus-induced asthma. Am J Respir Crit Care Med. 2005;172:433–439. doi: 10.1164/rccm.200412-1621OC. [DOI] [PubMed] [Google Scholar]
- Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, et al. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc Natl Acad Sci U S A. 2008;105:13562–13567. doi: 10.1073/pnas.0804181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380:651–659. doi: 10.1016/S0140-6736(12)60988-X. [DOI] [PubMed] [Google Scholar]
- Hansbro PM, Kaiko GE, Foster PS. Cytokine/anti-cytokine therapy – novel treatments for asthma? Br J Pharmacol. 2011;163:81–95. doi: 10.1111/j.1476-5381.2011.01219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw DE, Berry MA, Hargadon B, McKenna S, Shelley MJ, Green RH, et al. Association between neutrophilic airway inflammation and airflow limitation in adults with asthma. Chest. 2007;132:1871–1875. doi: 10.1378/chest.07-1047. [DOI] [PubMed] [Google Scholar]
- Vazquez-Tello A, Halwani R, Hamid Q, Al-Muhsen S. Glucocorticoid receptor-beta up-regulation and steroid resistance induction by IL-17 and IL-23 cytokine stimulation in peripheral mononuclear cells. J Clin Immunol. 2013;33:466–478. doi: 10.1007/s10875-012-9828-3. [DOI] [PubMed] [Google Scholar]
- Nakagome K, Matsushita S, Nagata M. Neutrophilic inflammation in severe asthma. Int Arch Allergy Immunol. 2012;158(Suppl 1):96–102. doi: 10.1159/000337801. [DOI] [PubMed] [Google Scholar]
- Aujla SJ, Alcorn JF. T(H)17 cells in asthma and inflammation. Biochim Biophys Acta. 2011;1810:1066–1079. doi: 10.1016/j.bbagen.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Zijlstra GJ, Ten Hacken NH, Hoffmann RF, van Oosterhout AJ, Heijink IH. Interleukin-17A induces glucocorticoid insensitivity in human bronchial epithelial cells. Eur Respir J. 2012;39:439–445. doi: 10.1183/09031936.00017911. [DOI] [PubMed] [Google Scholar]
- Roussel L, Houle F, Chan C, Yao Y, Berube J, Olivenstein R, et al. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J Immunol. 2010;184:4531–4537. doi: 10.4049/jimmunol.0903162. [DOI] [PubMed] [Google Scholar]
- Linden A. Role of interleukin-17 and the neutrophil in asthma. Int Arch Allergy Immunol. 2001;126:179–184. doi: 10.1159/000049511. [DOI] [PubMed] [Google Scholar]
- Bousquet J, Chanez P, Lacoste JY, White R, Vic P, Godard P, et al. Asthma: a disease remodeling the airways. Allergy. 1992;47:3–11. doi: 10.1111/j.1398-9995.1992.tb02242.x. [DOI] [PubMed] [Google Scholar]
- Howell CJ, Pujol JL, Crea AE, Davidson R, Gearing AJ, Godard P, et al. Identification of an alveolar macrophage-derived activity in bronchial asthma that enhances leukotriene C4 generation by human eosinophils stimulated by ionophore A23187 as a granulocyte-macrophage colony-stimulating factor. Am Rev Respir Dis. 1989;140:1340–1347. doi: 10.1164/ajrccm/140.5.1340. [DOI] [PubMed] [Google Scholar]
- Tliba O, Amrani Y. Airway smooth muscle cell as an inflammatory cell: lessons learned from interferon signaling pathways. Proc Am Thorac Soc. 2008;5:106–112. doi: 10.1513/pats.200705-060VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PJ, Bhavsar PK, Michaeloudes C, Khorasani N, Chung KF. Corticosteroid insensitivity of chemokine expression in airway smooth muscle of patients with severe asthma. J Allergy Clin Immunol. 2012;130:877–885. doi: 10.1016/j.jaci.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshane J, Zmijewski JW, Luther R, Gaggar A, Deshane R, Lai JF, et al. Free radical-producing myeloid-derived regulatory cells: potent activators and suppressors of lung inflammation and airway hyperresponsiveness. Mucosal Immunol. 2011;4:503–518. doi: 10.1038/mi.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AL, Maxwell PS, Pearce-Pinto G, Elliot JG, Carroll NG. The relationship of reticular basement membrane thickness to airway wall remodeling in asthma. Am J Respir Crit Care Med. 2002;166:1590–1595. doi: 10.1164/rccm.2108069. [DOI] [PubMed] [Google Scholar]
