

Abstract
Chemoattractant-induced reactive oxygen species (ROS) generation by adherent neutrophils occurs in two phases: the first is very rapid and transient, and the second one is delayed and lasts up to 30–40 min. We examined the role of phosphoinositide 3-kinases (PI3Ks) and Src-family kinases (SFKs) in these responses using human neutrophils treated with inhibitory compounds or murine neutrophils deficient of PI3Kγ or Hck, Fgr, and Lyn. Our studies show that PI3Kγ is indispensable for the early, fMLF-induced ROS generation and AKT and ERK phosphorylation, but is dispensable for the late response to fMLF. Additionally, the response to TNF, an agonist triggering only the delayed phase of ROS generation, was also unaffected in PI3Kγ-deficient neutrophils. In contrast, inhibition of SFKs by a selective inhibitor in human, or SFK deficiency in murine, neutrophils resulted in the inhibition of both the early and late phase of ROS generation, without affecting the early phase of AKT phosphorylation, but inhibiting the late one. Selective inhibitors of PI3Kα and PI3Kδ markedly reduced both the early and late response to fMLF and TNF in human neutrophils. These findings suggest that class IA PI3Ks may be activated by PI3Kγ via Ras in the early phase of the response and by SFKs in the late phase. The evidence that inhibition of SFKs in human, or SFK deficiency in murine, neutrophils results in suppression of Vav phosphorylation at all time points of the response to fMLF or TNF suggests that SFKs are indispensable for Vav phosphorylation.
Myeloid leukocytes generate reactive oxygen species (ROS) upon interaction of a wide variety of ligands with specific surface receptors due to the activation of a multimeric NADPH oxidase, also referred to as NOX2, that transfers electrons from NADPH to molecular oxygen (1). Besides exerting microbicidal actions, ROS are cytotoxic and, via oxidative inactivation of tyrosine and serine/threonine phosphatases, enhance protein kinase activities, leading to activation of transcription factors and amplification of inflammation (2).
Early studies by Nathan and Ding (3) made the paradigm that neutrophil ROS generation, in response to chemoattractants, occurs in two phases. The first phase is very rapid, transient, independent of cell adhesion to cell and extracellular-matrix integrin counterreceptors and enhanced by priming agents such as LPS, cytokines, and the F-actin depolimerizing mycotoxin cytochalasin B (CB). In contrast, the second phase ensues after several minutes, lasts up to 30–40 min, is strictly dependent on engagement of neutrophil integrins (4), and inhibited by CB (reviewed in Ref. 5). A few cytokines, such as TNF, are able to trigger the late integrin-dependent response selectively.
Studies in mice with the genetic deficiencies of various signal transduction components have established the paradigm that the early phase of neutrophil ROS generation in response to chemoattractants is absolutely dependent on the γ isoform of phosphoinositide 3-kinase (PI3Kγ) (6), whereas the adhesion, integrin-dependent late response requires Src-family kinases (SFKs) (7) and Syk (8), a cytoplasmic tyrosine kinase acting downstream of SFKs (9, 10). Later studies suggested a more complex scenario, providing evidence that in primed neutrophils, also the PI3K IA isoforms α, β, and δ may be stimulated by the chemoattractant fMLF, and SFKs may be implicated in this pathway (11, 12). Additionally, adhesion-dependent neutrophil ROS generation in response to TNF was reported to depend on class IA PI3Ks (13), and SFKs were implicated also in the early, adhesion-independent phase of fMLF-induced superoxide generation (14).
Mechanisms by which PI3Ks and SFKs regulate NADPH oxidase activity are only partly understood. Phosphatidylinositol phosphorylated at the 3 position of the inositol ring (PIP3), formed by PI3Ks, provides a binding site for the PX domain of cytosolic NADPH oxidase components (1). Activation of the Ser/Thr kinase AKT following its binding to PIP3 via its pleckstrin homology domain results in phosphorylation of p47phox in its autoinhibitory region (1, 15). Finally, PIP3 formation by PI3Ks regulates activation of at least three different guanine nucleotide exchange factors (GEFs) for the small GTP-binding protein Rac, a component essential for activation of NADPH oxidase (1, 16–18). Notably, neutrophils deficient of one of these three GEFs (i.e., P-Rex1, Vav proteins, and DOCK2) are markedly defective in NADPH oxidase activation (19–23).
Mechanisms of activation of NADPH oxidase by SFKs are poorly understood. In the context of the classical chemoattractant-induced early phase of ROS generation, SFKs have been reported to regulate activation of PI3Ks (11, 12, 24). Additionally, SFKs are required for chemoattractant-induced Vav phosphorylation and Rac activation and neutrophils deficient of Vav1 or Hck and Fgr have a defect in NADPH oxidase activation by fMLF (14, 19). Class IA PI3Ks and Vav proteins have been reported to also regulate the integrin-dependent phase of neutrophil activation (12, 13, 25–27). An additional important substrate of the integrin/SFKs/Syk signaling pathway regulating this late phase of ROS generation has been recently identified as phospholipase C (PLC) γ2 (28).
In this report, we addressed the role of PI3Ks and SFKs in the early and late phase of ROS generation by unprimed human and murine neutrophils in response to the chemoattractant fMLF and TNF. Using inhibitory compounds, we found that PI3Kγ and SFKs play essential and nonredundant roles in activation of the early and the late phases of ROS generation in human neutrophils. Deficiency of PI3Kγ resulted in a total suppression of the early phase of ROS generation in response to fMLF in mouse neutrophils but did not affect the adhesion-dependent late phase in response to different agonists. Conversely, the combined deficiency of Hck, Fgr, and Lyn resulted in an almost total or total suppression of both the early and the late phases of ROS generation, respectively, an effect that, based on the use of selective inhibitors, was largely dependent on the class IA PI3Ks PI3Kα and PI3Kδ, as well as the Rac GEF Vav. Collectively, these findings help define the mechanisms of regulation of ROS production by PI3Ks and SFKs.
Materials and Methods
Cell preparation
Generation and maintenance of hck−/−fgr−/−lyn−/− triple-knockout mice and PI3Kγ knockout mice in the C57BL/6J background were as described (6, 29). Wild-type and knockout animals used in the experiments were at 8–10 wk of age. Animals were housed at a pathogen-free facility at the University of Verona or Turin and treated according to protocols approved by the Minister of Health of Italy and the university animal care committee. Mouse bone marrow neutrophils were isolated by centrifugation of bone marrow cells flushed from femurs and tibias over a Percoll discontinuous density gradient (Amersham, Arlington Heights, IL) exactly as described previously (14). After isolation, neutrophils were washed in Ca2+/Mg2+-free HBSS then resuspended at 10 × 106/ml in HBSS supplemented with 0.5 mM CaCl2 and 5 mM D-glucose (HGCa). Routinely, cell suspensions were left at room temperature for 1 h before assay. Human PMNs were prepared from buffy coats of healthy volunteers by centrifugation through Ficoll Paque Plus (Amersham). Patients provided their informed consent before samples were taken. Contaminating erythrocytes were removed by Dextran 500 (Amersham) sedimentation followed by hypotonic lysis. After isolation, cells were suspended in HGCa at 10 × 106/ml and left at room temperature for 1 h before assay. Neutrophils were either left untreated or incubated for 10 min at 37°C with 10 μM 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) (Calbiochem, Darmstadt, Germany), 100 nM wortmannin (Calbiochem), 1 μM diphenyleneiodonium chloride (Calbiochem), or 50 U/ml superoxide dismutase (Sigma-Aldrich, St. Louis, MO). For experiments with selective inhibitors of different PI3K isoforms, human neutrophils were pretreated for 10 min at room temperature with the PI3Kγ inhibitor AS604850 (Alexis Biochemical, San Diego, CA), the PI3Kα inhibitor compound 15e (Alexis Biochemical), the PI3Kβ inhibitor TGX22 (Alexis Biochemical), the PI3Kδ inhibitor IC87114 (Symansis, Auckland, New Zealand), or the broad-specificity PI3K inhibitor compound PI103 (Cayman Chemical, Montigny-le-Bretonneux, France) at the concentration indicated in the text.
Measurement of ROS production
Isoluminol-based chemiluminescence assays (30) were performed in HGCa in the presence of HRP and isoluminol (both from Sigma-Aldrich) in a temperature-controlled Multilabel Count Victor X5 (PerkinElmer). Assays were run in dark 96-well plates precoated with 250 μg/ml human fibrinogen (14) in HGCa containing (final concentration): 100 μM isoluminol and 8 U/ml HRP. After addition of 1 × 105 human neutrophils or 7.5 × 105 mouse neutrophils, plates were left at 37°C for 5–10 min and the reaction started by addition of the stimulus (see text for concentrations). Light emission was recorded every minute.
Active Ras pulldown assay
Measurement of active RAS (Ras-guanosine triphosphate) was carried out using a RAS Activation assay kit (EMD Millipore; Merck, Darmstadt, Germany) according to the manufacturer's instructions. Neutrophils were stimulated in suspension with 5 μM fMLF, and, at different time points, cells were lysed in ice cold lysis buffer. Cell lysates were centrifuged at 4°C for 5 min at 13,000 rpm, and the supernatant was incubated with glutathione GST-RAF-RBD coupled to glutathione agarose for 1 h at 4°C. Proteins bound to the beads were washed three times in lysis buffer, and the amount of bound proteins was quantified by Western blot analysis.
