

Summary
Background and objectives
Src family kinases (SFKs) play a critical role in initiating and propagating signals in platelets. The aims of this study were to quantitate SFK members present in platelets and to analyze their contribution to platelet regulation using glycoprotein VI (GPVI) and intregrin αIIbβ3, and in vivo.
Methods and Results
Mouse platelets express four SFKs, Fgr, Fyn, Lyn and Src, with Lyn expressed at a considerably higher level than the others. Using mutant mouse models, we demonstrate that platelet activation by collagen-related peptide (CRP) is delayed and then potentiated in the absence of Lyn, but only marginally reduced in the absence of Fyn or Fgr, and unaltered in the absence of Src. Compound deletions of Lyn/Src or Fyn/Lyn, but not of Fyn/Src or Fgr/Lyn, exhibit a greater delay in activation relative to Lyn-deficient platelets. Fibrinogen-adherent platelets show reduced spreading in the absence of Src, potentiation in the absence of Lyn, but no change in the absence of Fyn or Fgr. In mice double-deficient in Lyn/Src or Fgr/Lyn, the inhibitory role of Lyn on spreading on fibrinogen is lost. Lyn is the major SFK-mediating platelet aggregation on collagen at arterial shear and its absence leads to a reduction in thrombus size in a laser injury model.
Conclusion
These results demonstrate that SFKs share individual and overlapping roles in regulating platelet activation, with Lyn having a dual role in regulating GPVI signaling and an inhibitory role downstream of αIIbβ3, which requires prior signaling through Src.
Keywords: GPVI, integrin αIIbβ3, Lyn, platelets, Src family kinases
Introduction
Src-family kinases (SFKs) are a family of eight structurally related tyrosine kinases, namely Lyn, Fyn, Src, Fgr, Blk, Hck, Yes and Lck, characterized by a SH2 and SH3 domain and a tyrosine kinase group. The greatest sequence diversity is found in the myristoylated N-terminal region which is sometimes called a SH4 domain [1,2]. In six of the SFKs, this region contains a cysteine residue which supports palmitoylation and localization to lipid rafts. The two exceptions are Src and Blk which are located outside of these domains [3,4]. SFKs are regulated through phosphorylation on inhibitory and activatory tyrosines [1,5]. Receptor tyrosine phosphatases regulate the activity of SFKs through dephosphorylation of the inhibitory site, with CD148 and PTP1B performing this function in platelets [6–8].
Src-family kinases have a critical role in many cellular processes, including proliferation, differentiation, motility and adhesion [1,5,9]. In platelets, SFKs are critical for initiating activation by the collagen receptor glycoprotein VI (GPVI) and the integrin αIIbβ3 [10–12]. They also contribute to signaling by G protein-coupled and cytokine receptors[13]. In addition, SFKs inhibit platelet activation via phosphorylation of immunoreceptor tyrosine-based inhibition motifs (ITIMs) in several membrane immunoglobulin receptors including PECAM-1 [14,15].
The GPVI–FcRγ-chain receptor complex has a conserved immunoreceptor tyrosine-based activation motif (ITAM), characterized by two YxxLs separated by seven amino acids. Clustering of GPVI leads to phosphorylation of the conserved tyrosines in the immunoreceptor tyrosine-based activation motif (ITAM) by SFKs and binding of Syk via its tandem SH2 domains. Syk then initiates a signaling cascade that consists of adapter and effector proteins, including further roles for SFKs [11,12]. GPVI signaling takes place in lipid rafts which are enriched in receptors and signaling proteins, including SFKs and the membrane adapter LAT, which nucleates a signalosome that provides a docking site for a variety of proteins including PLCγ2 [16–18].
The integrin αIIbβ3 is excluded from membrane rafts but also signals via SFKs and uses many of the same adapter and effector proteins as used by GPVI, with the notable exception of LAT [19]. In human platelets, αIIbβ3 also signals via the low-affinity immune receptor, FcγRIIA [20], but this is absent in the mouse genome and there is as yet no evidence of ITAM involvement in αIIbβ3 signaling in mouse platelets.
A critical question in regard to each of the SFKs is their net overall contribution to the regulation of platelets. Fyn, Lyn and Src are recognized as the major SFKs in human and mouse platelets. Expression of Fgr, Hck, Lck and Yes in platelets has also been reported but not in all studies, whereas the presence of Blk has not been described [21–23].
Lyn and Fyn are constitutively associated with a proline-rich domain (PRD) in the GPVI cytosolic tail via their SH3 domains with Lyn being held in an active conformation [24–26]. This arrangement positions the collagen receptor in a ‘ready-to-go’ state that can be triggered by clustering, with Lyn being the major kinase mediating activation [25]. Deletion of the PRD delays GPVI signaling in both transfected cell lines and in platelets revealing a second, yet to be characterized, route of SFK activation [25,27]. This delay in response is overcome at later times under static conditions but results in a marked inhibition in activation at arteriolar rates of shear [25,28]. A similar delay in activation by GPVI and inhibition of response under shear is observed in Lyn-deficient platelets [25]. The similar phenotype of the proline-rich mutant of GPVI and the Lyn-deficient platelets suggests a causative association. In contrast, platelet adhesion on collagen under flow is not altered in the absence of Fyn, even although a partial reduction in aggregation to collagen-related peptide (CRP) has been reported [25,28]. Mice double deficient in Fyn and Lyn have a greater defect response to CRP, demonstrating that the two SFKs work in synergy to regulate platelet activation [28].
Fyn and Src have been shown to associate with distinct regions of the β3-cytoplasmic tail [8,10,29]. Further, platelets deficient in Fyn or Lyn exhibit a mild defect or marked potentiation in spreading on fibrinogen, respectively [29,30]. On the other hand, spreading on fibrinogen is not altered in the absence of Fgr, Hck and Lyn but is blocked by the additional absence of Src revealing a critical role in αIIbβ3 signaling [31]. Together, these results reveal that several SFKs participate in integrin-mediated spreading, with individual SFKs performing distinct roles.
A full understanding of the role of each of the SFKs in platelet activation is essential for the understanding of thrombo-inflammatory processes and platelet regulation. In the present study, we have measured the levels of SFKs in mouse platelets and investigated their roles downstream of the collage receptor GPVI and the integrin αIIbβ3 through the use of mouse models deficient in one or more SFKs.
Methods
Materials, mutant mice [32-34], preparation of mouse platelets and aggregation [35], flow cytometry, western blotting and immunoprecipitation studies [34], quantitation of SFKs in mouse platelets, lipid raft isolation [36], static adhesion and spreading studies [37,38], whole blood flow studies on immobilized collagen [37,39], the laser-induced thrombus formation model [7,40] and statistical analysis are described in Data S1.
Results
Expression and localization of SFKs in mouse platelets
There are several reports of expression of Fyn, Lyn and Src in mouse platelets supported by specific antibodies, gene expression arrays on platelets and megakaryocytes, and functional studies using mutant mice platelets [25,28,29,31,34,41]. On the other hand, there are contrasting reports of expression of Fgr, Hck, Lck and Yes, and no reports of expression of Blk [21,23,42].
We re-investigated the presence of Blk, Fgr, Hck, Lck and Yes in mouse platelets by western blotting of platelets and control tissues that are known to express SFKs from wild-type (WT) and mutant mouse models. A band corresponding to Fgr was present in the spleen and platelets from WT mice but, as expected, not from Fgr-deficient mice (Fig. 1A). Robust expression of Hck and Yes was observed in the spleen and lung, respectively, and was abolished in mice deficient in the corresponding SFKs (Fig. 1A). In contrast, we did not observe a specific band corresponding to Hck or Yes by comparison of control and Hck- and Yes-deficient platelets, respectively, even after long exposure times (Fig. 1A). Robust expression of Blk and Lck was also observed in the spleen but not in mouse platelets (Fig. 1A). Thus, four of the eight SFK members, Fgr, Fyn, Lyn and Src, were detected in mouse platelets by western blotting (Fig. 1A and S1).
Fig. 1.
