

Abstract
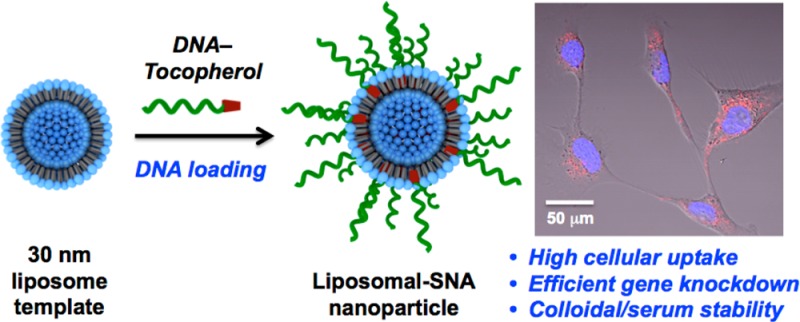
A novel class of metal-free spherical nucleic acid nanostructures was synthesized from readily available starting components. These particles consist of 30 nm liposomal cores, composed of an FDA-approved 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) lipid monomer. The surface of the liposomes was functionalized with DNA strands modified with a tocopherol tail that intercalates into the phospholipid layer of the liposomal core via hydrophobic interactions. The spherical nucleic acid architecture not only stabilizes these constructs but also facilitates cellular internalization and gene regulation in SKOV-3 cells.
In 1996, we introduced the concept of spherical nucleic acid (SNA) nanoparticle conjugates, structures typically synthesized from inorganic nanoparticle templates and shells of highly oriented nucleic acid ligands immobilized on the surface of such particles.1 Since then, SNAs have been prepared by many research groups in a variety of different forms.2 Core compositions including gold, silica,3 iron oxide,4 and Ag,5 with shell compositions consisting of DNA, RNA, LNA,6 and PNA,7 have all been prepared and explored. Hollow SNA structures consisting of cross-linked oligonucleotides8 along with DNA-block copolymer micelles9 have been synthesized. Although there is now a tremendous structural and compositional diversity among the known SNAs, they all share some common properties and features. Their polyvalent architectures allow them to cooperatively bind oligonucleotides and form duplex structures that exhibit very narrow melting transitions.10 These properties have been exploited in the development of high-sensitivity, high-selectivity, and massively multiplexed genomic detection systems.11 While linear nucleic acids do not enter cells well without polymer, peptide, or viral transfection agents, the three-dimensional SNA structure is recognized by Class A scavenger receptors12 and is rapidly taken up into over 60 different cell types without the need for an ancillary transfection agent.13 This property has made such structures important elements in strategies for both intracellular detection14 and gene regulation via antisense or siRNA pathways.15 Consequently, many groups are now exploring the potential of such structures for therapeutic applications. As with any new chemical construct, the barrier to therapeutic use is high, especially when the structures are made from materials that have known problems with clearance or unknown biodistribution characteristics. Ideally, one would like an SNA structure that is made from readily available starting materials, can be synthesized at scale, and consists of components that have been a part of FDA-approved pharmaceuticals.2a,16 Herein, we report the first strategy for making such structures, which consist of 30 nm liposomal cores stabilized with a dense shell of oligonucleotides with a hydrophobic tail that can intercalate between the phospholipids that define the liposomal structure (Scheme 1). As with conventional SNAs, these novel liposomal structures rapidly enter multiple cell lines and can be used to effectively knock down gene expression via antisense pathways. Unlike conventional liposome structures, the oligonucleotide cargo in these novel SNAs is deliberately arranged on the surface of the liposomal entity, which is stabilized in the sub-100 nm size range. Larger DNA-functionalized liposomal entities,17 along with ≥200 nm diameter DNA lipid vesicles (with DNA strands facing outward and inward),18 have been explored in the context of programmable materials assembly.
Scheme 1. Assembly of Liposomal SNAs from DOPC SUVs and Tocopherol-Modified DNA.