Western blotting
Murine or human neutrophils (10 × 106/ml) in HGCa were plated in 24-well plates precoated with human fibrinogen (Sigma-Aldrich) and stimulated with fMLF (see text) or human (10 ng/ml) (PeproTech, Rocky Hill, NJ) or murine TNF (5 ng/ml) (BioSource International, Camarillo, CA) for the time indicated in the Results. At the appropriate time, cell activation was stopped by addition of a half volume of ice-cold HBSS (without Ca2+ and glucose) containing a 3-fold concentration of protease inhibitors (Roche Molecular Biochemicals, Mannheim, Germany) supplemented with 3 mM Na3VO4, 30 μM phenylarsine oxide (Sigma-Aldrich), and 3 mM diisopropyl-fluorophosphate (Sigma-Aldrich). Samples were kept in ice for 10 min before lysis with 4× Sample Buffer. Samples were separated on SDS-PAGE gels and transferred to nitrocellulose Hybond C (Amersham). After quenching with 3% BSA in TBS for 1 h, blots were incubated overnight at 4°C with primary Abs, followed by HRP-conjugated donkey anti-rabbit or goat anti-mouse Abs (Amersham). Immunoreactivity was detected using Immobilon Western detection reagent (Millipore) and analyzed with a Quant Image LAS400 (GE Healthcare). Abs used in this study were as follows: anti-phosphospecific Ab directed against Akt (Ser473), p38 MAPK (Thr180/Tyr182), and ERK 1/2 MAPK (Thr202/Tyr204) were from Cell Signaling Technology (Beverly, MA). Anti-protein Abs directed against Akt and p38 were from Cell Signaling Technology. Anti-protein Abs directed against ERK1/2 were from Santa Cruz Biotechnology. Anti-Vav Abs were from Upstate Biotechnology, and anti-phosphospecific Ab directed against Vav (Y174) was from EnoGene Biotech (New York, NY).
Cell spreading
Human or murine neutrophils in HGCa were plated in 24-well tissue-culture plastic in the absence of any stimulus. Photos were taken with a ×40 phase-contrast objective with a Digital Compact Olympus (Olympus, Hamburg, Germany) camera.
Statistics
The Student t test has been applied to examine the statistical significance of differences between most of the data with the exception of the Ras activation data, which were examined by the two-way ANOVA with Bonferroni post hoc test. Values of *p < 0.05 or **p < 0.01 were taken as significant.
Results
ROS generation in response to fMLF by unprimed human neutrophils occurs in two distinct phases that require PI3K and SFK activities
In this study, ROS generation was assayed with a very sensitive chemiluminescence assay (30) that allowed us to monitor neutrophil ROS generation in the absence of any priming agent. fMLF-stimulated ROS generation by human unprimed neutrophils plated in fibrinogen-coated well plates occurred in two phases (Supplemental Fig. 1A–D). One was very rapid and, after a peak at ∼0.5–1 min, declined rapidly. The second phase became detectable within 5–10 min and steadily increased up to 25–30 min when a plateau was reached. The detected chemiluminescence signal was due mainly to the extracellular generation of superoxide anion by the NADPH oxidase because addition of superoxide dismutase, which catalizes the dismutation of superoxide to hydrogen peroxide, significantly suppressed the response (Supplemental Fig. 1A). Addition of diphenyleneiodonium chloride, an NADPH oxidase inhibitor, completely blocked the response.
As predicted from previous studies (see Ref. 14 and references contained therein), inhibition of SFKs by PP2 (Supplemental Fig. 1B) or PI3Ks using broad specificity inhibitors such as wortmannin (Supplemental Fig. 1C) or PI-103 (Supplemental Fig. 1D) totally inhibited both the early and the late adhesion-dependent response of human neutrophils to fMLF. Notably, the dependence of these responses on both SFKs and PI3Ks seems to be a general feature of signal transduction pathways ensuing from chemotactic peptide receptors, because similar results were obtained using C5a instead of fMLF (Supplemental Fig. 1E). Consistent with previous findings, PMA-induced ROS generation was not inhibited by PP2 or wortmannin (Supplemental Fig. 1F).
SFKs are not implicated in activation of class IA PI3Ks in the course of the early response to fMLF
Recent studies have demonstrated that class IA PI3Ks may be activated, either directly or indirectly, by trimeric G protein–coupled receptors (see Ref. 31). Notably, in TNF-primed neutrophils, fMLF triggers two phases of PIP3 formation, the second of which peaks at 1 min, is dependent on class IA PI3Ks, and inhibited by the SFK inhibitor PP1 (11). We therefore examined the effect of a SFK inhibitory drug or SFK genetic deficiency on phosphorylation of the PI3K downstream target AKT in human or murine neutrophils, respectively (Fig. 1). In our assay conditions, fMLF triggered a robust AKT phosphorylation on the S473 residue within 1 min, which then declined but remained detectable up to 30 min in human neutrophils. Whereas in wortmannin-treated neutrophils AKT phosphorylation was totally blunted at all time points tested (Fig. 1A), the SFK inhibitor PP2 only slightly decreased AKT phosphorylation at 1 min, but had a strong inhibitory effect at later time points (Fig. 1B). In three distinct experiments (Fig. 1C), AKT phosphorylation in PP2-treated neutrophils was 69.0 ± 6.9% at 1 min, compared with untreated cells; however, this difference was not statistically significant. Consistently, deficiency of Hck, Fgr, and Lyn, the three SFKs expressed at the highest levels in neutrophils, had a minor effect on fMLF-induced AKT phosphorylation at 1 min (Fig. 1D), but resulted in a significantly reduced AKT phosphorylation at later time points. We conclude that activation of PI3Ks occurring in the first phase of oxidant generation (within 1 min) is mainly independent of SFK activity. However, PI3K activity preceding, or concomitant with, the second adhesion-dependent phase of oxidant generation requires SFKs.
Figure 1.
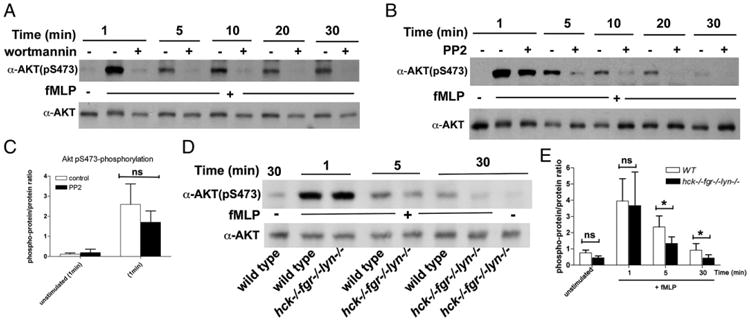
Different roles of PI3K and SFK activities in fMLF-induced AKT phosphorylation. Human (A–C) or murine (D) neutrophils were plated in fibrinogen-coated wells and stimulated with 1 or 5 μM fMLF for human or mouse cells, respectively, in the presence or the absence of PP2 or wortmannin as described in Materials and Methods. After different times of incubation, cells were lysed and lysates subjected to immunoblot analysis with Abs of the indicated specificity as described in Materials and Methods. (C) reports densitometric analysis, expressed as ratio between the phosphoprotein versus the total specific protein signal, of AKT phosphorylation at 1 min after the addition of fMLF in neutrophils treated or not with 10 μM PP2. Mean results ± SD of three independent experiments are reported. In (E), histograms at the right of the immunoblot report densitometric analysis, expressed as ratio between the phosphoprotein versus the total specific protein signal, of AKT phosphorylation in wild-type versus hck−/−fgr−/−lyn−/− neutrophils. Mean results ± SD of three independent experiments are reported. *p < 0.05.
PI3Kγ regulates activation of Ras and phosphorylation of the Ras downstream target ERK proteins
Because class IA PI3Ks may also be activated via an increase in the GTP-bound form of Ras (31), and fMLF has been demonstrated to activate GDP/GTP exchange on Ras (32), we examined fMLF-stimulated phosphorylation of the Ras downstream target ERK proteins and the dependence of these response on PI3K and SFK activities in human and murine neutrophils. fMLF triggered a rapid, but transient, phosphorylation of ERK proteins that peaked at 1 min and then declined to background levels in human neutrophils (Fig. 2). Consistent with previous reports (12, 14), ERK phosphorylation was not affected by PP2 (Fig. 2A), but markedly inhibited by wortmannin (Fig. 2B). fMLF also induced tyrosine phosphorylation of another MAP kinase member, p38, which peaked, similar to ERK phosphorylation, at 1 min of stimulation; however, neither PP2 nor wortmannin had any effect on this early p38 phosphorylation (Supplemental Fig. 2A, 2B).
Figure 2.
