

Abstract
Increasing evidence suggests that androgen independent prostate cancer maintains a functional androgen receptor (AR) pathway despite the low levels of circulating androgen following androgen withdrawal, the molecular mechanisms of which are not well defined yet. To address this question, we investigated the effects of ET-1 on AR expression. Western analysis and RT-PCR revealed that in the presence of ET-1, levels of AR significantly increased in a time- and dose- dependent manner in LNCaP cells. Pre-treatments with inhibitors of Src and Phosphoinositide Kinase 3 (PI-3K) suppressed ET-1-induced AR expression. As ET-1 was reported to cause a transient increase in c-Myc mRNA levels, we examined the involvement of c-Myc in ET-1-mediated AR expression. Transient transfection of c-Myc siRNA neutralized ET-1-induced AR expression, suggesting that AR induction by ET-1 is c-Myc dependent. AR can regulate the transcription of its own gene via a mechanism in which c-Myc plays a crucial role. Therefore, we assessed if ET-1-induced-c-Myc leads to the enhancement of AR transcription. Reporter gene assays using the previously identified AR gene enhancer containing a c-Myc binding site were conducted in LNCaP cells. We found that ET-1 induced reporter gene activity from the construct containing the wild type but not mutant c-Myc binding site. Chromatin immunoprecipitation assays confirmed that ET-1 increased interaction between c-Myc and c-Myc binding sites in AR enhancer, suggesting that ET-1-induced AR transcription occurs via c-Myc-mediated AR transcription. Together, these data support the notion that ET-1, via Src/PI-3K signaling, augments c-Myc expression leading to enhanced AR expression in prostate cancer.
INTRODUCTION
The prostate gland is regulated by androgen, the action of which is mediated by the androgen receptor (AR). Increasing evidence demonstrates that the majority of androgen independent PCs expresses AR and other androgen-regulated genes such as PSA. We have observed that LNCaP cells surviving in culture in androgen-depleted medium exhibit up-regulation of AR expression [1]. Increased levels of AR protein has been implicated in enabling cells to more effectively use low levels of androgens [2, 3]. Visakorpi et al. reported AR gene amplification and over-expression in one-third of hormone-refractory, recurrent PCs [4]. To determine whether enhanced AR expression, following androgen withdrawal results from increased gene copy number, Holzbeierlein et al compared AR levels in androgen independent PC patients with androgen dependent primary PC patients by microarray analysis [5]. A significant increase in the level of the AR mRNA was detected in all androgen independent PC samples tested. Immunohistochemistry and fluorescent in situ hybridization revealed that only 8 of 29 androgen independent PC with high levels of AR had increased gene copy number, indicating that strong expression of the AR may occur by mechanisms other than gene amplification [5]. To identify these other possible mechanisms, we have examined the microenvironment after androgen withdrawal in PC. One of the major pathological characteristics in PC following androgen withdrawal is development of neuroendocrine (NE) differentiation [6]. A large number of recent studies suggest that NE differentiation, as reflected by increased tissue expression and/or blood levels of neuroendocrine secretory products such as Endothelin-1 (ET-1), correlates with poor prognosis, tumor progression, and androgen-independence [7, 8]. Our previous studies have also demonstrated that neuropeptides can regulate the AR pathway by transactivating AR and its coactivator p300 [9]. In this report, we investigated the possibility that neuropeptides contribute to enhanced AR expression in androgen-independent PC [10].
Endothelin-1 is a 21-amino acid peptide that is a cleavage product of the less potent 39-amino acid prohormone big ET-1 [11]. ET-1 protein is highly expressed by PC cell lines and PC tumor specimens, and elevated levels of plasma ET-1 are present in men with androgen-independent PC. Moreover, ET-1 significantly potentiates androgen-independent PC cell growth mediated by polypeptide growth factors such as IGF-I, IGF-II and EGF [12]. ET-1 is normally produced by prostate epithelial cells, which express ET-1 receptor subtypes A and B (ETA and ETB receptors) [13]. The mitogenic effects of ET-1 can be blocked by the addition of a selective antagonist of the ETA but not the ETB receptor, suggesting that the effects of ET-1 are mediated through the ETA receptor [12]. On activation by ET-1, ETA interacts with and activates a G-protein coupled receptor (GPCR) that triggers a parallel activation of several signal-transducing pathways.
The human AR gene contains at least four androgen response elements (ARE) and is itself regulated by AR [14]. This androgen-mediated up-regulation of AR mRNA is transcriptional and cell specific [14, 15, 16]. Deletion and mutational analysis indicated that one c-Myc binding site in the AR gene is species conserved and required for AR transcription. Aside from regulation by androgen, it has also been reported that IL-6 increases AR mRNA and protein expression, suggesting that factors other than androgen can also enhance androgen activity by up-regulating AR [17].
In the present study, we examined the effect of ET-1 on AR expression. We report that in the presence of ET-1, levels of AR protein and mRNA significantly increase and ET-1-induced AR expression is suppressed by inhibitors of Src and PI-3 K or by knock down of c-Myc. A construct containing a mutant c-Myc binding site in AR gene abrogated ET-1-induced AR transcription. Together these results suggest that ET-1 can augment AR expression via the Src/PI-3K/c-Myc pathway in PC cells.
MATERIALS AND METHODS
Chemicals and materials
Endothelin 1 (ET-1) was purchased from Sigma-Aldrich (St. Louis, MO). Src kinase inhibitor PP2 and Akt/PI-3K inhibitor, LY294002 were purchased from Calbiochem Inc. (La Jolla, CA). ET-1 receptor antagonists, BQ123 and BQ788 were purchased from Sigma-Aldrich (St. Louis, MO).
Cell culture
PC cell lines were maintained as described previously [9]. LNCaP cells (a kind gift from Dr. WD Heston, Cleveland Clinic, OH) and PC3 cells were maintained in RPMI1640 supplemented with 2 mM glutamine, 1% nonessential amino acids, 100 U/ml streptomycin and penicillin, and 10% FBS. Medium were replaced with MEM containing 5% charcoal stripped serum (CS) 24 hours before various treatments. Afterwards, cells were washed with PBS for three times and harvested. Cell pellets were frozen at −80 °C for future use.
Western blot analysis
Fifty μg of proteins were resolved by SDS-PAGE, and transferred onto nitrocellulose. The membrane was blocked, exposed to primary antibodies to AR and c-Myc (Santa Cruz biotechnology, Inc. Santa Cruz, CA) and β-actin (Sigma-Aldrich, St. Louis, MO), secondary antibody and ECL reagents (GE Healthcare, Buckinghamshire, UK) according to standard procedures provided by the supplier. Exposed and developed films were scanned for densitometric analysis using the gel scanner mode of ImageJ software v 1.33 (National Insititutes of Health, Bethesda, Maryland USA) and plotted in relative units of actin-normalized pixel counts using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego California USA).