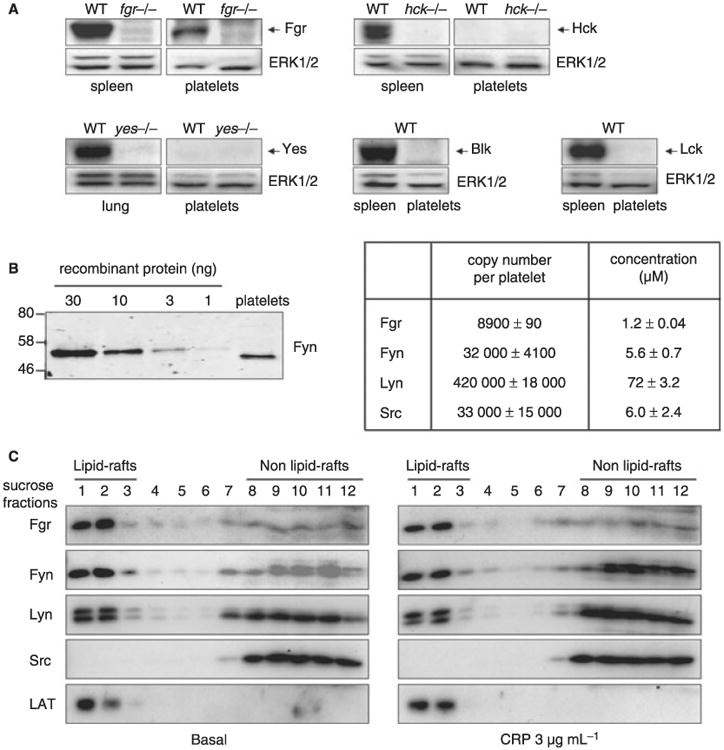
Expression and localization of Lyn, Fyn, Src and Fgr in mouse platelets. (A) Whole cell lysates were prepared from spleen, lung or washed platelets of control and different members of Src family kinase (Yes, Hck and Fgr)-deficient mouse as described in the Methods. Expression of the different Src family kinase members (Yes, Hck, Fgr, Blk and Lck) was detected by western blot using specific antibodies. Data are representative of at least three independents experiments. (B) The protein level of Fgr, Fyn, Lyn and Src were quantitated by densitometry in mouse platelets as described in the Methods. Representative western blot is shown in the left panel. (C) Resting and collagen-related peptide (CRP)-stimulated platelets were lysed in 1% Brij 58 lysis buffer and lipid raft were isolated by sucrose gradient ultracentrifugation. The lipid rafts fractions were detected using an anti-LAT and localization of Fgr, Fyn, Lyn and Src was detected using specific antibodies. Data are representative of two independents experiments.
A quantitative western blotting approach, illustrated in Fig. 1B for Fyn, was used to compare the levels of the four SFKs. Lyn is the most abundant with an estimated intracellular concentration of 72 ± 3.2 μmol L−1. The levels of Fyn, Src and Fgr were considerably lower at 5.6 ± 0.7, 6.0 ± 2.4 and 1.2 ± 0.04 μmol L−1, respectively (Fig. 1B). We also used a pan phospho-specific antibody to the highly conserved activation site of all SFKs (anti-SFK pY418) to investigate expression (Fig. S1). The pan phospho-specific antibody detected three bands in mouse platelets, with the lower two co-migrating with Lyn. These two bands were absent in Lyn-deficient mouse platelets (Fig. S1). The upper band migrates with Fgr, Fyn and Src and is reduced by 40%–50% in mice deficient in Fyn or Src, but is unaltered in mice deficient in Fgr (Fig. S1). This is consistent with the similar level of expression of Fyn and Src and the much lower level of expression of Fgr (with the caveat that the stoichiometry of phosphorylation is not known).
Lipid rafts are cholesterol-rich, plasma microdomains enriched in a variety of signlling proteins. We investigated the distribution of SFKs in lipid rafts in platelets using Brij-58 and sucrose gradient ultracentrifugation. Samples were western blotted for the different SFKs and for the membrane adapter LAT to identify the raft fraction [19] and analyzed by densitometry. Similar levels of Fgr, Fyn and Lyn co-localized with LAT in the raft fraction in resting and CRP-stimulated platelets, whereas Src was absent from this fraction (Fig. 1C). Fgr, Fyn and Lyn were also present in the non-raft fraction where Src is exclusively present. Densitometry analysis shows that there is no significant difference in expression and localization of Fgr, Fyn, Lyn and Src in resting and CRP-stimulated platelets (not shown). Thus, Fgr, Fyn and Lyn are capable of localizing to lipid rafts in platelets whereas Src is excluded from these domains.
Impact of deficiency of single SFKs on platelet activation by GPVI and integrin αIIbβ3
The functional role of Fyn, Lyn and to a limited extent Src has been previously investigated in platelets using mice deficient in the individual SFKs and in the case of Fyn and Lyn, mice deficient in both kinases [25,28,29,31,34,41]. Spreading of platelets on fibrinogen from mice deficient in Fgr, Hck and Lyn or Fgr, Hck, Lyn and Src has also been reported [31].
Mice deficient in Fgr, Fyn, Lyn or Src were born at Mendelian frequency and were viable, although Src-deficient mice tended to be smaller than their littermates most probably as a result of under nourishment as they had to be fed on a liquid diet as previously reported [43]. When measured at 8 weeks of age, the platelet count and size, and the corresponding parameters of other blood cells, was similar in the four SFK-deficient mice to their littermate controls (not shown). The platelet count was reduced by approximately 25% in Lyn-deficient mice of 12 weeks of age as previously reported [44] and thus experiments were conducted prior to this time. The level of expression of the glycoproteins GPVI, α2- and αIIb-integrin subunits and GPIbα were similar in platelets from the four SFK-deficient mice and controls, and deletion of one SFK had no significant effect on the level of other SFKs (not shown and [28,34]).
We and others have previously reported that aggregation and dense granule secretion of platelets by the GPVI-specific ligands, CRP or convulxin, is delayed and then potentiated in the absence of Lyn and partially reduced in the absence of Fyn [25,28]. These observations have been confirmed in the present study (the effect was less marked than previously observed, possibly because of batch variation in CRP) and extended to mice deficient in Fgr or Src (Fig. 2A). Aggregation and secretion of dense- and α-granules induced by low concentrations of CRP were inhibited in the absence of Fgr to a similar extent to those observed in the absence of Fyn (Fig. 2A). In contrast, responses to low concentrations of CRP were not altered in the absence of Src (Fig. 2A and not shown). Thus, Fgr and Fyn play minor roles in mediating activation by GPVI with Lyn being the major SFK initiating platelet activation by the collagen receptor as shown by the delay in onset of response.
Fig. 2.
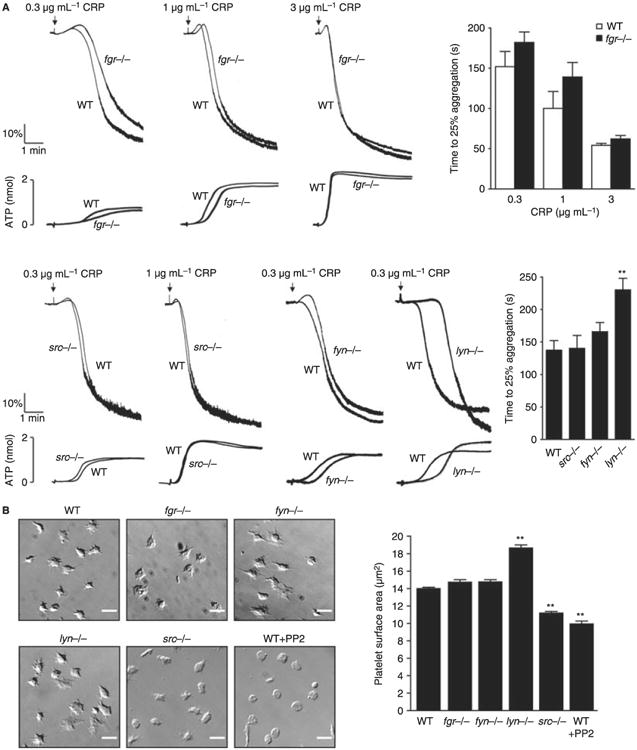
Impact of deficiency of Src family kinases (SFK) in platelet responses. (A) Washed platelets (2 × 108 mL−1) prepared from fyn−/−, lyn−/−, src−/− or fgr−/− mice and their respective litter-matched controls were stimulated with different doses of collagen-related peptide (CRP) (0.3, 1 and 3 μg mL−1). Platelet aggregation was measured as a change in light transmission and ATP secretion was measured as luciferin/luciferase-mediated luminescence, using a lumi-aggregometer. Representative images are shown (n = 3–6 mice per condition). The time to 25% of aggregation was calculated and data are shown as mean ± standard error of the mean (SEM). One-way anova test, **P < 0.01 vs. wild type (WT). (B) Washed platelets (2 × 107 mL−1) from fgr−/− fyn−/−, lyn−/− or src−/− and their respective litter-matched controls or WT platelets treated for 10 min with 10 μmol L−1 PP2 were placed on a fibrinogen-coated surface for 45 min. Platelets were fixed and images captured using differential interference contrast (DIC) microscopy (scale bar: 5 μm). Representative DIC images of platelets captured from four independent experiments for each genotype are shown. The surface area of platelets per condition were measured using ImageJ software (mean ± standard error of the mean [SEM]; n = 150–300 individual platelets per mice; four mice per genotype). Oneway anova test, *P < 0.05 vs. WT. (C) Anticoagulated blood from lyn−/−, fyn−/−, src−/− or fgr−/− and their respective litter-matched controls or WT platelets treated for 10 min with 3 μmol L−1 Dasatinib was perfused through collagen-coated capillary tubes at 1000 s−1 for 4 min. DIC images of fixed platelets on collagen fibers after being flowed through collagen-coated capillary tubes at 1000 s−1 for 4 min (scale bar: 20 μm) are shown. Representative images of thrombus formed from at least three independent experiments of each genotype are shown. Area covered by platelet thrombi and the percentage of thrombi with different surface were measured in each of at least three different experiments and mean ± SEM is shown. One-way anova test, *P < 0.05, **P < 0.01 vs. WT. (D) Platelets from WT or Lyn-deficient mice were fluorescently labeled ex vivo with rat anti–mouse αIIb primary antibody and Alex488-conjugated secondary antibody before being reintroduced into recipient mice. Arterioles in cremaster muscles of recipients were subsequently injured by laser, and the accumulation of platelets into the thrombi was assessed. Curves represent the median integrated thrombus fluorescence intensity in arbitrary units (pixels) for 23–25 thrombi induced in five mice of each genotype. Dot plot represents time to peak (seconds) and mean peak size (pixels) from between 23 and 27 thrombi from five mice.