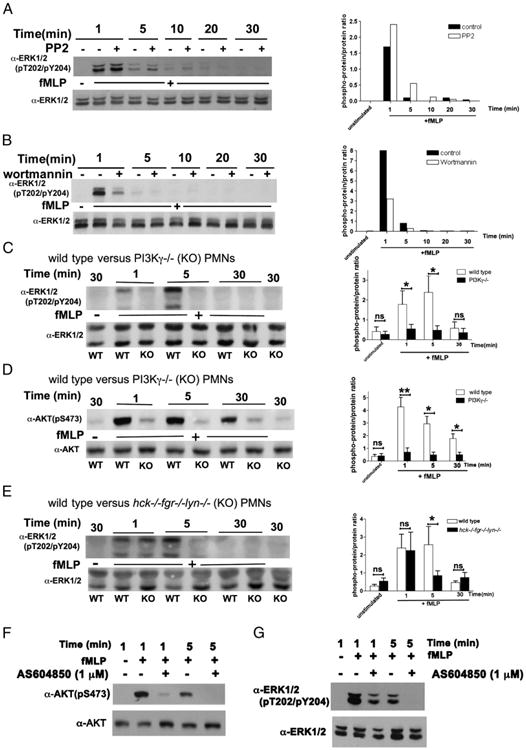
Different roles of PI3K and SFK activities in fMLF-induced ERK phosphorylation. Human or murine neutrophils were stimulated in the presence or the absence of the indicated inhibitors and lysed as described in Fig. 1 and Materials and Methods. Lysates were subjected to immunoblot analysis with Abs of the indicated specificity as described in Materials and Methods. Human neutrophils stimulated with 1 μM fMLF in the presence or the absence of PP2 (A) or wortmannin (B). Graphs to the right of the immunoblots report densitometric analysis, expressed as ratio between the phosphoprotein versus the total ERK protein signal. One representative experiment of three to four performed with identical results is reported. (C and D) Wild-type or PI3Kγ−/− murine neutrophils were left untreated or stimulated with 5 μM fMLF and lysed at the time points indicated. Graphs to the right of the immunoblots report densitometric analysis expressed as ratio between the phosphoprotein versus the total ERK (C) or AKT (D) protein signal. Mean results ± SD of three independent experiments are reported. (E) Wild-type or hck−/−fgr−/−lyn−/− neutrophils were stimulated as described above for analysis of ERK phosphorylation. Graphs to the right of the immunoblots report densitometric analysis expressed as ratio between the phosphoprotein versus the total ERK protein signal. Mean results ± SD of three independent experiments are reported. (F and G) Human neutrophils were stimulated with 1 μM fMLF in the presence or the absence of 1 μM AS604850 for analysis of AKT and ERK phosphorylation. One representative experiment of two to three performed with identical results is reported. *p < 0.05, **p < 0.01.
The time course of fMLF-stimulated ERK phosphorylation in murine neutrophils differed from that detected in human cells, displaying a maximum at 5 min (Fig. 2). ERK phosphorylation at 1 min was defective in PI3Kγ−/− neutrophils (Fig. 2C), but not in cells deficient of Fgr, Hck, and Lyn (Fig. 2E). Notably, at this time point, AKT phosphorylation was not affected by SFK deficiency (see Fig. 1D), but totally defective in PI3Kγ−/− neutrophils (Fig. 2D). At the 5-min time point after fMLF stimulation, both SFK and PI3Kγ deficiency resulted in a marked decrease of ERK phosphorylation, suggesting that they may play nonredundant roles in activation of the Ras pathway. Notably, we reproducibly found that PI3Kγ deficiency causes a marked decrease in AKT phosphorylation also at the later time points of 30 min (Fig. 2D). Although, we did not address this issue in detail, this finding suggests that, independently of the mechanism involved, prolonged AKT phosphorylation in response to fMLF requires PI3Kγ (see Discussion).
We also examined the effect of a selective inhibitor of PI3Kγ, the AS604850 compound, on ERK phosphorylation in human neutrophils. At 1 μM, AS604850 inhibited the early PI3Kγ-dependent phase of ROS generation, but not the late, adhesion-dependent, and PI3Kγ-independent one (see below). Notably, at this same concentration, AS605240 markedly inhibited (1-min time point) or totally suppressed (5-min time point) both AKT and ERK phosphorylation (Fig. 2F, 2G).
A very recent report demonstrated that fMLF activates PI3Kγ also downstream a PLCβ/diacylglycerol pathway that impinges on the Ras GEF RasGRP4, thus activating Ras (33). To strengthen the evidence emerging from our studies (Fig. 2) that positive feedback circuits of PI3K activation may also include a PI3Kγ/Ras, besides a Ras/PI3Kγ, pathway (33), we addressed whether fMLF-induced Ras activation is defective in PI3Kγ−/− neutrophils (Fig. 3). We found that deficiency of PI3Kγ resulted in a marked inhibition of GTP loading on Ras at 30 s after fMLF stimulation (i.e., at a time point preceding the peak of the early wave of ROS generation).
Figure 3.
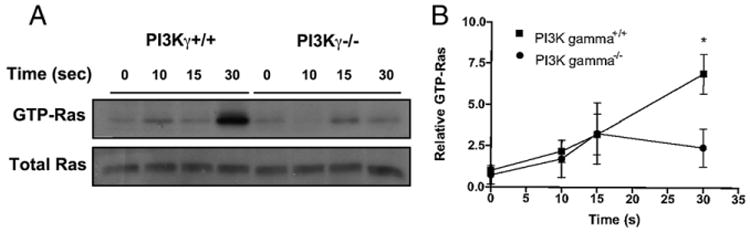
PI3Kγ deficiency results in reduced Ras activation. Neutrophils were isolated from wild-type or PI3γ knockout mice and Ras activation after fMLF (5 μM) stimulation assayed as described in Materials and Methods. (A) Representative detection of active Ras and total Ras is shown. (B) Mean results of Ras activation obtained in four independent experiments. Values were normalized by setting the percentage stimulation of PI3Kγ+/+ cells at time 0 s to 1. *p < 0.05.
Taken together, the data reported in Figs. 1–3 suggest that PI3Kγ plays a major role in the early response to fMLF and, via activation of Ras, it may establish a positive-feedback loop of PI3K activation implicating the class IA isoforms. In contrast, SFKs play only a marginal role in the regulation of PI3K activity in response to fMLF that occurs within 1 min following stimulation, which is coincident with the first wave of ROS generation (see Fig. 4 and Supplemental Fig. 1).
Figure 4.
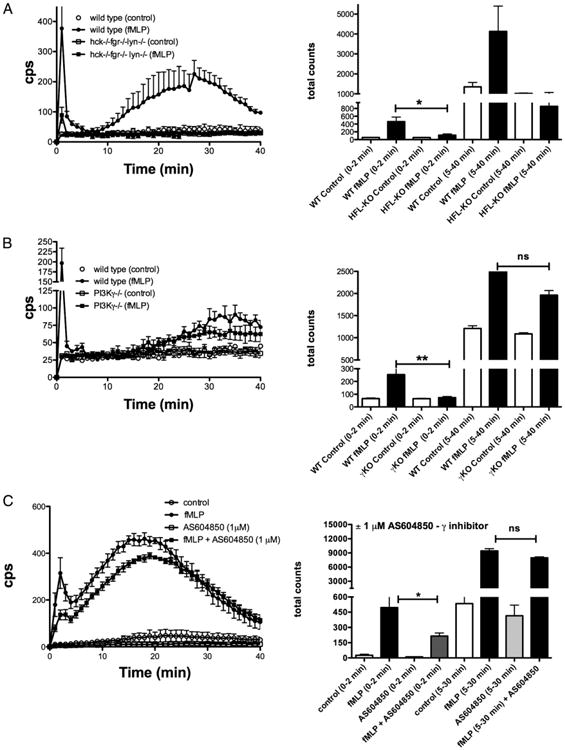
Different roles of SFKs and PI3Kγ in regulation of the early and late phase of fMLF-induced ROS generation in murine neutrophils. (A and B) Neutrophils were isolated from wild-type, hck−/−fgr−/−lyn−/− knockout or PI3γ knockout mice as described in Materials and Methods and plated in fibrinogen-coated wells. Cells were stimulated with 5 μM fMLF, and chemiluminescence was recorded every 1 min and up to 40 min. Mean results ± SD of three to four independent experiments are reported. Graphs in the right panel report the total chemiluminescence detected during the early (0–2 min) or the late (5–40 min) phase of ROS generation.(C) Human neutrophils were stimulated with 1 μM fMLF in the presence or the absence of 1 μM AS604850. Mean results ± SD of four independent experiments are reported. Graph in right panel reports the total chemiluminescence detected during the early (0–2 min) or the late (5–30 min) phase of ROS generation. *p < 0.05, **p < 0.01. KO, Knockout; WT, wild-type.
