

Abstract
This study introduces a flexible and compound targeted approach to Deplete and Enrich Select Ingredients to Generate Normalized Extract Resources, generating DESIGNER extracts, by means of chemical subtraction or augmentation of metabolites. Targeting metabolites based on their liquid–liquid partition coefficients (K values), K targeting uses countercurrent separation methodology to remove single or multiple compounds from a chemically complex mixture, according to the following equation: DESIGNER extract = total extract ± target compound(s). Expanding the scope of the recently reported depletion of extracts by immunoaffinity or solid phase liquid chromatography, the present approach allows a more flexible, single- or multi-targeted removal of constituents from complex extracts such as botanicals. Chemical subtraction enables both chemical and biological characterization, including detection of synergism/antagonism by both the subtracted targets and the remaining metabolite mixture, as well as definition of the residual complexity of all fractions. The feasibility of the DESIGNER concept is shown by K-targeted subtraction of four bioactive prenylated phenols, isoxanthohumol (1), 8-prenylnaringenin (2), 6-prenylnaringenin (3), and xanthohumol (4), from a standardized hops (Humulus lupulus L.) extract using specific solvent systems. Conversely, adding K-targeted isolates allows enrichment of the original extract and hence provides an augmented DESIGNER material. Multiple countercurrent separation steps were used to purify each of the four compounds, and four DESIGNER extracts with varying depletions were prepared. The DESIGNER approach innovates the characterization of chemically complex extracts through integration of enabling technologies such as countercurrent separation, K-by-bioactivity, the residual complexity concepts, as well as quantitative analysis by 1H NMR, LC-MS, and HiFSA-based NMR fingerprinting.
Botanical dietary supplements, and other natural health products in general, are highly complex chemical entities. Even well-authenticated products such as single herbal extracts originate from complex chemical–biological matrices that contain (many) thousands of metabolites. These metabolomes are formed by interactive biosynthetic pathways from a combination of common building blocks that bring about a vast array of chemodiversity. The inherent complexity of the metabolome is the origin of residual complexity (RC),1 a phenomenon encountered ubiquitously in natural product research. The residual complexity concept explains many of the persistent challenges associated with natural product drug discovery as well as projects directed at the identification of (multiple) biological activities and active lead compounds. One example of a well-studied botanical, which has evaded the reductionist models of scientific investigations, is preparations derived from hops (Humulus lupulus L., Cannabaceae). As detailed below, the plethora of known H. lupulus constituents, including those more recently discovered, still does not provide a persuasive explanation of numerous, traditionally well-founded beneficial uses of H. lupulus for human health.2,3
The majority of studies on botanicals and complex natural (health) products take a reductionist approach and are directed at finding single or a few actives from the metabolome. More importantly, focusing on a single or very few of their constituents may not necessarily unravel relevant biology. In addition, this approach bears a similar risk of failing to explain the observed biological activity, both qualitatively and quantitatively, as does the approach of bioassay-guided fractionation. One highly influential concept in modern biomedical research is the targeted deletion of specific genes and the generation of “knockout” strains of organisms. This concept is widely applied to microbes (e.g., knockout Escherichia coli) and animals (e.g., knockout mice). Considering the similarities in the complexity of genomes and metabolomes, the process of knocking out single entity can be an important concept in studying the overwhelming complexity of both genes and metabolomes, respectively. The approach of targeted removal (“knockout”) of a single, or several selected, chemical entities represents a compelling alternative to studying metabolomic natural (health) products. However, while the complete and selective removal of a single entity may be desirable, doing so with very high specificity is rather challenging and/or quite laborious (see knockout concept below). The present work introduces the concept of Depletion and Enrichment of Select Ingredients Generating Normalized Extract Resources (DESIGNER) as a novel approach to exploring the biology of complex extracts.
The concept integrates advanced countercurrent separation (CS; includes countercurrent chromatography [CCC] and centrifugal partition chromatography [CPC]) methodology with metabolomic analysis by LC-MS, UHPLC-UV, and quantitative 1H NMR (qHNMR) for the targeted design of selectively prepared extracts. This process yields DESIGNER materials that are derived from otherwise unaltered metabolomic mixtures such as natural product extracts (Figure 1). The concept of DESIGNER extracts utilizes the flexibility of possible adjustments of chromatographic selectivity, polarity, and orthogonality and can thus target single and multiple metabolites, producing single and multiple knockout, knock-down, and knock-in extracts. The present study elaborates the DESIGNER concept for single and multiple modifications of an extract of hops (H. lupulus) developed for in vivo studies.2 Hops consist of the dried strobili of Humulus lupulus, and they have been shown to contain a plethora of phytochemical constituents such as essential oil, di-, and triprenylated phloroglucinol derivatives, chalcones, and other prenylated flavonoids, such as isoxanthohumol (1), 8-prenylnaringenin (2), 6-prenylnaringenin (3), and xanthohumol (4). Hops have been associated with a variety of biological activities,2−7 and the prenylphenols 1–4 (Chart 1) are widely considered bioactive marker compounds (S1, Supporting Information, provides an overview of the activities). Moreover, as materials that contain complex patterns of both chalcones and flavanones, hops extracts represent known cases of both static and dynamic residual complexity,8−10 from which possible instances of synergism or antagonism have not been explored rigorously.