Spreading of different single SFK-deficient platelets on fibrinogen was monitored over 45 min. During this time course, platelets continuously bind and spread on the matrix protein such that the final picture is a net summary of capture of platelets at various stages of spreading as determined by the time of contact with the surface. Monitoring of spreading in this way therefore provides a kinetic capture of spreading from various times up to 45 min. Spreading but not adhesion of platelets on fibrinogen is abolished in the presence of the SFK inhibitor PP2 but is not altered in the presence of its inactive analogue PP3 (Fig. 2B and not shown). The degree of spreading is reduced by over 70% in Src-deficient platelets, with adhesion again being unaffected (Fig. 2B). In comparison, there was no difference in spreading of Fgr- or Fyn-deficient platelets on fibrinogen (Fig. 2B), whereas Lyn-deficient platelets show an approximate doubling in surface area but no change in adhesion (Fig. 2B). These observations establish Src as the major SFK mediating spreading on fibrinogen and demonstrate that Lyn has a net inhibitory role. The greater inhibitory effect of PP2 relative to the reduction in the Src-deficient platelets suggests that one or more of the other SFKs contributes to spreading.
Platelet aggregation in single SFK-deficient mice under flow conditions
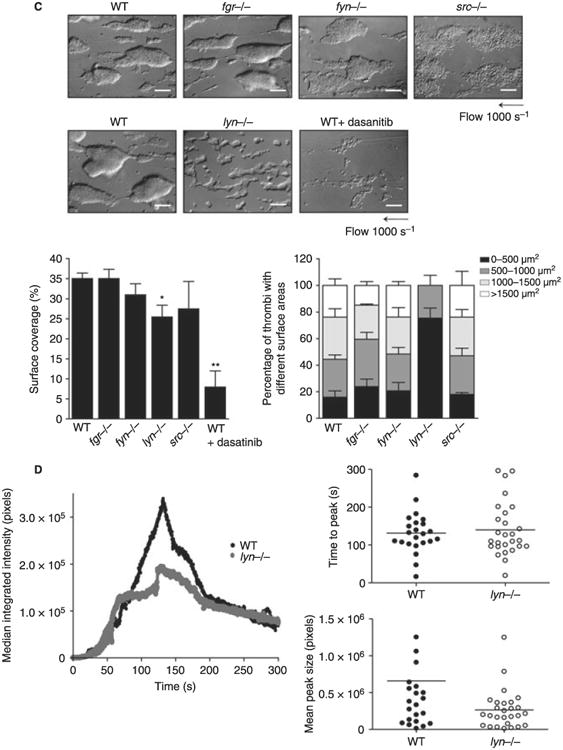
Platelet aggregation on collagen under flow conditions involves signaling through a combination of platelet surface receptors including GPVI, integrin αIIbβ3, ADP and thromboxane receptors, all which signal through SFKs to varying extents. The net contribution of SFKs to aggregation of platelets on fibrillar collagen was monitored at a shear rate of 1000 s−1 as, under these conditions, adhesion and aggregation are critically dependent on signaling through GPVI [39]. Blood from Fgr, Fyn or Src-deficient mice formed robust aggregates on collagen fibers that were similar in size and surface area to each other and to those of litter-matched controls (Fig. 2C). In contrast, blood from Lyn-deficient mice formed only small aggregates along the length of the collagen fibers resulting in a significant decrease in surface area (Fig. 2C). However, this effect was less than that observed in the presence of Dasatinib, a SFK inhibitor which (unlike PP2) is bioavailable in blood [8,29,45], suggesting that one or more other SFKs mediates this response in association with Lyn. Interestingly, there was no significant change in blood loss in mice deficient in Lyn or any of the other SFKs relative to littermate controls as measured by a tail bleeding assay (not shown). In contrast, tail bleeding was increased from 2.83 ± 1.2 mg in non-treated mice to 69.8 ± 21.9 mg in mice treated with the pan-SFK inhibitor Dasatinib, in agreement with previous reports [8,29,45].
However, a role for Lyn in supporting thrombus formation in vivo was observed in a laser-induced vessel injury model. The overall time course of thrombus formation in the Lyn-deficient platelets was similar to that in littermate controls but the magnitude was significantly reduced (Fig. 2D), which may reflect a role for Lyn in platelet activation by several receptors which contribute to thrombus growth including GPIb and PAR-4 [46,47]. It is unclear whether the observed role for Lyn in the laser injury model relative to the tail bleeding assay is as a result of a fundamental difference in these two assays or if the former is simply a more sensitive assay given that the time course of thrombus formation was the same to that of control platelets with only height being affected.
Impact of double deficiency of SFK on platelet activation by GPVI
The above studies confirm Lyn and Src as the major SFKs mediating platelet activation by the collagen receptor GPVI and the integrin αIIbβ3, respectively, but suggest that additional SFKs also play a role for both receptors in view of the more pronounced inhibitory effect of the pan SFK inhibitors such as Dasatinib and PP2. To investigate this, we generated various combinations of mice deficient in two SFKs, namely mice deficient in Fyn/Lyn (fyn−/−lyn−/−), Fgr/Lyn (fgr−/−lyn−/−) and Lyn/Src (lyn−/−src−/−). In addition, we generated mice deficient in Fyn/Src (fyn−/−src−/−) as these two SFKs are constitutively associated with the β3-integrin tail [8,29]. Mice deficient in Lyn/Src and Fyn/Src and their matched controls were generated as radiation chimeras by transplantation of fetal liver cells because of the high level of lethality during gestation as previously reported [48]. Bone marrow depletion was confirmed by western blotting of radiation chimeric platelets with appropriate antibodies as previously described [34].
The platelet counts of the fyn−/−lyn−/− and fyn−/−src−/− mice were not significantly different to their respective controls, whereas the platelet counts of the lyn−/−src−/− and fgr−/−lyn−/− mice were reduced to approximately 40% and 70% of littermate controls at 8 weeks, respectively (Table 1). A significant increase in the level of monocytes was also observed in these two mice models (not shown). Platelet size was increased by approximately 15% (P < 0.01) in the fyn−/−lyn−/− and the fgr−/−lyn−/− mice but was not altered in the two other double-deficient mice (Table 1). The level of expression of GPVI, α2- and αIIb-integrin subunits and GPIbα were similar in all double-deficient platelets compared with WT platelets. GPIbα was reduced by approximately 15% in platelets from fgr−/−lyn−/− and lyn−/−src−/− mice (P < 0.05) (Table 1). The level of the remaining SFKs, assessed by western blot, was similar in all double-deficient and WT platelets (not shown and [34]).
Table 1. Platelet count, volume and surface glycoprotein expression in control and deficient platelets.
| Control | fyn−/−lyn−/− | fgr−/−lyn−/− | Chimeric control | fyn−/−src−/− | lyn−/−src−/− | |
|---|---|---|---|---|---|---|
| PC, 103 mm−3 | 366 ± 42 | 823 ± 47 | 267 ± 49** | 744 ± 52 | 813 ± 32 | 481 ± 49** |
| MPV, μm3 | 5.5 ± 0.1 | 6.2 ± 0.2** | 6.3 ± 0.2** | 5.4 ± 0.1 | 5.2 ± 0.1 | 5.7 ± 0.1 |
| GPIb | 50.4 ± 1.0 | 50.8 ± 4.9 | 37.9 ± 3.5* | 41.2 ± 2.5 | 44.5 ± 3.3 | 36.2 ± 1.9* |
| GPIIb | 89.5 ± 12.8 | 108.9 ± 15.6 | 104 ± 23 | 103.5 ± 17.0 | 118.3 ± 12.3 | 89.6 ± 7.9 |
| GPVI | 16.7 ± 1.0 | 22.2 ± 1.6 | 15.9 ± 0.5 | 19.9 ± 2.4 | 18.5 ± 2.5 | 16.7 ± 0.4 |
| α2 | 3.6 ± 0.5 | 3.3 ± 0.4 | 3.7 ± 1.0 | 3.4 ± 0.3 | 3.6 ± 0.1 | 3.1 ± 0.4 |
Whole blood platelet count and mean platelet volume (MPV) for control and deficient mice platelets were analyzed with an automated blood analyzer (ABX Pentra 60). Results are expressed as means ± standard error of the mean (SEM) for at least six mice per group. Expression of glycoproteins on the platelet surface was determined by flow cytometry using specific fluorescein isothiocyanate-labeled antibodies. Results are expressed as median fluorescence intensity ± SEM for at least four mice per group. One-way anova.