The γ isoform of class I PI3Ks is indispensable for the early phase of ROS generation in response to fMLF, but plays no role in the late, adhesion-, and SFK-dependent one
To better elucidate the role of class IA and class IB PI3Ks and SFKs in signal transduction pathways implicated in the two waves of ROS generation in response to fMLF, we examined this response in neutrophils with the genetic deficiency of PI3Kγ and SFKs. As reported in Fig. 4A, and in accord with previous studies in CB– primed hck−/−fgr−/− neutrophils (14), the early, fMLF-induced wave of ROS generation was markedly reduced in triple-knockout hck−/−fgr−/−lyn−/− neutrophils and, as shown in human cells (Supplemental Fig. 1B), totally suppressed by PP2 (data not shown). Notably, the integrin-dependent wave of oxidant generation displayed a more stringent dependency on SFK activities, and, according to previous data with hck−/−fgr−/− neutrophils (7), it was totally suppressed in hck−/−fgr−/−lyn−/− neutrophils (Fig. 4A).
Examining PI3Kγ-deficient neutrophils, we found that, as expected from previous studies (6), there was a total lack of the early response to fMLF (Fig. 4B). In contrast, the late phase of ROS generation in wild-type and PI3Kγ−/− neutrophils was comparable. Consistently, treatment of human neutrophils with the selective PI3Kγ inhibitor AS604850 at 1 μM concentration significantly inhibited the early, but not the late, adhesion-dependent phase of ROS generation (Fig. 4C).
Adhesion-dependent, TNF-stimulated ROS generation by neutrophils requires SFKs but is independent of PI3Kγ
To strengthen the above-described findings suggesting that SFKs, but not PI3Kγ, are indispensable for the late adhesion-dependent phase of oxidant generation, we examined the response to TNF, an agonist that triggers only the late, adhesion-dependent phase of neutrophil activation (3, 5, 7).
Experiments with human neutrophils treated with SFK or PI3K inhibitors showed a total dependence of the response to TNF on these kinases (Supplemental Fig. 3A, 3B). Despite some variability at the early time points (compare Supplemental Fig. 3C–E), the time course of this response was coincident with the phosphorylation of the PI3K downstream target AKT, being detectable at least after 5 min of stimulation (Supplemental Fig. 3C), a finding confirming previous studies (13), but undetectable at earlier time points (see Supplemental Fig. 3C–E). This delayed TNF-induced AKT phosphorylation was almost totally suppressed by PP2 and wortmannin in human neutrophils (Supplemental Fig. 3D, 3E). In murine neutrophils, TNF-induced AKT phosphorylation was less strong than that induced by fMLF (Supplemental Fig. 3F, 3E, compare with Figs. 1D, 2D, respectively). However, whereas deficiency of PI3Kγ did not substantially reduce AKT phosphorylation at 5 and 30 min from stimulation, deficiency of Hck, Fgr, and Lyn reduced AKT phosphorylation at these time points (Supplemental Fig. 3G).
We then examined the response of murine neutrophils plated on fibrinogen and stimulated with TNF or plated on plain tissue-culture plastic in the absence of any stimulus (Fig. 5). In these assay conditions, ROS generation by SFK-deficient neutrophils is totally defective (7 and data not shown). In contrast, deficiency of PI3Kγ did not significantly affect ROS generation by neutrophils stimulated with TNF (Fig. 5A) or plated on plain plastic in the absence of any stimulus (Fig. 5B).
Figure 5.
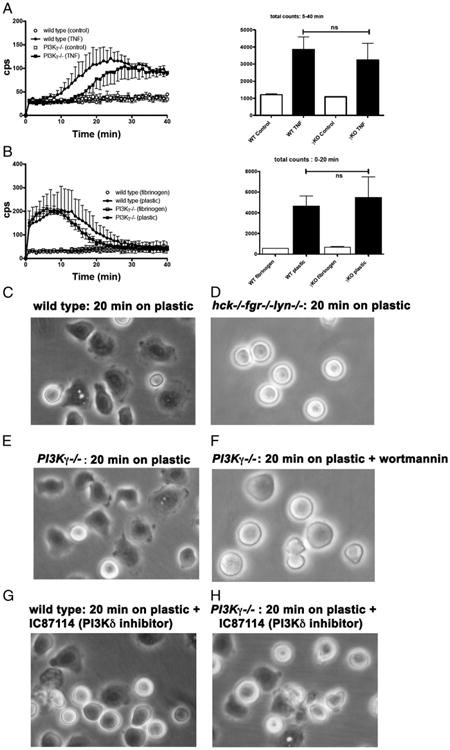
Late, TNF-induced ROS generation is independent of PI3Kγ. Neutrophils were isolated from wild-type or PI3γ knockout mice as described in Materials and Methods and plated in fibrinogen-coated wells in the presence of 5 ng/ml TNF (A) or in tissue-culture plastic wells in the absence of any stimulus (B). Mean results ± SD of four independent experiments are reported. (C–F) Neutrophils were isolated from wild-type, hck−/−fgr−/−lyn−/−knockout, or PI3γ knockout mice as described in Materials and Methods and plated in tissue-culture plastic wells in the absence of any stimulus. Note that whereas both wild-type and PI3γ knockout neutrophils spread on plastic, either deficiency of Hck, Fgr, and Lyn or treatment with wortmannin totally suppressed spreading. Wild-type (G) or PI3γ knockout (H) neutrophils were plated in tissue-culture plastic wells in the absence of any stimulus and in the presence of 0.5 μM IC 87114. Photos were taken with a ×40 objective after 20 min. KO, Knockout; WT, wild-type.
Because ROS generation by neutrophils plated on fibrinogen in the presence of TNF or on plain plastic is accompanied by cell spreading over the adherence surface, a response that was reported to depend on β2 integrins (34), we also examined neutrophil spreading on plastic. Wild-type murine neutrophils spread nicely on plastic (Fig. 5C), and deficiency of Hck, Fgr, and Lyn resulted in a total absence of this response (Fig. 5D). Notably, spreading of PI3Kγ-deficient neutrophils on plastic was comparable to that of wild-type cells (Fig. 5E), but totally suppressed by wortmannin (Fig. 5F). The selective PI3Kδ inhibitor IC87114 reduced neutrophils spreading in wild-type (Fig. 5G) and PI3Kγ-deficient (Fig. 5H) neutrophils, suggesting that this class IA PI3K isoform, together with other wortmannin-sensitive PI3Ks, regulates neutrophil spreading. A quantitation of surface area of murine neutrophils plated on tissue-culture plastic confirmed that selective PI3Kα and δ inhibitors reduced spreading of both wild-type and PI3Kγ−/− murine neutrophils (Table I). In contrast, a PI3Kβ inhibitor did not inhibit or only marginally inhibited spreading of wild-type or mutant neutrophils, respectively.
Table I. PI3Kα and δ inhibitors reduce murine neutrophil spreading on uncoated tissue-culture plastic.
| Mouse Strain | Untreated | + IC87114 (PI3Kδ Inhibitor) | + Compound 15e (PI3Kα Inhibitor) | + TGX-221 (PI3Kβ Inhibitor) |
|---|---|---|---|---|
| Wild-typea | 98.6 ± 24.3 | 69.7 ± 15.0*** | 57.1 ± 7.5*** | 96.7 ± 29.7 (NS) |
| PI3Kγ−/−a | 114.5 ± 22.0 | 86.9 ± 25.0** | 63.0 ± 6.5*** | 89.0 ± 21.3* |
Mean areas (± SD) from ≥50 analyzed cells in two separate experiments are reported. Data are expressed in μm2.
Mean area of both wild-type and PI3Kγ−/− neutrophils plated on plastic in the presence of wortmannin [i.e., in condition totally inhibiting cell spreading (see Fig. 5)] was ∼50 μm2.
p < 0.05,
p < 0.01,
p < 0.001.
The dependence on class IA, but not class IB, PI3Ks of integrin-dependent spreading was confirmed by studies with human neutrophils treated with selective PI3K inhibitors (Supplemental Fig. 4). In fact, spreading of human neutrophils on plastic in the absence of any stimulus or on fibrinogen in the presence of TNF was markedly inhibited by PI3Kα and δ inhibitors. In contrast, a PI3Kγ inhibitor did not affect neutrophil spreading on plastic or only marginally affected it on fibrinogen in the presence of TNF.
PI3Kα and δ are implicated in adhesion-dependent ROS generation
To identify PI3Ks responsible for the late, adhesion-dependent phase of ROS generation, we used inhibitors that have been reported to display some selectivity for different class IA PI3Ks (35). At 40 nM concentration, the compound TGX-221, which should selectively inhibit the PI3Kβ isoform by ∼85% (26), had some inhibitory effect on the response to TNF (Fig. 6A) and both the acute and the late response to fMLF (Fig. 6B). However, data of total light units integrated over 2 min for the acute and 5–30 min for the late adhesion-dependent response showed that differences between untreated and drug-treated neutrophils were not statistically significant (Fig. 6C). This finding agrees with the evidence that TGX-221 only marginally inhibited murine (Table I) or human (data not shown) neutrophil spreading. In contrast, compound 15e, a relative selective inhibitor of PI3Kα, and compound IC87114, a relatively selective inhibitor of PI3Kδ, used at the low doses of 0.1 and 0.5 μM, respectively, markedly suppressed the response to TNF (Fig. 6D, 6G). Data of total light units integrated over 5–30 min (Fig. 6F, 6I) showed that the response to TNF was inhibited by 76 ± 16% (n = 6) and 87 ± 14% (n = 6) by the compound 15e and IC87114, respectively.