Real-time PCR and RT-PCR
Total RNAs from cell samples were extracted using the Qiagen RNeasy Mini kit (Valencia, CA). One Og RNA served as the template for first strand synthesis using a random primer and 125 U of MultiScribe™-reverse transcriptase (TaqMan Reverse Transcription Reagents, Roche, Branchburg, NJ). For real-time PCR, the primers used were human androgen receptor (from RealTimePrimers.com, Elkins Park, PA) 5′ CCT GGC TTC CGC AAC TTA CAC 3′ (forward) and 5′GGA CTT GTG CAT GCG GTA CTC 3′; 18S rRNA, 5′CGA GCC TGG ATA CC3′ (forward) and 5′ GCC GTC CCT CTT AAT CAT GG 3′ (reverse). Oligonucleotides were synthesized commercially (Invitrogen, Carlsbad, CA). Quantitative real time PCRs were performed with the ABI 7000 system and using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). Thermocycling was carried out in a final volume of 20 μl, containing 2 μl of cDNA sample, 4 mM MgCl2, 0.1 μM primers and 10 μl SYBR green I Master mix. After a 10-min initial denaturation at 95°C, the 50 cycles run consists of a 15-s denaturation step at 95°C and an annealing and extension step at 58°C for 1 min. The mean of three repeated PCR values was used in the statistical analysis. Results were normalized to 18S to reduce variability between RNA amounts introduced into the RT-PCR reactions.
Transfection and Measurement of CAT Activities
For PC-3 cells, 10 μg of plasmid DNA were transfected with either p350CAT (c-myc wild type and mutant) plasmid DNAs with or without cotransfection of 2 μg of pFLAGAR expression vector DNA using Lipofectamine (Life Technologies, Inc.) according to the manufacturer’s recommendations. Cells grown in phenol-free medium containing CS serum for 24 h were then treated with different reagents for an additional 24 h. Cells were harvested, and cell lysates were prepared for performing CAT assays using a CAT assay system (Promega Corp., Madison, WI). Each transfection experiment was performed in duplicate or triplicate on at least three separate occasions. Results represent an average of independent experiments with data presented as relative CAT activity using means of untreated controls as standards.
Chromatin immunoprecipitation (Chip) assays
ChIP assays were performed using the CHIP-IT TM KIT according to the manufacturer’s recommendation (Active Motif, Inc., Carlsbad, CA). Briefly, LNCaP cells were plated in 150 mm dishes overnight, re-fed with phenol red-free PRMI medium containing charcoal stripped serum for 48 hr, and then treated with 50 nM ET-1. Eighteen hours later, cells were fixed with formaldehyde, which cross-links and preserves protein/DNA interactions. DNA was then sonicated into small uniform fragments and the DNA/protein complexes were immunoprecipitated using either rabbit normal IgG or a rabbit antibody against c-myc (Santa Cruz Corp., CA). Following immunoprecipitation, DNA cross-linking was reversed, and proteins removed with proteinase K treatment. After DNA purification, PCR analysis was performed to compare the enriched amounts of immunoprecipitated DNA. Negative control primers (Forward: 5′-ATGGTTGCCACTGGGGATCT-3′ and Reverse: 5′-TGCCAAAGCCTAGGGGAAGA-3′) were provided by the manufacturer to amplify a region between glyceraldehydes-3-phosphate dehydrogenase (GAPDH) gene and the chromosome condensation-related SMC-associated protein (CNAP1) gene. In CHIP assay, c-myc primers were used to amplify a 246bp DNA fragment in androgen receptor genomic DNA. This region includes partial DNA sequence in exon D (Sense primer: 5′-ACTGAGGAGACAACCCAG) and partial DNA sequence in intron between exon D and E (Antisense primer: 5′-TCTCATGCTCCCACTTCC) in AR gene. PCR products were loaded on 2.0% agarose gel for separation.
RESULTS
ET-1 induces AR RNA and protein expression
Increased levels of AR protein have been implicated in progression to androgen independent PC, enabling cells to more effectively use low levels of androgens [2, 3]. To determine whether products of neuroendocrine differentiation may affect AR expression, we measured the effects of ET-1 on the expression of AR in LNCaP cells cultured in CS medium by Western analysis. AR protein levels increased 2.4–2.9 fold compared to untreated control in dose- and time- dependent manner (Figure 1 A & B). As shown in Figure 1C & D, RT-PCR confirmed that treatment with 50 nM ET-1 for 18 hours resulted in increased AR transcripts (2.7 fold) which paralleled the increase in AR protein. These data demonstrate that ET-1 increases AR mRNA and protein in LNCaP cells under low testosterone conditions.
Figure 1. Induction of AR by ET-1 is on both protein and mRNA levels.