Figure 1.
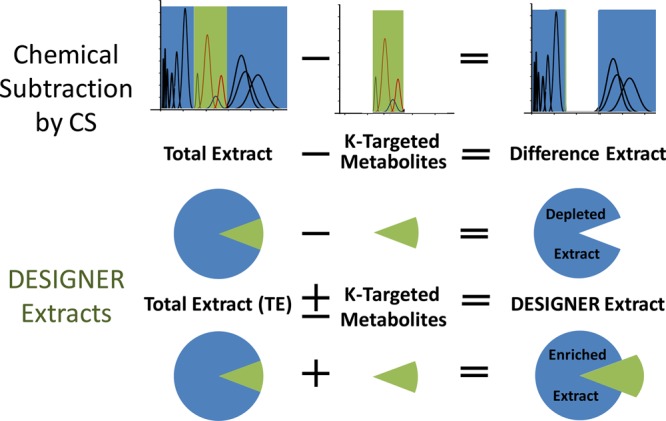
Schematic representation of chemical subtraction and the production of DESIGNER extracts. Chemical subtraction is a result of the K-targeted depletion of metabolites from a total extract with countercurrent separation (CS). DESIGNER extracts are more widely described as Depletion and Enrichment of Select Ingredients Generates Normalized Extract Resources and, thus, yield materials that are designed to evaluate the biological activity of natural products both within and outside of their natural matrices.
Chart 1. Structures of 1–4.

The generation of knockout extracts has received attention since it represents a direct way to study the biological contribution of a given compound to the overall activity of its original extract. Recently, the production of knockout extracts by immunoaffinity chromatography has been described.11,12 This technique uses monoclonal antibodies to “selectively” remove a single phytoconstituent from a complex mixture to produce an extract without the component, in some ways analogous to a knockout mouse lacking a specific gene. In an early report (1998),13 Tanaka and Shoyama used an ELISA assay to quantify specific compounds in herbal medicines.13 They further expanded this technique to using monoclonal immunoaffinity chromatography to concentrate active components from Panax ginseng,12,14,15Panax japonicus,16Coleus forskohlii,15 and Cannabis sativa.15 In a 2007 paper,11 the authors proposed the use of the term “knockout extracts” for the eluents of these monoclonal antibody columns. The authors applied this concept to remove “selectively” the triterpene saponin, gingenoside Re, from Panax quinquefolius,11 and glycyrrhizin from Glycyrrhiza spp.,17 respectively, to better understand the impact of single compounds on the potential overall antidiabetes,11 antiobesity,11 and anti-inflamatory17 activities. This methodology, while superficially very specific, does suffer from cross-reactivity with nontargeted metabolites, as observed in the cases of 1-deoxyforskolin, 1,9-dideoxyforskolin, 6-acetyl-7-deacetylforskolin, and 7-deacetylforskolin binding to the antiforskolin monoclonal antibody,13,15 cannabidiol, cannabigenoravin, 7-hydroxy-Δ6-THC, 7-oxo-Δ6-THC, and other cannabinoids binding to antitetrahydrocannabinolic acid (THCA) monoclonal antibody,13,15 and also gingenoside Rc, and gingenoside Rd binding to antigingenoside Rb1 monoclonal antibody.14 In addition, this methodology has two other drawbacks: the time involved and the uncertainty due to generating the antibodies and the low loading capacity of the immunoaffinity chromatography columns. Molecularly imprinted polymers may take the place of antibodies to remove target metabolites from complex mixtures.18,19 This method, however, has not been used to study bioactivity. A variation in this approach was reported by Liu et al. in 2010, in which they employed acetylcholinesterase to remove metabolites from extracts of Lycoris radiata referring to the bound metabolites as “fishings” and the unbound eluents as “knockouts”.20 This technique has recently been used to extract epimedins A-C and icariin from Epimedium brevicornum, and to examine their individual contributions to the bioactivity.21 The authors also used the “fishing” term introduced by Liu et al. Given the numerous drawbacks of the antibody approach such as the unpredictable specificity, limited practical feasibility, time involved, and low capacity, there is a definite need for another approach. The present study has developed a chemical alternative and explored its potential and limitations with regard to the metabolomic content of total plant extracts from hops.
In 2008, the concept of chemical subtraction was introduced using countercurrent separation (CS) for the targeted removal of a single compound from a bioactive mixture of metabolites.22 The term “chemical subtraction” reflects the similarity with arithmetic subtraction and applies to a chemical operation aimed at removing or deleting certain components, but also implies that no chemical modifications are made, by virtue of the liquid-only advantage of CS (see also “Designing Extracts” under “Discussion”). Chemical subtraction was initially developed to study E. coli antiadherent phytochemicals that may be active against urinary tract infections.22 A known antibacterial component, benzoic acid, was selectively removed by CS from cranberry (Vaccinium macrocarpon) juice, with a purity of 97.47% measured by qHNMR.22 Since then, this chromatographic technique has evolved further with regard to the analyte targeting based on partition coefficients (K; K-targeting), as in the case of preparative and/or analytical separation of bilobalide and ginkgolides A, B, C, and J, from G. biloba.23 It is rarely possible to remove a single metabolite in a single step. Therefore, multiple orthogonal countercurrent separation steps may be performed with coeluents of the target compound from the first countercurrent separation added to the depleted extract. As a result, the biological properties of the extract can be examined by chemical subtraction. Herein, the bioactive prenylated hop phenols, 1–4, were chosen as examples to demonstrate this methodology for a botanical extract that has been the subject of extensive chemical and biological investigation in the UIC/NIH Botanical Center. Applying the elution–extrusion method in countercurrent separation,24 quantitative recovery of a DESIGNER extract has been accomplished. Chemical subtraction enables further chemical and biological characterization of both the DESIGNER extracts and the residual complexity of the purified target compound(s). As a result, the interaction of the target(s) and the DESIGNER extracts permit the evaluation of synergistic/antagonistic relationships.