P < 0.05,
P < 0.01 vs. respective control.
An increased delay in aggregation and dense-granule secretion to CRP was observed in fyn−/−lyn−/− and lyn−/−src−/− platelets relative to lyn−/− platelets and, for a low concentration of the GPVI-specific peptide, the magnitude of aggregation and secretion also dramatically reduced. The defect in response was considerably greater in lyn−/−src−/− platelets than in fyn−/−lyn−/− platelets (Fig. 3A,B). A slight delay in aggregation and dense-granule secretion to CRP was also seen in fyn−/−src−/− platelets but this was less than that observed in the absence of Lyn (Fig. 3A,B). Interestingly, fgr−/−lyn−/− platelets show a reduced delay in aggregation and dense-granule secretion in comparison to WT or lyn−/− platelets, which could reflect a weak inhibitory role for Fgr in signaling by GPVI (Fig. 3A,B). These results demonstrate that, in the absence of Lyn, Src and Fyn play compensatory roles in supporting aggregation and secretion by GPVI, with Src having the more significant role. In the presence of Lyn, Fyn and Src have only a minor role in GPVI signaling.
Fig. 3.
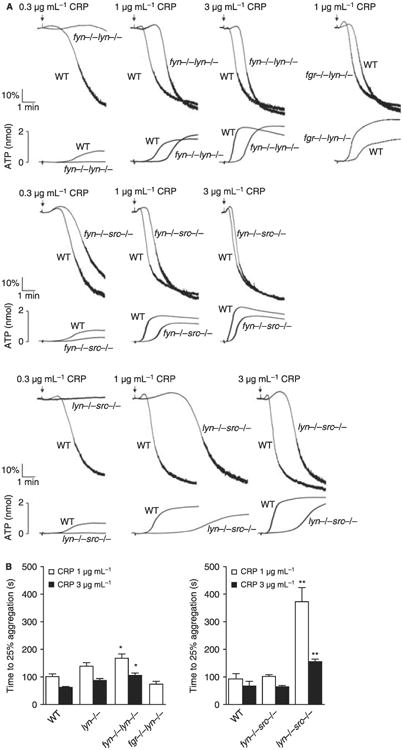
Impact of double deficiency of Src family kinases (SFK) in glycoprotein VI (GPVI)-induced platelet aggregation. (A) Washed platelets (2 × 108 mL−1) prepared from fyn−/−lyn−/− (top), fgr−/−lyn−/− (top), fyn−/−src−/− (middle) or lyn−/−src−/− (bottom) mice and their respective litter-matched control were stimulated with different doses of CRP (0.3, 1 and 3 μg mL−1). Platelet aggregation was measured as a change in light transmission and ATP secretion was measured as luciferin/luciferase-mediated luminescence, using a lumi-aggregometer. Representative images are shown (n = 4 mice per condition). (B) Time to 25% of aggregation was calculated and data are shown as mean ± SEM. One-way anova test, *P < 0.05, **P < 0.01 vs. wild type (WT).
The molecular basis of the difference in response to CRP in the double SFK-deficient platelets was investigated by measurement of tyrosine phosphorylation in whole cell lysates and of key signaling proteins in the GPVI cascade, namely FcR γ-chain, Syk and PLCγ2. A direct comparison of the phosphorylation results is complicated by the different time course of response in the various double SFK-deficient models. In consideration of this, we have focused on the pattern of tyrosine phosphorylation and on the level of phosphorylation of key signaling proteins relative to each other. A representative set of blots from platelets that have been stimulated by CRP (3 μg mL−1) for 3 min is shown in Fig. 4. CRP stimulates a similar pattern of increase in tyrosine phosphorylation in whole cell lysates in mice deficient in Lyn/Fyn, Lyn/Src and Fyn/Src-deficient platelets, but with quantitative differences in individual bands. There is a significant reduction in tyrosine phosphorylation of the FcR γ-chain in all three double-deficient platelets relative to WT mice, consistent with a critical role of SFKs in mediating phosphorylation of the FcR γ-chain ITAM. On the other hand, a significant reduction in phosphorylation of Syk and PLCγ2 was only observed in platelets deficient in Fyn/Src and Lyn/Src, whereas potentiation of phosphorylation of PLCγ2 was observed in Fyn/Lyn double-deficient platelets relative to controls, consistent with a negative feedback role of Lyn that is more pronounced in the Fyn/Lyn relative to Lyn/Src-deficient platelets. Thus, these results demonstrate qualitatively distinct roles of SFKs and emphasize that the interpretation of protein phosphorylation measurements needs to be considered in the context of the time course and pattern of response. It is noteworthy that potentiation of PLCγ2 phosphorylation in Fyn/Lyn-deficient platelets was not seen in our previous study [28] using a lower concentration of CRP (1 μg mL−1). This could reflect the relative contribution of the opposing actions of Lyn in GPVI signaling at the two different concentrations of the synthetic collagen peptide, although it is likely that other factors have also contributed to this difference including the use of lotrafiban rather than EGTA and the more mild nature of the defect in both the Fyn- and Lyn-deficient platelets relative to that seen in our previous study [28] as mentioned above.
Fig. 4.
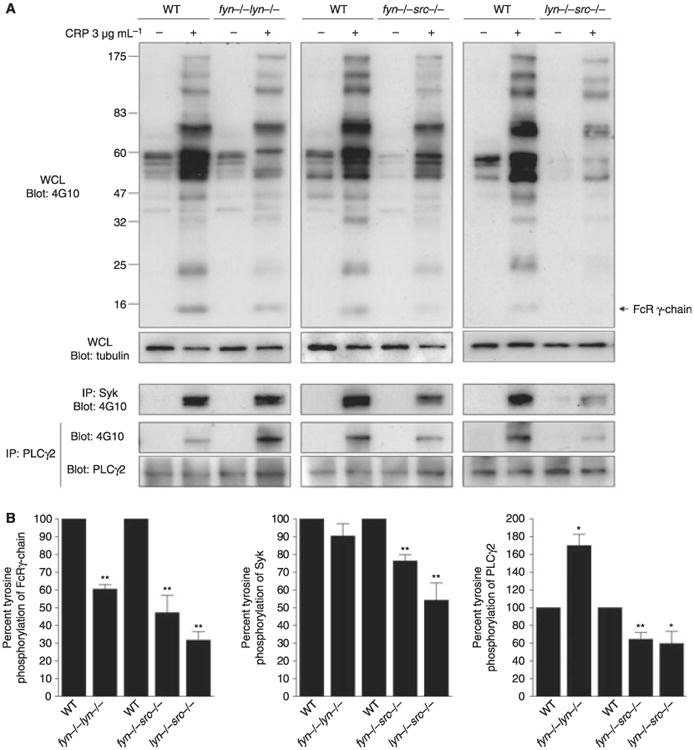
Impact of double deficiency of Src family kinases (SFK) in protein tyrosine phosphorylation in response to glycoprotein VI (GPVI) stimulation. Washed platelets (2 × 108 mL−1) from fyn−/−src−/−, lyn−/−src−/− or from fyn−/−lyn−/− mice in comparison to their respective litter-matched controls were activated for 3 min with collagen-related peptide (CRP) (3 μg mL−1) in the presence of lotrafiban (10 μmol L−1) and lysed. Whole platelet lysates were immunobloted with anti-phosphotyrosine antibody. The FcRγ-chain was observed on long exposure of whole cell lysates. Syk and PLCγ2 were immunoprecipitated from whole platelet lysates, and immunoprecipitates were immunoblotted with an anti-phosphotyrosine antibody and Syk or PLCγ2 antibodies (A) Representative blots of three independent experiments are shown. (B) Data represent mean ± standard error of the mean (SEM). One-way anova test, *P < 0.05, **P < 0.01 vs. respective controls.
