

MOLECULAR MECHANISMS UNDERLYING ACUTE AND CHRONIC ALCOHOLISM
Introduction
Alcohols are organic compounds containing a hydroxyl (-OH) group attached to a carbon atom and the aliphatic alcohols are described by the general formula CnH2n+1OH. Ethanol, the psychoactive constituent of alcohol, has been used recreationally for tens of thousands of years (Hanson, 1995) and is one of the largest health burdens on society (Cargiulo, 2007). For the remainder of this chapter alcohol will refer to ethanol, and alcoholism is the functional equivalent of alcohol use disorder, as outlined by the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-V) (American Psychiatric Association, 2013).
The maladaptive behaviors of alcoholism rely on changes in the brain that lead to compulsive and excessive drinking, afflicting all organs, with damage as a secondary consequence of alcoholism. While acute use of alcohol, such as binge drinking and intoxication, causes cellular changes in the brain that last for hours, chronic alcohol use induces widespread neuroadaptations in the nervous system that can last a lifetime. This involves the remodeling of synapses that are dependent upon changes in gene expression in the presence of chronic alcohol use (Wilke et al., 1994), and is illustrated in Figure 6.1.
Fig. 6.1.

A schematic illustrating the dynamic interactions between alcohol and the nervous system. (Adapted from Spanagel, 2009.)
In order to fully understand the effects of alcohol on behavior and thus enable the development of efficacious treatments, it is necessary to understand the actions of alcohol at the molecular level. It is remarkable how little is known about alcohol’s molecular targets, in view of alcohol’s burden on public health and its long-term and widespread use. One reason for this might be alcohol’s low binding affinity to proteins, reflected by the fact that clinically relevant intoxication levels of alcohol are measured in millimolar concentrations, whereas most other drugs of abuse are measured in nanomolar concentrations. A consequence of the high concentrations in the body is that there are a large number of potential molecular targets.
In the early days of alcohol research it was thought that, because of the small size and non-specific nature of the alcohol molecule, it would likely not have a specific binding domain on proteins, but would instead interfere with the lipid membranes of the central nervous system (CNS). However, alcohol has only been shown to interact with the lipid bilayer at concentrations much higher than clinically relevant (Pang et al., 1980; Goldstein, 1984; Peoples et al., 1996). For this reason, researchers have focused their search on other molecular targets of alcohol. Most of the examples below come from animal models and postmortem tissue analysis of alcoholics, animal models, and cell cultures. Many of the molecular pathways that are sensitive to alcohol are highly conserved across species such as humans, mice, rats, worms, and fruit flies (Dick and Foroud, 2002, Lewohl et al., 2011). In addition, considerable progress has been made in defining binding cavities for alcohol in several proteins, including ion channels (Harris et al., 2008; Howard et al., 2011, Sauguet et al., 2013).
This chapter will discuss the known and proposed molecular targets of alcohol in the brain that may be important for behaviors meeting the DSM-V criteria for alcohol use disorder. Additionally, the molecular mechanisms of approved and prospective treatments for alcoholism will be discussed.
Primary targets
Alcohol’s effects on the brain are diverse and include changes in levels and function of neurotransmitters, receptors, enzymes, and other molecules, culminating in synaptic changes in brain circuitry regulating compulsivity and inhibition. Changes in these molecular systems lead to tolerance and withdrawal when alcohol is removed from the system.
Alcohol metabolism and distribution, which are discussed in depth in Chapter 4, can be summarized as follows: After consumption, alcohol is absorbed into the blood stream through the stomach and intestines, and it readily crosses the blood–brain barrier. Alcohol is distributed with body water and is found at approximately the same concentration in all tissues, including the nervous system. Most of the metabolism of alcohol occurs in the liver, and the brain has limited metabolism by the mitochondrial cytochrome P450 (CYP2E1), catalase, and other pathways (Zakhari, 2006).
CYP2E1 metabolism of alcohol produces acetaldehyde and is a source of acetaldehyde in brain. Additional acetaldehyde may enter the brain from peripheral conversion of alcohol to acetaldehyde by an enzyme found in the liver called alcohol dehydrogenase (ADH). In fact, ADH can be considered a “target” of alcohol since it binds alcohol through a zinc atom on ADH and the hydroxyl group of alcohol. Details on the proposed receptor-binding sites of alcohol are beyond the scope of this chapter. Interested readers may wish to explore this subject further in these reviews (Harris et al., 2008, Howard et al., 2011).
In the periphery, acetaldehyde is the primary metabolite of alcohol and is responsible for the flushing effect that encompasses face flushing, nausea, vomiting, headache, tachycardia, and sweating. Disulfiram (Antabuse), the first of three approved treatments for alcoholism, employs the aversive nature of acetaldehyde by inhibiting aldehyde dehydrogenase enzyme, thereby allowing for an accumulation of acetaldehyde. Disulfiram is a rather non-specific enzyme inhibitor and it may be useful in the treatment of cocaine dependence and even cancer chemotherapy due to actions on sites other than aldehyde dehydrogenase (Shorter and Kosten, 2011, Schmitt et al., 2012). Although disulfiram decreases drinking when taken regularly, it has low patient compliance because of the aversive effects (Moriarty, 1950; Barth and Malcolm, 2010). In addition to the peripheral flushing effect, acetaldehyde may have actions in the brain relevant to the acute effects of alcohol (Quertemont et al., 2005) and even the development of alcoholism (Deng and Deitrich, 2008). Acetaldehyde is self-administered by rodents through intravenous and intracerebral ventricular routes (Amit et al., 1977, Brown et al., 1979; Myers et al., 1984). Rats show preference for the physical place where they received central or peripheral administration of acetaldehyde over places where they received saline (conditioned place preference) (Quintanilla and Tampier, 2003, Spina et al., 2010). Although more research is necessary to fully understand these mechanisms, the neurobiological – and consequently the behavioral – actions of alcohol most likely depend on central contributions from both alcohol and its metabolites, acetaldehyde and acetate (Jiang et al., 2013). The development of alcohol tolerance and dependence comes from alterations in brain structure and function over time, and is long-lasting. This involves the remodeling of synapses that are dependent upon changes in gene expression in the presence of chronic alcohol (Rhodes and Crabbe, 2005). We will review the molecular adaptations at the acute and chronic stages of alcohol use that may underlie the hallmarks of alcoholism: withdrawal, tolerance, relapse, and craving.
It is important to note that these long-lasting drug-responsive alterations are unlikely to be encoded in RNAs or proteins due to the fast turnover rate of those molecules. This suggests that DNA is in charge of cellular memory, and that epigenetic mechanisms may be critical components of long-term learning and memory processes (Levenson and Sweatt, 2005), as well as chronic dependence on drugs (Renthal and Nestler, 2008).
DNA
Both the sequence and structure of DNA molecules control all downstream processes such as RNA transcription and protein translation, and both can contribute to the development, progression, and persistence of alcoholism. A great deal of work has been done in the field of genes and molecular pathways involved in fetal alcohol syndrome and alcohol-related developmental disorders (Goodlett et al., 2005; Sulik, 2005) and will be covered in other chapters of this book. Also covered in another chapter of this book are results from genome-wide association and quantitative trait loci studies that reveal mutations that may be important for predicting future alcoholism in certain groups or individuals.
Recent research has emphasized the notion that epigenetic mechanisms (which exert lasting control over gene expression through structural modifications of the DNA without altering the sequence) could mediate stable changes in brain function associated with alcoholism (Waddington, 2012). DNA methylation, histone acetylation, and phosphorylation are three common epigenetic-enzymatic modifications to chromatin structure that make the DNA less or more accessible to transcription factors and enzymes, thus changing the transcriptional activity of the target genes. Chronic exposure to alcohol was found to induce changes in the chromatin structure, specifically on gene promoters causing changes in gene expression in alcoholics (Guerri and Pascual, 2010; Ponomarev et al., 2012). This suggests that epigenetic mechanisms are involved in both biochemical and behavioral responses to alcohol.
Histone deacetylase (HDAC) is an enzyme that removes the acetyl group from histone proteins on DNA, making the DNA less accessible to transcription factors. Acute alcohol was found to decrease HDAC activity and increase acetylation of histones (H3 and H4) in the rat amygdala. Conversely, withdrawal from chronic alcohol in rats was found to increase HDAC activity and decrease H3 and H4 acetylation in the rat amygdala (Pandey et al., 2008). An HDAC inhibitor (trichostatin A) blocks the increase in HDAC activity and rescues the deficits in H3 and H4 acetylation in the amygdala. This change prevents the development of withdrawal-related anxiety in rats, suggesting a potential role for HDAC inhibitors as therapeutic agents in treating alcohol withdrawal symptoms (Pandey et al., 2008). CREB-binding protein (CBP) is a histone acetylase (HAT) that acetylates nearby histones, and allows for subsequent transcriptional activation. Acute alcohol exposure increases levels of CBP and neuropeptide Y (NPY), while withdrawal produces the opposite effect. Moreover, the withdrawal-induced anxiety behavior was found to correlate with the levels of CBP and NPY in the amygdala (Pandey et al., 2008). CBP was found to be important for normal learning and memory mechanisms in the brain since mutations of CBP cause a form of mental retardation in humans (Tsankova et al., 2007). This finding suggests an important mechanism in alcoholism, since addiction is thought to be an aberrant learning and memory process (Hyman et al., 2006). Figure 6.2 summarizes the epigenetic changes associated with alcohol.
Fig. 6.2.

Epigenetic modifications associated with alcoholism. CREB-binding protein (CBP) and histone deacetylase (HDAC) activity are altered by acute alcohol exposure. Chronic alcohol exposure results in neuroadaptations opposing the acute effects of alcohol in order to maintain a homeostatic state. Withdrawal after chronic alcohol exposure is associated with increased HDAC activity and decreased levels of CBP and the associated histone acetylation (Starkman et al., 2012). DNMT, DNA methyltransferase; Ac, acetyl; Me, methyl; HAT, histone acetylase.
Mice lacking a specific HDAC, HDAC5, display normal rewarding responses to acute cocaine exposure and become hypersensitive when chronically exposed to cocaine (Renthal et al., 2007). Chronic cocaine administration inactivates HDAC5 by exporting it out of the nucleus, resulting in histone hyperacetylation and increased mRNA expression of HDAC5 target genes. An example of a target gene of HDAC5 that is found to be increased during both chronic cocaine and alcohol exposure is neurokinin 1 receptor (NK1R). This receptor is also known as the substance P receptor, due to its importance in pain transmission. Silencing the translation of the NK1R using a technique known as RNA interference was found to reduce alcohol drinking in mice, emphasizing the role that NK1R plays in alcoholism (Baek et al., 2010). In fact, an NK1R antagonist (L822429) decreased voluntary alcohol consumption, suppressed stress-induced reinstatement of alcohol seeking, and increased sensitivity to the sedative effects of alcohol in rats (Schank et al., 2011). Similar effects were also seen in detoxified alcoholic inpatients, as an NK1R antagonist suppressed spontaneous alcohol cravings, improved overall well-being, blunted cravings induced by a challenge procedure, and attenuated concomitant cortisol responses. An analysis of brain function in human alcoholics compared with controls while performing behavioral emotional tasks was performed using functional magnetic resonance imaging and corroborated the above results, suggesting NK1R antagonism as a potential therapeutic target for the treatment of alcoholism (George et al., 2008).
DNA is the master regulator in the cell and is possibly a molecular implementation of the persistence of drug effects (or even memories) in the brain. DNA exerts its control through gene expression, which includes: RNA transcription, protein translation, and regulatory processes like microRNA and RNA splicing. In addition to alcohol’s indirect effect on gene expression via epigenetic actions on chromatin, alcohol can directly target the gene expression machinery, which will be explored below and is detailed in Figure 6.3.
Fig. 6.3. Synaptic transmission mechanism and the synaptic neuroadaptations occurring after chronic alcohol use.

(A) Synaptic transmission is the process by which the brain communicates information. Depending on the inputs to the pre-synaptic neuron’s dendrites, an action potential (i.e., nerve impulse) will be generated at the axon hillock and propagate down the axon through the movement of charged particles (i.e., the ions – Na+ and K+). The synapse is the gap between the axon of the presynaptic neuron and the dendrite of the postsynaptic neuron. In order for the information to cross the synapse, the electrical signal of the action potential must be converted into a chemical signal. This is achieved by releasing chemicals (neurotransmitters) from the presynaptic nerve terminal in a voltage-dependent (and therefore calcium-dependent) manner. The presynaptic nerve terminal contains the neurotransmitter release machinery needed for this to occur. Once released the neurotransmitter will diffuse across the synapse and bind to receptors on the postsynaptic nerve’s dendrites. The two major types of receptors of concern here are ligand-gated ion channels and G-protein-coupled receptors. Ligand-gated ion channels undergo a conformational change when a ligand (i.e., neurotransmitter) is bound and allow a particular ion (e.g. Cl−, Na+, Ca2+, etc.) to flow into or out of the cell. When G-protein-coupled receptors are activated they affect secondary messengers and molecular cascades, resulting in changes in the postsynaptic neuron. It is necessary to understand the basics of synaptic transmission because acute and chronic alcohol exposure causes changes in many molecules important for transmission and changes transmission properties. (Adapted from Clapp et al., 2008.)