A key aspect of chemical subtraction refers to the purity assessment of the target compounds (Ts) as well as the evaluation of their residual complexity (RC).1,22 Its detection and quantitation in the original and depleted extracts establishes the efficiency and selectivity of the subtraction method. Residual complexity is associated with the impurity profile of isolated compounds, which can either be static residual complexity (SRC),1 relating to the presence of stable minor impurities, or dynamic residual complexity (DRC),1 relating to chemical reactivity and instability. The conversion of chalcones to flavanones through an intramolecular Michael addition reaction,25 and the chemical degradation of bioactive (Z)-ligustilide to (Z)-butylidenephthalide and phthalic acid anhydride as well as other components,26 are clear cases of dynamic residual complexity. Residual complexity, in either of its two forms, may be evaluated by qHNMR,27,28 allowing efficient quantification of subtracted compounds. This approach is ideally applied to both the DESIGNER extracts and the removed target compounds in order to characterize the chemical subtraction process.
The present study extends the chemical subtraction method exploited in recent years29,30 for the subtraction of single or multiple target components prepared by K-targeted countercurrent separation using multiple orthogonal solvent systems and steps. In this manner, unique botanical DESIGNER extracts, depleted of, or enriched in, 1, 2/3, and/or 4, were produced for further biological and chemical characterization of hops constituents (Figure 1). In parallel, this study describes the development of K-targeted metabolomic profiling. Targeted metabolomics refers to the specific analysis of a group of selected metabolites (markers) contained in the extract or metabolomic sample.31−33 As each metabolite reveals a characteristic behavior in a specific countercurrent separation solvent system(s), expressed as partition coefficients (K), the target metabolites can be selectively withdrawn or subtracted based on their K values. Therefore, this method combines selective depletion of a metabolite, or a group of metabolites, from a complex extract by means of countercurrent separations, with exhaustive chemical characterization of the end-products, namely, the target metabolites and the DESIGNER (depleted and/or enriched) extract.
Results and Discussion
Enabling Analytical Technology
Two interacting technologies enable the present chemical approach to dissecting the biological effects of individual components in complex natural product extracts. First, newer quantitative methods such as UHPLC/HRMS carry the limits of detection and quantitation into the nanogram or even femtogram range. Their requirement of authentic standards for calibration is a drawback not shared by quantitative 1H NMR (qHNMR), where sensitivity is lower, but in practice often not limiting as recently shown by assays of individual components in complex mixtures by taking advantage of computational full spin analysis.34,35 Combining 1H NMR iterative Full Spin Analysis (HiFSA) with qHNMR has the distinct advantages that HiFSA profiles only need to be developed once per analyte and can then be applied to any field strength to quantitate a compound even in complex mixtures. Moreover, this technology is orthogonal to LC-MS methodology and requires neither chromatography nor the development of a standard curve. Second, countercurrent separation, both preparative and scalable method, is an ideal procedure for producing DESIGNER extracts. As this chromatographic method is known for practically 100% sample recovery, the original extract can be reconstituted at any time as a check on the stability of the constituents. This methodology is further complemented by qHNMR, which allows calculation of the partition coefficients (K values) of all major constituents of a crude extract, in any solvent system.23 With “K-by-NMR”, the researcher can choose a solvent system that will place the chosen compound(s) in the “sweet spot” of a countercurrent separation run for targeted collection. It is worth noting that a determination of “K-by-bioactivity” in the same solvent system can provide a strong indication that the compound sought is the principal active principle, especially if there is identity between K-by-NMR and K-by-bioactivity in multiple orthogonal solvent systems. The suitability of K-targeting has recently been exemplified by the development of a sensitive qHNMR assay for ginkgotoxin, a negative marker produced by the widely used botanical, Ginkgo biloba. The involved countercurrent separation improved the sensitivity of the assay by 282-fold.36
Starting Material, Chemical Characterization, and Pilot Study
The study material was a clinical extract37 of spent hops (total extract), which was profiled and characterized by UHPLC-UV, LC-MS-MS, and quantitative 1H NMR aided with 1H iterative Full Spin Analysis (qHNMR-HiFSA).38,39 The percentages of the four markers or target compounds were determined as isoxanthohumol (1) 1.00–1.20% w/w, 8-prenylnaringenin (2) 0.30–0.35% w/w, 6-prenylnaringenin (3) 1.05–1.15% w/w, and xanthohumol (4) 32.0–35.0% w/w.