Figure 6.
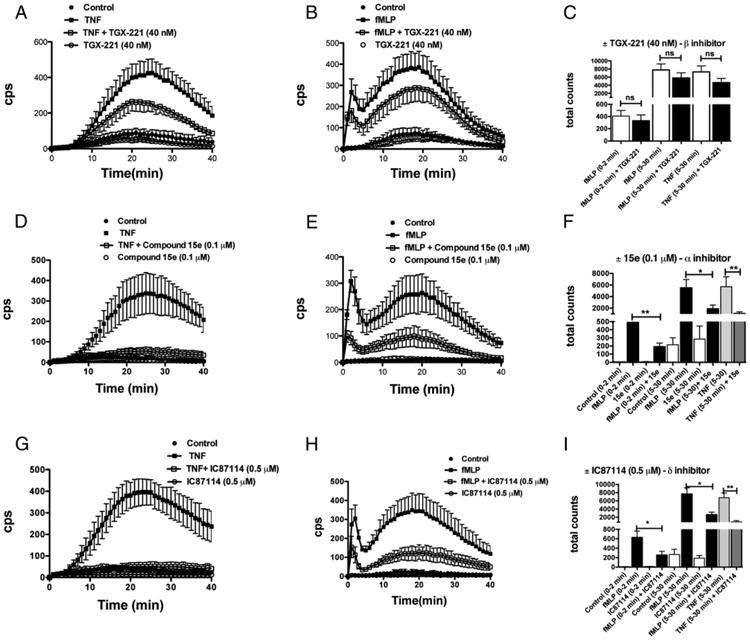
Inhibitors of the α and δ isoforms of class IA PI3Ks inhibit the early and late phase of ROS generation by human neutrophils. Human neutrophils were plated in fibrinogen-coated wells and stimulated with 10 ng/ml TNF (A, D, G) or 1 μM fMLF (B, E, H) in the absence or the presence of 40 nM TGX-221 (A–C), 0.1 μM compound 15e (D–F), or 0.5 μM IC87114 (G–I). Chemiluminescence was recorded every 1 min and up to 40 min. Mean results ± SD of five experiments are reported. (C, F, and I) Histograms report the total chemiluminescence detected during the early (0–2 min) or late (5–30 min) phase of ROS generation in the absence or the presence of the indicated inhibitor. *p < 0.05, **p < 0.01.
Examining the effects of these two inhibitors on the response to fMLF (Fig. 6E, 6H), we found that both the early and the late response were significantly inhibited. Using different doses of compound 15e and IC87114 on fMLF-induced ROS generation did not allow us to find a concentration at which only the late adhesion-dependent response was inhibited (data not shown). We conclude that, also in our experimental conditions, PI3Kα and/or PI3Kδ are implicated in the early response to fMLF.
SFKs are essential for the early and late phase of Vav phosphorylation in response to fMLF and TNF
Results described so far suggest that PI3Ks and SFKs play a coordinated but nonredundant role in regulation of neutrophil ROS generation. Because GTP loading on Rac is indispensable for activation of the NADPH oxidase and, among the three Rac GEFs implicated in Rac activation in neutrophils (see Introduction), only Vav is known to be directly activated downstream SFKs (36), we addressed the effects of SFK inhibitors and SFK deficiency on Vav phosphorylation in human and murine neutrophils, respectively (Fig. 7). Specifically, we examined phosphorylation of Y174, the Vav1 residue that in its unphosphorylated form dictates intramolecular inhibitory interaction between the Vav acidic (Ac) motif and the DH domain; phosphorylation of Y174, or analogous residues of Vav 2/3, releases this interaction and allows access of the DH domain to its target (36). As shown in Fig. 7A, fMLF triggered a rapid Y174 phosphorylation of Vav that increased up to 30 min [i.e., within the time frame of ROS generation (see Supplemental Fig. 1) in human neutrophils]. In contrast, TNF-induced Vav phosphorylation was delayed (Fig. 7B), but, similar to the fMLF-induced response, coincident with the time course of ROS generation (see Supplemental Fig. 3). Notably, PP2 totally suppressed Vav tyrosine phosphorylation in response to fMLF or TNF in human neutrophils at all of the time points tested. Time courses of Vav phosphorylation in response to fMLF or TNF in wild-type murine neutrophils were similar to those of human cells, but neutrophils deficient of Hck, Fgr, and Lyn were totally defective in Vav phosphorylation at all time points tested and independently of the stimulus used (Fig. 7C).
Figure 7.
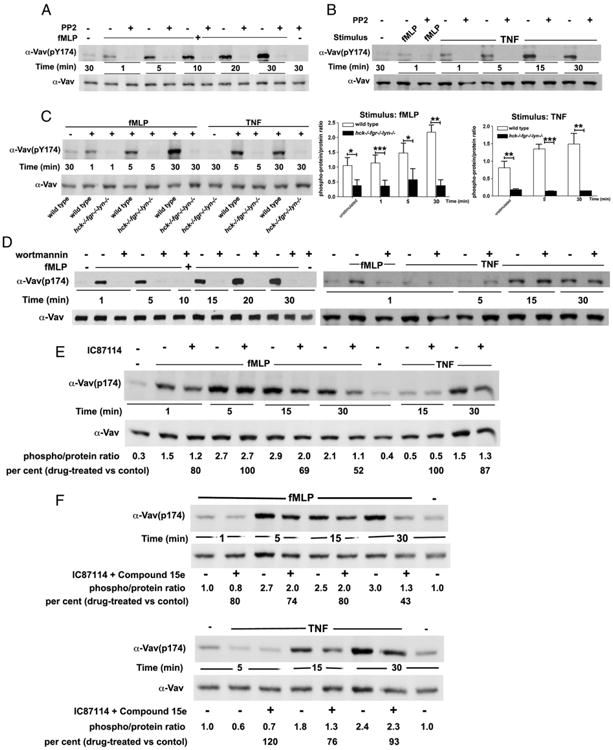
SFKs are indispensable for phosphorylation of Vav during the early and the late phase of ROS generation. Human neutrophils were plated in fibrinogen-coated wells and stimulated with 1 μM fMLF (A) or 10 ng/ml TNF (B) in the presence or the absence of PP2 as described in Materials and Methods. After different times of incubation, cells were lysed and lysates subjected to immunoblot analysis with Abs of the indicated specificity as described in Materials and Methods. One representative experiment of three performed with identical results is reported. (C) Neutrophils were isolated from wild-type or hck−/−fgr−/−lyn−/− knockout mice as described in Materials and Methods and plated in fibrinogen-coated wells. Cells were stimulated with 5 μM fMLF or 5 ng/ml TNF. After different times of incubation, cells were lysed and lysates subjected to immunoblot analysis with Abs of the indicated specificity. Graphs to the right of the immunoblot report densitometric analysis, expressed as ratio between the phosphoprotein versus the total Vav protein signal. Mean results ± SD of three independent experiments are reported. *p < 0.05, **p < 0.01, ***p < 0.001 (D–F). Human neutrophils were plated in fibrinogen-coated wells and stimulated with 10 ng/ml TNF or 1 μM fMLF in the absence or the presence of 100 nM wortmannin (D), 0.5 μM IC87114 alone, (E) or in combination with 0.1 μM compound 15e (F). After different times, cells were lysed and processed for immunoblot analysis as described in Materials and Methods. One representative experiment of two to three performed is reported.
Having established that SFKs are essential for Vav phosphorylation in both the early adhesion-independent and the late adhesion-dependent phase of neutrophil responses, we addressed the role of PI3Ks in the regulation of Vav phosphorylation using broad specificity and selective PI3K inhibitors. As shown in Fig. 7D, wortmannin suppressed fMLF-induced Vav phosphorylation in the course of both the early and late response; in contrast, the late TNF-induced response was unaffected by wortmannin. Studies with selective class IA inhibitors showed that both PI3Kα and PI3Kδ regulate the late adhesion-dependent phase of Vav phosphorylation in response to fMLF. In fact, the PI3Kδ inhibitor IC87114 alone partially reduced Vav phosphorylation at the 15-and 30-min time points after fMLF stimulation (Fig. 7E), and this late fMLF-induced response was more markedly reduced when it was used in combination with the PI3Kα inhibitor compound 15e (Fig. 7F). However, consistent with the data obtained with wortmannin, the PI3Kδ inhibitor alone or in combination with the PI3Kα inhibitor did not affect the late response to TNF. These findings suggest that in human neutrophils, Vav phosphorylation is regulated by PI3Ks in a stimulus-dependent manner, and TNF-induced Vav phosphorylation is largely independent of PI3K activities. The clear dissociation between inhibition of ROS generation by class IA PI3K inhibitors (Fig. 6) and Vav phosphorylation suggest that PIP3 formation is required for activation of additional Rac GEFs (see Ref. 23 and references contained therein).