(B) An example of a synapse stimulated by alcohol use and the molecular cellular cascade associated with alcohol use. GABA, γ-aminobutyric acid; NMR, NMDA (N-methyl-D-asparatate) receptor; VGCC, voltage-gated calcium channel; Camp, cyclic adenosine monophosphate; PKA, protein kinase A; PKC, protein kinase C; caMK, calmoldulin kinase; MAPk, mitogen-activated protein kinase; CREB, cyclic AMP response element-binding protein. (Adapted from Koob et al., 2005.)
Neurotransmitter systems
Glutamate
Glutamate is the primary excitatory neurotransmitter in the brain. Glutamatergic (glutamate-using) systems contain both ionotropic (ligand-gated receptors coupled to the flow of charged particles – N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), kainate) and metabotropic (receptors coupled to G proteins – mGluR) receptors. The glutamatergic system plays an important role in long-term potentiation and long-term depression and therefore has physiologic implications that are important for learning and memory. Generally, all glutamate receptors are inhibited by acute alcohol treatment, although some subtypes are only affected by very high concentrations. Alcohol’s acute actions on the glutamatergic system have been implicated in tolerance, withdrawal, craving, relapse, and dependence.
Alcohol acts as a non-competitive inhibitor of the AMPA/kainate receptors at high concentrations (Dildy-Mayfield and Harris, 1992, 1995, Akinshola et al., 2003). Out of all the glutamatergic receptors, NMDA receptors are the most sensitive to the effects of alcohol. Alcohol’s binding site on the NMDA receptor is not known, and there has been some evidence to suggest that alcohol exerts its effect on the NMDA receptor through protein kinase C (Li et al., 2005). Acute application of alcohol to hippocampal neuronal slices reduces NMDA activity at a concentration that produces intoxication in humans and is linearly related to alcohol’s intoxicating potency (Lovinger et al., 1989).
In a drug discrimination test, MK-801, an NMDA antagonist, elicited the same response that animals were trained to give in the presence of alcohol, indicating that alcohol’s NMDA-antagonizing effects are important for mediating the subjective effects of alcohol (Butelman et al., 1993). In recently detoxified alcoholics, ketamine, an NMDA antagonist, was able to mimic the behavioral effects of alcohol (Krystal et al., 1998). The production of alcohol-like effects by ketamine supports the clinical role of NMDA receptor function facilitating alcohol’s effects on humans.
The response to alcohol depends on the subunit composition of the glutamatergic receptor. A study using rat NMDA receptor subunits expressed in Xenopus oocytes suggests that NMDA receptors containing certain subunits are more sensitive to alcohol than others (Raeder et al., 2008). Alterations in subunit composition of glutamate receptors were seen after chronic use of alcohol in mice (Ortiz et al., 1995). Chronic alcohol was also found to increase the expression of AMPA1 receptor subunit in the brains of alcoholics (Lewohl et al., 2000).
“Acute tolerance” is the term used to describe the decreased inhibitory effects of alcohol over a short period of time. NMDA receptor activity in cultured neurons returns to baseline levels after less than 1 hour of alcohol exposure (Lai et al., 2004). Once alcohol is removed from the system, the hyperglutamatergic state can produce a severe withdrawal syndrome characterized by agitation, anxiety, and disorientation and is associated with a susceptibility to seizures and excitotoxic cell death. NMDA receptor antagonists can be applied to protect cells from this type of death (Hoffman et al., 1995; al Qatari et al., 2001). The acute withdrawal syndrome is clinically relevant and well characterized. The question remains as to whether the neuroadaptations leading to acute withdrawal syndrome contribute to the propensity of relapse or lead to alcoholism.
Acute alcohol inhibits the function of the NMDA receptor, while chronic use of alcohol seems to upregulate NMDA receptor expression in the brain (Qiang and Ticku, 2005). This upregulation is thought to counteract the acute inhibition of the glutamatergic system and also the sedative effects of increased γ-aminobutyric acid receptor A (GABA(A)) activity, as discussed below. Figure 6.4 presents an illustration of the main neuro-transmitter systems and brain regions involved in the neuroadaptations associated with acute and chronic alcohol use.
Fig. 6.4. The molecular adaptations in the mesocorticolimbic pathway occurring with alcoholism.

(A) The mesocorticolimbic system includes several brain areas: prefrontal cortex (PFC), nucleus accumbens (NAc), ventral tegmental area (VTA), and amygdala (Amg). Alcohol consumption has a major effect on the dopaminergic, glutamatergic, and GABAergic systems in this pathway. Acute effects of alcohol include: increased dopamine release (associated with reward), increased GABA receptor activity (associated with anxiolysis, sedation, and motor incoordination), and decreased glutamate receptor activity. DA, dopamine; EtOH, ethanol; NMDA-R, N-methyl-D-aspartate receptor; GABA-R, γ-aminobutyric acid receptor.
(B) Chronic alcohol consumption causes neuroadaptations to oppose the effects of acute alcohol and include: decreased dopamine release and increased dopamine receptor expression, decreased GABA receptor expression, increased glutamate release and increased NMDA receptor expression.
Acamprosate, one of the three approved treatments for alcoholism, is proposed to exert at least part of its effect by altering glutamatergic function (De Witte et al., 2005), though the exact mechanism by which it interacts is unclear. Acamprosate was found to reduce relapse rate, increase abstinence rate, and decrease excessive drinking in alcohol-dependent rats, and had no effect in non-dependent rats (Spanagel et al., 1996a, b, c). In dozens of clinical trials conducted in Europe, about half of the alcoholics treated with acamprosate maintained sobriety compared to the placebo group (Mason and Ownby, 2000; Mann et al., 2004), but results of US clinical trials have shown less beneficial effects of acamprosate (Anton et al., 2006; Mason et al., 2006). Several studies seeking to identify acamprosate’s mechanism of action have failed to show direct modulation of NMDA-Rs at clinically relevant concentrations (Reilly et al., 2008) and a recent study suggests that calcium is the active moiety of acamprosate (Spanagel et al., 2013). Application of acamprosate on GABA(A), glycine, vanilloid-1 receptors and voltage-gated Na+ channels did not exert any effect (Reilly et al., 2008). On the other hand, knocking out some of the mGluR subunits of the metabotropic glutamate receptors in mice or by using an mGluR antagonist reduces the ability of acamprosate to affect behavior (Blednov and Harris, 2008). Taken together, the evidence indicates that alcohol’s modulation of the glutamatergic system is an important molecular mechanism by which alcohol exerts its behavioral effects and a potential target for the treatment of alcoholism.
Dopamine
Dopamine is a neurotransmitter in the CNS and binds to several different types of receptors (D1 and D2 families). Dopamine is thought to contribute to alcoholism by signaling in the midbrain dopaminergic system, a brain circuit involved in associative learning, incentive salience, and reward prediction (Gonzales et al., 2004). The midbrain dopaminergic system originates in the ventral tegmental area and projects to regions of the brain such as the striatum, nucleus accumbens, and prefrontal cortex (PFC). Alcohol and other drugs of abuse increase dopaminergic activity in the midbrain region of rodents and humans (Boileau et al., 2003). Dopamine release in the midbrain partially mediates the positive-reinforcing properties of acute alcohol exposure necessary for the development of alcoholism (Raeder et al., 2008). Moreover, preference for alcohol has been directly correlated with alcohol-induced dopamine release in the midbrain. This is illustrated by the fact that rats which are bred to prefer alcohol release more dopamine than wild-type rats in an alcohol self-administration study (Weiss et al., 1993). Also, mice lacking different dopamine receptors and transporters show modified alcohol preference compared with controls, further illustrating dopamine’s involvement in alcohol-related behaviors (Crabbe et al., 2006).
Acute alcohol exposure activates dopamine reward pathways, whereas chronic treatment produces a hypodopaminergic state associated with dysphoria, which can lead to craving and relapse (Koob and Volkow, 2010). A 1-year-long chronic alcohol treatment in rats decreases dopamine and its metabolite in the striatum, decreases tyrosine hydroxylase protein levels, and increases dopamine transporter protein levels compared to controls (Rothblat et al., 2001). Positron emission tomography scans show that chronic alcoholics have fewer D2 receptors when compared with non-alcoholics. D2 receptors in the striatum are mainly localized on GABA-synthesizing cells. These results provide evidence of GABAergic involvement in the dopaminergic abnormalities seen in alcoholics (Volkow et al., 2002). These results also reflect the brain’s homeostatic mechanism to adapt to initial increases in dopamine levels after chronic exposure to alcohol.
The dopamine receptor antagonist fluphenazine will block alcohol self-administration when injected into the nucleus accumbens (Rassnick et al., 1992). Additionally, several clinical trials have shown that aripiprazole, an atypical antipsychotic, reduces craving and increases positive subjective feelings in alcoholics (Martinotti et al., 2007; Brunetti et al., 2012). Although these clinical trials are promising, the mechanism by which dopamine malfunction caused by alcohol contributes to clinical alcoholism is currently unknown.
GABA(A)
GABA is the primary inhibitory neurotransmitter, and the GABA(A) receptor, a ligand-gated chloride channel, is the most abundant inhibitory receptor in the mammalian brain. Acute alcohol exposure enhances GABA(A) function. The molecular actions by which alcohol may exert its effects on GABAergic activity are by directly binding to the receptor, increasing presynaptic release of GABA or releasing GABAergic steroids (Lobo and Harris, 2008). This acute action of alcohol on the GABAergic system, along with the effects of chronic alcohol use, is illustrated in Figure 6.5. Alcohol intoxication is characterized by anxiolysis, sedation, impaired cognitive function, hypnosis, and impaired motor function. Many of these behaviors are mimicked by the administration of pharmacologic GABA(A) agonists like muscimol and benzodiazepines (Grobin et al., 1998). Moreover, the effects of alcohol can be diminished by using pharmacologic GABA antagonists like bucucilline and picrotoxin (Hyytia and Koob, 1995), suggesting that GABA(A) signaling is directly involved in the acute actions of alcohol. Electrophysiologic data support alcohol’s actions on GABA(A) receptors, showing potentiation of GABA-mediated chloride influx following alcohol administration in a variety of preparations. Mihic et al. (1997) identified a 45-amino-acid residue necessary and sufficient for the enhancement of GABA(A) receptor function by alcohol, suggesting that alcohol’s binding site is between the transmembrane 2 and the transmembrane 3 regions of the receptor (Mihic et al., 1997). This is supported by crystallographic analysis of alcohol binding to a related ligand-gated ion channel, GLIC (Sauget et al., 2013). Chronic alcohol use causes changes in GABA receptor subunit composition in human alcoholics (Lewohl et al., 1997), and it should be noted that alcohol’s pharmacologic action on GABA(A) receptors (and consequently behavioral responses) strongly depends on the subunit composition of the receptor. Alcohol increases GABAergic neuro-transmission over a wide range of concentrations, with some studies showing that delta-containing receptors are more sensitive than other GABA(A) receptors (Wallner et al., 2003). Use of mutant mice either lacking a subunit or containing an ethanol-resistant subunit demonstrated that the alpha-2 and alpha-3 subunits take part in mediating the motor-impairing effects of alcohol and that the alpha-2 subunit takes part in the aversive properties of alcohol (Blednov et al., 2011b, 2013).
Fig. 6.5. The γ-aminobutyric acid (GABA) system and the effects of acute (A) and chronic (B) alcohol exposure.
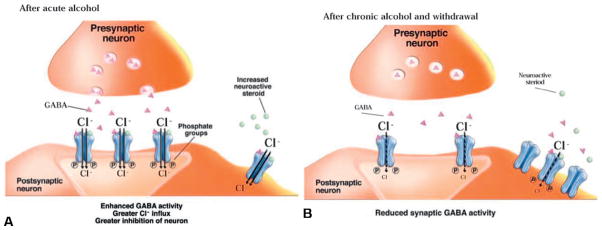
(A) GABA acts in part through the activation of GABA(A) receptors, which are ligand-gated ion channels that allow chloride ions (Cl−) to flow into the neuron after GABA binds to it. Greater influx of Cl− into the neuron makes it more difficult for the cell to generate a new action potential (i.e., nerve impulse). This is why GABA is considered to be an inhibitory neurotransmitter. The molecular actions by which alcohol may exert its effects on GABAergic activity is by directly binding to the receptor, increasing presynaptic release of GABA, or releasing GABAergic steroids that activate extrasynaptic receptors (Clapp et al., 2008).
(B) After chronic alcohol exposure and during withdrawal, GABA activity at the synapse is reduced, leading to reduced inhibition of the postsynaptic neuron. This results in development of anxiety and hyperexcitability. (Adapted from Clapp et al., 2008.)
Chronic alcohol use causes the downregulation of GABA(A) receptors due to the initial overstimulation by the alcohol. In turn, glutamate receptors are upregulated to counteract the sedative effects of increased GABA(A). During withdrawal from alcohol, the excess of glutamate receptors combined with the lack of GABA(A), causes anxiety, dysphoria, and in severe cases seizures. Topiramate, an anticonvulsant seizure medication, was found to reduce voluntary alcohol intake in humans with alcohol dependence in recent clinical trials (Johnson et al., 2008). Alcoholic patients achieved a rate of continuous abstinence much higher than those in a placebo group. They also reported fewer cravings compared to a placebo group but complained of other side-effects. Topiramate is associated with glutamate system inhibition, although the exact mechanism by which topiramate promotes abstinence is not yet understood.