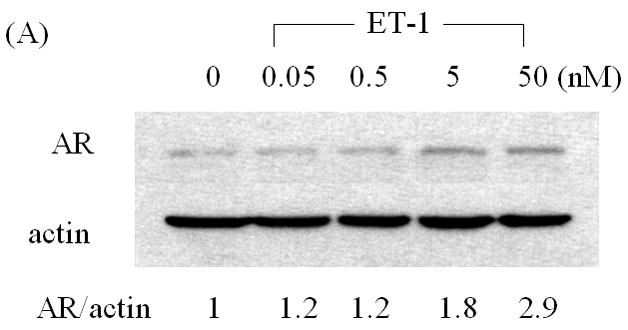
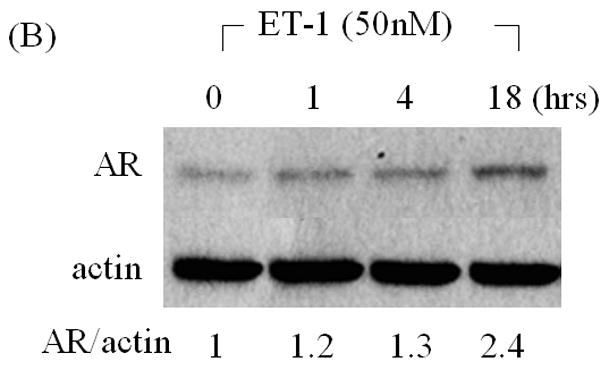
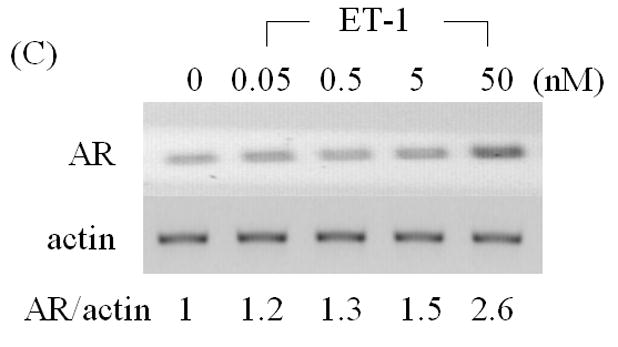
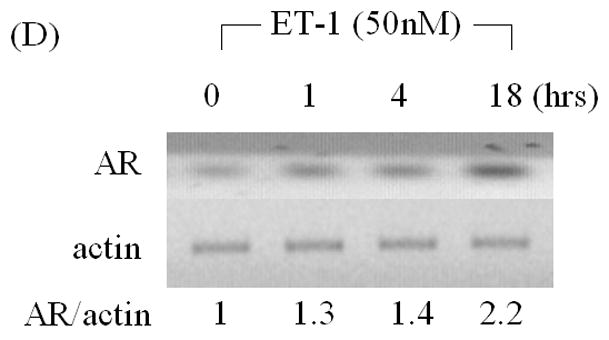
(A) LNCaP cells were cultured in MEM supplemented with 5% charcoal stripped (CS) serum. Twenty-four hours later, treatments of various concentrations of ET-1 (0, 0.05, 0.5, 5.0 and 50 nM) were carried out for 18 hours. Western blots of cell lysates were probed with AR and actin antibodies respectively. (B) Lysates from LNCaP cells cultured in MEM containing 5% CS and treated with 50 nM ET-1 at various time points (0, 1, 4, 18 hrs) were subjected to Western analysis as described in (A). (C) & (D) RNAs extracted from LNCaP cells with the same treatments as shown in (A) & (B) were analyzed by RT-PCR using primer pairs specific to AR and actin and resolved on a 1% agarose gel. All experiments were repeated at least three times with two independent preparations of cell lysates with similar results. Relative density of band was calculated by taking control as 1.
We next assessed the effects of inhibitors of ET-1 signaling on AR expression. Neutral endopeptidase (NEP) specifically catalyzes the degradation of ET-1. Treatment with recombinant NEP blocked the ET-1 mediated increase in AR protein (Figure 2A). Similarly, incubation with ET-1 receptor antagonists (ETA receptor antagonists BQ123 and ETB receptor antagonist BQ788) showed that ETA receptor antagonists BQ123, but not ETB receptor antagonist BQ788, neutralized ET-1-induced AR expression (Figures 2B). Furthermore, we conducted real time PCR with ET-1 treatment following pre-exposure to ETA receptor antagonist BQ123 or neutral endopeptidase (NEP). The results of real time PCR shown in Figure 2C, demonstrated that ET-1 resulted in a 2 fold increased of the levels of androgen receptor mRNA which was consistent with that of RT-PCR and Western Blot analyses (Figure 2A & B). Suppression of ETA receptor or degradation of ET-1 blocked ET-1-induced up-regulation of AR mRNA. These data demonstrate that ET-1-induced AR expression is specific to ET-1 treatment and its cognate ETA receptor.
Figure 2. Induction of AR by ET-1 is specific to GPCR pathway.
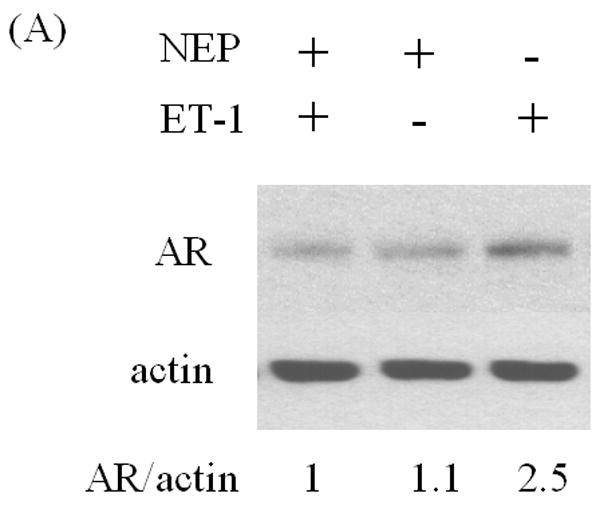
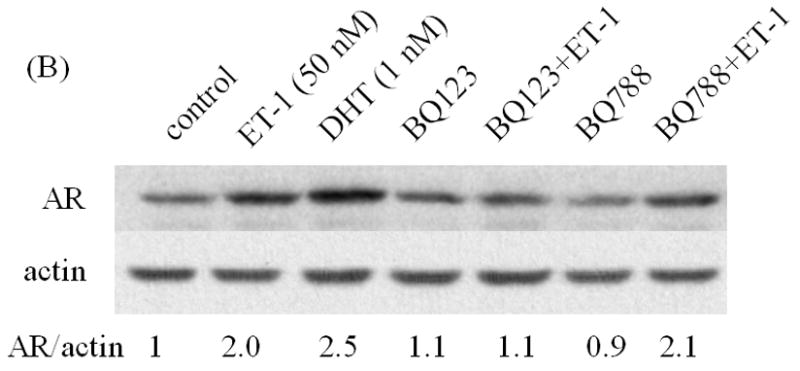
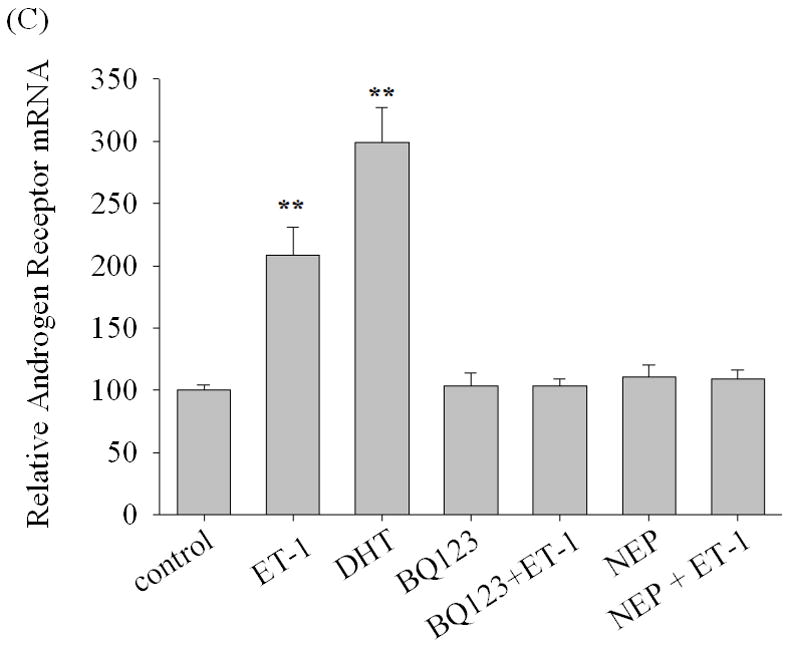
LNCaP cells cultured in medium containing 5% CS were pre-treated with recombinant neutral endopeptidase (NEP) (50 ug/ml) which degrades ET-1 (A), ET-1 receptor antagonists BQ123 (1 uM) and BQ788 (1 uM) (B), and then treated with 50 nM ET-1 for 18 hrs. Western blots based on these cell lysates were conducted and probed with AR and actin antibodies. (C) Real time PCR analysis of the up-regulation of androgen receptor mRNAs by ET-1. Message RNAs prepared from LNCaP cells cultured in medium containing 5% CS and treated with 50 nM of ET-1 for 18 hours following pre-exposure to BQ123, an ETA receptor antagonist, or neutral endopeptidase (NEP) were subjected to real time PCR analysis with a pair of primers specific to AR and normalized with 18s rRNA. The treatment of 10 nM DHT was used as positive control. Relative levels of androgen receptor mRNAs were calculated by taking control (no treatment) as 100. Experiments were repeated at least twice with similar results. All data represent the average of one experiment and are presented as mean ± SEM, ** p < 0.01 represent statistical significance compared to the value of control. All experiments were repeated at least two times with three independent preparations of cell lysates with similar results.