Impact of double deficiency of SFK on platelet activation by GPVI and integrin αIIbβ3
Investigation of adhesion/spreading on fibrinogen was complicated by a significant increase in spreading of WT radiation chimeric platelets relative to those from non-irradiated mice (P < 0.05). To account for this, spreading was normalized by comparison of the WT radiation chimeric platelet response (= 100%) to that in the presence of the SFK inhibitor PP2 (= 0%), which is assumed to represent complete inhibition. The surface area of platelets on fibrinogen from Lyn/Src- and Fyn/Src-deficient mice was reduced by over 70% and 50%, respectively, relative to litter-matched controls (Fig. 5A), whereas Fyn/Lyn-deficient platelets exhibited a significant increase in spreading of 135% which is similar to that seen in Lyn-deficient platelets (Figs 2B and 5B). The surface area of platelets from mice deficient in Fgr/Lyn was not significantly different to controls (Fig. 5B), although there was a reduction in the number of platelets with filopodia (41.3 ± 1.9% vs. 73.4 ± 3.4%; P < 0.05). These results highlight the differential role of Src and Lyn in the regulation of the integrin αIIbβ3 outside-in signaling, with Lyn having an inhibitory role and Src a positive role. In platelets double deficient in Lyn/Src or Fgr/Lyn, spreading is abolished or similar to that of control platelets, respectively, suggesting that the potentiation induced in the absence of Lyn is mediated downstream of Src and Fgr (Fig. 5A,B).
Fig. 5.
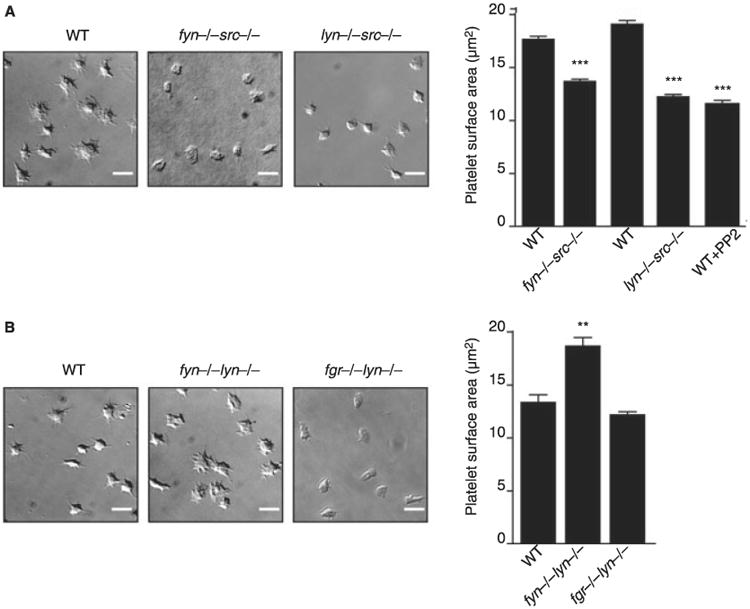
Impact of double deficiency of Src family kinases (SFK) in αIIbβ3 outside-in signaling. Washed platelets (2 × 107 mL−1) from fyn−/−src−/− or lyn−/−src−/− radiation chimeric mice (A) or from fyn−/−lyn−/− or fgr−/−lyn−/− mice (B) in comparison to their respective litter-matched controls or wild-type (WT) platelets treated for 10 min with 20 μmol L−1 PP2 were placed on a fibrinogen-coated surface for 45 min and then fixed. Images captured by differential interference contrast (DIC) microscopy (scale bar: 5 μm). Representative DIC images of platelets captured from four independent experiments for each genotype are shown. The surface area of platelets per condition were measured using ImageJ software (mean ± standard error of the mean [SEM]; n = 150–250 individual platelets per mice; four mice per genotyping). One-way anova test, **P < 0.01, ***P < 0.005 vs. respective controls.
Platelet aggregation in SFK double-deficient mice under flow conditions
The increase in spreading in the radiation chimeric platelets relative to controls did not result in a significant change in adhesion and aggregation of platelets on collagen at 1000 s−1 (not shown). On the other hand, a significant reduction in surface coverage relative to controls was observed in the absence of Fyn/Lyn or Lyn/Src, which was similar in magnitude to that in Lyn-deficient platelets but less than that seen in the presence of Dasatinib (Figs 2C and 6). In addition, there was also a significant reduction in aggregate size in all three sets of platelets (Figs 2C and 6), with the net size of lyn−/−src−/− platelets being less than that of lyn−/− platelets at 300 ± 30 and 390 ± 15 μm2, respectively, although this is also is influenced by the decrease in platelet count in the double-deficient platelets. The surface coverage of fyn−/−src−/− platelets on collagen at 1000 s−1 was similar to that in control platelets, although the size of the aggregates was also significantly reduced (Fig. 6). A reduction in aggregation was also observed in Fgr/Lyn-deficient platelets but again this is likely to also reflect the reduction in platelet count (not shown). There was no significant increase in blood loss using a tail bleeding assays in the Fyn/Lyn or Fyn/Src double-deficient mice relative to their matched controls (not shown).
Fig. 6.
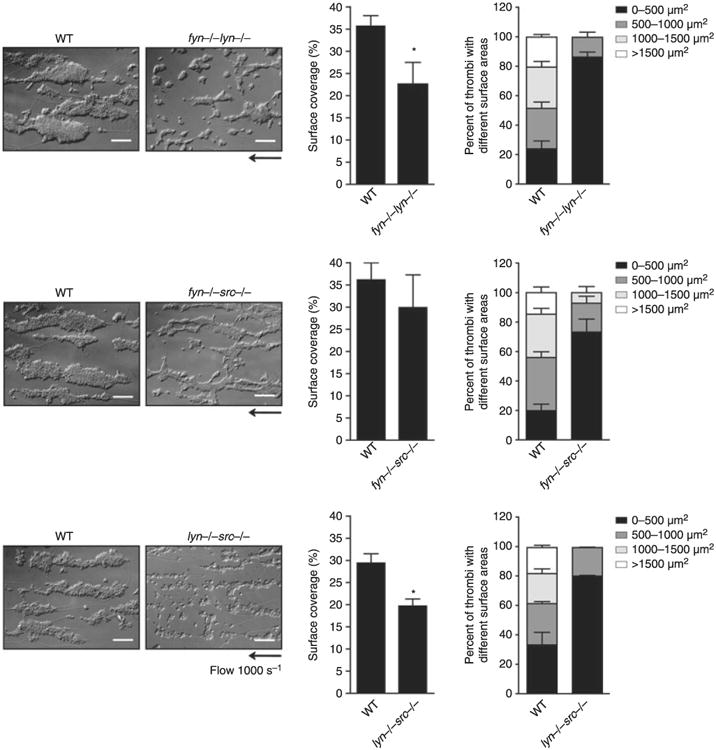
Impact of double deficiency of Src family kinases (SFK) in thrombus formation on collagen fibers under flow conditions. Anticoagulated blood from lyn−/−fyn−/−, fyn−/−src−/− or lyn−/−src−/− and their respective litter-matched controls was perfused through collagen-coated capillary tubes at 1000 s−1 for 4 min. Differential interference contrast (DIC) images of fixed platelets on collagen fibers after being flowed through collagen-coated capillary tubes at 1000 s−1 for 4 min (scale bar: 20 μm) are representative pictures of thrombus formed from at least three independent experiments of each genotype. Area covered by platelet thrombi and the percentage of thrombi with a different surface were measured in each of at least three different experiments and mean ± standard error of the mean (SEM) is shown. Student's t-test, *P < 0.05 vs. wild-type (WT) control.
Discussion
The present study demonstrates that mouse platelets express four SFKs, Fgr, Fyn, Lyn and Src, and establishes their roles in regulating platelet activation after engagement of the collagen receptor GPVI and the integrin αIIbβ3. Lyn is the major SFK regulating GPVI signaling and its absence causes a delay in activation and a marked reduction in aggregation on collagen under shear and in thrombus formation after laser injury in vivo. Mice deficient in Lyn/Src and, to a lesser extent, mice deficient in Lyn/Fyn have a further delay in activation suggesting that all three SFKs participate in GPVI signaling. In addition, Lyn has a powerful inhibitory role which results in a delayed potentiation of aggregation to low concentrations of GPVI agonists and increased spreading on the αIIbβ3-ligand, fibrinogen, which signals mainly via Src. Fgr plays a minor role in signaling by both receptors, although it abrogates the inhibitory effect of Lyn on fibrinogen in accordance with a previous report on Fgr, Hck and Lyn-deficient platelets [31] (Hck is undetectable in mouse platelets). Overall, these results demonstrate that SFKs have distinct and overlapping roles in regulating platelet activation by GPVI and integrin αIIbβ3.