GABA(B)
GABA(B) receptors are metabotropic receptors responsible for mediating slow inhibitory responses in the brain and have been implicated in alcoholism. Acute alcohol was seen to increase inhibitory GABA(B) signaling in a concentration-dependent manner in rat midbrain dopaminergic neurons in a current and single-electrode voltage clamp electrophysiologic experiment (Federici et al., 2009). Silencing the GABA(B) receptor in fruit flies using RNA interference decreases acute motor impairment after alcohol exposure (Dzitoyeva et al., 2003). Similarly, in mice, cerebellar injections of GABA(B) agonists accentuated and antagonists attenuated motor impairment following acute alcohol (Dar, 1996). Conversely, recordings in CA1 pyramidal neurons have indicated that GABA(B) signaling was immune to the acute effects of alcohol but became modulated after chronic exposure, perhaps because of neuroadaptations from dependence (Frye et al., 1991). Baclofen (b-parachlorophenol GABA), a GABA(B) agonist prescribed to treat muscle spasticity, reduces relapse in dependent humans and decreases alcohol consumption in rats (Addolorato et al., 2002a b; Maccioni and Colombo, 2009, Agabio et al., 2012). Dr. Olivier Ameisen describes attending over 5000 AA meetings over the course of several years in an attempt to reduce his alcohol craving and abstain from drinking. Despite all efforts he continued drinking and eventually treated himself with high doses of baclofen; he was able to “cure” his disease (Ameisen, 2005, 2012). Preclinical studies show that baclofen is able to suppress withdrawal symptoms in alcoholics, promote alcohol abstinence, and reduce withdrawal-related anxiety and alcohol craving (Addolorato et al., 2002a). There is also evidence for baclofen having no significant effect on alcoholics (Garbutt et al., 2010), suggesting that baclofen may be effective in treating only certain subtypes of alcoholics. There is still research to be done on baclofen and its side-effects, and therefore it is not yet approved by the Food and Drug Administration (FDA) for the treatment of alcoholism (Leggio et al., 2010).
RNA splicing is a process that removes the intervening, non-coding sequences of genes (introns) from pre-mRNA and joins the protein-coding sequences (exons) together in order to enable translation of mRNA into a protein. Sometimes exons are extended or skipped, or introns are retained during the splicing process, leading to “alternative splicing.” Alternative splicing of the pre-mRNA creates different splice variants for the same gene, which results in a functional diversity of proteins (Matlin et al., 2005). Eventually, the mRNA transcripts are exported to the cytoplasm and translated into different isoforms of the same protein that vary in their functional properties (Coetzee et al., 1999; Dredge et al., 2001). Drugs of abuse such as alcohol can affect alternative splicing. For example, chronic alcohol consumption enhances the complexity of GABA(B) receptor splicing (Lee et al., 2010, 2013). Human alcoholics show differences in GABA(B) receptor splice variants when compared to non-alcoholics, which may contribute to the pharmacologic effects of GABA(B) agonists such as baclofen in the treatment of alcoholism (Lee et al., 2013).
In summary, pharmacologic, behavioral, and electro-physiologic evidence supports a role for GABA(B) in mediating the effects of alcohol, and that many of the behaviors associated with alcoholism result from the direct alcohol-induced modulation of GABAergic neurotransmission. It should be noted that the specific behaviors elicited by alcohol depend highly on receptor subtype expression and cellular location. These behaviors will hopefully become evident in the future through behavioral characterization of genetically manipulated mice.
Serotonin (5-HT)
Serotonin is a neurotransmitter that serves many functions, including regulating mood, sleep, appetite, learning, memory, and other phenomena. The supply of neuronal serotonin originates mostly from neurons in the raphe nuclei whose axons innervate almost the entire brain, reflecting serotonin’s vast physiologic roles. There are many types of receptors for serotonin in the CNS and all are metabotropic, except for 5-HT3, which is a ligand-gated ion channel similar to nicotinic acetylcholine receptors. Evidence indicates that serotonin is involved in mediating both acute and chronic alcohol action and perhaps the development and maintenance of alcoholism.
Alcohol facilitates serotonergic transmission in part by increasing the potency with which the agonist can activate the 5-HT3 receptor (Sung et al., 2000) and the time spent in the open state (Zhou et al., 1998). In general an inverse relationship between serotonin transmission and alcohol drinking has been established. Increased serotonergic transmission is associated with less alcohol consumption and less serotonergic transmission is linked to more alcohol consumption, in animal models of drinking and in human alcoholics (for review, see Lovinger, 1997).
Electrophysiologic recordings in neuroblastoma cells show that acute alcohol potentiates 5-HT3 receptor-mediated ion currents (Lovinger, 1991). Acute alcohol was also found to activate the 5-HT3 receptor in oocytes (Harris et al., 1995), ganglion neurons (Lovinger and White, 1991), frontal cortex neurons (Sung et al., 2000), and human embryonic kidney 293 cells (Lovinger and Zhou, 1994).
Serotonin levels in animal brains are elevated after acute alcohol exposure (Murphy et al., 1982; LeMarquand et al., 1994). Mice lacking the 5-HT1B serotonin receptor consume larger amounts of alcohol compared to wild-type. Mice lacking the receptor show much higher incoordination compared to wild-type mice (after just one injection of alcohol), suggesting this receptor is involved in the intoxication process (Crabbe et al., 1996). The 5-HT2 receptor seems to be important for the reinforcing properties of alcohol because antagonists selectively decrease acute alcohol reinforcement (Roberts et al., 1998). Blockade of the serotonin transporter (5-HTT) with fluoxetine, a selective serotonin reuptake inhibitor (SSRI) (a widely used antidepressant), or by genetic knockout, decreases alcohol consumption in rodents (Kelai et al., 2003). Decreased consumption of alcohol following SSRI treatment has been observed in almost every rat model of alcoholism (Brown et al., 1979; Sellers et al., 1992; Ciccocioppo et al., 2006). Human studies have given less consistent results, but it appears that SSRIs may be effective in some subpopulations of alcoholics, such as late-onset individuals (Kranzler et al., 2012).
In addition to enhancing 5-HT activity in the brain using SSRIs, another approach widely used to understand serotonin’s effect on alcohol consumption has been to selectively block the 5-HT3 receptor. 5-HT3 antagonists decrease alcohol self-administration (Fadda et al., 1991; Hodge et al., 1993) and consumption in rodents (Sellers et al., 1994).
In humans, the drug ondansetron, a 5-HT3 antagonist that is FDA-approved for the treatment of chemotherapy-induced nausea, is a promising treatment for alcoholics. In a clinical trial, alcoholics were randomly selected to receive either ondansetron or a placebo for 11 weeks. The ondansetron patients with early-onset alcoholism had fewer drinks per day and reported more days of total abstinence than the placebo group (Johnson et al., 2000). In another clinical trial with 71 alcoholic men, almost four times as many people receiving a 6-week treatment with a low dose of ondansetron (0.25 mg) showed significant levels of decreased drinking when compared to placebo (Sellers et al., 1994). In a double-blind placebo-controlled, Latin square cross-over design study analyzing 5-HT3 receptor’s function in mediating the reinforcing properties of alcohol (Johnson et al., 1993), 16 healthy social drinkers received ondansetron treatment and the resulting subjective psychologic effects to a small amount of alcohol were recorded. Ondansetron treatment decreased the subjective pleasurable effects of alcohol and the desire to drink (Johnson et al., 1993). Since the serotonin transporter is important for regulating the serotonergic system, alleles at the gene encoding 5-HTT might predict the severity of the alcoholism and the therapeutic response to treatment with ondansetron. A clinical trial with 283 alcoholics found that ondansetron recipients had fewer drinks per day and more days spent totally abstinent than those who received placebo. The effect was greater in alcoholics with the LL genotype than the SS or LS genotype of the 5′-regulatory region of the serotonin transporter gene (Johnson et al., 2011). Moreover, animal studies have demonstrated that ondansetron might also be useful for treating opioid withdrawal symptoms (Pinelli et al., 1997).
Big potassium (BK) channels
The BK channel is a high-conductance calcium- and voltage-dependent potassium channel (Atkinson et al., 1991) and plays a dominant role in shaping neuronal activity and, unlike other voltage-gated ion channels, is strongly affected by alcohol (Treistman and Martin, 2009). Tolerance, which is the loss of drug effectiveness over time, is an important component of addiction. Acute tolerance can have a rapid onset within minutes of alcohol exposure, whereas tolerance after prolonged alcohol exposure develops over the course of days or weeks. The degree of acute behavioral tolerance to alcohol exhibited by a naïve subject can predict the likelihood of alcoholism (Treistman and Martin, 2009). Thus, the determinants of acute tolerance are important to understand. The BK channel is a key target in the development of tolerance in invertebrates and mammals (Davies et al., 2003; Martin et al., 2008). There are several variables that influence the response of the BK channel to alcohol, including subunit composition, splice variant, and post-translational mechanisms (see section on microRNA, below). BK channels from neurons of wild-type mice (in which the β4 subunit is well represented) exhibit little tolerance. By contrast, neuronal BK channels from β4 knockout mice do display acute tolerance. In addition to displaying tolerance, the β4 knockout mice drink more than their wild-type counterparts in an alcohol self-administration assay (Martin et al., 2008).
Drugs of abuse such as alcohol affect alternative splicing. The development of acute tolerance to alcohol provides another example that demonstrates the effects of alternative splicing of mRNAs on alcohol-related behavior (seen previously with the GABA(B) receptors). Tolerance was found to involve the splicing of the mRNA of the alpha subunit of the BK channel. The BK channel was found to have a sensitive and non-sensitive splice variant to alcohol. Within minutes of exposure to alcohol, the expression of the more alcohol-sensitive variant was selectively decreased, therefore producing tolerance (Treistman and Martin, 2009).
Transcription factors
Transcription factors serve as a key mechanism by which distinct gene programs are controlled, because they bind to highly specific DNA-regulatory sequences (control elements) (Renthal and Nestler, 2008; Rahman, 2012). Activation of alcohol-responsive transcription factors is also likely to result in changes in the expression of those genes with the corresponding control elements.
Heat shock factor protein 1 (HSF1) is a transcription factor which was found to be involved in the effects of acute alcohol application. Acute alcohol was found to increase GABA-R a4 subunit mRNA by increasing the binding of HSF1 to the promoter regions of GABA, increasing its transcription. A small interfering RNA (siRNA), used to block expression of HSF1, reduced HSF1 protein and reduced the alcohol-induced increases in the GABA-R a4 subunit, while introducing an active form of HSF1-induced GABA-R a4 transcription in the absence of alcohol (Pignataro et al., 2007). Acute alcohol also facilitates activation of HSF1 to the promoter region of synaptotagmin-1, a protein involved in synaptic transmission and release, suggesting that alcohol has a direct control of neurotransmitter release in an acute fashion (Pignataro et al., 2007, 2009). HSF family of mRNA was found to be different between alcoholic and control postmortem frontal cortices (Lewohl et al., 2000), as well as in cultured cortical neurons exposed to chronic alcohol treatment (Wang et al., 2007). These results indicate that these transcription factors are also involved in alcohol dependence.
The activation of several signaling pathways involving cAMP, Ca2+ and extracellular signal regulated kinase lead to the phosphorylation of CREB, a transcription factor cyclic AMP (cAMP) response element-binding protein (CREB) (Winstanley et al., 2007). Some chromatin-remodeling enzymes target chromatin by interacting with specific transcription factors by guiding them to a specific locus on the DNA. When phosphorylated, the transcription factor CREB interacts with CBP, a HAT that helps facilitate target gene activation by acetylating neighboring histones. This process was found to play a major role in behavioral responses to cocaine. Long-term treatment with alcohol was found to increase CREB transcription factor activation, which in turn increases the binding of CREB to the promoter of the glutamate receptor, which then increases the transcription of a specific glutamate receptor subunit NR2B. Site-directed mutation in the sequence where CREB binds abolished the stimulatory effect by alcohol, suggesting that CREB is involved in mediating alcohol-induced upregulation of the NR2B gene (Rani et al., 2005). This is a great example of an alcohol-responsive transcription factor affecting downstream expression of receptors which are also involved in mediating the response to alcohol.
Another example suggesting that the downstream transcription of genes is selective to either the acute or chronic stages of addiction is that of the immediate-early genes, such as c-Fos and FosB. These immediate-early genes are induced rapidly in the brain by acute use of drugs such as cocaine and alcohol, whereas the transcription of genes such as Cdk5 and Bdnf is induced by chronic use of cocaine (Taylor et al., 2007). Chronic and compulsive use of drugs also causes the accumulation of delta fosB, a member of the Fos family of transcription factors. Delta fosB accumulates in specific regions of the brain in response to many chronic stimuli and persists for several weeks after the end of the stimulus (Watanabe et al., 2009; Damez-Werno et al., 2012). Delta fosB represents a molecular mechanism that can initiate and then sustain changes in gene expression that persist long after drug exposure ceases (Nestler et al., 2001).
microRNAs
MicroRNAs are short, non-coding RNA strands that regulate the translation of target mRNA into protein posttranscriptionally by binding to complementary sequences of the mRNA 3′-untranslated region (Schratt et al., 2006). MicroRNAs are highly abundant in the brain and have also been found to mediate the neuroadaptations induced by exposure to drugs of abuse, such as alcohol (Lewohl et al., 2011; Mayfield and Nunez, 2012). Chronic alcohol use causes global changes in microRNA expression in human alcoholic brains as well as in a variety of animal models (Li and van der Vaart, 2011).