- Chetta A, Foresi A, Del Donno M, Bertorelli G, Pesci A, Olivieri D. Airways remodeling is a distinctive feature of asthma and is related to severity of disease. Chest. 1997;111:852–857. doi: 10.1378/chest.111.4.852. [DOI] [PubMed] [Google Scholar]
- Reed CE. The natural history of asthma in adults: the problem of irreversibility. J Allergy Clin Immunol. 1999;103:539–547. doi: 10.1016/s0091-6749(99)70221-6. [DOI] [PubMed] [Google Scholar]
- Ten Hacken NH, Postma DS, Timens W. Airway remodeling and long-term decline in lung function in asthma. Curr Opin Pulm Med. 2003;9:9–14. doi: 10.1097/00063198-200301000-00002. [DOI] [PubMed] [Google Scholar]
- Fixman ED, Stewart A, Martin JG. Basic mechanisms of development of airway structural changes in asthma. Eur Respir J. 2007;29:379–389. doi: 10.1183/09031936.00053506. [DOI] [PubMed] [Google Scholar]
- Pascual RM, Peters SP. The irreversible component of persistent asthma. J Allergy Clin Immunol. 2009;124:883–890. doi: 10.1016/j.jaci.2009.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron C, Al-Ramli W, Hamid Q. Remodeling in asthma. Proc Am Thorac Soc. 2009;6:301–305. doi: 10.1513/pats.200808-089RM. [DOI] [PubMed] [Google Scholar]
- Carroll N, Elliot J, Morton A, James A. The structure of large and small airways in nonfatal and fatal asthma. Am Rev Respir Dis. 1993;147:405–410. doi: 10.1164/ajrccm/147.2.405. [DOI] [PubMed] [Google Scholar]
- Liew FY. IL-33: a Janus cytokine. Ann Rheum Dis. 2012;71(Suppl 2):i101–i104. doi: 10.1136/annrheumdis-2011-200589. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Fujita J, Kawaguchi M, Kokubu F, Ohara G, Ota K, Huang SK, et al. Interleukin-33 induces interleukin-17F in bronchial epithelial cells. Allergy. 2012;67:744–750. doi: 10.1111/j.1398-9995.2012.02825.x. [DOI] [PubMed] [Google Scholar]
- Saglani S, Lui S, Ullmann N, Campbell GA, Sherburn RT, Mathie SA, et al. IL-33 promotes airway remodeling in pediatric patients with severe steroid-resistant asthma. J Allergy Clin Immunol. 2013;132:676–685. doi: 10.1016/j.jaci.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauad T, Silva LF, Santos MA, Grinberg L, Bernardi FD, Martins MA, et al. Abnormal alveolar attachments with decreased elastic fiber content in distal lung in fatal asthma. Am J Respir Crit Care Med. 2004;170:857–862. doi: 10.1164/rccm.200403-305OC. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131:636–645. doi: 10.1016/j.jaci.2012.12.1564. [DOI] [PubMed] [Google Scholar]
- Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010;75:1–12. doi: 10.1016/j.steroids.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Glucocorticosteroids: current and future directions. Br J Pharmacol. 2011;163:29–43. doi: 10.1111/j.1476-5381.2010.01199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgate ST, Polosa R. The mechanisms, diagnosis, and management of severe asthma in adults. Lancet. 2006;368:780–793. doi: 10.1016/S0140-6736(06)69288-X. [DOI] [PubMed] [Google Scholar]