C-Myc siRNA suppresses ET-1-mediated AR expression
ET-1 can cause a transient increase in c-Myc mRNA levels [19]. C-Myc has been reported to mediate AR transcription, suggesting that ET-1-included AR may be mediated via c-Myc. We found that c-Myc protein increases in the presence of 50 nM ET-1 in LNCaP cells (Figure 3A). Transfection of c-Myc siRNA, which decreased c-Myc protein (data not shown), eliminated induction of AR by ET-1 (Figure 3B). These data suggest that ET-1-induced AR expression is mediated via c-Myc.
Figure 3. Induction of AR by ET-1 is mediated by the c-Myc pathway.
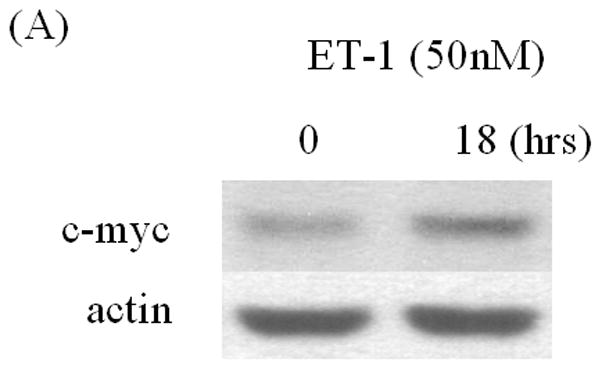
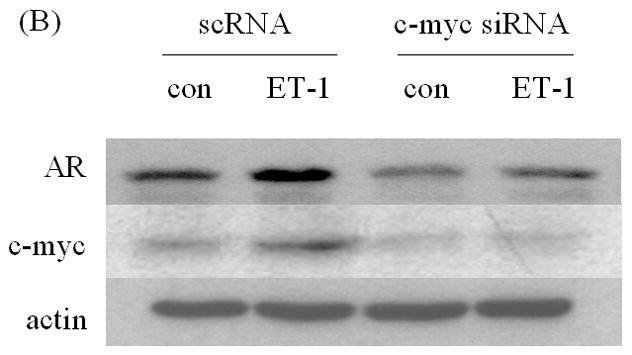
(A) LNCaP cells cultured in medium containing 5% CS were treated with 50 nM ET-1 for 18 hrs. Cell lysates were analyzed by Western blotting using c-Myc and actin antibodies respectively. (B) LNCaP cells transfected with c-Myc siRNA by lipofectamine method were lysed and subjected to Western analysis using AR, c-Myc and actin antibodies. All experiments were repeated at least two times with three independent preparations of cell lysates with similar results.
Src and PI-3K are responsible for ET-1-induced AR expression
Studies from our lab and others have shown that ET-1 activates Src kinase in PC cells, and that Src kinase causes significant transcriptional activation of Myc [20, 21]. Pre-incubation with the Src inhibitor PP2 eliminated ET-1-induced AR expression (Figure 4A) [20]. Dominguez-Caceres et al. (2004) reported that Akt activation was required for Src initiated activation of c-Myc expression [22]. Therefore, we next tested the effect of PI3-K inhibitor LY294002 (LY) in ET-1-mediated AR expression and showed that ET-1-induced AR expression was suppressed by Akt inhibitors (Figure 4 B). As expected, PP2 and LY also inhibited ET-1-induced c-Myc expression (Figure 4C). Together, these data suggest that ET-1-induced AR expression is mediated by the Src/PI-3K/c-Myc signaling pathway.
Figure 4. The effects of inhibitors of Src and PI-3 K on ET-1-induced expression of AR and c-Myc in LNCaP cells.
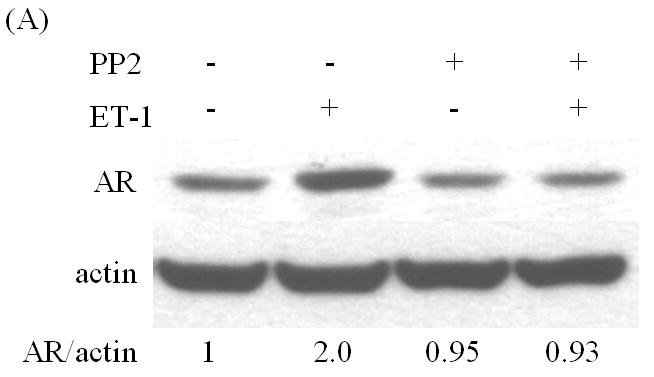
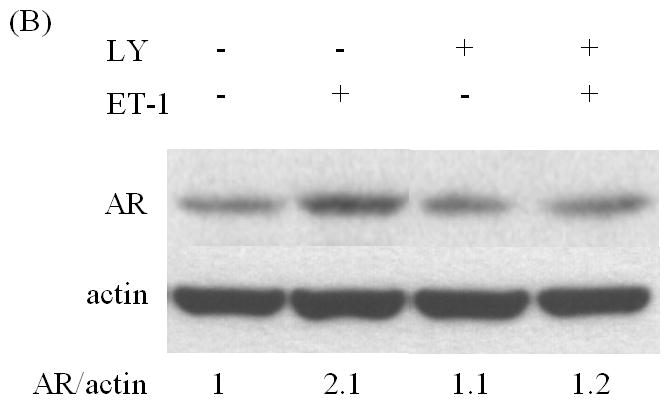
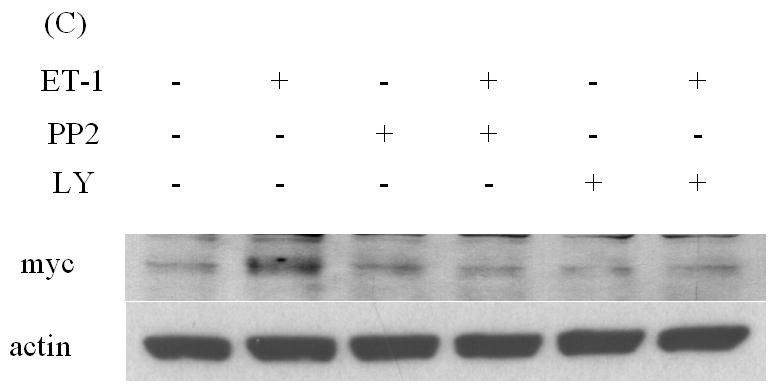
LNCaP cells cultured in medium containing 5% CS serum were pre-treated with various kinase inhibitors: PP2 (inhibitor of Src kinase) (100 nM) (A) and LY (inhibitor of PI-3 K) (100 nM) (B) for 30 minutes. Following treatments with 50 nM ET-1 for 18 hours, cells were lysed to conduct Western analysis using AR and actin antibodies. (C) LNCaP cells pre-treated with LY and PP2 were treated with 50 nM ET-1 for 18 hrs. Cell lysates were analyzed by Western blotting and probed using c-Myc and actin antibodies. All experiments were repeated at least two times with three independent preparations of cell lysates with similar results.