The mechanism by which alcohol produces changes in microRNA expression is unknown. As discussed previously in this chapter, alcohol has the ability to affect receptor tyrosine kinases and mitogen-activated protein kinases, which in turn regulate transcription factor activity and thus microRNA gene expression. Alcohol can also alter the epigenetics on the DNA sites for micro-RNA, thereby altering their expression.
The mechanism contributing to the development of molecular tolerance to alcohol specifically involving the BK channel (discussed above) was found to be controlled by a microRNA (Pietrzykowski et al., 2008). In rodent neurons alcohol upregulates a particular micro-RNA 9 (miR9) which subsequently affects the expression of specific splice variants of the BK channel, some of which are less or more sensitive to alcohol. This mechanism suggests that alcohol-sensitive microRNAs are regulatory master-switches for the development of tolerance, a crucial component of alcohol addiction.
Many studies use computational approaches to identify potential interactions of alcohol with gene expression. Networks of highly correlated RNAs enable the drawing of potential cellular pathways of alcohol-responsive RNAs (Wolstenholme et al., 2011; Ponomarev et al., 2012). In this study Dicer was found to be a downregulated alcohol-responsive gene with the greatest overrepresentation of microRNA-targeting effects in human alcoholics. This suggests a negative-feedback loop between microRNAs and Dicer to control their own availability (Lewohl et al., 2011).
Synaptic mRNA translation and microRNAs
While most mRNAs are restricted to the neuronal soma, significant amounts of mRNA are found in synaptic compartments of the cell and translated into protein locally (Steward and Levy, 1982; Steward and Banker, 1992). RNAs – specifically in the synaptic compartments of the cell – are found to play a key role in the different states of addiction such as dependence, tolerance, withdrawal and craving (Russo et al., 2010; Mayfield and Nunez, 2012). Similar to mRNA, microRNAs have also been found in synaptic compartments of the cell, suggesting a local form of mRNA regulation by microRNA (Schuman et al., 2006; Schratt, 2009). Some studies examined the composition of microRNAs in the synapse and revealed many synaptically enriched and depleted microRNA families (Lugli et al., 2005; Eipper-Mains et al., 2011; Pichardo-Casas et al., 2012). Lugli et al. (2005) have elegantly shown that the microRNA-processing enzymes such as Dicer and elf2c are also enriched in the synapse and that their activity is modulated by neuronal activity. Other studies from this group show the synaptic enrichment of microRNA precursor forms, suggesting that neuronal activity might activate the Dicer enzyme to process the synaptic microRNA precursor into the mature microRNA form, thus rendering it active in the synapse. Recent findings in Aplysia californica indicate that synaptic microRNA-124 (miR-124) takes part in regulating long-term facilitation, suggesting a mechanism by which microRNAs serve as a dynamic brake on synaptic mRNA translation and emphasizing the importance of this highly conserved mechanism (Rajasethupathy et al., 2009).
A single microRNA has the potential to target many genes, and multiple microRNAs can cooperate to target the same genes (Grimson et al., 2007; Lewohl et al., 2011). microRNA-7 (miR-7) and microRNA-153 (miR-153) were found to be differentially expressed between human alcoholic brain tissue and control group brain tissue. Interestingly, miR-7 and miR-153 were found to both regulate the expression of α-synuclein (Doxakis, 2010). α-synuclein is a protein that plays a major role in neurotransmitter release in presynaptic terminals (Liu et al., 2004; Greten-Harrison et al., 2010) and is involved in dopaminergic neurotransmission and neuro-degenerative disorders (Doxakis, 2010). Studies show that alcohol dependence in humans, as well as in rodents, is related to levels of α-synuclein (Bonsch et al., 2005a, b). Interestingly, overexpression of mir-7 and mir-153 significantly reduces endogenous alpha-synuclein levels, whereas inhibition of mir-7 and mir-153 enhances translation of alpha-synuclein. These findings illustrate a mechanism by which alcohol changes the expression of two microRNAs and how they can cooperate to target an mRNA that is known to be involved in alcoholism as well. This cooperation between the microRNAs allows regulation of gene expression with a reduced number of active microRNAs. Since alcoholism is a complex trait with global changes in gene expression, microRNAs serve as good targets for treatment, since they control global cellular changes in gene expression.
Neuroimmune
Substantial evidence has accumulated that implicates an unlikely target, the neuroimmune system, in the development, progression, and persistence of alcoholism. The term neuroimmune refers to signaling molecules that were first associated with innate immunity but are also commonly found in the brain. The brain uses immune signaling systems as neuromodulators that have functions distinct from their role in the immune system and are critical for normal brain functions like neuronal plasticity (Boulanger et al., 2001). One example of this is the major histocompatibility complex class I molecule, which serves as a primary mediator of immune response in the periphery but modulates activity-dependent refinement and plasticity in cortical synapses and the developing visual system in the CNS (Glynn et al., 2011; Elmer and McAllister, 2012). Alcohol-related behavior can be affected by immune signaling that originates in the brain or immune signaling derived from the periphery that crosses the blood–brain barrier to act on the brain.
Ingesting alcohol activates the neuroimmune system (Crews et al., 2011), which is proposed to lead to further increases in alcohol consumption, producing an escalating feedforward loop not conducive to a homeostatic state. Alcohol consumption compromises the tight junctions in gut epithelium, allowing lipopolysaccharide (LPS), a Gram-negative bacterial endotoxin that is normally confined to the gut, to leak into the blood stream, where it binds to and activates Toll-like receptors (TLRs) expressed on liver Kupffer cells and other tissues. This initiates a signaling cascade that culminates in the release of proinflammatory cytokines in the blood stream that can then cross the blood–brain barrier and interact with the brain, thus affecting behavior. Researchers have found that alcohol increases TLR expression in the brain and increases its sensitivity to LPS (Crews et al., 2012; Vetreno and Crews, 2012). To test how peripheral release of LPS would affect drinking behavior, Blednov et al. (2011a) injected mice with LPS and found that a single injection produces long-lasting increases in alcohol consumption consistent with neuroimmune signaling mediating the reinforcing properties of alcohol. The single injection of LPS also increased the firing rate of dopamine neurons in the ventral tegmental area, providing an example of how neuroimmune activation following peripheral LPS administration modulates brain reward circuitry (Blednov et al., 2011a). Indeed, peripheral immune function has been found to be important for other mental illnesses such as schizophrenia, depression, and autism (Dantzer et al., 2008; Kelley and Dantzer, 2011; Derecki et al., 2012; Jones and Thomsen, 2013; Takao et al., 2013); however, the mechanism by which peripheral immune activation exerts its effects on the brain is unknown and presents a field to be explored.
Immune signaling molecules are also found within the brain, where they can be used for normal, non-immune signaling, but can also reflect pathology and lead to neurodegeneration if proinflammation is left unchecked (El Khoury, 2010). Neurodegenerative effects of immune signaling have been demonstrated in Alzheimer’s and Parkinson’s disease (Carta and Pisanu, 2013; Hickman and El Khoury, 2013). Neurodegeneration has been observed in alcoholics and is especially prominent in the PFC (Fadda and Rossetti, 1998). Neurodegeneration in the PFC can affect judgment and reasoning capabilities and further exacerbate chronic alcohol consumption. It is possible that neuroimmune activation from alcohol consumption could contribute to the neurodegeneration seen in alcoholics (Blanco et al., 2005; Hua et al., 2007; Alfonso-Loeches et al., 2010; Qin and Crews, 2012).
Other evidence linking neuroimmune function with alcoholism is that immune gene expression in the brain was found to be altered in human alcoholics (Liu et al., 2006; Crews et al., 2012; Ponomarev et al., 2012), mice, and fruit flies after alcohol exposure (Qin et al., 2008; Kong et al., 2010), and rodent genetic models of high alcohol consumption (Mulligan et al., 2006; Saba et al., 2006). Furthermore, mice lacking genes related to immune function show decreased alcohol consumption compared to littermate controls (Blednov et al., 2012). Acute and chronic alcohol use activates microglia, the resident macrophages of the brain, and increases proinflammatory cytokines via nuclear factor κ B (NFκB) in the brain (Crews et al., 2011). Behavioral responses to acute alcohol were altered in mice that are lacking TLR receptors, TLR2 or TLR4 (Wu et al., 2011). In a key publication, the knockdown of TLR4 in the rat amygdala using siRNA decreased alcohol self-administration, demonstrating that neuroimmune signaling independent of input from peripheral cytokines was sufficient in regulating alcohol behavior (Liu et al., 2011).
Pioglitazone and rosiglitazone, agonists of the peroxisome proliferator activator receptor (PPAR) type gamma, have anti-inflammatory properties largely mediated through their ability to inhibit the transcription factor NFκB and thus decrease proinflammatory cytokine production (Daynes and Jones, 2002). Pioglitazone was found to reduce alcohol drinking, abolish reinstatement of alcohol seeking, reduce alcohol self-administration, and decrease the severity of physical withdrawal symptoms in rats (Stopponi et al., 2011). Another PPAR agonist, clofibrate, prevents the acquisition of nicotine self-administration in naïve rats and monkeys and decreases nicotine self-administration in nicotine-dependent rats and monkeys, suggesting that the PPAR agonist might be viable options for treating the neuroimmune pathologies in alcoholism and other forms of addiction. The pharmacologic blockade of neuroimmune activation reduces alcohol reward and decreases consumption using many types of anti-inflammatory drugs like minocycline, doxycycline, topiramate, anakinra, indomethacin, and CAPE (Agarwal, 2001; Pascual et al., 2007; Breslin et al., 2010; Wu et al., 2011; McIver et al., 2012; Blednov et al., 2013; Zalewska-Kaszubska et al., 2013). Taken together, these findings have built substantial evidence for neuroimmune modulation of acute and chronic alcohol consumption and offer unique unexplored targets for therapeutic intervention.
SUMMARY AND FUTURE DIRECTIONS
Excessive alcohol consumption causes widespread persistent changes throughout the brain. These molecular and cellular adaptations are thought to be the mechanisms by which neurons adapt to chronic alcohol use. These changes can eventually lead to alcoholism, depending on several factors, such as genetic predisposition, sex and environmental factors such as stress, age of drinking onset, and access to alcohol (Fig. 6.6). Once a person becomes an alcoholic, drinking becomes increasingly compulsive and seems to escapes voluntary control. The heterogeneous nature of alcoholism reflects the complex and multifaceted nature of alcohol’s molecular effects on the CNS. The fact that multiple neuro-transmitter systems are affected by alcohol makes it difficult to pinpoint the molecular mechanisms that are primarily responsible for the disease. Indeed, alcohol and its metabolites’ molecular targets are many and varied, and include modifications of the genome, transcriptome, and proteome. Alcoholism is most likely the result of the cumulative interactions within and between all three systems. Nevertheless, identifying the molecular targets is a crucial step in understanding alcoholism and developing new treatments. Tables 6.1–6.3 summarize all the pharmaceutic treatments, genes, and neurotransmitter systems discussed in this chapter.
Fig. 6.6.

Alcoholism is a complex disease caused by genetic and environmental factors. The neural changes characterized by alcoholism result from a complex dynamic system with a plethora of contributing factors. These factors are both environmental and internal. These internal factors depend on complex genetic states and molecular interactions. Since the normal brain is one of recursive feedback loops and regulatory controls, a drug like alcohol (whose effects are global) demonstrates radical alterations with a variety of entry points. Articulating the causes of alcoholism thus relies on research at a variety of scales and modalities. (Adapted from Starkman et al., 2012.)
Table 6.1.
Summary table of all pharmaceutic treatments discussed in this chapter
| Drug | Description | Advantages | Disadvantages |
|---|---|---|---|
| Disulfiram (Antabuse)* | Inhibitor of aldehyde dehydrogenase enzyme | Prevents alcohol consumption | Aversive therapy and does not reduce alcohol cravings, leading to poor patient compliance |
| Acamprosate (Campral)* | Mechanism of action is unclear. Proposed to be altering glutamatergic system | Few side-effects and high patient compliance | Only for patients who had already been withdrawn from alcohol. Abstinence rates are lower than naltrexone and the combination of naltrexone and acamprosate |
| Naltrexone* | Mu-opioid receptor antagonist | Reduces craving. Highest abstinent rates among FDA-approved drugs for alcoholism | Anxiety, trouble sleeping, nausea. A better response to naltrexone has been associated with possession of the G allele of the A118G polymorphism of the μ-opioid receptor gene (OPRM1) |
| Benzodiazepines† | GABA(A)-positive modulators | Treats withdrawal symptoms | Has abuse potential. Causes sedation. Cross-tolerance with alcohol |
| Ondansetron (Zofran)† | 5-HT3 receptor antagonist | May reduce alcohol consumption | |
| Fluoxetine (Prozac)† | Selective serotonin reuptake inhibitor (SSRI) | May reduce alcohol consumption in some patients (late-onset individuals) | Human studies show inconsistent results. SSRI side-effects |
| Fluphenazine† | Dopamine receptor antagonist | Reduced drinking in rodents | No human clinical trial. Typical antipsychotic side-effects |
| Aripiprazole (Abilify)† | Dopamine receptor antagonist | Aripiprazole reduces craving and is well tolerated | Atypical antipsychotic side-effects |
| Topiramate (Topamax)† | Mechanism of action is unclear. Associated with glutamatergic system inhibition | Successful in treating volunteers who were drinking heavily | May effect memory/thinking and cause sedation |
| Baclofen (Kemstro, Lioresal, Liofen, Gablofen, Beklo and Baclosan)† | GABA(B) agonist | Effectiveness is controversial | |
| Pioglitazone (Actos)† Rosiglitazone (Avandia)† Clofibrate (Atromid-S)† |
Peroxisome proliferator activator receptor (PPAR) gamma agonists (pioglitazone and rosiglitazone) PPAR alpha agonist (clofibrate) |
Drugs have been found to be hepatoprotective and neuroprotective in rodents | No published human data |
| Minocycline (Minocin)† Doxycycline (Vibramycin, Monodox, Oracea, Doryx, Vibrox, Adoxa, Doxyhexal, Doxylin, Doxoral, Doxy-1 and Atridox)† Anakinra (Kineret)† Indomethacin (Indocin)† CAPE |
Anti-inflammatory antibiotics | Well tolerated | No human data. Alters metabolism of oral contraceptives |
| Gabapentin† | GABA analog, calcium channel and GABA modulator | Increases sleep quality in alcoholics. Analgesic and anxiolytic | Causes sedation. Does not affect mood or craving |
Food and Drug Administration (FDA)-approved for treatments of alcoholism.