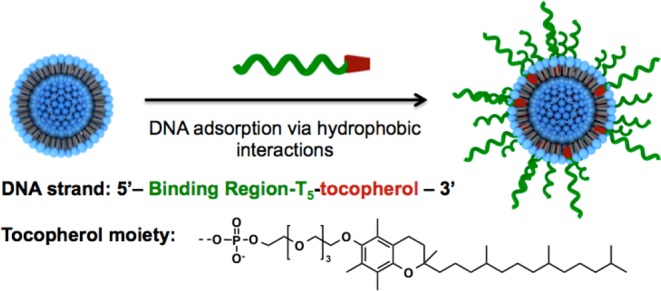
A typical liposomal SNA was synthesized in two steps. The first step involves the preparation of 30 nm diameter unilamellar vesicles from lipid monomers. This size particle is ideal from the standpoint of SNA transfection19 and is in the appropriate range for maximizing higher blood circulation and preventing clearance through the kidneys.16a Unfortunately, liposomes in this size regime are often unstable and fuse to form larger structures. Therefore, a goal of this work was to determine a way of synthesizing such structures and avoiding such fusion pathways. To prepare small unilamellar vesicles (SUVs), we selected commercially available 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) monomer. In a typical experiment, a suspension of DOPC monomers in 20 mM HEPES buffer saline (HBS) was sonicated to produce on average 30 nm SUV particles that were then isolated by centrifugation (100000g). Further extrusion of this material through a polycarbonate membrane with 30 nm pores gave particles with a polydispersity index (PDI) of 0.11 in 70% overall yield (Figure 1A). The average diameter of the particles was also confirmed by TEM analysis using negative staining (Figure S2 in the Supporting Information).
Figure 1.
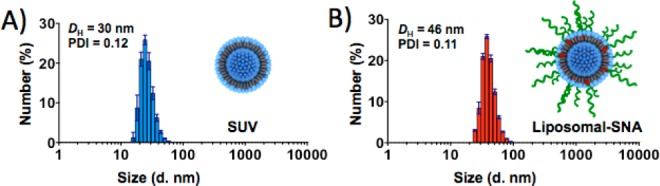
(A) DLS of SUVs after purification. (B) DLS of liposomal SNAs after purification.
The second step of the synthesis involves surface functionalization of the liposome with a nucleic acid derivative possessing a hydrophobic tocopherol moiety, which effectively inserts into the lipid bilayer defining the SUV. Although a variety of hydrophobic head groups might be suitable,17,20 α-tocopherol (a form of vitamin E) was chosen because of its biocompatibility and low cost. The liposomal SNAs were synthesized by incubating a suspension of SUVs (1.3 mM by lipid) with the nucleic acid–tocopherol conjugates (16 μM) using a lipid-to-nucleic acid ratio of 80:1 for 12 h at room temperature. The unbound tocopherol–nucleic acid was then removed from the sample by size-exclusion chromatography on a sepharose column. After modification with T30-DNA, a significant drop in the zeta potential from −1 to −23 mV occurred, indicating liposome surface functionalization with the negatively charged nucleic acid. In addition, DLS analysis of the final nanoparticle samples showed an increase in particle size from 30 to 46 nm, which was consistent with the loading of the 30 bases long DNA strand (Figure 1B). The synthesized liposomal SNAs had on average 70 DNA strands per particle (page S5 in the Supporting Information). This surface coverage is sufficient to exhibit many of the cooperative properties of such structures (vide infra).