The feasibility of creating DESIGNER extracts (DEs) by countercurrent separation was evaluated through a pilot study using a 20 mL hydrodynamic countercurrent separation instrument. Initially, a K-targeted profile of the four bioactive prenylated phenols from hops was performed. The behavior of the target compounds in specific CS solvent systems was defined by their K-values to enable identification, subtraction, and quantification. This was followed by a series of CS steps applying orthogonal solvent systems as a means to establish the feasibility of depleting the targeted compounds from the extract, and thus obtaining both target compounds (Ts) and the DESIGNER or depleted extracts. Specifically, HEMWat 0 was employed for the first step followed by HEMWat −3 (for compounds 2–4) and HterAcWat +3 (for compound 1) in the second step. The total extract, DESIGNER extract, and K-targeted compounds (Ts) were analyzed by UHPLC-UV (Figure 2), and the residual content of the target compounds in the depleted extract was determined quantitatively by UHPLC-UV (Figure 2). The concentrations of 1–4 were reduced to 0.040, 0.035, 0.014, and 0.070% w/w, respectively. Therefore, in the K-targeted metabolomic profiling, depletion of the four botanical K-targeted markers by countercurrent separation decreased their content by 28-fold for isoxanthohumol (1) 9-fold for 8-prenylnaringenin (2), 78-fold for 6-prenylnaringenin (3), and 478-fold for xanthohumol (4).
Figure 2.
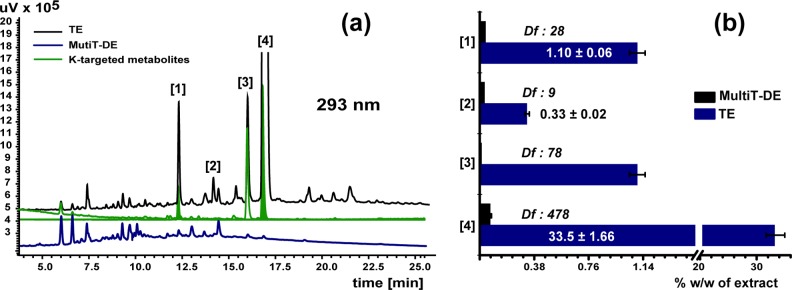
Panel (a) UHPLC-UV chromatograms of total extract (TE), DESIGNER extract (MultiT-DE, in blue), and K-targeted subtracted metabolites (in green) generated in the pilot study. The retention times of 1–4 were 12.2, 14.2, 16.0, and 16.9 min, respectively. Due to its low abundance, 2 is not assigned in the K-targeted trace. Panel (b): Quantitative results obtained for compounds 1 to 4 expressed as mean ± standard deviation of three independent analyses. Df stands for depletion fold and was calculated for each targeted metabolite, T, after a two-step CS as follows: Df = (T% [w/w])TE/(T% [w/w])DE.
Target Compound 1 and Isoxanthohumol-Depleted DESIGNER Extract (1-DE)
After the first countercurrent separation subtraction step with HEMWat 0 (0.54 ≤ K ≤ 0.61) as solvent system, the target compound 1 showed a purity of 54.6% w/w (qHNMR profile; Figure S2, Supporting Information). After a second subtraction step, when using HterAcWat +3 (2.08 ≤ K ≤ 2.18) as the solvent system, the purity of 1 was shown to be (94.7% w/w). When the same separation was done with HEMWat −3, the purity was only (86.8% w/w). It is observed from the NMR profiles of the total extract, 1-DE and subtracted target compound 1, that metabolite 1 was selectively removed (qHNMR profiles; Figure S3, Supporting Information). The quantitative residual complexity1 of 1 in the 1-DE, defined by UHPLC-UV, corresponded to 0.070% w/w, which implies that compound 1 was depleted 17.1-fold from its original concentration in the extract (Table 1). The dynamic residual complexity of 1 was also investigated since the rearrangement of 4 to 1 is known to occur.37 In the LC-MS profile of subtracted 1, the chalcone isomer 4 was not detected (LC-MS profile; Figure S5, Supporting Information), but it was present in the corresponding 1-DE (UHPLC-UV profile; Figure S9, Supporting Information). When the UHPLC-UV profiles of the total extract and the respective 1-DE are compared, the amount of 4 was nearly the same. This highlighted the capacity of the CS method to selectively remove compound 1.
Table 1. Summary of the Chemical Subtraction of Individual Metabolites from Hops Extract, Forming the final DESIGNER Extracts and Reflecting Both the Purities of the Subtracted Metabolites and Their Depletion in the DESIGNER Extract.
| subtracted compound | initial concn [%] | 1st CS stepaK-values | qHNMR purity [%] | S1b | 2nd CS stepaK-values | qHNMR purity [%] | S2b | Dfd,c | DESIGNER extracts |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.10 | 0.54–0.61 | 54.6 | 108 | 2.08–2.18 | 94.7 | 14.8 | 17 | 1-DE |
| 2 | 0.33 | 2.69–2.76 | 0.74 | 2.3 | 0.75–0.90 | 1.28 | 1.7 | 6 | 2/3-DE |
| 3 | 1.10 | 2.73–2.90 | 4.26 | 4.0 | 0.88–1.26 | 66.0 | 44 | 11 | 2/3-DE |
| 4 | 33.5 | 2.50–2.73 | 90.1 | 18 | 0.75–0.90 | 97.4 | 4.1 | 457 | 4-DE |
| 1 | 1.10 | 0.54–0.61 | 58.8 | 128 | 2.08–2.18 | 86.8 | 4.6 | 15 | multiT-DE |
| 2 | 0.33 | 2.69–2.76 | 1.97 | 6.1 | 0.75–0.90 | 8.18 | 4.4 | 7.5 | multiT-DE |
| 3 | 1.10 | 2.73–2.90 | 4.11 | 4.0 | 0.88–1.26 | 59.5 | 34 | 11 | multiT-DE |
| 4 | 33.5 | 2.50–2.73 | 93.0 | 26 | 0.75–0.90 | 97.4 | 2.8 | 326 | multiT-DE |
Countercurrent separation.