FDA-approved for the treatment of other diseases.
GABA, γ-aminobutyric acid.
Table 6.3.
Summary of neurotransmitter systems discussed in this chapter
| Glutamate | |
| Acute alcohol inhibits NMDA receptor function, while chronic use of alcohol upregulates NMDA receptor expression in the brain | Lovinger et al., 1989; Qiang and Ticku, 2005 |
| Alcohol acts as a non-competitive inhibitor of the AMPA/kainate receptors at high concentrations | Dildy-Mayfield and Harris, 1992, 1995; Akinshola et al., 2003 |
| MK-801, an NMDA antagonist, mimics the subjective effects of alcohol in animals | Butelman et al., 1993 |
| Ketamine, an NMDA antagonist, mimics the behavioral effects of alcohol in detoxified alcoholics | Krystal et al., 1998 |
| Chronic alcohol increases the expression of AMPA1 receptor subunit in the brains of alcoholics | Lewohl et al., 2000 |
| NMDA receptors containing specific subunits are more sensitive to alcohol than others | Raeder et al., 2008 |
| Alterations in subunit composition of glutamate receptors were seen after chronic use of alcohol in mice | Ortiz et al., 1995 |
| Dopamine | |
| Acute alcohol exposure activates dopamine reward pathways, whereas chronic treatment produces a hypodopaminergic state associated with dysphoria, which could lead to craving and relapse | Koob and Volkow, 2010 |
| Alcohol increases dopaminergic activity in the midbrain region of rodents and humans | Boileau et al., 2003 |
| Dopamine release in the midbrain partially mediates the positive reinforcing properties of acute alcohol exposure | Raeder et al., 2008 |
| Alcohol-preferring rats release more DA than wild-type rats in an alcohol self-administration study | Weiss et al., 1993 |
| Mice lacking different DA receptors and transporters show modified alcohol preference compared with controls | Crabbe et al., 2006 |
| Chronic alcohol treatment decreases DA and its metabolite in the striatum, decreases tyrosine hydroxylase protein levels, and increases DA transporter protein levels in rats compared to controls | Rothblat et al., 2001 |
| PET scans show that chronic alcoholics have fewer D2 receptors when compared with non-alcoholics | Volkow et al., 2002 |
| The DA receptor antagonist fluphenazine will block alcohol self-administration when injected into the nucleus accumbens | Rassnick et al., 1992 |
| Aripiprazole, an atypical antipsychotic, reduces craving and increases positive subjective feelings in alcoholics | Martinotti et al., 2007, Brunetti et al., 2012 |
| GABA(A) | |
| Behavioral intoxication is mimicked by the administration of pharmacologic GABA(A) agonists like muscimol and benzodiazepines | Grobin et al., 1998 |
| The effects of alcohol can be diminished by using pharmacologic GABA antagonists like bicuculline and picrotoxin | Hyytia and Koob, 1995 |
| Chronic alcohol use causes changes in GABA receptor subunit composition in human alcoholics | Lewohl et al., 1997 |
| Alcohol increases GABAergic neurotransmission over a wide range of concentrations, with some studies showing that delta-containing receptors are more sensitive than other GABA(A) receptors | Wallner et al., 2003 |
| Alpha-2 and alpha-3 subunits take part in mediating the motor-impairing effects of alcohol | Blednov et al., 2011b |
| Alpha-2 subunit takes part in the aversive properties of alcohol | Blednov et al., 2013 |
| GABA(B) | |
| Acute alcohol increases inhibitory GABA(B) signaling in a concentration-dependent manner in rat midbrain dopaminergic neurons in a current and single-electrode voltage clamp electrophysiologic experiment | Federici et al., 2009 |
| Silencing the GABA(B) receptor in fruit flies using RNA interference decreases acute motor impairment after alcohol exposure | Dzitoyeva et al., 2003 |
| Cerebellar injections in mice of GABA(B) agonists accentuated and antagonists attenuated motor impairment following acute alcohol | Dar, 1996 |
| CA1 pyramidal neuron recordings indicated that GABA(B) signaling was immune to the acute effects of alcohol but became modulated after chronic exposure | Frye et al., 1991 |
| Baclofen, a GABA(B) agonist, reduces relapse in dependent humans and decreases alcohol consumption in rats | Addolorato et al., 2002a, b; Maccioni and Colombo, 2009; Agabio et al., 2012 |
| GABA(B) has complex alternative splicing leading to several splice variants in the alcoholic brain. Alternative splicing might contribute to the pharmacologic effects of GABA(B) agonists such as baclofen in the treatment of alcoholism | Lee et al., 2013 |
| Serotonin (5-HT) | |
| Acute alcohol potentiates 5-HT3 receptor | Lovinger, 1991; Lovinger and White, 1991; Lovinger and Zhou, 1994; Harris et al., 1995; Sung et al., 2000 |
| Serotonin levels in animal brains are elevated after acute alcohol exposure | Murphy et al., 1982; LeMarquand et al., 1994 |
| Mice lacking the 5-HT1B serotonin receptor consume larger amounts of alcohol compared to wild type | Crabbe et al., 1996 |
| 5-HT2 antagonists selectively decrease acute alcohol reinforcement | Roberts et al., 1998 |
| Blockade of the serotonin transporter (5-HTT) with selective serotonin reuptake inhibitors (SSRIs), or by genetic knockout, decreases alcohol consumption in rats and partially in humans | Brown et al., 1979; Sellers et al., 1992; Kelai et al., 2003; Ciccocioppo, Economidou et al., 2006; Kranzler et al., 2012 |
| Selective antagonists of the 5-HT3 receptor decrease alcohol self-administration and consumption in rodents | Fadda et al., 1991; Hodge et al., 1993; Sellers et al., 1994 |
| Selective antagonists of the 5-HT3 receptor decrease the amount of drinks per day and increase the amount of abstinence time in alcoholics. Also, decreases the subjective pleasurable effects of alcohol and the desire to drink | Johnson et al., 1993, 2000; Sellers et al., 1994 |
| The effect was greater in alcoholics with the LL genotype than the SS or LS genotype of the 5′-regulatory region of the serotonin transporter gene | Johnson et al., 2011 |
NMDA, N-methyl-D-aspartate; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; DA, dopamine; PET, positron emission tomography; GABA, γ-aminobutyric acid.
An outstanding question that remains is: what could be responsible for the persistent drug memory that is able to linger for years and causes relapse despite all the negative consequences of alcoholism? Molecular turnover (except for DNA) is relatively rapid. DNA sequence is not changed by drug exposure, but DNA protein (chromatin) structure can be changed by environmental events, including drugs, and these epigenetic mechanisms provide promising approaches to the long-lasting effects of drug dependence. Genomics, proteomics, and transriptomics are promising fields that will help elucidate the molecular mechanisms of alcoholism.
It is clear today that the GABA and glutamate systems play a major role in alcohol’s acute anxiolytic effects, as well as withdrawal-induced hyperexcitability that leads to seizures, neurotoxicity, and cellular death. It is hypothesized that dopamine partly mediates the acute rewarding effects of alcohol during intoxication, the anhedonic state during the development of alcoholism, and the fear of anhedonia during withdrawal, potentially leading to relapse. Cross-tolerance of alcohol with barbiturates suggests overlapping pharmacologic actions involving GABA(A) receptors, but the exact binding domain for alcohol on its primary targets remains to be discovered.
Alcohol’s propensity for modifying the neural landscape is reflected by its many responsive molecules and signaling pathways, such as receptors, kinases, scaffolding proteins, neurotransmitters, hormones, chaperones, transcription factors, and cytokines. Establishing causality between alcohol and the following cellular and behavioral adaptation has proven elusive, and identifying the small effects on each system that culminate in alcoholism is a complex challenge. The availability of a large variety of animal models (ranging from flies and worms to primates), as well as the advancement of neurobiologic techniques (including gene expression micro-array and whole-genome RNA sequencing proteomics) provides hope for discovering the molecular mechanisms underpinning the neurobiology of alcoholism and revealing new therapeutic targets for this societal health burden.
Table 6.2.