These novel liposomal SNA structures have several interesting properties. First, they are remarkably stable compared to the native 30 nm liposome constructs from which they derive. For example, if the SUVs without an oligonucleotide surface layer are stored for 4 days at 37 °C, they fuse and form larger polydisperse structures (on average >100 nm structures with some micrometer-sized entities, Figure 2A). In contrast, the liposomal SNAs show no evidence of particle degradation or fusion over the same time period under nearly identical conditions (Figure 2B). This increase in stability for the liposomal SNA system is likely a result of the repulsive forces between the negatively charged nucleic acid strands that comprise the liposomal SNA’s surface, which both stabilize the individual particles and inhibit particle–particle fusion interactions.21 Moreover, the negatively charged DNA corona on the liposomal SNA serves as a protecting layer for the liposomal core and inhibits its degradation in the presence of serum proteins.22 For example, the serum stability of the liposomal SNAs was investigated by measuring the release of a sulforhodamine dye physically incorporated within the core of a liposomal SNA at a self-quenching concentration of 20 mM (i.e., the dye concentration in core = 20 mM). In this experiment, rupture of the liposomal core results in a release of the sulforhodamine dye from the interior of the particle and a subsequent elimination of self-quenching, giving rise to an increase in fluorescence (Figure 2C).23 In a typical experiment, rhodamine-containing liposomal SNAs were incubated in 10% fetal bovine serum (FBS) at 37 °C, and the fluorescence spectra were recorded continuously for 3 h. A similar stability study was performed for non-functionalized particles. Similar to our thermal stability studies, DNA-functionalized particles remained stable in serum for the duration of the experiment (Figure 2D).23 No release of the dye was observed during 3 h of incubation. In contrast, incubation of the bare DOPC liposomes led to a significant release of the rhodamine fluorophore (Figure 2D), indicating rapid decomposition of the liposomal structure in serum.
Figure 2.
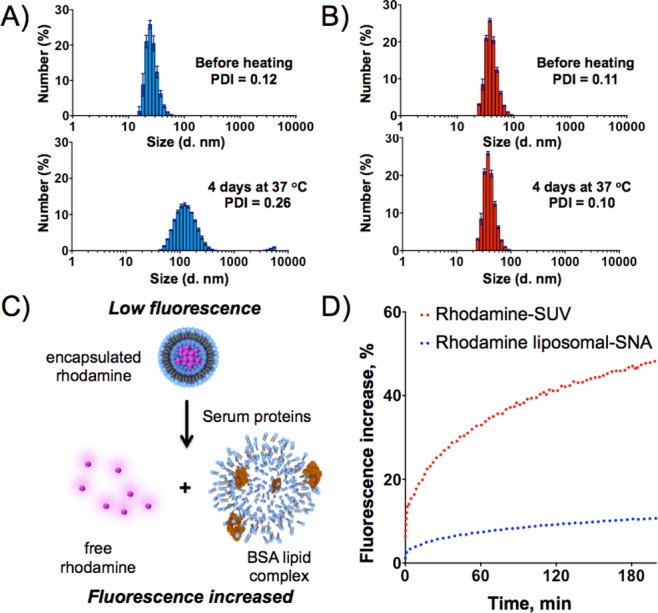
Stability studies of SUV and liposomal SNAs. Change in average diameter of SUVs (A) and liposomal SNAs (B) before (top) and after (bottom) heating in buffer, as measured by DLS. (C) Schematic representation of the decomposition of rhodamine-encapsulated liposome in the presence of bovine serum albumin, a major component of FBS. (D) Degradation of SUVs (red traces) and liposomal SNAs (blue traces) in the presence of 10% FBS, as measured by increases in the fluorescence intensity of rhodamine.
A second property of liposomal SNAs is their ability to cooperatively bind complementary nucleic acids. To explore the binding and subsequent melting properties of the liposomal SNA constructs, we synthesized two sets of liposomal SNA nanoparticles, each made with different DNA sequences (particle A and particle B, Figure 3A). A DNA linker sequence that is complementary to the oligonucleotide sequences of both liposomal SNAs was used to facilitate polymerization through hybridization. Upon addition of the linker sequence to an equimolar mixture of the two liposomal SNA particles, aggregation occurred as evidenced by (1) increased extinction at 260 nm due to light scattering and (2) eventual formation of a flaky white precipitate.17a These aggregates were re-suspended in 20 mM HBS (150 mM NaCl), and a melting analysis was performed by monitoring the extinction at 260 nm. Importantly, a remarkably narrow melting transition was observed at 47.5 °C (full width at half-maximum of the first derivative is ∼2 °C), which is highly diagnostic of an SNA structure with a high surface density of nucleic acids.
Figure 3.