Enrichment factor.
Fold depletion.
Values for the final DESIGNER extracts are very similar to but not identical with Df values determined during the pilot study (Figure 2).
Target Compounds 2 and 3 – 8-Prenylnaringenin/6-Prenylnaringenin-Depleted DESIGNER Extract (2/3-DE)
Compounds 2 and 3 were subtracted with qHNMR assays of 0.74% (2.69 ≤ K ≤ 2.76) and 4.26% (2.73 ≤ K ≤ 2.90), respectively (qHNMR profiles; Figure S2, Supporting Information), during the first countercurrent separation step using HEMWat 0 as a solvent system. Compound 4 (91.1% w/w) was the major “impurity”, because coelution of the three prenylated phenols occurred. After the second countercurrent separation step with HEMWat −3 as solvent system, compound 4 was only 1.24% w/w in the subtracted mixture, while compound 2 was 1.28% w/w and compound 3 was 66.0% w/w. A comparison of the total extract, the 2/3-DE, and the subtracted fraction, is shown in Figure S3, Supporting Information. The chalcone α,β-dihydroxanthohumol, was also identified in the subtracted mixture by LC-MS and 1H NMR, and accounted for 8.37% w/w in the finally subtracted compound mixture.
Subtraction of metabolite 2, per se, represents an analytical challenge as it is a minor component which elutes between 3 and 4 with some overlap. The elution overlap of 4 and 2 accounted for the decrease in 2 after the second separation step. Metabolites 2 and 3 are regioisomers, differentiated only in the position of the prenyl moiety on ring A. Additionally, 2 may also be present as the racemic isomerization product of desmethylxanthohumol.1
Target Compound 4 – Xanthohumol-Depleted DESIGNER Extract (4-DE)
The qHNMR purity of 4 was 90.1% w/w (qHNMR profile; Figure S2, Supporting Information) after the first countercurrent separation subtraction using HEMWat 0 (2.5 ≤ K ≤ 2.73). A second countercurrent separation procedure using HEMWat −3 as solvent system (0.75 ≤ K ≤ 0.90) was applied, and compound 4 was obtained with a purity of 97.4% w/w (Table 1). As can be seen from the NMR profiles of the total extract, 4-DE, and subtracted target compound 4 (Figure 3), target compound 4 was selectively removed from the total extract. The residual of 4 in the 4-DE, determined by UHPLC-UV, corresponded to 0.070% w/w. The UHPLC-UV profile of the total extract compared to the 4-DE and LC-MS profile of the subtracted 4 (Supporting Information, Figures S8 and S5, respectively) showed that 4 was selectively removed. The presence of compound 1 could be detected in the subtracted xanthohumol fraction (LC-MS profile; Figure S5, Supporting Information) due to an intramolecular Michael addition reaction in which compound 4 tended to form compound 1; an example of dynamic residual complexity.1
Figure 3.

NMR profiles of total extract (TE), DESIGNER extracts, and K-targeted subtracted metabolites. The NMR measurements were conducted in MeOH-d4 under quantitative conditions (qHNMR). Panel (a): Comparison of NMR profiles of total extract (in black) vs xanthohumol (4) DESIGNER extract or 4-DE (in blue), and 4 (in green) after the second CS step. Subtracted metabolite 4 is shown to have been subtracted from the TE when compared to its respective DESIGNER (4-DE). The qHNMR final purity of subtracted metabolite 4 corresponds to 97.38% w/w, calculated by applying the 100% method. Panel (b): Comparison of NMR profiles of total extract (in black), MultiT-DE (in blue), and subtracted metabolites 1 (in cyan blue), 2/3, and 4 (in green) after the second CS step. Based on the NMR profile, the K-targeted metabolites are shown to have been subtracted from the TE to produce the MultiT-DE.
Target Compounds 1–4 – Multiple Target Depleted DESIGNER Extracts (1–4–DEs)
The NMR profile comparison of the total extract vs the Multiple Target-DE (MultiT-DE, Figure 4) showed a depletion of the four target compounds. For the simultaneous subtraction of the four prenylated phenols, the HEMWat 0 solvent system was used in the first countercurrent separation step. The qHNMR percentage of 1 in the subtracted fraction was 58.8% w/w. In a separate fraction, the percentages of 2–4, were 1.97, 4.11, and 93.0% w/w, respectively (Table 1). The congeneric α,β-dihydroxanthohumol accounted for 1.16% w/w of the 2–4 fraction after the first CS step. The qHNMR profiles of subtracted metabolites after the first step can be seen in Figure S4, Supporting Information. A second step was performed with the subtracted metabolites using HEMWat −3 as the solvent system for both fractions. The resulting qHNMR percentage of 1 was 86.8% w/w. A separate fraction contained 2 and 3 at levels of 8.18 and 59.5% w/w, respectively, along with 11.5% w/w of α,β-dihydroxanthohumol. Finally, the fraction containing 4 was at 97.4% w/w purity. In this case, coelution of 2–4 is advantageous, as the aim in this case was to subtract them all from the total extract.