ET-1-induced AR transcription is blocked by mutated c-Myc
To more thoroughly elucidate the mechanisms by which ET-1-induced c-Myc expression leading to a transcriptional increase in AR, we examined the effect of ET-1 on AR transcription. Dai JL, et al., (1996) identified a 350-bp enhancer containing two AREs (ARE-1 and ARE-2 located on exons D and E, respectively) that confer androgen-specific regulation of AR messenger RNA [14, 15, 16]. A Myc consensus site is located within the 350-bp fragment and is required for androgen regulation. Mutation at this c-Myc site has previously been shown to decrease physiologic androgen induction (Figure 5A). We therefore compared the effect of ET-1 on AR transcription between plasmids p350wtMycCAT and p350mMycCAT [15]. Following co-transfection of p350wtMycCAT or p350mMycCAT with the AR expression vector pFLAG-AR into PC-3 cells. Cells were cultured in the presence and absence of 50 nM ET-1 and CAT activities measured. The results demonstrated that in the absence of ET-1, CAT activities of both vectors were similarly low. However, in the presence of ET-1, CAT activities were increased to 1.5 fold (p<0.001, with treatment of 50 nM ET-1 vs. without treatment) in cells containing p350wtMycCAT, whereas no enhanced CAT activities were detected in cells containing p350mMycCAT (Figure 5B). These data suggest that treatment with ET-1 enhances c-Myc expression that sequentially interacts with the c-Myc site in the AR gene leading to an increase in AR expression. The results from CHIP assays further verified that after pull-down of DNA-protein complexes with c-Myc antibody and amplification of chromatin DNAs with c-Myc primers, the PCR products of LNCaP cells with ET-1 treatment were enriched in comparison with that of PBS treatment, or the pull-down with control IgG (lanes 7 vs. 6 and 4, Figure 6A). In contrast, when the control primers and DNA templates (see Methods) were used, no differences could be detected between treatments of ET-1 and PBS (Figure 6B). The data suggested that ET-1 treatment augmented c-Myc expression and consequently led to increased interaction between c-Myc and c-Myc binding sites in AR enhancer. In order to examine whether the enhanced expression of AR leads to increased expression of AR regulatory genes, we conduced Western blot analysis to determine the expression of prostate specific antigen (PSA) in the presence of ET-1. As shown in Figure 6C, the treatment of 50 nM ET-1 resulted in an increased of PSA expression, which was eliminated by pre-treatment of BQ123 or NEP. The data suggest that ET-1 increases the expression of AR target gene.
Figure 5. CAT activities of a construct containing c-Myc binding site in AR gene.
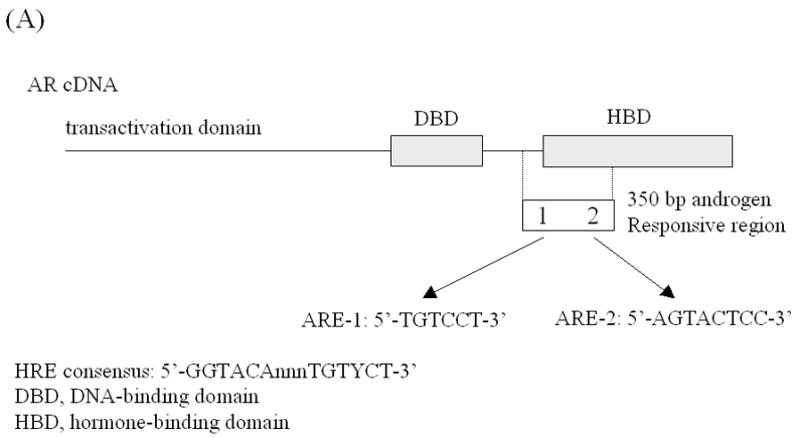

(A) Schematic illustrating the structure of the plasmids p350wtMycCAT and p350mMycCAT. The plasmid p350wtMycCAT is a vector containing the 350 bp regulatory fragment of AR gene inserted upstream of tk the promoter and a CAT reporter. This 350 bp region contains two AREs and one c-Myc binding site. Plasmid p350mMycCAT possesses the identical construct except that the c-Myc binding site is mutated [15, 16]. (B) P350wtMycCAT or p350mMycCAT were co-transfected with AR expression vector (pFLAG-AR) into PC-3 cells by lipofectamine method. Twenty-four hours after transfection, treatments with or without 50 nM ET-1 were carried out. DHT (1 nM) was used as a positive control. Cells co-transfected with AR and p350wtMycCAT or p350mMycCAT with PBS treatments were used as controls (lane Control). Cell lysates were harvested and CAT activities measured. Relative CAT activities were calculated by values of various treatments vs. Control (without treatment). P values were calculated.
Figure 6.