Summary table of genes discussed in this chapter
| Gene symbol | Gene name | Function | Reference |
|---|---|---|---|
| CYP2E1 | Cytochrome P450 | Involved in ethanol metabolism | Zakhari, 2006 |
| ADH | Alcohol dehydrogenase | Involved in ethanol metabolism. A primary target of alcohol. Binds alcohol through a zinc atom on ADH and the hydroxyl group of alcohol | Zakhari, 2006 |
| CBP | CREB-binding protein | Allows for transcriptional activation through histone acetylase activity. Acute alcohol exposure increases levels of CBP. Withdrawal produces the opposite effect | Pandey et al., 2008 |
| NPY | Neuropeptide Y | Acute alcohol exposure increases levels of NPY. Withdrawal produces the opposite effect | Pandey et al., 2008 |
| HDAC5 | Histone deacetylase 5 | Mice lacking HDAC5 become hypersensitive when chronically exposed to cocaine. Chronic cocaine administration inactivates HDAC5 by exporting it out of the nucleus, resulting in histone hyperacetylation and increased mRNA expression of HDAC5 target genes | Renthal et al., 2007 |
| NK1R | Neurokinin 1 receptor | NK1R is increased during both chronic cocaine and alcohol exposure. Silencing the translation of the NK1R using a technique known as RNA interference was found to reduce alcohol drinking in mice | Baek et al., 2010 |
| NK1R antagonist (L822429) decreased voluntary alcohol consumption, suppressed stress-induced reinstatement of alcohol seeking, and increased sensitivity to the sedative effects of alcohol in rats | Schank et al., 2011 | ||
| An NK1R antagonist suppressed spontaneous alcohol cravings, improved overall well-being, blunted cravings induced by a challenge procedure, and attenuated concomitant cortisol responses in detoxified alcoholic inpatients. An analysis of brain function in human alcoholics compared with controls, while performing behavioral emotional tasks, was performed using functional magnetic resonance imaging and corroborated the above results, suggesting NK1R antagonism as a potential therapeutic target for the treatment of alcoholism | George et al., 2008 |
References
- Addolorato G, Caputo F, Capristo E, et al. Baclofen efficacy in reducing alcohol craving and intake: a preliminary double-blind randomized controlled study. Alcohol Alcohol. 2002a;37:504–508. doi: 10.1093/alcalc/37.5.504. [DOI] [PubMed] [Google Scholar]
- Addolorato G, Caputo F, Capristo E, et al. Rapid suppression of alcohol withdrawal syndrome by baclofen. Am J Med. 2002b;112:226–229. doi: 10.1016/s0002-9343(01)01088-9. [DOI] [PubMed] [Google Scholar]
- Agabio R, Maccioni P, Carai MA, et al. The development of medications for alcohol-use disorders targeting the GABAB receptor system. Recent Pat CNS Drug Discov. 2012;7:113–128. doi: 10.2174/157488912800673137. [DOI] [PubMed] [Google Scholar]
- Agarwal DP. Genetic polymorphisms of alcohol metabolizing enzymes. Pathol Biol (Paris) 2001;49:703–709. doi: 10.1016/s0369-8114(01)00242-5. [DOI] [PubMed] [Google Scholar]
- Akinshola BE, Yasuda RP, Peoples RW, et al. Ethanol sensitivity of recombinant homomeric and heteromeric AMPA receptor subunits expressed in Xenopus oocytes. Alcohol Clin Exp Res. 2003;27:1876–1883. doi: 10.1097/01.ALC.0000098874.65490.52. [DOI] [PubMed] [Google Scholar]
- al Qatari M, Khan S, Harris B, et al. Acamprosate is neuroprotective against glutamate-induced excitotoxicity when enhanced by ethanol withdrawal in neocortical cultures of fetal rat brain. Alcohol Clin Exp Res. 2001;25:1276–1283. doi: 10.1097/00000374-200109000-00006. [DOI] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, et al. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci. 2010;30:8285–8295. doi: 10.1523/JNEUROSCI.0976-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameisen O. Complete and prolonged suppression of symptoms and consequences of alcohol-dependence using high-dose baclofen: a self-case report of a physician. Alcohol Alcohol. 2005;40:147–150. doi: 10.1093/alcalc/agh130. [DOI] [PubMed] [Google Scholar]
- Ameisen O. Suppressing addiction using high-dose baclofen, rather than perpetuating it using substitution therapy. J Psychopharmacol. 2012;26:1042–1043. doi: 10.1177/0269881111430734. author reply 1044. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5. American Psychiatric Publishing; Arlington, VA: 2013. [Google Scholar]
- Amit Z, Brown ZW, Rockman GE. Possible involvement of acetaldehyde, norepinephrine and their tetrahydroisoquinoline derivatives in the regulation of ethanol seld-administration. Drug Alcohol Depend. 1977;2:495–500. doi: 10.1016/0376-8716(77)90049-7. [DOI] [PubMed] [Google Scholar]
- Anton RF, O’Malley SS, Ciraulo DA, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA. 2006;295:2003–2017. doi: 10.1001/jama.295.17.2003. [DOI] [PubMed] [Google Scholar]
- Atkinson NS, Robertson GA, Ganetzky B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science. 1991;253:551–555. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- Baek MN, Jung KH, Halder D, et al. Artificial microRNA-based neurokinin-1 receptor gene silencing reduces alcohol consumption in mice. Neurosci Lett. 2010;475:124–128. doi: 10.1016/j.neulet.2010.03.051. [DOI] [PubMed] [Google Scholar]
- Barth KS, Malcolm RJ. Disulfiram: an old therapeutic with new applications. CNS Neurol Disord Drug Targets. 2010;9:5–12. doi: 10.2174/187152710790966678. [DOI] [PubMed] [Google Scholar]
- Blanco AM, Valles SL, Pascual M, et al. Involvement of TLR4/type I IL-1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J Immunol. 2005;175:6893–6899. doi: 10.4049/jimmunol.175.10.6893. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Harris RA. Metabotropic glutamate receptor 5 (mGluR5) regulation of ethanol sedation, dependence and consumption: relationship to acamprosate actions. Int J Neuropsychopharmacol. 2008;11:775–793. doi: 10.1017/S1461145708008584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Geil C, et al. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun. 2011a;25 (Suppl 1):S92–S105. doi: 10.1016/j.bbi.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Borghese CM, McCracken ML, et al. Loss of ethanol conditioned taste aversion and motor stimulation in knockin mice with ethanol-insensitive alpha2-containing GABA(A) receptors. J Pharmacol Exp Ther. 2011b;336:145–154. doi: 10.1124/jpet.110.171645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Ponomarev I, Geil C, et al. Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addict Biol. 2012;17:108–120. doi: 10.1111/j.1369-1600.2010.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Black M, et al. Linking GABA(A) receptor subunits to alcohol-induced conditioned taste aversion and recovery from acute alcohol intoxication. Neuropharmacology. 2013;67:46–56. doi: 10.1016/j.neuropharm.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau I, Assaad JM, Pihl RO, et al. Alcohol promotes dopamine release in the human nucleus accumbens. Synapse. 2003;49:226–231. doi: 10.1002/syn.10226. [DOI] [PubMed] [Google Scholar]
- Bonsch D, Greifenberg V, Bayerlein K, et al. Alpha-synuclein protein levels are increased in alcoholic patients and are linked to craving. Alcohol Clin Exp Res. 2005a;29:763–765. doi: 10.1097/01.alc.0000164360.43907.24. [DOI] [PubMed] [Google Scholar]
- Bonsch D, Lenz B, Kornhuber J, et al. DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport. 2005b;16:167–170. doi: 10.1097/00001756-200502080-00020. [DOI] [PubMed] [Google Scholar]
- Boulanger LM, Huh GS, Shatz CJ. Neuronal plasticity and cellular immunity: shared molecular mechanisms. Curr Opin Neurobiol. 2001;11:568–578. doi: 10.1016/s0959-4388(00)00251-8. [DOI] [PubMed] [Google Scholar]
- Breslin FJ, Johnson BA, Lynch WJ. Effect of topiramate treatment on ethanol consumption in rats. Psychopharmacology (Berl) 2010;207:529–534. doi: 10.1007/s00213-009-1683-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown ZW, Amit Z, Rockman GE. Intraventricular self-administration of acetaldehyde, but not ethanol, in naive laboratory rats. Psychopharmacology (Berl) 1979;64:271–276. doi: 10.1007/BF00427509. [DOI] [PubMed] [Google Scholar]
- Brunetti M, Di Tizio L, Dezi S, et al. Aripiprazole, alcohol and substance abuse: a review. Eur Rev Med Pharmacol Sci. 2012;16:1346–1354. [PubMed] [Google Scholar]
- Butelman ER, Baron SP, Woods JH. Ethanol effects in pigeons trained to discriminate MK-801, PCP or CGS-19755. Behav Pharmacol. 1993;4:57–60. [PubMed] [Google Scholar]
- Cargiulo T. Understanding the health impact of alcohol dependence. Am J Health Syst Pharm. 2007;64 (Suppl 3):S5–S11. doi: 10.2146/ajhp060647. [DOI] [PubMed] [Google Scholar]
- Carta AR, Pisanu A. Modulating microglia activity with PPAR-gamma agonists: a promising therapy for Parkinson’s disease? Neurotox Res. 2013;23:112–123. doi: 10.1007/s12640-012-9342-7. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Economidou D, Cippitelli A, et al. Genetically selected Marchigian Sardinian alcohol-preferring (msP) rats: an animal model to study the neuro-biology of alcoholism. Addict Biol. 2006;11:339–355. doi: 10.1111/j.1369-1600.2006.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapp P, Bhave SV, Hoffman PL. How adaptation of the brain to alcohol leads to dependence: a pharmacological perspective. Alcohol Res Health. 2008;31:310–339. [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, et al. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Phillips TJ, Feller DJ, et al. Elevated alcohol consumption in null mutant mice lacking 5-HT1B serotonin receptors. Nat Genet. 1996;14:98–101. doi: 10.1038/ng0996-98. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Phillips TJ, Harris RA, et al. Alcohol-related genes: contributions from studies with genetically engineered mice. Addict Biol. 2006;11:195–269. doi: 10.1111/j.1369-1600.2006.00038.x. [DOI] [PubMed] [Google Scholar]
- Crews FT, Zou J, Qin L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun. 2011;25 (Suppl 1):S4–S12. doi: 10.1016/j.bbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Qin L, Sheedy D, et al. High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry. 2012;73:602–612. doi: 10.1016/j.biopsych.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damez-Werno D, LaPlant Q, Sun H, et al. Drug experience epigenetically primes Fosb gene inducibility in rat nucleus accumbens. J Neurosci. 2012;32:10267–10272. doi: 10.1523/JNEUROSCI.1290-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar MS. Mouse cerebellar GABAB participation in the expression of acute ethanol-induced ataxia and in its modulation by the cerebellar adenosinergic A1 system. Brain Res Bull. 1996;41:53–59. [PubMed] [Google Scholar]
- Davies AG, Pierce-Shimomura JT, Kim H, et al. A central role of the BK potassium channel in behavioral responses to ethanol in C. elegans. Cell. 2003;115:655–666. doi: 10.1016/s0092-8674(03)00979-6. [DOI] [PubMed] [Google Scholar]
- Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–759. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- De Witte P, Littleton J, Parot P, et al. Neuroprotective and abstinence-promoting effects of acamprosate: elucidating the mechanism of action. CNS Drugs. 2005;19:517–537. doi: 10.2165/00023210-200519060-00004. [DOI] [PubMed] [Google Scholar]
- Deng XS, Deitrich RA. Putative role of brain acetaldehyde in ethanol addiction. Curr Drug Abuse Rev. 2008;1:3–8. doi: 10.2174/1874473710801010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Foroud T. Genetic strategies to detect genes involved in alcoholism and alcohol-related traits. Alcohol Res Health. 2002;26:172–180. [PMC free article] [PubMed] [Google Scholar]
- Dildy-Mayfield JE, Harris RA. Acute and chronic ethanol exposure alters the function of hippocampal kainate receptors expressed in Xenopus oocytes. J Neurochem. 1992;58:1569–1572. doi: 10.1111/j.1471-4159.1992.tb11380.x. [DOI] [PubMed] [Google Scholar]
- Dildy-Mayfield JE, Harris RA. Ethanol inhibits kainate responses of glutamate receptors expressed in Xenopus oocytes: role of calcium and protein kinase C. J Neurosci. 1995;15:3162–3171. doi: 10.1523/JNEUROSCI.15-04-03162.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285:12726–12734. doi: 10.1074/jbc.M109.086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dredge BK, Polydorides AD, Darnell RB. The splice of life: alternative splicing and neurological disease. Nat Rev Neurosci. 2001;2:43–50. doi: 10.1038/35049061. [DOI] [PubMed] [Google Scholar]
- Dzitoyeva S, Dimitrijevic N, Manev H. Gamma-aminobutyric acid B receptor 1 mediates behavior-impairing actions of alcohol in Drosophila: adult RNA interference and pharmacological evidence. Proc Natl Acad Sci U S A. 2003;100:5485–5490. doi: 10.1073/pnas.0830111100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eipper-Mains JE, Kiraly DD, Palakodeti D, et al. microRNA-Seq reveals cocaine-regulated expression of striatal microRNAs. RNA. 2011;17:1529–1543. doi: 10.1261/rna.2775511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J. Neurodegeneration and the neuroimmune system. Nat Med. 2010;16:1369–1370. doi: 10.1038/nm1210-1369. [DOI] [PubMed] [Google Scholar]
- Elmer BM, McAllister AK. Major histocompatibility complex class I proteins in brain development and plasticity. Trends Neurosci. 2012;35:660–670. doi: 10.1016/j.tins.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadda F, Rossetti ZL. Chronic ethanol consumption: from neuroadaptation to neurodegeneration. Prog Neurobiol. 1998;56:385–431. doi: 10.1016/s0301-0082(98)00032-x. [DOI] [PubMed] [Google Scholar]
- Fadda F, Garau B, Marchei F, et al. MDL 72222, a selective 5-HT3 receptor antagonist, suppresses voluntary ethanol consumption in alcohol-preferring rats. Alcohol Alcohol. 1991;26:107–110. doi: 10.1093/oxfordjournals.alcalc.a045088. [DOI] [PubMed] [Google Scholar]
- Federici M, Nistico R, Giustizieri M, et al. Ethanol enhances GABAB-mediated inhibitory postsynaptic transmission on rat midbrain dopaminergic neurons by facilitating GIRK currents. Eur J Neurosci. 2009;29:1369–1377. doi: 10.1111/j.1460-9568.2009.06700.x. [DOI] [PubMed] [Google Scholar]
- Frye GD, Taylor L, Trzeciakowski JP, et al. Effects of acute and chronic ethanol treatment on pre- and postsynaptic responses to baclofen in rat hippocampus. Brain Res. 1991;560:84–91. doi: 10.1016/0006-8993(91)91218-p. [DOI] [PubMed] [Google Scholar]