Lyn is the most highly expressed of the SFKs and is present at over 10 times the level of Fyn and Src, and over 50 times that of Fgr. Lyn is localized to membrane rafts and is the major SFK mediating activation by GPVI. Src, on the other hand, is the major SFK mediating signaling by integrin αIIbβ3, with both Src and the integrin being localized outside of lipid rafts. However, the role of SFKs in platelet activation cannot be explained solely on the basis of their localization to rafts. For example, a significant proportion of Lyn is present in the non-raft fraction and yet functions as the major inhibitory SFK downstream of both GPVI and integrin αIIbβ3, presumably through phosphorylation of platelet ITIM receptors, including PECAM-1 and G6b. This class of receptor recruit the SH2 domain-containing tyrosine phosphatases, SHP-1 and SHP-2, and the inositol phosphatase, SHIP1, to mediate inhibition [15,49,50]. Similarly, mice deficient in Lyn and Src show an enhanced delay in activation relative to Lyn-deficient platelets even although Src is localized to the non-raft fraction.
Src family kinases are also known to participate in signaling by G protein-coupled receptors and it is therefore important to consider to what extent the results could reflect this role, bearing in mind that ADP and TxA2 are critical positive feedback agonists in platelet activation, and that thrombin also supports platelet activation in vivo. The general consensus is that SFKs play a minor role in aggregation and secretion to low concentrations of Gi and Gq protein-coupled receptor agonists [46,47,51–57]. Consistent with this, platelet activation in the individual and double-deficient SFK mice was unaltered to thrombin (Fig. S2), TxA2 mimetic, U46619 and ADP (not shown). Spreading of mouse platelets on fibrinogen under the conditions used is independent of ADP and TxA2 [58] and so results cannot reflect a role downstream of either of these receptors. Nevertheless, it is possible that observations at high shear on a collagen surface or in vivo could reflect a role for SFKs in signaling by G protein-coupled receptors, as well as by other receptors including GPIb [46,47].
The observation that the SFK inhibitors, PP2 and Dasatinib, have a more powerful inhibitory effect than loss of any of the four SFKs against GPVI and integrin αIIbβ3 signaling strongly suggests that one or more SFKs work in combination with Lyn and Src to mediate platelet activation by GPVI and integrin αIIbβ3. In the case of GPVI, the additional role is played by Src and to a lesser extent by Fyn, and in the case of αIIbβ3, this role is played by Lyn and to a lesser extent by Fyn. Fgr also appears to play a minor role in signaling by GPVI and integrin αIIbβ3. The redundancy between SFKs is further illustrated by the greater level of inhibition induced by the pan-SFK inhibitor Dasatinib on aggregation under flow conditions relative to the single and double SFK-deficient platelets and by the observation that Dasatinib causes a significant increase in tail bleeding times whereas these are not altered in the various SFK-mutant mouse models.
The conclusion that mice platelets express only four SFKs needs to be considered in light of previous studies which have reported expression of the SFKs, Hck, Lck and Yes in mouse platelets. We have been unable to detect any expression of these three kinases in spite of demonstration of the efficacy and specificity of each antibody for different members through western blotting of deficient control tissues of different SFK members. Explanations for the contrasting results in the literature include contamination with other cell types (notably in the genomic studies) and limitations in the specificity of available antibodies given the close sequence homology of SFKs. While the sensitivity limits for detection by western blotting is antibody dependent, the strong signals observed in control tissues and the detection range of western blotting strongly suggest that none of these kinases are likely to be expressed in platelets at a significant level. This is further underscored by the observation that an antibody against the phosphorylated conserved activatory site in SFKs detects three bands which appear to correspond to Fgr, Fyn, Lyn and Src as described above. The lack of expression of Blk, Lck, Hck and Yes is corroborated by a SAGE library from mouse megakaryocyte generated within our laboratory [49]. We found no tags for Blk, Lck, Hck or Yes, consistent with a lack of expression of these kinases within mouse platelets. In contrast to this, Lyn, Fyn and Src all have multiple SAGE tags. A recent expression database published by Rowley et al. [59] also demonstrates high expression of Lyn, Src and Fyn, low expression of Fgr and very little to no expression of Yes or Lck. In addition, Yes-deficient platelets show normal platelet aggregation in response to CRP stimulation and normal spreading on fibrinogen, which correlate with no or little expression of Yes in mouse platelets (not shown).
This study has investigated the roles of Fgr, Fyn, Lyn and Src in hemostasis and thrombosis in mouse platelets using both single-deficient mice and several combinations of double mutants. We show for the first time that Lyn plays a dominant role in regulating aggregation on collagen at arteriolar shear rates, as blood from Lyn-deficient mice forms only small aggregates along the length of the collagen fibers. This is presumably the consequence of the delay in adhesion that occurs in the absence of Lyn, as similar results are seen in the presence of αIIbβ3-blockade in platelets expressing a proline-depleted form of GPVI which is unable to bind to Lyn [see Introduction; 25]. Lyn-deficient mice have defective thrombus formation as shown using a laser injury model, although presumably sufficient thrombus is formed to prevent an increase in bleeding at least in a tail bleeding assay.
One of the striking observations in this study was the marked reduction in platelet counts in mice deficient in Fgr/Lyn and Lyn/Src that was evident by 8 weeks after birth. Lyn-deficient mice have been shown to develop a weak myeloproliferative disease (MPD) which begins after 8 weeks and is associated with increased myeloid progenitors and a reduced number of mature B cells [44,60]. These effects are believed to be due to altered ITIM phosphorylation and loss of membrane recruitment of the inositol 5-phosphatase, SHIP1 [44]. The onset of the MPD is associated with a mild reduction in platelet count in the Lyn-deficient platelets with age and may be mediated at the level of the hematopoietic stem cell. The combined defect of loss of Lyn and several other SFKs also induces profound effects on the development and function of hematopoietic lineages. These include a marked increase in neutrophil and dendritic cell activation in mice deficient in Fgr/Hck [61], altered hematopoietic stem cells, differentiation, marked fibrosis and invasion of inflammatory cells in the lung which results in death within the first 2 months of life in Lyn/Hck-deficient mice [13]. The defect in platelet count in the Fgr/Lyn and Lyn/Src mice observed in this study is therefore likely to be as a result of altered HSC function and possibly to an early onset of a MPD.
In conclusion, this study demonstrates that although Lyn and Src are the major SFKs mediating platelet activation by the collagen receptor GPVI and the integrin αIIbβ3, Fyn and Fgr also have synergistic and complementary roles in regulating platelet function, and that each of the four SFKs plays a unique role in supporting platelet activation. Significantly, the present study also demonstrates that loss of any of the four SFKs does not give rise to excessive bleeding. Given the central role of SFKs in many biological processes, these observations have important implications for the development of inhibitors that target individual SFKs, in particular Lyn. However, the individual SFKs are unlikely to be targets for the development of anti-thrombotics because of their multiple roles in other tissues, notably on the immune system, unless irreversible inhibitors can be developed to selectively target platelets.
Supplementary Material
Figure S1. Identification of Src family kinases (SFKs) on phospho-tyrosine blots
Figure S2. Washed platelets (2 × 108/ml) prepared from fyn−/−, lyn−/−, src−/− or fgr−/−, fyn−/− lyn−/−, fyn−/−src−/− or lyn−/−src−/− mice and their respective litter-matched controls were stimulated with thrombin (0.5 UI mL−1).
Data S1. Methods.
Acknowledgments
We would like to thank C. Hughes and B. Grygielska for technical support, and I. Ricketts, C. Smith and J. Ullah in the Biomedical Service Unit for generating the radiation chimeric mice and maintaining the mouse colonies. This work was supported by the BHF (PG/06/129/21681). S.P.W. holds a BHF Chair (CH/03/003).
Footnotes
Disclosure of Conflict of Interest: The authors state that they have no conflict of interest.