- Jarjour NN, Erzurum SC, Bleecker ER, Calhoun WJ, Castro M, Comhair SA, et al. Severe asthma: lessons learned from the National Heart, Lung, and Blood Institute Severe Asthma Research Program. Am J Respir Crit Care Med. 2012;185:356–362. doi: 10.1164/rccm.201107-1317PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael J, Paterson IC, Diaz P, Crompton GK, Kay AB, Grant IW. Corticosteroid resistance in chronic asthma. Br Med J (Clin Res Ed) 1981;282:1419–1422. doi: 10.1136/bmj.282.6274.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang AS. Steroid response in refractory asthmatics. Korean J Intern Med. 2012;27:143–148. doi: 10.3904/kjim.2012.27.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel S. Severe/fatal asthma. Chest. 2003;123(Suppl 3):405S–410S. doi: 10.1378/chest.123.3_suppl.405s-a. [DOI] [PubMed] [Google Scholar]
- Silverpil E, Linden A. IL-17 in human asthma. Expert Rev Respir Med. 2012;6:173–186. doi: 10.1586/ers.12.12. [DOI] [PubMed] [Google Scholar]
- Robinson KM, Manni ML, Biswas PS, Alcorn JF. Clinical consequences of targeting IL-17 and T17 in autoimmune and allergic disorders. Curr Allergy Asthma Rep. 2013;13:587–595. doi: 10.1007/s11882-013-0361-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J Immunol. 1995;154:4719–4725. [PubMed] [Google Scholar]
- Fahy JV. Eosinophilic and neutrophilic inflammation in asthma: insights from clinical studies. Proc Am Thorac Soc. 2009;6:256–259. doi: 10.1513/pats.200808-087RM. [DOI] [PubMed] [Google Scholar]
- Varga G, Ehrchen J, Tsianakas A, Tenbrock K, Rattenholl A, Seeliger S, et al. Glucocorticoids induce an activated, anti-inflammatory monocyte subset in mice that resembles myeloid-derived suppressor cells. J Leukoc Biol. 2008;84:644–650. doi: 10.1189/jlb.1107768. [DOI] [PubMed] [Google Scholar]
- Petersen BC, Lukacs NW. IL-17A and IL-25: therapeutic targets for allergic and exacerbated asthmatic disease. Future Med Chem. 2012;4:833–836. doi: 10.4155/fmc.12.39. [DOI] [PubMed] [Google Scholar]
- Zhang K, Bai X, Li R, Xiao Z, Chen J, Yang F, et al. Endogenous glucocorticoids promote the expansion of myeloid-derived suppressor cells in a murine model of trauma. Int J Mol Med. 2012;30:277–282. doi: 10.3892/ijmm.2012.1014. [DOI] [PubMed] [Google Scholar]
- Gregory LG, Jones CP, Walker SA, Sawant D, Gowers KH, Campbell GA, et al. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax. 2013;68:82–90. doi: 10.1136/thoraxjnl-2012-202003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliher-Beckley AJ, Williams JG, Cidlowski JA. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol Cell Biol. 2011;31:4663–4675. doi: 10.1128/MCB.05866-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel NL, Moore NL. Steroid receptor phosphorylation: a key modulator of multiple receptor functions. Mol Endocrinol. 2007;21:2311–2319. doi: 10.1210/me.2007-0101. [DOI] [PubMed] [Google Scholar]
- Irusen E, Matthews JG, Takahashi A, Barnes PJ, Chung KF, Adcock IM. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: role in steroid-insensitive asthma. J Allergy Clin Immunol. 2002;109:649–657. doi: 10.1067/mai.2002.122465. [DOI] [PubMed] [Google Scholar]
- Bhavsar P, Khorasani N, Hew M, Johnson M, Chung KF. Effect of p38 MAPK inhibition on corticosteroid suppression of cytokine release in severe asthma. Eur Respir J. 2010;35:750–756. doi: 10.1183/09031936.00071309. [DOI] [PubMed] [Google Scholar]
- Li LB, Goleva E, Hall CF, Ou LS, Leung DY. Superantigen-induced corticosteroid resistance of human T cells occurs through activation of the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK-ERK) pathway. J Allergy Clin Immunol. 2004;114:1059–1069. doi: 10.1016/j.jaci.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Adcock IM, Lane SJ, Brown CR, Lee TH, Barnes PJ. Abnormal glucocorticoid receptor-activator protein 1 interaction in steroid-resistant asthma. J Exp Med. 1995;182:1951–1958. doi: 10.1084/jem.182.6.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, et al. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 2006;203:7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1 beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–6903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To Y, Ito K, Kizawa Y, Failla M, Ito M, Kusama T, et al. Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:897–904. doi: 10.1164/rccm.200906-0937OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura H, Komaki Y, Koarai A, Ichinose M. Nitrative stress in refractory asthma. J Allergy Clin Immunol. 2008;121:355–360. doi: 10.1016/j.jaci.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Osoata GO, Hanazawa T, Brindicci C, Ito M, Barnes PJ, Kharitonov S, et al. Peroxynitrite elevation in exhaled breath condensate of COPD and its inhibition by fudosteine. Chest. 2009;135:1513–1520. doi: 10.1378/chest.08-2105. [DOI] [PubMed] [Google Scholar]
- Peters SP, Kunselman SJ, Icitovic N, Moore WC, Pascual R, Ameredes BT, et al. Tiotropium bromide step-up therapy for adults with uncontrolled asthma. N Engl J Med. 2010;363:1715–1726. doi: 10.1056/NEJMoa1008770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstahl GJ, Chen CW, Maykut R, Georgiou P, Peachey G, Bruce J. The eXpeRience registry: the ‘real-world’ effectiveness of omalizumab in allergic asthma. Respir Med. 2013;107:1141–1151. doi: 10.1016/j.rmed.2013.04.017. [DOI] [PubMed] [Google Scholar]
- Tomkinson A, Tepper J, Morton M, Bowden A, Stevens L, Harris P, et al. Inhaled vs subcutaneous effects of a dual IL-4/IL-13 antagonist in a monkey model of asthma. Allergy. 2010;65:69–77. doi: 10.1111/j.1398-9995.2009.02156.x. [DOI] [PubMed] [Google Scholar]
- Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–1431. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368:2455–2466. doi: 10.1056/NEJMoa1304048. [DOI] [PubMed] [Google Scholar]
- Garlisi CG, Kung TT, Wang P, Minnicozzi M, Umland SP, Chapman RW, et al. Effects of chronic anti-interleukin-5 monoclonal antibody treatment in a murine model of pulmonary inflammation. Am J Respir Cell Mol Biol. 1999;20:248–255. doi: 10.1165/ajrcmb.20.2.3327. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhang S, Li DW, Jiang SJ. Efficacy of anti-interleukin-5 therapy with mepolizumab in patients with asthma: a meta-analysis of randomized placebo-controlled trials. PLoS ONE. 2013;8:e59872. doi: 10.1371/journal.pone.0059872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JM, Oh CK, LaForce C, Miller SD, Pearlman DS, Le C, et al. Safety profile and clinical activity of multiple subcutaneous doses of MEDI-528, a humanized anti-interleukin-9 monoclonal antibody, in two randomized phase 2a studies in subjects with asthma. BMC Pulm Med. 2011;11:14. doi: 10.1186/1471-2466-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Arima M, Honda K, Hirata H, Eda F, Yoshida N, et al. Anti-interleukin-9 antibody treatment inhibits airway inflammation and hyperreactivity in mouse asthma model. Am J Respir Crit Care Med. 2002;166:409–416. doi: 10.1164/rccm.2105079. [DOI] [PubMed] [Google Scholar]
- Yang G, Li L, Volk A, Emmell E, Petley T, Giles-Komar J, et al. Therapeutic dosing with anti-interleukin-13 monoclonal antibody inhibits asthma progression in mice. J Pharmacol Exp Ther. 2005;313:8–15. doi: 10.1124/jpet.104.076133. [DOI] [PubMed] [Google Scholar]