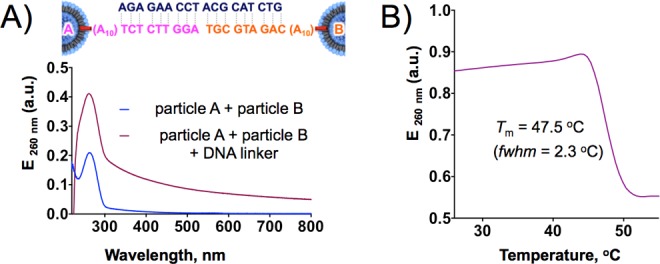
(A) Extinction spectra of liposomal SNAs before (blue) and after (red) aggregation in the presence of linker DNA strands. (B) Melting transition of liposomal SNA aggregates monitored as a change in extinction at 260 nm.
The most important property of SNAs pertains to their ability to enter cells without the need for ancillary transfection agents.2a To determine if liposomal SNAs exhibit this behavior, we incubated ovarian adenocarcinoma cells (SKOV-3, American Type Culture Collection) in the presence of the liposomal SNAs synthesized with a 5′-Cy5-labeled DNA in the absence of any transfection agents at different DNA concentrations. Remarkably, liposomal SNAs readily entered cells in high quantities even after a short 1 h incubation time (Figures 4A,B). No significant uptake of free DNA strand (5′-Cy5-labeled) in SKOV-3 cells was detected even after 36 h of incubation under identical conditions. Similar to the Au-SNAs,24 high uptake of liposomal SNAs in SKOV-3 cells did not cause any measurable toxicity, even at high concentrations (2 μM, Figure 4C). In contrast, when DharmaFECT 1 was used in an attempt to deliver a comparable amount of DNA, significant cytotoxicity, which reduced cell viability to 35% over a 24 h time period of incubation, was observed (Figure 4C).
Figure 4.
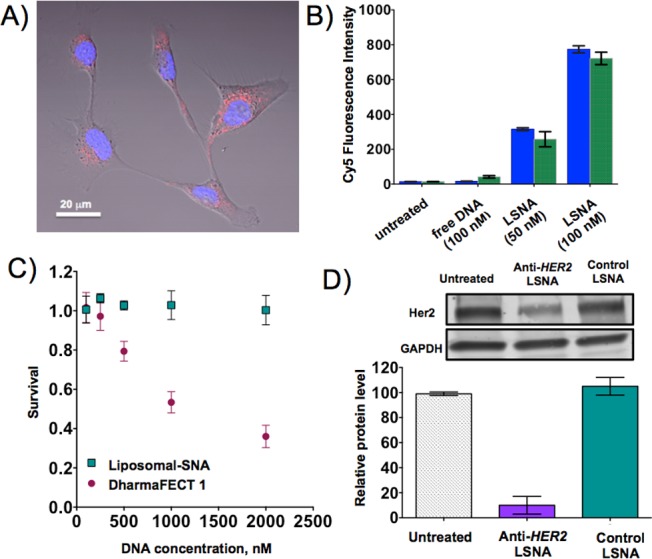
(A) Confocal micrograph of SKOV-3 cells incubated with 100 nM Cy5-labeled liposomal SNAs (red) for 24 h. (B) Flow cytometry analysis of uptake of 5-Cy5-labeled DNA strand and 5′-Cy5-labeled liposomal SNAs in SKOV-3 cells after 1 h (blue bars) and 36 h (green bars) of incubation. (C) Cytotoxicity (alamarBlue assay) of liposomal SNAs and DharmaFECT-delivered DNA in SKOV-3 cells. (D) HER2 gene knockdown in SKOV-3 cells using anti-HER2 liposomal SNA constructs at 1 μM DNA.
After establishing that liposomal SNAs are not cytotoxic, we synthesized a liposomal SNA capable of knocking down human epidermal growth factor receptor 2 (HER2). To compare the effectiveness of the antisense activity of liposomal SNAs to that of conventional transfection systems, SKOV-3 cells were incubated in the presence of anti-HER2 liposomal SNAs and control liposomal SNAs with a scrambled sequence (Figure 4D). Importantly, HER2 protein levels were reduced by 85% in the presence of anti-HER2 liposomal SNAs compared to the internal reference gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Figure 4D). Collectively, these results demonstrate the potential to use the liposomal SNAs to effect both cellular transfection and gene regulation.