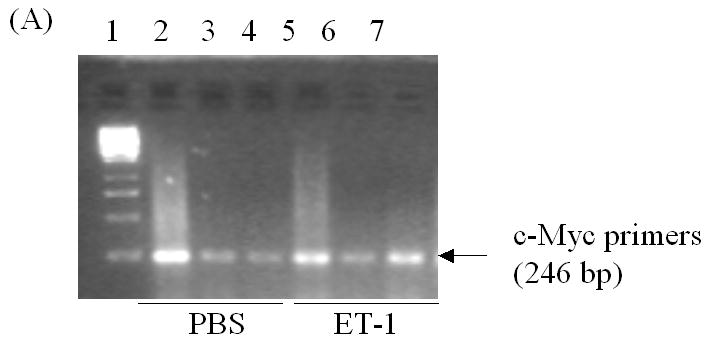
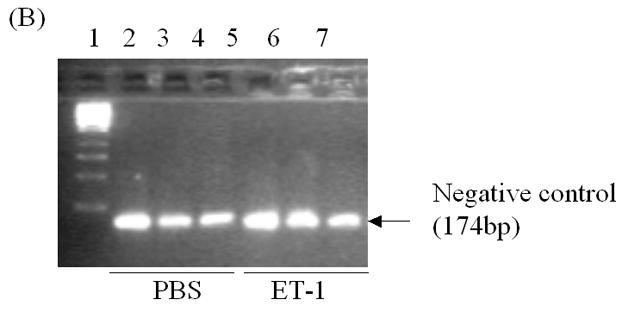
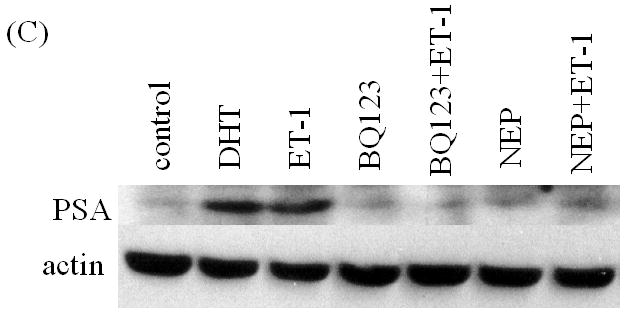
In vivo interaction of c-Myc with the c-Myc site in AR gene and ET-1-induced expression of AR regulatory genes. (A) & (B) Chromatin immunoprecipitation (CHIP) assays showing in vivo interaction of c-Myc and c-myc binding region. Complexes of DNAs and transcription factors from LNCaP cells treated without and with ET-1 were subjected to immunoprecipitated with antibodies of c-Myc and control IgG. The purified DNAs in the supernatants (input) and imunnoprecipitates were amplified by PCR with primers specific to c-Myc (A) and control primers (B) (see Methods). Lane 1 is 1 kb DNA marker (Fermentas life science, MD). Lanes 2, 3 and 4 are the CHIP products from input, rabbit IgG control and c-Myc antibody with PBS treatment. Lanes 5, 6 and 7 are the CHIP products from input, rabbit IgG control and c-Myc antibody with 50 nM ET-1 treatment for 18 hrs. The data represent one experiment performed on three separate occasions with similar results. (C) Cell lysates of LNCaP cells treated with 50 nM ET-1 or pre-exposure to NEP and ET-1 antagonists BQ123 or BQ 788 were subjected to Western analysis and probed with antibodies of PSA and actin.
DISCUSSION
Increasing evidence demonstrates that neuroendocrine (NE) differentiation in prostate cancer (PC) contributes to androgen independent progression. Recent studies have focused more closely on AR regulation, expression, and function to explain the development of androgen-independent PC [2]. We have previously studied the involvement of neuropeptides in PC progression and shown that neuropeptides can induce activation of Src, ligand-independent phosphorylation of the IGF-1 receptor and Akt, and rapid PKCδ degradation [20, 23]. Furthermore, we have also shown that neuropeptides lead to aberrant regulation of glucocorticoid receptor splicing, and attenuate apoptosis via the Akt/survivin pathway [24, 25]. In the present study, we studied neuropeptide regulation of the AR in PC cells. We demonstrate that: 1) ET-1 increases AR protein and mRNA levels; 2) Inhibitors of Src and PI-3K suppresses ET-1 induced AR expression; and 3) ET-1-induced AR expression is suppressed by c-Myc siRNA (Figure 7). These data are consistent with c-Myc acting via the intragene enhancer of AR. Our findings are the first to demonstrate neuropeptide-mediated AR up-regulation and provide a possible mechanism by which AR expression increases in androgen independent PCs after androgen withdrawal.
Figure 7. Schematic illustrating the signaling pathways mediating of ET-1 effect on AR expression.
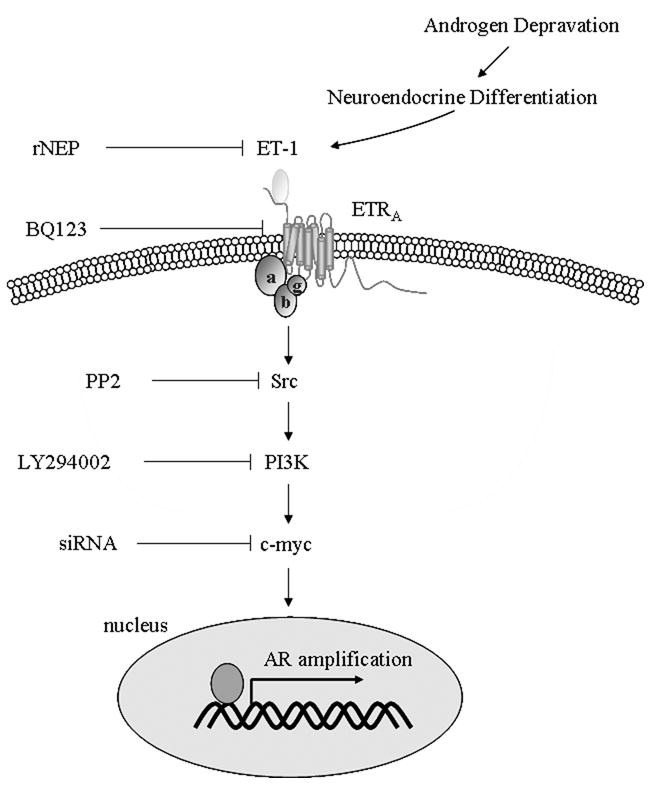
After androgen withdrawal, ET-1 enhanced c-Myc expression, via activation of Src and PI-3 K signaling, leading to increased level of AR transcription.