Discussion
In this report, we addressed signal transduction pathways regulating neutrophil ROS generation in an experimental setting mirroring cell responses occurring in the context of neutrophil adhesion to the vascular endothelium and/or the interstitium. Agonists of trimeric G protein–coupled receptors trigger a very rapid and transient generation of ROS. These same agonists, or some cytokines, induce a slower, but prolonged ROS generation, which depends on adhesive interactions mediated by integrins (see Introduction and Ref. 5). This modality of ROS generation is believed to play an important role in the induction of tissue damage in inflammatory pathology. Our findings show that different PI3K isoforms and SFKs play an essential and not redundant role in regulating both phases of ROS generation (see Fig. 8 for a model).
Figure 8.
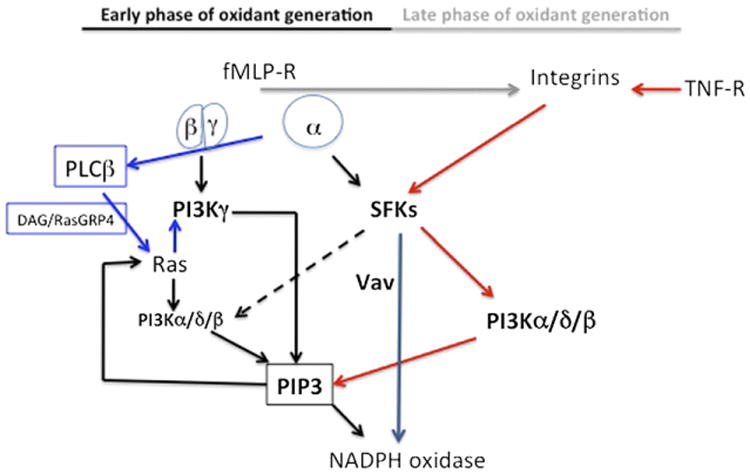
Model for signal transduction mechanisms regulating early, adhesion-independent, and late, adhesion-dependent neutrophil ROS generation. Trimeric G proteins coupled to fMLF receptor(s) (fMLF-R) dissociate in a β/γ heterodimer and a Gαi subunit upon ligand–receptor interaction. The β/γ heterodimer activates PI3Kγ that promotes formation of PIP3. The evidence that Ras activation and phosphorylation of the Ras downstream target ERK is virtually absent in PI3Kγ-deficient murine neutrophils (Figs. 2, 3) suggests that activation of GEFs for Ras by PIP3 may result in activation of class IA PI3Ks. However, this scenario is complicated by the recent demonstration that G protein–coupled receptors impinge on PI3Kγ via a PLCβ/diacylglycerol/RasGRP4 pathway (blue arrows) that plays a key role in PI3K-dependent neutrophil responses (33). Taken together, these findings point to the existence of a positive-feedback circuit of PI3K activation implicating Ras/PI3Kγ/Ras. Gαi, β-arrestin bound to the receptor–G protein complex or the G protein– coupled receptor directly, impinges on SFKs (43, 44). For the sake of simplicity, only the first possibility is highlighted in the drawing. Although mechanism of activation of SFKs by trimeric G protein–coupled receptors in neutrophils is still unknown, several reports pointed to a role of SFKs in signal transduction by this class of receptors (14, 38, 45–49). Activation of PI3Kγ and SFKs are both essential for the early activation of NADPH oxidase by fMLF, and either deficiency or inhibition of these kinases results in a marked or total suppression of ROS formation in murine or human neutrophils, respectively (Fig. 4, Supplemental Fig. 1). Because inhibitors of the class IA PI3K isoforms PI3Kα and PI3Kδ inhibit the early phase of ROS generation (Fig. 6), it is likely that a positive-feedback circuit triggered by PI3Kγ/Ras results in activation of class IA PI3Ks. Albeit, using selective inhibitors, we found that PI3Kβ plays a minor role in the early response to fMLF, this PI3K isoform has been implicated in the prolonged, late phase of ROS generation by neutrophils adhering to Aspergillus hyphae or immune complexes (26, 50). It is likely that class IA PI3Ks play a highly redundant role in regulation of this response. SFKs play a minor role in activation of class IA PI3Ks (broken arrow) in the early response to fMLF. In fact, inhibition of SFKs by PP2 in human, or deficiency of Hck, Fgr, and Lyn in murine, neutrophils only slightly inhibits, or has no effect on, phosphorylation of the PI3K downstream target AKT, respectively (Fig. 1). SFK inhibition (Supplemental Fig. 1) or deficiency (Fig. 4) result in a total or marked reduction of ROS generation. One important target of SFKs is the Vav family of GEFs for Rac. Inhibition of SFKs by PP2 in human, or deficiency of Hck, Fgr, and Lyn in murine, neutrophils results in a total inhibition of Vav phosphorylation (Fig. 7). Following interaction with chemoattractants, TNF, or other cytokines, neutrophils generate ROS in a fashion characterized by lateness and β2 integrin dependency (6). This response is totally independent of PI3Kγ (Figs. 4, 5), and requires class IA PI3Ks and SFKs (Figs. 5, 6). SFKs regulate class IA PI3Ks in the course of this response (Fig. 1, Supplemental Fig. 3) and are essential for phosphorylation of Vav proteins (Fig. 7).
As previously demonstrated (6), the class IB PI3K isoform PI3Kγ plays an essential role in stimulation of neutrophil ROS generation by fMLF in murine neutrophils. Neutrophils deficient of PI3Kγ (Fig. 4) are virtually unable to generate ROS in response to fMLF. Together with previous evidence with TNF-primed neutrophils (11), this finding points to the conclusion that PI3Kγ play an indispensable role in the early phase of ROS generation triggered by chemoattractants. However, relatively selective inhibitors of PI3Kα and PI3Kδ reduced the early fMLF-induced phase of ROS generation (Fig. 6), confirming previous reports (11, 25, 37) on the implication of class IA PI3Ks in the response to chemoattractants.
Also SFKs are indispensable for activating ROS generation by fMLF. Early and more recent studies (14, 38) demonstrated a clear defect in responses to fMLF by neutrophils deficient of Hck and Fgr or Hck, Fgr, and Lyn. The early fMLF-induced ROS generation is suppressed by the SFK inhibitor PP2 in human neutrophils (Supplemental Fig. 1) and markedly defective in Hck, Fgr, and Lyn-deficient neutrophils (Fig. 4). Combined with previous findings (11), one possible explanation of these results is that SFKs regulate class IA PI3Ks, establishing a positive-feedback circuit of PIP3 formation. However, examination of the PI3K downstream target AKT led us to conclude that the role played by SFKs in regulation of this response is mainly independent of activation of class IA PI3Ks. Indeed, PP2 only slightly inhibited AKT phosphorylation at an early time point after addition of the stimulus, and this was not defective in hck−/−fgr−/−lyn−/− neutrophils (Fig. 1).
An alternative mechanism of triggering of a positive-feedback circuit of PI3K activation is based on activation of Ras and its direct interaction with PI3K catalytic subunits (31). Examining phosphorylation of a Ras downstream target, the ERK proteins, we found that this is almost totally suppressed by the broad-specificity PI3K inhibitor wortmannin and the PI3Kγ selective inhibitor A604850 in human neutrophils and by the lack of PI3Kγ expression in murine neutrophils (Fig. 2). Together with the evidence that fMLF activates GDP/GTP exchange on Ras (32) and this is defective in PI3Kγ-deficient neutrophils (Fig. 3), these findings suggest that PI3Kγ may trigger activation of class IA PI3Ks via Ras activation (Fig. 8). In light of the recent evidence that PI3Kγ lies downstream of Ras in neutrophils stimulated with fMLF (33) (Fig. 8), our findings point to the existence of a complex, positive-feedback circuit of Ras/PI3Kγ/Ras activation. Also this mechanism of amplification of PI3K activities is likely independent of SFKs. In fact, whereas early (1 min) phosphorylation of ERKs requires PI3Kγ, a defect of ERKs phosphorylation is detectable only at later time points in hck−/−fgr−/−lyn−/− neu-trophils (Fig. 2) (i.e., when the early burst of ROS generation is ceased) (Fig. 3). We conclude that SFKs play a nonredundant role in activation of the early phase of neutrophil ROS generation in response to fMLF and play a minor role in the activation of class IA PI3Ks in this phase of the response (Fig. 8).