In summary, we have developed a scalable synthetic route for novel metal-free liposomal SNAs. Such structures can be assembled rapidly from readily available, non-toxic starting materials. The SNA architecture not only stabilizes these small liposomal structures but also facilitates their internalization by SKOV3-3 cells. Consequently, such structures show promise as new biocompatible gene regulation constructs that exhibit many of the attractive properties of the more conventional gold nanoparticle-based SNAs.
Acknowledgments
This work was supported by the Defense Advanced Research Projects Agency (agreement HR0011-13-2-0018), the Center for Cancer Nanotechnology Excellence (CCNE) initiative of the National Institutes of Health (grant U54 CA151880), and the Air Force Office of Scientific Research (agreement FA-9550-11-1-0275).
Supporting Information Available
General synthesis procedures, material information, and instrumentation. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ R.J.B. and N.C. contributed equally.
The authors declare the following competing financial interest(s): C.A.M. is a founder and stockholder in AuraSense Therapeutics, a company that has licensed SNA technology.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mirkin C. A.; Letsinger R. L.; Mucic R. C.; Storhoff J. J. Nature 1996, 382, 607. [DOI] [PubMed] [Google Scholar]
- a Cutler J. I.; Auyeung E.; Mirkin C. A. J. Am. Chem. Soc. 2012, 134, 1376. [DOI] [PubMed] [Google Scholar]; b Briley W.; Halo T. L.; Randeria P. S.; Alhasan A. H.; Auyeung E.; Hurst S. J.; Mirkin C. A. In Nanomaterials for Biomedicine; Nagarajan R., Ed.; ACS Symposium Series1119; American Chemical Society: Washington, DC, 2012; pp 1–20. [Google Scholar]
- Young K. L.; Scott A. W.; Hao L.; Mirkin S. E.; Liu G.; Mirkin C. A. Nano Lett. 2012, 12, 3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler J. I.; Zheng D.; Xu X.; Giljohann D. A.; Mirkin C. A. Nano Lett. 2010, 10, 1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-S.; Lytton-Jean A. K. R.; Hurst S. J.; Mirkin C. A. Nano Lett. 2007, 7, 2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seferos D. S.; Giljohann D. A.; Rosi N. L.; Mirkin C. A. ChemBioChem 2007, 8, 1230. [DOI] [PubMed] [Google Scholar]
- Lytton-Jean A. K. R.; Gibbs-Davis J. M.; Long H.; Schatz G. C.; Mirkin C. A.; Nguyen S. T. Adv. Mater. 2009, 21, 706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler J. I.; Zhang K.; Zheng D.; Auyeung E.; Prigodich A. E.; Mirkin C. A. J. Am. Chem. Soc. 2011, 133, 9254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li Z.; Zhang Y.; Fullhart P.; Mirkin C. A. Nano Lett. 2004, 4, 1055. [Google Scholar]; b Alemdaroglu F. E.; Alemdaroglu N. C.; Langguth P.; Herrmann A. Adv. Mater. 2008, 20, 899. [Google Scholar]; c Liu H.; Zhu Z.; Kang H.; Wu Y.; Sefan K.; Tan W. Chem.—Eur. J. 2010, 16, 3791. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chien M.-P.; Thompson M. P.; Gianneschi N. C. Chem. Commun. 2011, 47, 167. [DOI] [PubMed] [Google Scholar]