Our data indicated that ET-1 activates c-Myc via Src/PI-3K signaling. The neuropeptides bombesin and ET-1 stimulate cSrc kinase activity and induce rapid degradation of PKCδ protein in PC cells [23]. Prolactin activates c-Myc via Src and PI-3K dependent Akt signaling in W53 cells [22], suggesting PI-3K is a downstream event of Src activation. ET-1 has also been shown to modulate insulin signaling through PI3-K pathway in vascular smooth muscle cells and induces phosphorylation of IGF-IRβ and Akt, independent of IGF-I in TSU-Pr1, DU145, and PC-3 cells [19, 25, 26]. Therefore, our observation is consistent with these earlier studies. However, the downstream events of PI-3K leading to activation of c-Myc are not clear, although FKHRL1-GSK dependent events may bridge PI-3K signaling to c-Myc activation [22]. The previous studies demonstrated that c-Jun and c-Myc mRNAs reached maximal levels 2 and 4 hours, respectively, after the addition of ET-1 [27]. In our study, induction of AR expression required 18 hours (Figure 1). Therefore, c-Myc induction precedes AR induction following ET-1 treatment.
The action of ET-1 on the prostate may be paracrine. Two receptors have been identified for members of the endothelin family: endothelin receptor A (ETA) and endothelin receptor B (ETB). ETA has a higher affinity to ET-1 and ET-2, and less for ET-3. ETB binds all three endothelin ligands identically, and may play an important role in ligand clearance. ETA and ETB receptors are both found in the normal prostate, whereas no ETB binding sites could be detected in human prostate cancer cell lines. Unlike the loss of ETB in prostate cancer, increased ETA expression has been associated with progression of prostate cancer [12, 28]. Our data that BQ123 (an antagonist of ETA receptor), but not BQ788 (an antagonist of ETB) suppresses ET-1’s effect demonstrate that it is ETA that mediates the effect of ET-1 to trigger downstream cascade of events by which nuclear transcription of several proto-oncogenes, including c-Myc and AR is induced.
Grad et al (1999) previously identified four functional AREs and a Myc consensus site in the AR coding region [15]. It is of interest that these AREs and Myc site appear to be well conserved (0 or 1 amino acid mismatches) among a number of species from rodents and monkeys to birds and frogs. Conservation of these sequences is consistent with a role for these regulatory elements in transcription of AR mRNA [15]. Our results showed that ET-1 induced c-Myc expression and consequently up-regulate AR transcription. The CAT activities resulting from our positive control (DHT) in p350wtMycCAT (2 fold increase in the presence of 10 nM DHT for 18 hours, Figure 5B) are lower than the previous report (7–8 fold changes in the presence of 50 nM of R1881 for 40 hours). This difference may be due to our usage of different type and dose of androgen, as well as shorter incubation times from the previous studies [15]. The induced fold of CAT activities by ET-1 is lower than the enhanced fold of AR mRNA for RT-PCR analysis. The explanation could be that: 1) the enhancer region (350 bp) selected for our study may be too short to include all cis-elements that are required for ET-1 effect; or 2) the presence of other mechanisms by which ET-1 regulates AR mRNA stability.
Lin HK & Chang C et al, reported that “PI3-K/Akt pathway can suppress AR activity in LNCaP cells with low passage numbers. In contrast, it can also enhance AR activity in LNCaP cells with high passage numbers”. “In LNCaP cells with low passage numbers, IGF-1 can activate PI-3K/Akt pathway that results in phosphorylation at Ser210 and Ser790, consequently changes AR protein stability” [29]. In our present study, we used LNCaP cells with high passage number. In addition, these LNCaP cells were cultured in CS-containing medium, instead of FCS-containing medium. The changes in the conditions may alter phosphorylation status of AR and also explained no suppression of AR activity by ET-1-mediated activation of PI-3K/Akt pathway was observed in our system. Our previous studies demonstrated that neuropeptides affect androgen receptor (AR) mediated transcription and the activation of histone acetyltransferase activity of AR coactivator p300 in PC cells [9, 30]. ET-1 and bombesin share several similar signaling, implying that ET-1 may also possess the function like bombesin to transactivate AR and activation of AR cofactor p300, aside from affecting AR expression. Several studies reported that Src kinase could phosphorylate tyrosine sites of AR and promote LNCaP cell proliferation in androgen depletion condition [31]. ET-1 can activate Src kinase, and may also induce tyrosine phosphorylation of AR, implying that ET-1 might also activate AR activity. Therefore, it is worthy to further explore ET-1-induced activation of Src kinase and PI-3K/Akt and its influence on AR activity. Our future investigations will focus on studies to explore ET-1 effect on transactivation of AR and its cofactors and decipher the intact profile of ET-1 effect on the AR signaling pathway.
Acknowledgments
This work was supported by NIH grants RO1 DK060908-02, RO1 CA80240, R01 HD33000, the Robert H. McCooey Memorial Cancer Research Fund, Ronald and Susan Lynch Professorship in Urologic Oncology and Brady Urology Foundation of the Department of Urology (to Lee, J.). We acknowledge Drs. Oscar Goodman and Katsuyuki Iida for helpful discussion and technical assistance of Ms. Samrina Kahlon.