- Singh D, Kane B, Molfino NA, Faggioni R, Roskos L, Woodcock A. A phase 1 study evaluating the pharmacokinetics, safety and tolerability of repeat dosing with a human IL-13 antibody (CAT-354) in subjects with asthma. BMC Pulm Med. 2010;10:3. doi: 10.1186/1471-2466-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365:1088–1098. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- Jia G, Erickson RW, Choy DF, Mosesova S, Wu LC, Solberg OD, et al. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J Allergy Clin Immunol. 2012;130:647–654. doi: 10.1016/j.jaci.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper E, Brightling C, Niven R, Oh C, Faggioni R, Poon K, et al. A phase II placebo-controlled study of tralokinumab in moderate-to-severe asthma. Eur Respir J. 2013;41:330–338. doi: 10.1183/09031936.00223411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, et al. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28:42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet. 2008;372:1107–1119. doi: 10.1016/S0140-6736(08)61452-X. [DOI] [PubMed] [Google Scholar]
- Park SJ, Lee YC. Interleukin-17 regulation: an attractive therapeutic approach for asthma. Respir Res. 2010;11:78. doi: 10.1186/1465-9921-11-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–4790. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- Pease JE. Targeting chemokine receptors in allergic disease. Biochem J. 2011;434:11–24. doi: 10.1042/BJ20101132. [DOI] [PubMed] [Google Scholar]
- Fryer AD, Stein LH, Nie Z, Curtis DE, Evans CM, Hodgson ST, et al. Neuronal eotaxin and the effects of CCR3 antagonist on airway hyperreactivity and M2 receptor dysfunction. J Clin Invest. 2006;116:228–236. doi: 10.1172/JCI25423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd CM, Brown Z. Chemokine receptors: therapeutic potential in asthma. Treat Respir Med. 2006;5:159–166. doi: 10.2165/00151829-200605030-00002. [DOI] [PubMed] [Google Scholar]
- Mercado N, Hakim A, Kobayashi Y, Meah S, Usmani OS, Chung KF, et al. Restoration of corticosteroid sensitivity by p38 mitogen activated protein kinase inhibition in peripheral blood mononuclear cells from severe asthma. PLoS ONE. 2012;7:e41582. doi: 10.1371/journal.pone.0041582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin AA, Lukacs NW. Treatment of cockroach allergen asthma model with imatinib attenuates airway responses. Am J Respir Crit Care Med. 2005;171:35–39. doi: 10.1164/rccm.200403-385OC. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Biochemical basis of asthma therapy. J Biol Chem. 2011;286:32899–32905. doi: 10.1074/jbc.R110.206466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. Theophylline. Am J Respir Crit Care Med. 2013;188:901–906. doi: 10.1164/rccm.201302-0388PP. [DOI] [PubMed] [Google Scholar]
- Kidney J, Dominguez M, Taylor PM, Rose M, Chung KF, Barnes PJ. Immunomodulation by theophylline in asthma. Demonstration by withdrawal of therapy. Am J Respir Crit Care Med. 1995;151:1907–1914. doi: 10.1164/ajrccm.151.6.7767539. [DOI] [PubMed] [Google Scholar]
- Castro M, Rubin AS, Laviolette M, Fiterman J, De Andrade Lima M, Shah PL, et al. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med. 2010;181:116–124. doi: 10.1164/rccm.200903-0354OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler ME, Laviolette M, Rubin AS, Fiterman J, Lapa ESJR, Shah PL, et al. Bronchial thermoplasty: long-term safety and effectiveness in patients with severe persistent asthma. J Allergy Clin Immunol. 2013;132:1295–1302. doi: 10.1016/j.jaci.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakonarson H, Bjornsdottir US, Halapi E, Bradfield J, Zink F, Mouy M, et al. Profiling of genes expressed in peripheral blood mononuclear cells predicts glucocorticoid sensitivity in asthma patients. Proc Natl Acad Sci USA. 2005;102:14789–14794. doi: 10.1073/pnas.0409904102. [DOI] [PMC free article] [PubMed] [Google Scholar]