- Hurst S. J.; Hill H. D.; Mirkin C. A. J. Am. Chem. Soc. 2008, 130, 12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhasan A. H.; Kim D. Y.; Daniel W. L.; Watson E.; Meeks J. J.; Thaxton C. S.; Mirkin C. A. Anal. Chem. 2012, 84, 4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Patel P. C.; Giljohann D. A.; Daniel W. L.; Zheng D.; Prigodich A. E.; Mirkin C. A. Bioconjugate Chem. 2010, 21, 2250. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Choi C. H. J.; Hao L.; Narayan S. P.; Auyeung E.; Mirkin C. A. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang K.; Fang H.; Wang Z.; Li Z.; Taylor J.-S. A.; Wooley K. L. Biomaterials 2010, 31, 1805. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McAllister K.; Sazani P.; Adam M.; Cho M. J.; Rubinstein M.; Samulski R. J.; DeSimone J. M. J. Am. Chem. Soc. 2002, 124, 15198. [DOI] [PubMed] [Google Scholar]; c Whitehead K. A.; Langer R.; Anderson D. G. Nat. Rev. Drug Discov. 2009, 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zheng D.; Seferos D. S.; Giljohann D. A.; Patel P. C.; Mirkin C. A. Nano Lett. 2009, 9, 3258. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Prigodich A. E.; Seferos D. S.; Massich M. D.; Giljohann D. A.; Lane B. C.; Mirkin C. A. ACS Nano 2009, 3, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rosi N. L.; Giljohann D. A.; Thaxton C. S.; Lytton-Jean A. K. R.; Han M. S.; Mirkin C. A. Science 2006, 312, 1027. [DOI] [PubMed] [Google Scholar]; b Giljohann D. A.; Seferos D. S.; Prigodich A. E.; Patel P. C.; Mirkin C. A. J. Am. Chem. Soc. 2009, 131, 2072. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jensen S. A.; Day E. S.; Ko C. H.; Hurley L. A.; Luciano J. P.; Kouri F. M.; Merkel T. J.; Luthi A. J.; Patel P. C.; Cutler J. I.; Daniel W. L.; Scott A. W.; Rotz M. W.; Meade T. J.; Giljohann D. A.; Mirkin C. A.; Stegh A. H. Sci. Transl. Med. 2013, 5, 209ra152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Farokhzad O. C.; Langer R. Adv. Drug Delivery Rev. 2006, 58, 1456. [DOI] [PubMed] [Google Scholar]; b Della Rocca J.; Liu D.; Lin W. Nanomedicine 2012, 7, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dave N.; Liu J. ACS Nano 2011, 5, 1304. [DOI] [PubMed] [Google Scholar]; b Granéli A.; Edvardsson M.; Höök F. ChemPhysChem 2004, 5, 729. [DOI] [PubMed] [Google Scholar]; c Stengel G.; Zahn R.; Höök F. J. Am. Chem. Soc. 2007, 129, 9584. [DOI] [PubMed] [Google Scholar]; d Gunnarsson A.; Jönsson P.; Marie R.; Tegenfeldt J. O.; Höök F. Nano Lett. 2007, 8, 183. [DOI] [PubMed] [Google Scholar]
- Thompson M. P.; Chien M.-P.; Ku T.-H.; Rush A. M.; Gianneschi N. C. Nano Lett. 2010, 10, 2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giljohann D. A. Ph.D. Dissertation, Northwestern University, 2009. [Google Scholar]
- McMahon K. M.; Mutharasan R. K.; Tripathy S.; Veliceasa D.; Bobeica M.; Shumaker D. K.; Luthi A. J.; Helfand B. T.; Ardehali H.; Mirkin C. A.; Volpert O.; Thaxton C. S. Nano Lett. 2011, 11, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Zhang H.; Dever B.; Li X.-F.; Le X. C. Bioconjugate Chem. 2013, 24, 1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Senior J.; Gregoriadis G. Life Sci. 1982, 30, 2123. [DOI] [PubMed] [Google Scholar]; b Kim C.-K.; Kim H.-S.; Lee B.-J.; Han J.-H. Arch. Pharmacal. Res. 1991, 14, 336. [Google Scholar]; c Sułkowski W. W.; Pentak D.; Nowak K.; Sułkowska A. J. Mol. Struct. 2005, 744–747, 737. [Google Scholar]
- Versluis F.; Voskuhl J.; van Kolck B.; Zope H.; Bremmer M.; Albregtse T.; Kros A. J. Am. Chem. Soc. 2013, 135, 8057. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Hao L.; Hurst S. J.; Mirkin C. A. J. Am. Chem. Soc. 2012, 134, 16488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.