References
- 1.Shen R, Sumitomo M, Dai J, Harris A, Kaminetzky D, Gao M, Burnstein K, Nanus DM. Androgen-induced growth inhibition of androgen receptor expressing androgen-independent prostate cancer cells is mediated by increased levels of neutral endopeptidase. Endocrinol. 2000;141:1699–1704. doi: 10.1210/endo.141.5.7463. [DOI] [PubMed] [Google Scholar]
- 2.Culig Z, Hobisch A, Bartsch G, Klocker H. Expression and function of androgen receptor in carcinoma of the prostate. Microsc Res Tech. 2000;51:447–455. doi: 10.1002/1097-0029(20001201)51:5<447::AID-JEMT7>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 3.Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–3555. [PubMed] [Google Scholar]
- 4.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 5.Holzbeierlein J, Lai P, LaTulippe E, Smith A, Satagopan J, Zhang L, Ryan C, Smith S, Scher H, Scardino P, Reuter V, Gerald WL. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–27. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Sant’Agnese PA. Neuroendocrine differentiation in prostatic carcinoma: an update. Prostate Suppl. 1998;8:74–79. [PubMed] [Google Scholar]
- 7.Berruti A, Dogliotti L, Mosca A, Bellina M, Mari M, Torta M, Tarabuzzi R, Bollito E, Fontana D, Angeli A. Circulating neuroendocrine markers in patients with prostate carcinoma. Cancer. 2000;88:2590–2597. doi: 10.1002/1097-0142(20000601)88:11<2590::aid-cncr23>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 8.Wu JT, Erickson AJ, Tsao KC, Wu TL, Sun CF. Elevated serum chromogranin A is detectable in patients with carcinomas at advanced disease stages. Ann Clin Lab Sci. 2000;30:175–178. [PubMed] [Google Scholar]
- 9.Gong J, Zhu J, Goodman OB, Jr, Pestell RG, Schlegel PN, Nanus DM, Shen R. Activation of p300 histone acetyltransferase activity and acetylation of the androgen receptor by bombesin in prostate cancer cells. Oncogene. 2006;25:2011–2021. doi: 10.1038/sj.onc.1209231. [DOI] [PubMed] [Google Scholar]
- 10.Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma. Prostate. 1999;39:135–148. doi: 10.1002/(sici)1097-0045(19990501)39:2<135::aid-pros9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 11.Levin ER. Endothelins. N Engl J Med. 1995;333:356–363. doi: 10.1056/NEJM199508103330607. [DOI] [PubMed] [Google Scholar]
- 12.Nelson JB. Endothelin receptor antagonists. World J Urol. 2005;23:19–27. doi: 10.1007/s00345-004-0478-9. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi S, Tang R, Wang B, Opgenorth T, Stein E, Shapiro E, Lepor H. Localization of endothelin receptors in the human prostate. J Urol. 1994;151:763–766. doi: 10.1016/s0022-5347(17)35083-8. [DOI] [PubMed] [Google Scholar]
- 14.Grad JM, Lyons LS, Robins DM, Burnstein KL. The androgen receptor (AR) amino-terminus imposes androgen-specific regulation of AR gene expression via an exonic enhancer. Endocrinology. 2001;142:1107–1116. doi: 10.1210/endo.142.3.8049. [DOI] [PubMed] [Google Scholar]
- 15.Grad JM, Dai JL, Wu S, Burnstein KL. Multiple androgen response elements and a Myc consensus site in the androgen receptor (AR) coding region are involved in androgen-mediated up-regulation of AR messenger RNA. Mol Endocrinol. 1999;13:1896–1911. doi: 10.1210/mend.13.11.0369. [DOI] [PubMed] [Google Scholar]
- 16.Dai JL, Burnstein KL. Two androgen response elements in the androgen receptor coding region are required for cell-specific up-regulation of receptor messenger RNA. Mol Endocrinol. 1996;10:1582–1594. doi: 10.1210/mend.10.12.8961268. [DOI] [PubMed] [Google Scholar]
- 17.Smith PC, Hobisch A, Lin DL, Culig Z, Keller ET. Interleukin-6 and prostate cancer progression. Cytokine Growth Factor Rev. 2001;12:33–40. doi: 10.1016/s1359-6101(00)00021-6. [DOI] [PubMed] [Google Scholar]
- 18.Druckenthaner M, Schwarzer C, Ensinger C, Gabriel M, Prommegger R, Riccabona G, Decristoforo C. Evidence for Somatostatin receptor 2 in thyroid tissue. Regul Pept. 2007;138:32–39. doi: 10.1016/j.regpep.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Bravo R, Macdonald-Bravo H, Muller R, Hubsch D, Almendral JM. Bombesin induces c-fos and c-myc expression in quiescent Swiss 3T3 cells. Comparative study with other mitogens. Exp Cell Res. 1987;170:103–115. doi: 10.1016/0014-4827(87)90120-0. [DOI] [PubMed] [Google Scholar]
- 20.Sumitomo M, Milowsky M, Shen R, Navarro D, Dai J, Asano T, Hayakawa M, Nanus DM. Neutral endopeptidase inhibits neuropeptide-mediated transactivation of the insulin-like growth factor receptor-Akt cell survival pathway. Cancer Res. 2001;61:3294–3298. [PubMed] [Google Scholar]
- 21.Courtneidge SA. Role of SRC in signal transduction pathways. Biochem Soc Trans. 2002;30:11–17. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 22.Dominguez-Caceres MA, Garcia-Martinez JM, Calcabrini A, Gonzalez L, Porque PG, Leon J, Martin-Perez J. Prolactin induces c-Myc expression and cell survival through activation of Src/Akt pathway in lymphoid cells. Oncogene. 2004;23:7378–7390. doi: 10.1038/sj.onc.1208002. [DOI] [PubMed] [Google Scholar]
- 23.Sumitomo MR, Shen Goldberg JS, Dai J, Navarro D, Nanus DM. Neutral endopeptidase promotes phorbol ester-induced apoptosis in prostate cancer cells by inhibiting neuropeptide-induced protein kinase Cδ degradation. Cancer Res. 2000;60:6590–6596. [PubMed] [Google Scholar]
- 24.Zhu J, Gong JY, Goodman OB, Jr, Cartegni L, Nanus DM, Shen R. Bombesin attenuates pre-mRNA splicing of glucocorticoid receptor by regulating the expression of serine-arginine protein p30c (SRp30c) in prostate cancer cells. Biochim Biophys Acta. 2007;1773:1087–1094. doi: 10.1016/j.bbamcr.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong J, Lee J, Akio H, Schlegel PN, Shen R. Attenuation of apoptosis by chromogranin A-induced Akt and survivin pathways in prostate cancer cells. Endocrinology. 2007;148:4489–4499. doi: 10.1210/en.2006-1748. [DOI] [PubMed] [Google Scholar]
- 26.Jiang ZY, Zhou QL, Chatterjee A, Feener EP, Myers MG, Jr, White MF, King GL. Endothelin-1 modulates insulin signaling through phosphatidylinositol 3-kinase pathway in vascular smooth muscle cells. Diabetes. 1999;48:1120–1130. doi: 10.2337/diabetes.48.5.1120. [DOI] [PubMed] [Google Scholar]
- 27.Ergul A, Glassberg MK, Majercik MH, Puett D. Endothelin-1 promotes steroidogenesis and stimulates protooncogene expression in transformed murine Leydig cells. Endocrinol. 1993;132:598–603. doi: 10.1210/endo.132.2.8425480. [DOI] [PubMed] [Google Scholar]
- 28.Motte S, McEntee K, Naeije R. Endothelin receptor antagonists. Pharmacol Ther. 2005;110:386–414. doi: 10.1016/j.pharmthera.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 29.Lin H, Hu Y, Yang L, Altuwaijri S, Chen Y, Kang H, Chang C. Suppression Versus Induction of Androgen Receptor Functions by the Phosphatidylinositol 3-Kinase/Akt Pathway in Prostate Cancer LNCaP Cells with Different Passage Numbers. J Biol Chem. 2003;278:50902. doi: 10.1074/jbc.M300676200. [DOI] [PubMed] [Google Scholar]
- 30.Dai J, Shen R, Sumitomo M, Stahl R, Navarro D, Marvin C, GershengornNanus DM. Synergistic activation of the androgen receptor by bombesin and low-dose androgen. Clin Cancer Res. 2002;8:2399–2405. [PubMed] [Google Scholar]
- 31.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, Njar VCO, Brodie AMH, Yu L, Veenstra TD, Chen H, Qiu Y. Regulation of Androgen Receptor Activity by Tyrosine Phosphorylation. Cancer Cell. 2006;10:309–319. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]