The late, adhesion-dependent response to two independent stimuli [i.e., fMLF (Fig. 4) and TNF (Fig. 5)] and upon stimulus-independent interaction with plastic, a β2 integrin–mediated phenomenon (34, 39) (Fig. 5), is independent of PI3Kγ expression. However, the broad-specificity inhibitor wortmannin (Supplemental Figs. 1, 2) and more selective PI3Kα or PI3Kδ (Fig. 6) inhibitory compounds totally or markedly suppressed adhesion-dependent ROS generation by human neutrophils, respectively. These inhibitors also hampered both stimulus-independent neutrophil spreading on plastic and TNF-induced human and murine neutrophil spreading on fibrinogen (Fig. 5, Supplemental Fig. 4, and data not shown). These findings show that class IA PI3Ks play an indispensable, albeit overlapping, role in adhesion-dependent neutrophil activation by adhesive surfaces. This is in concert with previous studies that implicated class IA, but not class IB, PI3Ks in integrin-dependent ROS generation induced by neutrophil binding of Aspergillus fumigatus hyphae (26). In this last study, PI3Kβ was shown to play a greater role than PI3Kα in the ROS and spreading response to Aspergillus hyphae, whereas the TNF-induced response (Fig. 6) was less sensitive to PI3Kβ rather than PI3Kα or PI3Kδ inhibitors. One possible reason for this discrepancy may be that the interaction of neutrophil with Aspergillus hyphae involves other surface receptors in addition to β2 integrins (see Ref. 26 and reference contained therein). Notably, although deficiency of PI3Kγ did not result in a lack of late adhesion-dependent responses (Figs. 4, 5), we found that AKT phosphorylation in response to fMLF (Fig. 2), but not TNF (Supplemental Fig. 3), was dependent on PI3Kγ expression. This finding suggests that at these late time points, there is not a simple relationship among PI3K activity, PIP3 formation, and AKT phosphorylation. It is tempting to speculate that chemoattractant signaling can, independently of the net effect on PIP3 formation, impinge on pathways regulating AKT dephosphorylation. Clearly, this issue deserves further investigation.
In the course of the late, adhesion-dependent phase of the neutrophil response, SFKs regulate class IA PI3Ks. In fact, in response to both fMLF and TNF, the late phase of phosphorylation of the PI3K downstream target AKT was totally suppressed by PP2 in human neutrophils and reduced in hck−/−fgr−/−lyn−/− neutrophils (Fig. 1, Supplemental Fig. 3). Hence, whereas in the early response to chemoattractants PI3Ks and SFKs play a largely distinct, nonreduntant role, in the late adhesion-dependent response, activation of SFKs is also implicated in activation of class IA PI3Ks.
Independently of their role in regulating PI3K activities, SFKs deliver signals that are indispensable for NADPH oxidase activation both in the early and late phase of ROS generation. Activation of the small GTP-binding protein Rac is essential for activation of NADPH oxidase (1, 16–18). Whereas PIP3 formation by PI3Ks regulates activation of at least three different GEFs for Rac (see Ref. 23 and references contained therein), Vav family proteins seem to be the only target of SFKs. Recent studies highlighted an essential requirement for cooperation of P-Rex1 and Vav proteins in the early response to chemoattractants (23) but confirmed previous studies assigning to Vav an exclusive role in the regulation of responses depending on prolonged adhesion to Igs and integrin ligands (8, 27, 40, 41). SFKs play a central role in Vav protein phosphorylation in both the early and delayed response to both fMLF and TNF (Fig. 7). This effect may be either direct or, at least for the late, adhesion-dependent response, occur downstream of Syk (8) or Pyk2 (42). In the light of the strong evidence implicating Vav proteins in activation of neutrophil ROS generation (19, 23), this suggests that one major mechanism by which SFKs regulate ROS generation is Vav protein phosphorylation. Interestingly, we found that Vav phosphorylation is not absolutely dependent on PI3Ks (Fig. 7). In fact, TNF-induced activation of delayed neutrophil responses is accompanied by Vav phosphorylation also in the presence of wortmannin or more specific class IA PI3K inhibitors. The clear dissociation between inhibition of ROS generation and Vav phosphorylation in response to TNF by PI3K inhibitors (Fig. 7, Supplemental Fig. 3) suggests that other PI3K/PIP3-dependent Rac GEFs are also implicated in this response (see Refs. 14, 23 and references contained therein), and SFK-dependent phosphorylation of Vav occurring downstream integrin engagement may be independent of PI3K activities.
Mechanisms regulating neutrophil activation are attracting increasing attention due to the high incidence of autoimmune diseases and the need to identify new targets to control inflammation. We identified SFKs as a key component of signal transduction ensuing both from G protein–coupled receptors for chemoattractants and from integrin receptors in the presence of TNF. PI3Ks act in a coordinated, nonredundant fashion with SFKs in regulating both type of the responses. Notably, PI3Kγ is implicated only in the chemoattractant-induced response, whereas class IA PI3Ks, especially the PI3Kα and PI3Kδ isoforms, are essential for the regulation of the adhesion-dependent response. This scenario highlights a rationale to define possible strategies to control different forms of inflammatory responses and inhibit sterile inflammation avoiding excessive depression of host defenses.
Supplementary Material
Acknowledgments
This work was supported by grants from Ministero dell'Istruzione, dell'Università e della Ricerca of Italy (2009 Progetti di ricerca di rilevante interesse nazionale grant to G.B. and E.H.) and Fondation Leducq 09CVD01 (to E.H.). The research leading to results concerning the role played by Src kinases in regulating neutrophil responses received funding from the European Community's Seventh Framework Programme (FP7-2007-2013) under Grant Agreement HEALTH-F4-2011-282095-TARKINAID (to G.B.).
Abbreviations used in this article
- CB
cytochalasin B
- GEF
guanine nucleotide exchange factor
- HGCa
HBSS supplemented with 0.5 mM CaCl2 and 5 mM d-glucose
- PI3K
phosphoinositide 3-kinase
- PIP3
phosphatidylinositol phosphorylated at the 3 position of the inositol ring
- PLC
phospholipase C
- PP2
4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- ROS
reactive oxygen species
- SFK
Src-family kinase
Footnotes
Disclosures: The authors have no financial conflicts of interest.
The online version of this article contains supplemental material.
References
- 1.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C, Ding A. SnapShot: Reactive Oxygen Intermediates (ROI) Cell. 2010;140:951–951.e2. doi: 10.1016/j.cell.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Nathan CF. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J Clin Invest. 1987;80:1550–1560. doi: 10.1172/JCI113241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berton G, Laudanna C, Sorio C, Rossi F. Generation of signals activating neutrophil functions by leukocyte integrins: LFA-1 and gp150/95, but not CR3, are able to stimulate the respiratory burst of human neutrophils. J Cell Biol. 1992;116:1007–1017. doi: 10.1083/jcb.116.4.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berton G, Lowell CA. Integrin signalling in neutrophils and macrophages. Cell Signal. 1999;11:621–635. doi: 10.1016/s0898-6568(99)00003-0. [DOI] [PubMed] [Google Scholar]
- 6.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 7.Lowell CA, Fumagalli L, Berton G. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J Cell Biol. 1996;133:895–910. doi: 10.1083/jcb.133.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mócsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002;16:547–558. doi: 10.1016/s1074-7613(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 9.Berton G, Mócsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol. 2005;26:208–214. doi: 10.1016/j.it.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Mócsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, Vanhaesebroeck B, Turner M, Webb L, Wymann MP, et al. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood. 2005;106:1432–1440. doi: 10.1182/blood-2005-03-0944. [DOI] [PubMed] [Google Scholar]
- 12.Nijhuis E, Lammers JW, Koenderman L, Coffer PJ. Src kinases regulate PKB activation and modulate cytokine and chemoattractant-controlled neutrophil functioning. J Leukoc Biol. 2002;71:115–124. [PubMed] [Google Scholar]
- 13.Gao XP, Zhu X, Fu J, Liu Q, Frey RS, Malik AB. Blockade of class IA phosphoinositide 3-kinase in neutrophils prevents NADPH oxidase activation- and adhesion-dependent inflammation. J Biol Chem. 2007;282:6116–6125. doi: 10.1074/jbc.M610248200. [DOI] [PubMed] [Google Scholar]
- 14.Fumagalli L, Zhang H, Baruzzi A, Lowell CA, Berton G. The Src family kinases Hck and Fgr regulate neutrophil responses to N-formyl-methionyl-leucyl-phenylalanine. J Immunol. 2007;178:3874–3885. doi: 10.4049/jimmunol.178.6.3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Tang H, Hay N, Xu J, Ye RD. Akt isoforms differentially regulate neutrophil functions. Blood. 2010;115:4237–4246. doi: 10.1182/blood-2009-11-255323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts AW, Kim C, Zhen L, Lowe JB, Kapur R, Petryniak B, Spaetti A, Pollock JD, Borneo JB, Bradford GB, et al. Deficiency of the hematopoietic cell-specific Rho family GTPase Rac2 is characterized by abnormalities in neutrophil function and host defense. Immunity. 1999;10:183–196. doi: 10.1016/s1074-7613(00)80019-9. [DOI] [PubMed] [Google Scholar]