- Garbutt JC, Kampov-Polevoy AB, Gallop R, et al. Efficacy and safety of baclofen for alcohol dependence: a randomized, double-blind, placebo-controlled trial. Alcohol Clin Exp Res. 2010;34:1849–1857. doi: 10.1111/j.1530-0277.2010.01273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George DT, Gilman J, Hersh A, et al. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science. 2008;319:1536–1539. doi: 10.1126/science.1153813. [DOI] [PubMed] [Google Scholar]
- Glynn MW, Elmer BM, Garay PA, et al. MHCI negatively regulates synapse density during the establishment of cortical connections. Nat Neurosci. 2011;14:442–451. doi: 10.1038/nn.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB. The effects of drugs on membrane fluidity. Annu Rev Pharmacol Toxicol. 1984;24:43–64. doi: 10.1146/annurev.pa.24.040184.000355. [DOI] [PubMed] [Google Scholar]
- Gonzales RA, Job MO, Doyon WM. The role of mesolimbic dopamine in the development and maintenance of ethanol reinforcement. Pharmacol Ther. 2004;103:121–146. doi: 10.1016/j.pharmthera.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Horn KH, Zhou FC. Alcohol teratogenesis: mechanisms of damage and strategies for intervention. Exp Biol Med (Maywood) 2005;230:394–406. doi: 10.1177/15353702-0323006-07. [DOI] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, et al. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobin AC, Matthews DB, Devaud LL, et al. The role of GABA(A) receptors in the acute and chronic effects of ethanol. Psychopharmacology (Berl) 1998;139:2–19. doi: 10.1007/s002130050685. [DOI] [PubMed] [Google Scholar]
- Guerri C, Pascual M. Mechanisms involved in the neurotoxic, cognitive, and neurobehavioral effects of alcohol consumption during adolescence. Alcohol. 2010;44:15–26. doi: 10.1016/j.alcohol.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Hanson DJ. Preventing alcohol abuse : alcohol, culture, and control. Praeger; Westport, CT: 1995. [Google Scholar]
- Harris RA, Mihic SJ, Dildy-Mayfield JE, et al. Actions of anesthetics on ligand-gated ion channels: role of receptor subunit composition. FASEB J. 1995;9:1454–1462. doi: 10.1096/fasebj.9.14.7589987. [DOI] [PubMed] [Google Scholar]
- Harris RA, Trudell JR, Mihic SJ. Ethanol’s molecular targets. Sci Signal. 2008;1:re7. doi: 10.1126/scisignal.128re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, El Khoury J. The neuroimmune system in Alzheimer’s disease: the glass is half full. J Alzheimers Dis. 2013;33 (Suppl 1):S295–S302. doi: 10.3233/JAD-2012-129027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge CW, Samson HH, Lewis RS, et al. Specific decreases in ethanol-but not water-reinforced responding produced by the 5-HT3 antagonist ICS 205–930. Alcohol. 1993;10:191–196. doi: 10.1016/0741-8329(93)90034-l. [DOI] [PubMed] [Google Scholar]
- Hoffman PL, Iorio KR, Snell LD, et al. Attenuation of glutamate-induced neurotoxicity in chronically ethanol-exposed cerebellar granule cells by NMDA receptor antagonists and ganglioside GM1. Alcohol Clin Exp Res. 1995;19:721–726. doi: 10.1111/j.1530-0277.1995.tb01573.x. [DOI] [PubMed] [Google Scholar]
- Howard RJ, Slesinger PA, Davies DL, et al. Alcohol-binding sites in distinct brain proteins: the quest for atomic level resolution. Alcohol Clin Exp Res. 2011;35:1561–1573. doi: 10.1111/j.1530-0277.2011.01502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua F, Ma J, Ha T, et al. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol. 2007;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Hyytia P, Koob GF. GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur J Pharmacol. 1995;283:151–159. doi: 10.1016/0014-2999(95)00314-b. [DOI] [PubMed] [Google Scholar]
- Jiang L, Gulanski BI, De Feyter HM, et al. Increased brain uptake and oxidation of acetate in heavy drinkers. J Clin Invest. 2013;123:1605–1614. doi: 10.1172/JCI65153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Campling GM, Griffiths P, et al. Attenuation of some alcohol-induced mood changes and the desire to drink by 5-HT3 receptor blockade: a preliminary study in healthy male volunteers. Psychopharmacology (Berl) 1993;112:142–144. doi: 10.1007/BF02247375. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Roaches JD, Favors MA, et al. Ondansetron for reduction of drinking among biologically predisposed alcoholic patients: a randomized controlled trial. JAMA. 2000;284:963–971. doi: 10.1001/jama.284.8.963. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Rosenthal N, Capac JA, et al. Improvement of physical health and quality of life of alcohol-dependent individuals with topiramate treatment: US multisided randomized controlled trial. Arch Intern Med. 2008;168:1188–1199. doi: 10.1001/archinte.168.11.1188. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Ait-Daoud N, Seneviratne C, et al. Pharmacogenetic approach at the serotonin transporter gene as a method of reducing the severity of alcohol drinking. Am J Psychiatry. 2011;168:265–275. doi: 10.1176/appi.ajp.2010.10050755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Thomsen C. The role of the innate immune system in psychiatric disorders. Mol Cell Neurosci. 2013;53:52–62. doi: 10.1016/j.mcn.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Kelai S, Aissi F, Lesch KP, et al. Alcohol intake after serotonin transporter inactivation in mice. Alcohol Alcohol. 2003;38:386–389. doi: 10.1093/alcalc/agg095. [DOI] [PubMed] [Google Scholar]
- Kelley KW, Dantzer R. Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders. Brain Behav Immun. 2011;25 (Suppl 1):S13–S20. doi: 10.1016/j.bbi.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong EC, Allouche L, Chapot PA, et al. Ethanol-regulated genes that contribute to ethanol sensitivity and rapid tolerance in Drosophila. Alcohol Clin Exp Res. 2010;34:302–316. doi: 10.1111/j.1530-0277.2009.01093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M, Koob G, et al. Neurobiology of Addiction. Elsevier; Burlington: 2005. [Google Scholar]
- Kranzler HR, Feinn R, Armeli S, et al. Comparison of alcoholism subtypes as moderators of the response to sertraline treatment. Alcohol Clin Exp Res. 2012;36:509–516. doi: 10.1111/j.1530-0277.2011.01609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Webb E, et al. Dose-related ethanol-like effects of the NMDA antagonist, ketamine, in recently detoxified alcoholics. Arch Gen Psychiatry. 1998;55:354–360. doi: 10.1001/archpsyc.55.4.354. [DOI] [PubMed] [Google Scholar]
- Lai CC, Chang MC, Lin HH. Acute tolerance to ethanol inhibition of NMDA-induced responses in rat rostral ventrolateral medulla neurons. J Biomed Sci. 2004;11:482–492. doi: 10.1007/BF02256097. [DOI] [PubMed] [Google Scholar]
- Lee C, Mayfield RD, Harris RA. Intron 4 containing novel GABAB1 isoforms impair GABAB receptor function. PLoS One. 2010;5:e14044. doi: 10.1371/journal.pone.0014044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Mayfield RD, Harris RA. Altered GABAB1 receptor splicing in alcoholics. Biol Psychiatry. 2013 doi: 10.1016/j.biopsych.2013.08.028. In peer review (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggio L, Garbutt JC, Addolorato G. Effectiveness and safety of baclofen in the treatment of alcohol dependent patients. CNS Neurol Disord Drug Targets. 2010;9:33–44. doi: 10.2174/187152710790966614. [DOI] [PubMed] [Google Scholar]
- LeMarquand D, Pihl RO, Benkelfat C. Serotonin and alcohol intake, abuse, and dependence: findings of animal studies. Biol Psychiatry. 1994;36:395–421. doi: 10.1016/0006-3223(94)91215-7. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Lewohl JM, Crane DI, Dodd PR. Expression of the alpha 1, alpha 2 and alpha 3 isoforms of the GABAA receptor in human alcoholic brain. Brain Res. 1997;751:102–112. doi: 10.1016/s0006-8993(96)01396-0. [DOI] [PubMed] [Google Scholar]
- Lewohl JM, Wang L, Miles MF, et al. Gene expression in human alcoholism: microarray analysis of frontal cortex. Alcohol Clin Exp Res. 2000;24:1873–1882. [PubMed] [Google Scholar]
- Lewohl JM, Nunez YO, Dodd PR, et al. Up-regulation of microRNAs in brain of human alcoholics. Alcohol Clin Exp Res. 2011;35:1928–1937. doi: 10.1111/j.1530-0277.2011.01544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MD, van der Vaart AD. MicroRNAs in addiction: adaptation’s middlemen? Mol Psychiatry. 2011;16:1159–1168. doi: 10.1038/mp.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HF, Mochly-Rosen D, Kendig JJ. Protein kinase Cgamma mediates ethanol withdrawal hyper-responsiveness of NMDA receptor currents in spinal cord motor neurons. Br J Pharmacol. 2005;144:301–307. doi: 10.1038/sj.bjp.0706033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Ninan I, Antonova I, et al. alpha-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004;23:4506–4516. doi: 10.1038/sj.emboj.7600451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lewohl JM, Harris RA, et al. Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology. 2006;31:1574–1582. doi: 10.1038/sj.npp.1300947. [DOI] [PubMed] [Google Scholar]
- Liu J, Yang AR, Kelly T, et al. Binge alcohol drinking is associated with GABAA alpha2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proc Natl Acad Sci U S A. 2011;108:4465–4470. doi: 10.1073/pnas.1019020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo IA, Harris RA. GABA(A) receptors and alcohol. Pharmacol Biochem Behav. 2008;90:90–94. doi: 10.1016/j.pbb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM. Ethanol potentiation of 5-HT3 receptor-mediated ion current in NCB-20 neuroblastoma cells. Neurosci Lett. 1991;122:57–60. doi: 10.1016/0304-3940(91)90192-v. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Serotonin’s role in alcohol’s effects on the brain. Alcohol Health Res World. 1997;21:114–120. [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, White G. Ethanol potentiation of 5-hydroxytryptamine3 receptor-mediated ion current in neuroblastoma cells and isolated adult mammalian neurons. Mol Pharmacol. 1991;40:263–270. [PubMed] [Google Scholar]
- Lovinger DM, Zhou Q. Alcohols potentiate ion current mediated by recombinant 5-HT3RA receptors expressed in a mammalian cell line. Neuropharmacology. 1994;33:1567–1572. doi: 10.1016/0028-3908(94)90131-7. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- Lugli G, Larson J, Martone ME, et al. Dicer and eIF2c are enriched at postsynaptic densities in adult mouse brain and are modified by neuronal activity in a calpain-dependent manner. J Neurochem. 2005;94:896–905. doi: 10.1111/j.1471-4159.2005.03224.x. [DOI] [PubMed] [Google Scholar]
- Maccioni P, Colombo G. Role of the GABA(B) receptor in alcohol-seeking and drinking behavior. Alcohol. 2009;43:555–558. doi: 10.1016/j.alcohol.2009.09.030. [DOI] [PubMed] [Google Scholar]
- Mann K, Lehert P, Morgan MY. The efficacy of acamprosate in the maintenance of abstinence in alcohol-dependent individuals: results of a meta-analysis. Alcohol Clin Exp Res. 2004;28:51–63. doi: 10.1097/01.ALC.0000108656.81563.05. [DOI] [PubMed] [Google Scholar]
- Martin GE, Hendrickson LM, Penta KL, et al. Identification of a BK channel auxiliary protein controlling molecular and behavioral tolerance to alcohol. Proc Natl Acad Sci U S A. 2008;105:17543–17548. doi: 10.1073/pnas.0801068105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinotti G, Di Nicola M, Janiri L. Efficacy and safety of aripiprazole in alcohol dependence. Am J Drug Alcohol Abuse. 2007;33:393–401. doi: 10.1080/00952990701313660. [DOI] [PubMed] [Google Scholar]
- Mason BJ, Ownby RL. Acamprosate for the treatment of alcohol dependence: a review of double-blind, placebo-controlled trials. CNS Spectr. 2000;5:58–69. doi: 10.1017/s1092852900012827. [DOI] [PubMed] [Google Scholar]
- Mason BJ, Goodman AM, Chabac S, et al. Effect of oral acamprosate on abstinence in patients with alcohol dependence in a double-blind, placebo-controlled trial: the role of patient motivation. J Psychiatr Res. 2006;40:383–393. doi: 10.1016/j.jpsychires.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- Mayfield RD, Nunez YO. Understanding alcoholism through microRNA signatures in brains of human alcoholics. Frontiers in Genetics. 2012;3:17–29. doi: 10.3389/fgene.2012.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIver SR, Muccigrosso MM, Haydon PG. The effect of doxycycline on alcohol consumption and sensitivity: consideration for inducible transgenic mouse models. Exp Biol Med (Maywood) 2012;237:1129–1133. doi: 10.1258/ebm.2012.012029. [DOI] [PubMed] [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, et al. Sites of alcohol and volatile anaesthetic action on GABA-A and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Moriarty JD. The use of antabus in the therapy of alcoholic patients. Calif Med. 1950;73:144–147. [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Ponomarev I, Hitzemann RJ, et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc Natl Acad Sci U S A. 2006;103:6368–6373. doi: 10.1073/pnas.0510188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, McBride WJ, Lumeng L, et al. Regional brain levels of monoamines in alcohol-preferring and -nonpreferring lines of rats. Pharmacol Biochem Behav. 1982;16:145–149. doi: 10.1016/0091-3057(82)90026-0. [DOI] [PubMed] [Google Scholar]
- Myers WD, Ng KT, Marzuki S, et al. Alteration of alcohol drinking in the rat by peripherally self-administered acetaldehyde. Alcohol. 1984;1:229–236. doi: 10.1016/0741-8329(84)90103-4. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, Self DW. DeltaFosB: a sustained molecular switch for addiction. Proc Natl Acad Sci U S A. 2001;98:11042–11046. doi: 10.1073/pnas.191352698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz J, Fitzgerald LW, Charlton M, et al. Biochemical actions of chronic ethanol exposure in the mesolimbic dopamine system. Synapse. 1995;21:289–298. doi: 10.1002/syn.890210403. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, et al. Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci. 2008;28:3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang KY, Braswell LM, Chang L, et al. The perturbation of lipid bilayers by general anesthetics: a quantitative test of the disordered lipid hypothesis. Mol Pharmacol. 1980;18:84–90. [PubMed] [Google Scholar]