Supporting Information: Additional Supporting Information may be found in the online version of this article
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–27. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- 2.Summy JM, Qian Y, Jiang BH, Guappone-Koay A, Gatesman A, Shi X, Flynn DC. The SH4-Unique-SH3-SH2 domains dictate specificity in signaling that differentiate c-Yes from c-Src. J Cell Sci. 2003;116:2585–98. doi: 10.1242/jcs.00466. [DOI] [PubMed] [Google Scholar]
- 3.Sato I, Obata Y, Kasahara K, Nakayama Y, Fukumoto Y, Yamasaki T, Yokoyama KK, Saito T, Yamaguchi N. Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. J Cell Sci. 2009;122:965–75. doi: 10.1242/jcs.034843. [DOI] [PubMed] [Google Scholar]
- 4.Levental I, Lingwood D, Grzybek M, Coskun U, Simons K. Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc Natl Acad Sci U S A. 2010;107:22050–4. doi: 10.1073/pnas.1016184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 6.Ellison S, Mori J, Barr AJ, Senis YA. CD148 enhances platelet responsiveness to collagen by maintaining a pool of active Src family kinases. J Thromb Haemost. 2010;8:1575–83. doi: 10.1111/j.1538-7836.2010.03865.x. [DOI] [PubMed] [Google Scholar]
- 7.Senis YA, Tomlinson MG, Ellison S, Mazharian A, Lim J, Zhao Y, Kornerup KN, Auger JM, Thomas SG, Dhanjal T, Kalia N, Zhu JW, Weiss A, Watson SP. The tyrosine phosphatase CD148 is an essential positive regulator of platelet activation and thrombosis. Blood. 2009;113:4942–54. doi: 10.1182/blood-2008-08-174318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ. Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc Natl Acad Sci U S A. 2003;100:13298–302. doi: 10.1073/pnas.2336149100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corey SJ, Anderson SM. Src-related protein tyrosine kinases in hematopoiesis. Blood. 1999;93:1–14. [PubMed] [Google Scholar]
- 10.Shattil SJ. Integrins and Src: dynamic duo of adhesion signaling. Trends Cell Biol. 2005;15:399–403. doi: 10.1016/j.tcb.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost. 2005;3:1752–62. doi: 10.1111/j.1538-7836.2005.01429.x. [DOI] [PubMed] [Google Scholar]
- 12.Watson SP, Herbert JM, Pollitt AY. GPVI and CLEC-2 in hemostasis and vascular integrity. J Thromb Haemost. 2010;8:1456–67. doi: 10.1111/j.1538-7836.2010.03875.x. [DOI] [PubMed] [Google Scholar]
- 13.Xiao W, Hong H, Kawakami Y, Lowell CA, Kawakami T. Regulation of myeloproliferation and M2 macrophage programming in mice by Lyn/Hck, SHIP, and Stat5. J Clin Invest. 2008;118:924–34. doi: 10.1172/JCI34013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cicmil M, Thomas JM, Sage T, Barry FA, Leduc M, Bon C, Gibbins JM. Collagen, convulxin, and thrombin stimulate aggregation-independent tyrosine phosphorylation of CD31 in platelets. Evidence for the involvement of Src family kinases. J Biol Chem. 2000;275:27339–47. doi: 10.1074/jbc.M003196200. [DOI] [PubMed] [Google Scholar]
- 15.Ming Z, Hu Y, Xiang J, Polewski P, Newman PJ, Newman DK. Lyn and PECAM-1 function as interdependent inhibitors of platelet aggregation. Blood. 2011;117:3903–6. doi: 10.1182/blood-2010-09-304816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Locke D, Chen H, Liu Y, Liu C, Kahn ML. Lipid rafts orchestrate signaling by the platelet receptor glycoprotein VI. J Biol Chem. 2002;277:18801–9. doi: 10.1074/jbc.M111520200. [DOI] [PubMed] [Google Scholar]
- 17.Pasquet JM, Gross B, Quek L, Asazuma N, Zhang W, Sommers CL, Schweighoffer E, Tybulewicz V, Judd B, Lee JR, Koretzky G, Love PE, Samelson LE, Watson SP. LAT is required for tyrosine phosphorylation of phospholipase cgamma2 and platelet activation by the collagen receptor GPVI. Mol Cell Biol. 1999;19:8326–34. doi: 10.1128/mcb.19.12.8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ragab A, Severin S, Gratacap MP, Aguado E, Malissen M, Jandrot-Perrus M, Malissen B, Ragab-Thomas J, Payrastre B. Roles of the C-terminal tyrosine residues of LAT in GPVI-induced platelet activation: insights into the mechanism of PLC gamma 2 activation. Blood. 2007;110:2466–74. doi: 10.1182/blood-2007-02-075432. [DOI] [PubMed] [Google Scholar]
- 19.Wonerow P, Obergfell A, Wilde JI, Bobe R, Asazuma N, Brdicka T, Leo A, Schraven B, Horejsi V, Shattil SJ, Watson SP. Differential role of glycolipid-enriched membrane domains in glycoprotein VI- and integrin-mediated phospholipase Cgamma2 regulation in platelets. Biochem J. 2002;364:755–65. doi: 10.1042/BJ20020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boylan B, Gao C, Rathore V, Gill JC, Newman DK, Newman PJ. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood. 2008;112:2780–6. doi: 10.1182/blood-2008-02-142125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pestina TI, Stenberg PE, Druker BJ, Steward SA, Hutson NK, Barrie RJ, Jackson CW. Identification of the Src family kinases, Lck and Fgr in platelets. Their tyrosine phosphorylation status and subcellular distribution compared with other Src family members. Arterioscler Thromb Vasc Biol. 1997;17:3278–85. doi: 10.1161/01.atv.17.11.3278. [DOI] [PubMed] [Google Scholar]
- 22.Stenberg PE, Pestina TI, Barrie RJ, Jackson CW. The Src family kinases, Fgr, Fyn, Lck, and Lyn, colocalize with coated membranes in platelets. Blood. 1997;89:2384–93. [PubMed] [Google Scholar]
- 23.Zhao YH, Krueger JG, Sudol M. Expression of cellular-yes protein in mammalian tissues. Oncogene. 1990;5:1629–35. [PubMed] [Google Scholar]
- 24.Ezumi Y, Shindoh K, Tsuji M, Takayama H. Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor gamma chain complex on human platelets. J Exp Med. 1998;188:267–76. doi: 10.1084/jem.188.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmaier AA, Zou Z, Kazlauskas A, Emert-Sedlak L, Fong KP, Neeves KB, Maloney SF, Diamond SL, Kunapuli SP, Ware J, Brass LF, Smithgall TE, Saksela K, Kahn ML. Molecular priming of Lyn by GPVI enables an immune receptor to adopt a hemostatic role. Proc Natl Acad Sci U S A. 2009;106:21167–72. doi: 10.1073/pnas.0906436106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki-Inoue K, Tulasne D, Shen Y, Bori-Sanz T, Inoue O, Jung SM, Moroi M, Andrews RK, Berndt MC, Watson SP. Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J Biol Chem. 2002;277:21561–6. doi: 10.1074/jbc.M201012200. [DOI] [PubMed] [Google Scholar]
- 27.Bori-Sanz T, Inoue KS, Berndt MC, Watson SP, Tulasne D. Delineation of the region in the glycoprotein VI tail required for association with the Fc receptor gamma-chain. J Biol Chem. 2003;278:35914–22. doi: 10.1074/jbc.M301826200. [DOI] [PubMed] [Google Scholar]
- 28.Quek LS, Pasquet JM, Hers I, Cornall R, Knight G, Barnes M, Hibbs ML, Dunn AR, Lowell CA, Watson SP. Fyn and Lyn phosphorylate the Fc receptor gamma chain downstream of glycoprotein VI in murine platelets, and Lyn regulates a novel feedback pathway. Blood. 2000;96:4246–53. [PubMed] [Google Scholar]
- 29.Reddy KB, Smith DM, Plow EF. Analysis of Fyn function in hemostasis and alphaIIbbeta3-integrin signaling. J Cell Sci. 2008;121:1641–8. doi: 10.1242/jcs.014076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maxwell MJ, Yuan Y, Anderson KE, Hibbs ML, Salem HH, Jackson SP. SHIP1 and Lyn kinase negatively regulate integrin alpha IIb beta 3 signaling in platelets. J Biol Chem. 2004;279:32196–204. doi: 10.1074/jbc.M400746200. [DOI] [PubMed] [Google Scholar]
- 31.Obergfell A, Eto K, Mocsai A, Buensuceso C, Moores SL, Brugge JS, Lowell CA, Shattil SJ. Coordinate interactions of Csk, Src, and Syk kinases with [alpha]IIb[beta]3 initiate integrin signaling to the cyto-skeleton. J Cell Biol. 2002;157:265–75. doi: 10.1083/jcb.200112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowell CA, Soriano P, Varmus HE. Functional overlap in the src gene family: inactivation of hck and fgr impairs natural immunity. Genes Dev. 1994;8:387–98. doi: 10.1101/gad.8.4.387. [DOI] [PubMed] [Google Scholar]