Figure 4.
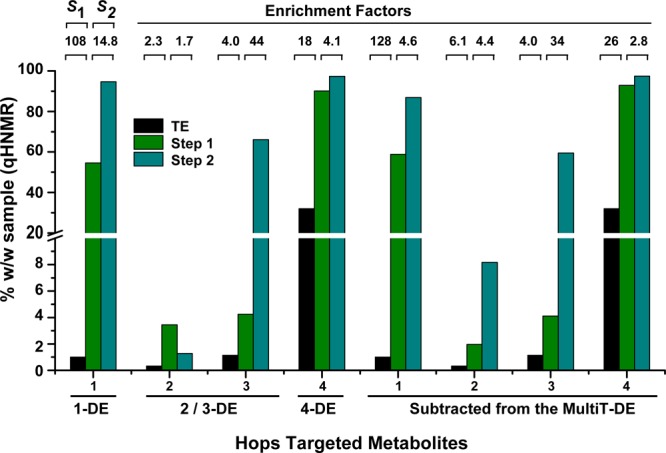
Concentrations of the target metabolites were determined by qHNMR in the initial crude extract (black bars) and for each subtraction step (green and blue bars). Both the concentration and final purity of each metabolite were expressed as mass percentage (% w/w). For each step, the enrichment factor (S) was calculated, with S1 corresponding to the enrichment factors after the first and S2 after the second CS step. A set of four DESIGNER extracts was prepared: extracts selectively depleted in metabolite 1 (1-DE), the metabolites 2 and 3 (2/3-DE), and metabolite 4 (4-DE); and an extract depleted in metabolites 1–4 extract (MultiT-DE). Metabolites 2 and 3 occurred in the same subtracted fraction; their respective enrichment factors (S1 and S2) are shown separately, as are the respective S values for MultiT-DE.
Enrichment Factors (S)
The enrichment factors (S) calculated for the four target metabolites obtained in creating the four different DESIGNER extracts are shown in Figure 4. The calculation is based on the procedure developed by the International Union of Pure and Applied Chemistry (IUPAC) in the Compendium of Chemical Terminology Gold Book,40 adapted to the concept of chemical subtraction, which describes that “S in liquid–liquid distribution is the factor by which the ratio of two substances in the feed must be multiplied to give their ratio after treatment”.41 The enrichment factor was, therefore, calculated with the following equation:
QA′ and QA correspond to the initial and final % w/w of species A. QB′ and QB correspond to the initial and final amounts of species B.40 In this case, “A” corresponds to the subtracted compound, and “B” to the remaining or residual components in the extract.
The calculated enrichment factors to create the 1-DE, 2/3-DE, and 4-DE DESIGNER extracts are summarized in Figure 4 and Table 1. After subtraction of compound 1 with HEMWat 0 as solvent system, its purity was enriched by a factor of 108 (1.10 to 54.6% w/w). Following the second countercurrent separation step with HterAcWat +3 as solvent system, the purity increased from 54.6 to 94.7% w/w, representing an enrichment of 1 by a factor of 14.8 (Figure 4). Compound 2 was enriched (S1 = 2.3) in the first step and less enriched (S2 = 1.7) in the second step due to coelution with 3 and 4 in both solvent systems. On the other hand, 3, although not highly enriched after the first step (S1 = 4.0), was more highly enriched after the second subtraction step (S2 = 44). The HEMWat −3 solvent system employed in the second step efficiently resolved 3 and 4, but did not completely separate 2 from 4. Compound 4 was enriched by a factor of 18 after the first subtraction step. After the second subtraction step, a lower enrichment factor was reached (S2 = 4.1) as purities from step one to step two (90.1 to 97.4% w/w) slightly increased (Figure 4). Therefore, the two-step countercurrent separation method using HEMWat 0 and HterAcWat +3 for compound 1 and HEMWat 0 and HEMWat −3 for compound 4 has been shown to be effective for performing chemical subtraction in total extracts of hops, as well as applying the capability of countercurrent separation to highly enrich selected metabolites, which results in the high purities attained.
The calculated enrichment factors observed when creating the multitarget DESIGNER extracts (multiT-DE) are summarized in Figure 4 and Table 1. These factors correlate well with the factors observed in the production of 1-DE, 2/3-DE, and 4-DE for 1, 3, and 4. In this case, compound 2 was enriched (S1 = 6.1 and S2 = 4.4) with each countercurrent separation step.