- Pascual M, Blanco AM, Cauli O, et al. Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. Eur J Neurosci. 2007;25:541–550. doi: 10.1111/j.1460-9568.2006.05298.x. [DOI] [PubMed] [Google Scholar]
- Peoples RW, Li C, Weight FF. Lipid vs protein theories of alcohol action in the nervous system. Annu Rev Pharmacol Toxicol. 1996;36:185–201. doi: 10.1146/annurev.pa.36.040196.001153. [DOI] [PubMed] [Google Scholar]
- Pichardo-Casas I, Goff LA, Swerdel MR, et al. Expression profiling of synaptic microRNAs from the adult rat brain identifies regional differences and seizure-induced dynamic modulation. Brain Res. 2012;1436:20–33. doi: 10.1016/j.brainres.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzykowski AZ, Friesen RM, Martin GE, et al. Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron. 2008;59:274–287. doi: 10.1016/j.neuron.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignataro L, Miller AN, Ma L, et al. Alcohol regulates gene expression in neurons via activation of heat shock factor 1. J Neurosci. 2007;27:12957–12966. doi: 10.1523/JNEUROSCI.4142-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignataro L, Varodayan FP, Tannenholz LE, et al. The regulation of neuronal gene expression by alcohol. Pharmacol Ther. 2009;124:324–335. doi: 10.1016/j.pharmthera.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinelli A, Trivulzio S, Tomasoni L. Effects of ondansetron administration on opioid withdrawal syndrome observed in rats. Eur J Pharmacol. 1997;340:111–119. doi: 10.1016/s0014-2999(97)01349-6. [DOI] [PubMed] [Google Scholar]
- Ponomarev I, Wang S, Zhang L, et al. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J Neurosci. 2012;32:1884–1897. doi: 10.1523/JNEUROSCI.3136-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang M, Ticku MK. Role of AP-1 in ethanol-induced N-methyl-D-aspartate receptor 2B subunit gene up-regulation in mouse cortical neurons. J Neurochem. 2005;95:1332–1341. doi: 10.1111/j.1471-4159.2005.03464.x. [DOI] [PubMed] [Google Scholar]
- Qin L, Crews FT. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J Neuroinflammation. 2012;9:5. doi: 10.1186/1742-2094-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, He J, Hanes RN, et al. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation. 2008;5:10. doi: 10.1186/1742-2094-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quertemont E, Tambour S, Tirelli E. The role of acetaldehyde in the neurobehavioral effects of ethanol: a comprehensive review of animal studies. Prog Neurobiol. 2005;75:247–274. doi: 10.1016/j.pneurobio.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Quintanilla ME, Tampier L. Acetaldehyde-reinforcing effects: differences in low-alcohol-drinking (UChA) and high-alcohol-drinking (UChB) rats. Alcohol. 2003;31:63–69. doi: 10.1016/j.alcohol.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Raeder H, Holter SM, Hartmann AM, et al. Expression of N-methyl-D-aspartate (NMDA) receptor subunits and splice variants in an animal model of long-term voluntary alcohol self-administration. Drug Alcohol Depend. 2008;96:16–21. doi: 10.1016/j.drugalcdep.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Rahman S. Epigenetic mechanisms: targets for treatment of alcohol dependence and drug addiction. CNS Neurol Disord Drug Targets. 2012;11:101. doi: 10.2174/187152712800269696. [DOI] [PubMed] [Google Scholar]
- Rajasethupathy P, Fiumara F, Sheridan R, et al. Characterization of small RNAs in Aplysia reveals a role for miR-124 in constraining synaptic plasticity through CREB. Neuron. 2009;63:803–817. doi: 10.1016/j.neuron.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani CS, Qiang M, Ticku MK. Potential role of cAMP response element-binding protein in ethanol-induced N-methyl-D-aspartate receptor 2B subunit gene transcription in fetal mouse cortical cells. Mol Pharmacol. 2005;67:2126–2136. doi: 10.1124/mol.104.007872. [DOI] [PubMed] [Google Scholar]
- Rassnick S, Pulvirenti L, Koob GF. Oral ethanol self-administration in rats is reduced by the administration of dopamine and glutamate receptor antagonists into the nucleus accumbens. Psychopharmacology (Berl) 1992;109:92–98. doi: 10.1007/BF02245485. [DOI] [PubMed] [Google Scholar]
- Reilly MT, Lobo IA, McCracken LM, et al. Effects of acamprosate on neuronal receptors and ion channels expressed in Xenopus oocytes. Alcohol Clin Exp Res. 2008;32:188–196. doi: 10.1111/j.1530-0277.2007.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ. Epigenetic mechanisms in drug addiction. Trends Mol Med. 2008;14:341–350. doi: 10.1016/j.molmed.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Crabbe JC. Gene expression induced by drugs of abuse. Curr Opin Pharmacol. 2005;5:26–33. doi: 10.1016/j.coph.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Roberts AJ, McArthur RA, Hull EE, et al. Effects of amperozide, 8-OH-DPAT, and FG 5974 on operant responding for ethanol. Psychopharmacology (Berl) 1998;137:25–32. doi: 10.1007/s002130050589. [DOI] [PubMed] [Google Scholar]
- Rothblat DS, Rubin E, Schneider JS. Effects of chronic alcohol ingestion on the mesostriatal dopamine system in the rat. Neurosci Lett. 2001;300:63–66. doi: 10.1016/s0304-3940(01)01548-8. [DOI] [PubMed] [Google Scholar]
- Russo SJ, Dietz DM, Dumitriu D, et al. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33:267–276. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba L, Bhave SV, Grahame N, et al. Candidate genes and their regulatory elements: alcohol preference and tolerance. Mamm Genome. 2006;17:669–688. doi: 10.1007/s00335-005-0190-0. [DOI] [PubMed] [Google Scholar]
- Sauguet L, Howard RJ, Malherbe L, et al. Structural basis for potentiation by alcohols and anaesthetics in a ligand-gated ion channel. Nat Commun. 2013;4:1697. doi: 10.1038/ncomms2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schank JR, Pickens CL, Rowe KE, et al. Stress-induced reinstatement of alcohol-seeking in rats is selectively suppressed by the neurokinin 1 (NK1) antagonist L822429. Psychopharmacology (Berl) 2011;218:111–119. doi: 10.1007/s00213-011-2201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt SM, Frezza M, Dou QP. New applications of old metal-binding drugs in the treatment of human cancer. Front Biosci (Schol Ed) 2012;4:375–391. doi: 10.2741/274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt G. microRNAs at the synapse. Nat Rev Neurosci. 2009;10:842–849. doi: 10.1038/nrn2763. [DOI] [PubMed] [Google Scholar]
- Schratt GM, Tuebing F, Nigh EA, et al. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–289. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Dynes JL, Steward O. Synaptic regulation of translation of dendritic mRNAs. J Neurosci. 2006;26:7143–7146. doi: 10.1523/JNEUROSCI.1796-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers EM, Higgins GA, Sobell MB. 5-HT and alcohol abuse. Trends Pharmacol Sci. 1992;13:69–75. doi: 10.1016/0165-6147(92)90026-3. [DOI] [PubMed] [Google Scholar]
- Sellers EM, Toneatto T, Romach MK, et al. Clinical efficacy of the 5-HT3 antagonist ondansetron in alcohol abuse and dependence. Alcohol Clin Exp Res. 1994;18:879–885. doi: 10.1111/j.1530-0277.1994.tb00054.x. [DOI] [PubMed] [Google Scholar]
- Shorter D, Kosten TR. Novel pharmacotherapeutic treatments for cocaine addiction. BMC Med. 2011;9:119. doi: 10.1186/1741-7015-9-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanagel R. Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol Rev. 2009;89:649–705. doi: 10.1152/physrev.00013.2008. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Holter SM, Allingham K, et al. Acamprosate and alcohol: I. Effects on alcohol intake following alcohol deprivation in the rat. Eur J Pharmacol. 1996a;305:39–44. doi: 10.1016/0014-2999(96)00174-4. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Putzke J, Stefferl A, et al. Acamprosate and alcohol: II. Effects on alcohol withdrawal in the rat. Eur J Pharmacol. 1996b;305:45–50. doi: 10.1016/0014-2999(96)00175-6. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Zieglgansberger W, Hundt W. Acamprosate and alcohol: III. Effects on alcohol discrimination in the rat. Eur J Pharmacol. 1996c;305:51–56. doi: 10.1016/0014-2999(96)00176-8. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Vengeliene V, Jandeleit B, et al. Acamprosate Produces its Anti-Relapse Effects via Calcium. Neuropsychopharmacology. 2013;39:783–791. doi: 10.1038/npp.2013.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina L, Longoni R, Vinci S, et al. Role of dopamine D1 receptors and extracellular signal regulated kinase in the motivational properties of acetaldehyde as assessed by place preference conditioning. Alcohol Clin Exp Res. 2010;34:607–616. doi: 10.1111/j.1530-0277.2009.01129.x. [DOI] [PubMed] [Google Scholar]
- Starkman BG, Sakharkar AJ, Pandey SC. Epigenetics-beyond the genome in alcoholism. Alcohol Res Health. 2012;34:293–305. doi: 10.35946/arcr.v34.3.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Banker GA. Getting the message from the gene to the synapse: sorting and intracellular transport of RNA in neurons. Trends Neurosci. 1992;15:180–186. doi: 10.1016/0166-2236(92)90170-d. [DOI] [PubMed] [Google Scholar]
- Steward O, Levy WB. Preferential localization of poly-ribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J Neurosci. 1982;2:284–291. doi: 10.1523/JNEUROSCI.02-03-00284.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopponi S, Somaini L, Cippitelli A, et al. Activation of nuclear PPARgamma receptors by the antidiabetic agent pioglitazone suppresses alcohol drinking and relapse to alcohol seeking. Biol Psychiatry. 2011;69:642–649. doi: 10.1016/j.biopsych.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Sulik KK. Genesis of alcohol-induced craniofacial dysmorphism. Exp Biol Med (Maywood) 2005;230:366–375. doi: 10.1177/15353702-0323006-04. [DOI] [PubMed] [Google Scholar]
- Sung KW, Engel SR, Allan AM, et al. 5-HT(3) receptor function and potentiation by alcohols in frontal cortex neurons from transgenic mice overexpressing the receptor. Neuropharmacology. 2000;39:2346–2351. doi: 10.1016/s0028-3908(00)00064-2. [DOI] [PubMed] [Google Scholar]
- Takao K, Kobayashi K, Hagihara H, et al. Deficiency of Schnurri-2, an MHC enhancer binding protein, induces mild chronic inflammation in the brain and confers molecular, neuronal, and behavioral phenotypes related to schizophrenia. Neuropsychopharmacology. 2013;38:1409–1425. doi: 10.1038/npp.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JR, Lynch WJ, Sanchez H, et al. Inhibition of Cdk5 in the nucleus accumbens enhances the locomotor-activating and incentive-motivational effects of cocaine. Proc Natl Acad Sci U S A. 2007;104:4147–4152. doi: 10.1073/pnas.0610288104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treistman SN, Martin GE. BK Channels: mediators and models for alcohol tolerance. Trends Neurosci. 2009;32:629–637. doi: 10.1016/j.tins.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, et al. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Vetreno RP, Crews FT. Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and Toll-like receptors in the adult prefrontal cortex. Neuroscience. 2012;226:475–488. doi: 10.1016/j.neuroscience.2012.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Maynard L, et al. Effects of alcohol detoxification on dopamine D2 receptors in alcoholics: a preliminary study. Psychiatry Res. 2002;116:163–172. doi: 10.1016/s0925-4927(02)00087-2. [DOI] [PubMed] [Google Scholar]
- Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41:10–13. doi: 10.1093/ije/dyr184. [DOI] [PubMed] [Google Scholar]
- Wallner M, Hanchar HJ, Olsen RW. Ethanol enhances alpha 4 beta 3 delta and alpha 6 beta 3 delta gamma-aminobutyric acid type A receptors at low concentrations known to affect humans. Proc Natl Acad Sci U S A. 2003;100:15218–15223. doi: 10.1073/pnas.2435171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Krishnan HR, Ghezzi A, et al. Drug-induced epigenetic changes produce drug tolerance. PLoS Biol. 2007;5:e265. doi: 10.1371/journal.pbio.0050265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Henriksson R, Ohnishi YN, et al. FOSB proteins in the orbitofrontal and dorsolateral prefrontal cortices of human alcoholics. Addict Biol. 2009;14:294–297. doi: 10.1111/j.1369-1600.2009.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss F, Lorang MT, Bloom FE, et al. Oral alcohol self-administration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther. 1993;267:250–258. [PubMed] [Google Scholar]
- Wilke N, Sganga M, Barhite S, et al. Effects of alcohol on gene expression in neural cells. EXS. 1994;71:49–59. doi: 10.1007/978-3-0348-7330-7_6. [DOI] [PubMed] [Google Scholar]
- Winstanley CA, LaPlant Q, Theobald DE, et al. DeltaFosB induction in orbitofrontal cortex mediates tolerance to cocaine-induced cognitive dysfunction. J Neurosci. 2007;27:10497–10507. doi: 10.1523/JNEUROSCI.2566-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolstenholme JT, Warner JA, Capparuccini MI, et al. Genomic analysis of individual differences in ethanol drinking: evidence for non-genetic factors in C57BL/6 mice. PLoS One. 2011;6:e21100. doi: 10.1371/journal.pone.0021100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Lousberg EL, Moldenhauer LM, et al. Attenuation of microglial and IL-1 signaling protects mice from acute alcohol-induced sedation and/or motor impairment. Brain Behav Immun. 2011;25 (Suppl 1):S155–S164. doi: 10.1016/j.bbi.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol Res Health. 2006;29:245–254. [PMC free article] [PubMed] [Google Scholar]
- Zalewska-Kaszubska J, Bajer B, Gorska D, et al. Effect of repeated treatment with topiramate on voluntary alcohol intake and beta-endorphin plasma level in Warsaw alcohol high-preferring rats. Psychopharmacology (Berl) 2013;225:275–281. doi: 10.1007/s00213-012-2812-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Verdoorn TA, Lovinger DM. Alcohols potentiate the function of 5-HT3 receptor-channels on NCB-20 neuroblastoma cells by favouring and stabilizing the open channel state. J Physiol. 1998;507:335–352. doi: 10.1111/j.1469-7793.1998.335bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]