- 33.Poole A, Gibbins JM, Turner M, van Vugt MJ, van de Winkel JG, Saito T, Tybulewicz VL, Watson SP. The Fc receptor gamma-chain and the tyrosine kinase Syk are essential for activation of mouse platelets by collagen. EMBO J. 1997;16:2333–41. doi: 10.1093/emboj/16.9.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Severin S, Pollitt AY, Navarro-Nunez L, Nash CA, Mourao-Sa D, Eble JA, Senis YA, Watson SP. Syk-dependent Phosphorylation of CLEC-2: a novel mechanism of hem-immunoreceptor tyrosine-based activation motif signaling. J Biol Chem. 2011;286:4107–16. doi: 10.1074/jbc.M110.167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki-Inoue K, Inoue O, Frampton J, Watson SP. Murine GPVI stimulates weak integrin activation in PLCgamma2-/- platelets: involvement of PLCgamma1 and PI3-kinase. Blood. 2003;102:1367–73. doi: 10.1182/blood-2003-01-0029. [DOI] [PubMed] [Google Scholar]
- 36.Pollitt AY, Grygielska B, Leblond B, Desire L, Eble JA, Watson SP. Phosphorylation of CLEC-2 is dependent on lipid rafts, actin polymerization, secondary mediators, and Rac. Blood. 2010;115:2938–46. doi: 10.1182/blood-2009-12-257212. [DOI] [PubMed] [Google Scholar]
- 37.McCarty OJ, Larson MK, Auger JM, Kalia N, Atkinson BT, Pearce AC, Ruf S, Henderson RB, Tybulewicz VL, Machesky LM, Watson SP. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem. 2005;280:39474–84. doi: 10.1074/jbc.M504672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCarty OJ, Zhao Y, Andrew N, Machesky LM, Staunton D, Frampton J, Watson SP. Evaluation of the role of platelet integrins in fibronectin-dependent spreading and adhesion. J Thromb Haemost. 2004;2:1823–33. doi: 10.1111/j.1538-7836.2004.00925.x. [DOI] [PubMed] [Google Scholar]
- 39.Auger JM, Kuijpers MJ, Senis YA, Watson SP, Heemskerk JW. Adhesion of human and mouse platelets to collagen under shear: a unifying model. Faseb J. 2005;19:825–7. doi: 10.1096/fj.04-1940fje. [DOI] [PubMed] [Google Scholar]
- 40.Kalia N, Auger JM, Atkinson B, Watson SP. Critical role of FcR gamma-chain, LAT, PLCgamma2 and thrombin in arteriolar thrombus formation upon mild, laser-induced endothelial injury in vivo. Microcirculation. 2008;15:325–35. doi: 10.1080/10739680701728822. [DOI] [PubMed] [Google Scholar]
- 41.Chari R, Kim S, Murugappan S, Sanjay A, Daniel JL, Kunapuli SP. Lyn, PKC-delta, SHIP-1 interactions regulate GPVI-mediated platelet-dense granule secretion. Blood. 2009;114:3056–63. doi: 10.1182/blood-2008-11-188516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lannutti BJ, Shim MH, Blake N, Reems JA, Drachman JG. Identification and activation of Src family kinases in primary megakaryocytes. Exp Hematol. 2003;31:1268–74. doi: 10.1016/j.exphem.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 43.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 44.Harder KW, Quilici C, Naik E, Inglese M, Kountouri N, Turner A, Zlatic K, Tarlinton DM, Hibbs ML. Perturbed myelo/erythropoiesis in Lyn-deficient mice is similar to that in mice lacking the inhibitory phosphatases SHP-1 and SHIP-1. Blood. 2004;104:3901–10. doi: 10.1182/blood-2003-12-4396. [DOI] [PubMed] [Google Scholar]
- 45.Gratacap MP, Martin V, Valera MC, Allart S, Garcia C, Sie P, Recher C, Payrastre B. The new tyrosine-kinase inhibitor and anticancer drug dasatinib reversibly affects platelet activation in vitro and in vivo. Blood. 2009;114:1884–92. doi: 10.1182/blood-2009-02-205328. [DOI] [PubMed] [Google Scholar]
- 46.Cho MJ, Pestina TI, Steward SA, Lowell CA, Jackson CW, Gartner TK. Role of the Src family kinase Lyn in TxA2 production, adenosine diphosphate secretion, Akt phosphorylation, and irreversible aggregation in platelets stimulated with gamma-thrombin. Blood. 2002;99:2442–7. doi: 10.1182/blood.v99.7.2442. [DOI] [PubMed] [Google Scholar]
- 47.Yin H, Liu J, Li Z, Berndt MC, Lowell CA, Du X. Src family tyrosine kinase Lyn mediates VWF/GPIb-IX-induced platelet activation via the cGMP signaling pathway. Blood. 2008;112:1139–46. doi: 10.1182/blood-2008-02-140970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stein PL, Vogel H, Soriano P. Combined deficiencies of Src, Fyn, and Yes tyrosine kinases in mutant mice. Genes Dev. 1994;8:1999–2007. doi: 10.1101/gad.8.17.1999. [DOI] [PubMed] [Google Scholar]
- 49.Senis YA, Tomlinson MG, Garcia A, Dumon S, Heath VL, Herbert J, Cobbold SP, Spalton JC, Ayman S, Antrobus R, Zitzmann N, Bicknell R, Frampton J, Authi KS, Martin A, Wakelam MJ, Watson SP. A comprehensive proteomics and genomics analysis reveals novel transmembrane proteins in human platelets and mouse megakaryocytes including G6b-B, a novel immunoreceptor tyrosine-based inhibitory motif protein. Mol Cell Proteomics. 2007;6:548–64. doi: 10.1074/mcp.D600007-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pasquet JM, Quek L, Stevens C, Bobe R, Huber M, Duronio V, Krystal G, Watson SP. Phosphatidylinositol 3,4,5-trisphosphate regulates Ca(2+) entry via btk in platelets and megakaryocytes without increasing phospholipase C activity. EMBO J. 2000;19:2793–802. doi: 10.1093/emboj/19.12.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hardy AR, Jones ML, Mundell SJ, Poole AW. Reciprocal cross-talk between P2Y1 and P2Y12 receptors at the level of calcium signaling in human platelets. Blood. 2004;104:1745–52. doi: 10.1182/blood-2004-02-0534. [DOI] [PubMed] [Google Scholar]
- 52.Jin J, Quinton TM, Zhang J, Rittenhouse SE, Kunapuli SP. Adenosine diphosphate (ADP)-induced thromboxane A(2) generation in human platelets requires coordinated signaling through integrin alpha(IIb)beta(3) and ADP receptors. Blood. 2002;99:193–8. doi: 10.1182/blood.v99.1.193. [DOI] [PubMed] [Google Scholar]
- 53.Dorsam RT, Kim S, Jin J, Kunapuli SP. Coordinated signaling through both G12/13 and G(i) pathways is sufficient to activate GPIIb/IIIa in human platelets. J Biol Chem. 2002;277:47588–95. doi: 10.1074/jbc.M208778200. [DOI] [PubMed] [Google Scholar]
- 54.Dorsam RT, Kim S, Murugappan S, Rachoor S, Shankar H, Jin J, Kunapuli SP. Differential requirements for calcium and Src family kinases in platelet GPIIb/IIIa activation and thromboxane generation downstream of different G-protein pathways. Blood. 2005;105:2749–56. doi: 10.1182/blood-2004-07-2821. [DOI] [PubMed] [Google Scholar]
- 55.Minuz P, Fumagalli L, Gaino S, Tommasoli RM, Degan M, Cavallini C, Lecchi A, Cattaneo M, Lechi Santonastaso C, Berton G. Rapid stimulation of tyrosine phosphorylation signals downstream of G-protein-coupled receptors for thromboxane A2 in human platelets. Biochem J. 2006;400:127–34. doi: 10.1042/BJ20061015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nash CA, Severin S, Dawood BB, Makris M, Mumford A, Wilde J, Senis YA, Watson SP. Src family kinases are essential for primary aggregation by G(i) -coupled receptors. J Thromb Haemost. 2010;8:2273–82. doi: 10.1111/j.1538-7836.2010.03992.x. [DOI] [PubMed] [Google Scholar]
- 57.Garcia A, Shankar H, Murugappan S, Kim S, Kunapuli SP. Regulation and functional consequences of ADP receptor-mediated ERK2 activation in platelets. Biochem J. 2007;404:299–308. doi: 10.1042/BJ20061584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thornber K, McCarty OJ, Watson SP, Pears CJ. Distinct but critical roles for integrin alphaIIbbeta3 in platelet lamellipodia formation on fibrinogen, collagen-related peptide and thrombin. FEBS J. 2006;273:5032–43. doi: 10.1111/j.1742-4658.2006.05500.x. [DOI] [PubMed] [Google Scholar]
- 59.Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, Nix DA, Yost CC, Zimmerman GA, Weyrich AS. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood. 2011;118:e101–11. doi: 10.1182/blood-2011-03-339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harder KW, Parsons LM, Armes J, Evans N, Kountouri N, Clark R, Quilici C, Grail D, Hodgson GS, Dunn AR, Hibbs ML. Gain- and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity. 2001;15:603–15. doi: 10.1016/s1074-7613(01)00208-4. [DOI] [PubMed] [Google Scholar]
- 61.Zhang H, Meng F, Chu CL, Takai T, Lowell CA. The Src family kinases Hck and Fgr negatively regulate neutrophil and dendritic cell chemokine signaling via PIR-B. Immunity. 2005;22:235–46. doi: 10.1016/j.immuni.2005.01.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Identification of Src family kinases (SFKs) on phospho-tyrosine blots
Figure S2. Washed platelets (2 × 108/ml) prepared from fyn−/−, lyn−/−, src−/− or fgr−/−, fyn−/− lyn−/−, fyn−/−src−/− or lyn−/−src−/− mice and their respective litter-matched controls were stimulated with thrombin (0.5 UI mL−1).
Data S1. Methods.