Designing Extracts with Countercurent-Based Chemical Subtraction
This study also demonstrates the interdependence of preparative chromatography and analytical assessment. The choice of CS enabled chemical subtraction with complete sample recovery and allows repeated fractionation via polarity adjusted orthogonal conditions. CS-based chemical subtraction generated both the purified target metabolites 1 and 4 and selectively depleted DESIGNER extracts. Limitations in chromatographic selectivity limited the chemical subtraction of 2 and 3, which could only be removed together. The exploration of new orthogonal solvent systems and/or alternative chromatographic techniques such as gel permeation might be required to separate 2 from 3, keeping in mind that adsorption-based chromatography is intrinsically flawed as a chemical subtraction technique. In addition, it must be ascribed to interactions between (congeneric?) analytes in the crude extracts that the enrichment of these two relatively low abundance metabolites was limited, whereas CS typically achieves high enrichment factors (S) for low-level constituents (e.g., S1 of 1 was 108; see Table 1). This might be in part due to the near coelution of 2 and 4, which has been observed in earlier studies6 and resulted in low enrichment of 2 in the subtracted fractions. Overall, while the presence of multiple prenylchalcone and flavanone congeners in the extract presented a separation challenge, the resulting depleted DESIGNER extracts were all greatly depleted in their respective target metabolites, regardless of their concentrations in original extract.
The creation of DESIGNER extracts (DEs) involved careful monitoring of the residual complexity of both the depletion of the target metabolite(s) in each DE, as well as the purity and composition of each of the removed fractions containing the target compound(s). This process used a combination of orthogonal methods: qHNMR, UHPLC with UV detection, and LC-MSn. The ability of qHNMR to reveal both relative molar abundances and structural information is a key strength of this technique. UHPLC-UV is particularly useful in comparing the relative abundance of the same analyte in different preparations. The capacity of LC-MSn to scan for a single molecular species enables high sensitivity assays for isomeric metabolites.
The demonstrated subtraction (“knockout”) of major and minor bioactive phytochemicals from hops enables the evaluation of the biological input of single metabolites on the overall activity of the total extract. The selective removal of target metabolites from total extracts, producing depleted DESIGNER extracts, may become a useful tool for the study of other pharmacological interaction studies of complex mixtures. DESIGNER extracts may also serve as new functional materials for drug discovery, as the biological impact of a specific metabolite or metabolites can be evaluated before and after chemical subtraction. This can facilitate the identification of low-level bioactive principles, including potent impurities. The process can design extracts that are reduced in, or free of, cytotoxic metabolites; deficient in inactive major compounds; and/or enriched in beneficial secondary metabolites. This can be of great benefit in cell-based bioassays, drug discovery, and botanical extracts development. The same methods used for depletion may be employed to add target metabolites to total extracts and develop enriched DESIGNER extracts. The latter can be achieved, for example, by returning the subtracted metabolites to the original metabolomic mixture. The DESIGNER technology is also applicable to combinatorial chemistry libraries, especially where the protocol leads to complex mixtures such as in the chemical engineering approach of López et al.41,42 Figure S13, Supporting Information, summarizes reports on the chemical engineering of metabolomic mixtures.
However, a DESIGNER extract should not be mistaken for chemically engineered extracts. The DESIGNER concept expressly maintains the chemistry of the starting material, which is rooted in the biosynthetic origin, albeit two or more entities are depleted, removed (“knocked out”), or enriched (“knocked in”). In contrast, chemically engineered extracts have undergone a synthetic chemical transformation. Accordingly, DESIGNER extracts are akin to their natural precursors and represent both useful tools for biological research and innovative potential intervention material for clinical applications. However, it is important to keep in mind that, even under the very gentle conditions of liquid-only CS and solvent evaporation in vacuo, dynamic residual complexity can still occurr and effect, for example, the chalcone/flavanone equilibrium of natural extracts, as shown recently for licorice extracts.43
Finally, the field of natural products research can benefit broadly and directly from the DESIGNER concept, because CS-based chemical subtraction is highly adaptable: any contemporary CS instrumentation (e.g., HSCCC, CPC), all separation conditions for previously investigated plants and other organisms reported in the primary CS literature and reviews,44,45 as well as all existing knowledge of solvent system suitability46−48 can be readily implemented for other natural products to design depleted and/or enriched “knockout or “knock-in” extracts, respectively. In other words: “designing” a depleted or enriched DESIGNER extract for a specific purpose by chemical subtraction is equally feasible as the successful “guessing” of suitable CS separation conditions by the use of the GUESS methodology introduced earlier.48
Experimental Section
General Experimental Procedures
The following instruments were used to generate the DESIGNER extracts and to obtain physical data: CherryOne automated operating system with a Tauto TBE 20A high-speed countercurrent chromatography equipped with a Foxy Jr. HPLC fraction collector (Teledyne Isco, Lincoln, NE, U.S.A.). A high-speed countercurrent chromatograph Model CCC-1000 Pharma-Tech Research Corp (Baltimore, MD) equipped with a set of three coils and a Series III ISO-2000 pump, and a fraction collector LKB BROMMA 2111 Multirac. 1H NMR spectra were measured at 600.13 MHz on a Bruker AVANCE-600 NMR spectrometer equipped with a 5 mm TXI cryoprobe. Offline 1D data processing was performed using Mnova NMR software package (v.6.0.2, MestreLab Research S.L., A Coruña, Spain), applying a Lorentzian-to-Gaussian window function (lb = −0.3 Hz, Gaussian factor = 0.05), as well as double zero filling (32K). Samples were analyzed on a Shimadzu Nexera UHPLC equipped with a Waters Acquity UPLC BEH C18 (2.1 × 5.0 mm, 1.7 μm) column and using a UV detection mode. Quantitative UHPLC-MS-MS analyses were carried out using a Shimadzu LCMS 8030 triple quadrupole mass spectrometer equipped with a Shimadzu Nexera UHPLC system and a Shimadzu XR-ODS III C18 column (2.0 × 50 mm, 1.6 μm). Qualitative LC-MS analysis were carried out using Waters (Milford, MA) 2695 solvent delivery system connected to a Waters SYNAPT quadrupole/time-of-flight mass spectrometer operated in the positive ion electrospray mode. HPLC separations were carried out using a Waters XBridge C18 reversed phase column (2.0 × 50 mm, 2.5 μm).