- 17.Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, Gonzalez-Aller C, Hiester A, deBoer M, Harbeck RJ, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci USA. 2000;97:4654–4659. doi: 10.1073/pnas.080074897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim C, Dinauer MC. Rac2 is an essential regulator of neutrophil nicotinamide adenine dinucleotide phosphate oxidase activation in response to specific signaling pathways. J Immunol. 2001;166:1223–1232. doi: 10.4049/jimmunol.166.2.1223. [DOI] [PubMed] [Google Scholar]
- 19.Kim C, Marchal CC, Penninger J, Dinauer MC. The hemopoietic Rho/Rac guanine nucleotide exchange factor Vav1 regulates N-formyl-methionyl-leucyl-phenylalanine-activated neutrophil functions. J Immunol. 2003;171:4425–4430. doi: 10.4049/jimmunol.171.8.4425. [DOI] [PubMed] [Google Scholar]
- 20.Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, Okkenhaug K, Coadwell WJ, Andrews SR, Thelen M, et al. P-Rex1 regulates neutrophil function. Curr Biol. 2005;15:1867–1873. doi: 10.1016/j.cub.2005.09.050. [DOI] [PubMed] [Google Scholar]
- 21.Dong X, Mo Z, Bokoch G, Guo C, Li Z, Wu D. P-Rex1 is a primary Rac2 guanine nucleotide exchange factor in mouse neutrophils. Curr Biol. 2005;15:1874–1879. doi: 10.1016/j.cub.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 22.Kunisaki Y, Nishikimi A, Tanaka Y, Takii R, Noda M, Inayoshi A, Watanabe K, Sanematsu F, Sasazuki T, Sasaki T, Fukui Y. DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. J Cell Biol. 2006;174:647–652. doi: 10.1083/jcb.200602142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawson CD, Donald S, Anderson KE, Patton DT, Welch HC. P-Rex1 and Vav1 cooperate in the regulation of formyl-methionyl-leucyl-phenylalanine-dependent neutrophil responses. J Immunol. 2011;186:1467–1476. doi: 10.4049/jimmunol.1002738. [DOI] [PubMed] [Google Scholar]
- 24.Stephens L, Eguinoa A, Corey S, Jackson T, Hawkins PT. Receptor stimulated accumulation of phosphatidylinositol (3,4,5)-trisphosphate by G-protein mediated pathways in human myeloid derived cells. EMBO J. 1993;12:2265–2273. doi: 10.1002/j.1460-2075.1993.tb05880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadhu C, Dick K, Tino WT, Staunton DE. Selective role of PI3K delta in neutrophil inflammatory responses. Biochem Biophys Res Commun. 2003;308:764–769. doi: 10.1016/s0006-291x(03)01480-3. [DOI] [PubMed] [Google Scholar]
- 26.Boyle KB, Gyori D, Sindrilaru A, Scharffetter-Kochanek K, Taylor PR, Mócsai A, Stephens LR, Hawkins PT. Class IA phosphoinositide 3-kinase b and d regulate neutrophil oxidase activation in response to Aspergillus fumigatus hyphae. J Immunol. 2011;186:2978–2989. doi: 10.4049/jimmunol.1002268. [DOI] [PubMed] [Google Scholar]
- 27.Gakidis MA, Cullere X, Olson T, Wilsbacher JL, Zhang B, Moores SL, Ley K, Swat W, Mayadas T, Brugge JS. Vav GEFs are required for beta2 integrin-dependent functions of neutrophils. J Cell Biol. 2004;166:273–282. doi: 10.1083/jcb.200404166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jakus Z, Simon E, Frommhold D, Sperandio M, Mócsai A. Critical role of phospholipase Cgamma2 in integrin and Fc receptor-mediated neutrophil functions and the effector phase of autoimmune arthritis. J Exp Med. 2009;206:577–593. doi: 10.1084/jem.20081859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng F, Lowell CA. Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. J Exp Med. 1997;185:1661–1670. doi: 10.1084/jem.185.9.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lundqvist H, Dahlgren C. Isoluminol-enhanced chemiluminescence: a sensitive method to study the release of superoxide anion from human neutrophils. Free Radic Biol Med. 1996;20:785–792. doi: 10.1016/0891-5849(95)02189-2. [DOI] [PubMed] [Google Scholar]
- 31.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 32.Cadwallader KA, Condliffe AM, McGregor A, Walker TR, White JF, Stephens LR, Chilvers ER. Regulation of phosphatidylinositol 3-kinase activity and phosphatidylinositol 3,4,5-trisphosphate accumulation by neutrophil priming agents. J Immunol. 2002;169:3336–3344. doi: 10.4049/jimmunol.169.6.3336. [DOI] [PubMed] [Google Scholar]
- 33.Suire S, Lécureuil C, Anderson KE, Damoulakis G, Niewczas I, Davidson K, Guillou H, Pan D, Clark J, Hawkins PT, Stephens L. GPCR activation of Ras and PI3Kc in neutrophils depends on PLCb2/b3 and the RasGEF RasGRP4. EMBO J. 2012;31:3118–3129. doi: 10.1038/emboj.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson DC, Schmalstieg FC, Arnaout MA, Kohl S, Tosi MF, Dana N, Buffone GJ, Hughes BJ, Brinkley BR, Dickey WD, et al. Abnormalities of polymorphonuclear leukocyte function associated with a heritable deficiency of high molecular weight surface glycoproteins (GP138): common relationship to diminished cell adherence. J Clin Invest. 1984;74:536–551. doi: 10.1172/JCI111451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 36.Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol. 2002;2:476–486. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- 37.Boulven I, Levasseur S, Marois S, Paré G, Rollet-Labelle E, Naccache PH. Class IA phosphatidylinositide 3-kinases, rather than p110 gamma, regulate formyl-methionyl-leucyl-phenylalanine-stimulated chemotaxis and superoxide production in differentiated neutrophil-like PLB-985 cells. J Immunol. 2006;176:7621–7627. doi: 10.4049/jimmunol.176.12.7621. [DOI] [PubMed] [Google Scholar]
- 38.Zhu JW, Doan K, Park J, Chau AH, Zhang H, Lowell CA, Weiss A. Receptor-like tyrosine phosphatases CD45 and CD148 have distinct functions in chemoattractant-mediated neutrophil migration and response to. S aureus Immunity. 2011;35:757–769. doi: 10.1016/j.immuni.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crowley CA, Curnutte JT, Rosin RE, André-Schwartz J, Gallin JI, Klempner M, Snyderman R, Southwick FS, Stossel TP, Babior BM. An inherited abnormality of neutrophil adhesion. Its genetic transmission and its association with a missing protein. N Engl J Med. 1980;302:1163–1168. doi: 10.1056/NEJM198005223022102. [DOI] [PubMed] [Google Scholar]
- 40.Zheng L, Sjölander A, Eckerdal J, Andersson T. Antibody-induced engagement of beta 2 integrins on adherent human neutrophils triggers activation of p21ras through tyrosine phosphorylation of the protooncogene product Vav. Proc Natl Acad Sci USA. 1996;93:8431–8436. doi: 10.1073/pnas.93.16.8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham DB, Robertson CM, Bautista J, Mascarenhas F, Diacovo MJ, Montgrain V, Lam SK, Cremasco V, Dunne WM, Faccio R, et al. Neutrophil-mediated oxidative burst and host defense are controlled by a Vav-PLCgamma2 signaling axis in mice. J Clin Invest. 2007;117:3445–3452. doi: 10.1172/JCI32729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kamen LA, Schlessinger J, Lowell CA. Pyk2 is required for neutrophil degranulation and host defense responses to bacterial infection. J Immunol. 2011;186:1656–1665. doi: 10.4049/jimmunol.1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luttrell DK, Luttrell LM. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene. 2004;23:7969–7978. doi: 10.1038/sj.onc.1208162. [DOI] [PubMed] [Google Scholar]
- 44.McGarrigle D, Huang XY. GPCRs signaling directly through Src-family kinases. Sci STKE. 2007;2007:e35. doi: 10.1126/stke.3922007pe35. [DOI] [PubMed] [Google Scholar]
- 45.Ptasznik A, Traynor-Kaplan A, Bokoch GM. G protein-coupled chemoattractant receptors regulate Lyn tyrosine kinase.Shc adapter protein signaling complexes. J Biol Chem. 1995;270:19969–19973. doi: 10.1074/jbc.270.34.19969. [DOI] [PubMed] [Google Scholar]
- 46.Mócsai A, Ligeti E, Lowell CA, Berton G. Adhesion-dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J Immunol. 1999;162:1120–1126. [PubMed] [Google Scholar]
- 47.Gutkind JS, Robbins KC. Translocation of the FGR protein-tyrosine kinase as a consequence of neutrophil activation. Proc Natl Acad Sci USA. 1989;86:8783–8787. doi: 10.1073/pnas.86.22.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barlic J, Andrews JD, Kelvin AA, Bosinger SE, DeVries ME, Xu L, Dobransky T, Feldman RD, Ferguson SS, Kelvin DJ. Regulation of tyrosine kinase activation and granule release through beta-arrestin by CXCRI. Nat Immunol. 2000;1:227–233. doi: 10.1038/79767. [DOI] [PubMed] [Google Scholar]
- 49.Scapini P, Morini M, Tecchio C, Minghelli S, Di Carlo E, Tanghetti E, Albini A, Lowell C, Berton G, Noonan DM, Cassatella MA. CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J Immunol. 2004;172:5034–5040. doi: 10.4049/jimmunol.172.8.5034. [DOI] [PubMed] [Google Scholar]
- 50.Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, Hirose M, Juss J, Oxley D, Chessa TA, et al. PI3Kβ plays a critical role in neutrophil activation by immune complexes. Sci Signal. 2011;4:ra23. doi: 10.1126/scisignal.2001617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.