Plant Material
A xanthohumol-enriched hops extract (Humulus lupulus), provided by Hopsteiner (Mainburg, Germany, and New York, NY, USA), was used as the original extract. A reference specimen of the hops extract is deposited in the UIC Botanical Center (College of Pharmacy, UIC, Chicago, IL) under number BC #402. The percentages of four markers or target compounds in this enriched extract were determined by UHPLC-UV and quantitative 1H NMR aided with 1H iterative Full Spin Analysis (qHNMR-HiFSA).
Chemicals
All the solvents and reagents, hexanes (Hex), ethyl acetate (EtOAc), methanol (MeOH), tert-butyl methyl ether (MTBE), acetonitrile (MeCN) for CS, acetonitrile (MeCN), water (H2O), formic acid (HCO2H) for HPLC, and methanol-d4 (D, 99.8%) for NMR, were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). DMSO-d6 (D, 99.98%) for NMR was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA, U.S.A.). Double-deionized water from EMD Millipore Milli-Q system (Billerica, MA, U.S.A.) was used for CS.
Experimental Procedures Used for the Preparation and Characterization of DESIGNER Extracts
The following describes the preparative and analytical methods developed to perform the K-targeted chemical subtraction of target metabolites as well as the metabolomic profiling and quantitative assessment of the starting materials and DESIGNER products.
Countercurrent Separation (CS) Pilot Study
Countercurrent separation was performed using a CherryOne automated operating system with a Tauto TBE 20A high speed countercurrent chromatography instrument for a pilot study. The coil volume was 20 mL, the flow rate 0.3 mL/min, and the revolution speed 1900 rpm. Fraction collection was accomplished with a Foxy Jr. HPLC fraction collector (Teledyne Isco, Lincoln, NE, U.S.A.). Fraction collection was set to 90 s per tube. Elution was run in a head-to-tail mode for a VCM = 36 mL, followed by elution–extrusion. The stationary phase retention volume ratios (Sf) for the solvent systems HEMWat 0 (hexane/ethyl acetate/methanol/water 5:5:5:5), HEMWat −3 (6:4:6:4), and HterAcWat +3 (hexane/methyl tert-butylether/acetonitrile/water 4:6:4:6) were 0.62, 0.73, and 0.50, respectively. K values were calculated according to previous work.49
Methods for the Stepwise Chemical Subtraction by CS
Nine CS fractionations were conducted to perform the K-targeted chemical subtraction of target metabolites of the starting materials in order to produce six purified target metabolite fractions and four DESIGNER extracts (see Figure S14, Supporting Information, for the extraction scheme). The phytochemical operations were carried out under gentle conditions, using liquid-only preparative CS and rotary or speedvac evaporation of solvents in vacuo at <45 °C.
First CS Subtraction
A high-speed countercurrent chromatograph (HSCCC) Model CCC-1000 Pharma-Tech Research Corp (Baltimore, MD) was equipped with a set of three coils and a Series III ISO-2000 pump. The coil volume was 320 mL, the flow rate was 1.5 mL/min, and the revolution speed was 800 rpm. Fraction collection was set to 3 min per tube. The eluent was connected to a fraction collector LKB BROMMA 2111Multirac. Elution was run in a head-to-tail mode for a VCM = 450 mL, followed by elution–extrusion. Premixed HEMWat 0 was used as the solvent system.46−48 The Sf values for the individual subtraction of 1, 2/3, and 4, to produce the depleted DESIGNER extracts (DEs), 4-DE, 1-DE, 2/3-DE, and MultiT-DE, were 0.73, 0.73, 0.80 and 0.75, respectively. The depleted extracts are generated after the first countercurrent separation step.
Second CS Subtraction
Five separate CS fractionations were performed to purify the subtracted mixtures of targeted metabolites, and the fractions collected that lacked the targeted metabolites were subsequently added to their respective depleted DESIGNER extracts: 1-DE, 2/3-DE, 4-DE, and MultiT-DE. In the purification of 1 to complete 1-DE, the conditions and equipment used were as described in section “First CS Subtraction”, but HterAcWat +3 was used as a solvent system.47 Elution was run in a head-to-tail mode for a VCM = 225 mL, followed by elution–extrusion. The Sf value was 0.70. The Tauto TBE 20A instrument and CherryOne operating system was used for purification of the 2/3 metabolite mixture and the completion of 2/3-DE with a HEMWat −3 solvent system. The flow rate was 0.5 mL/min, the rotation speed was 1600 rpm and the fraction collection interval was 2.5 min. Elution was run in a head-to-tail mode for a VCM = 90 mL, followed by elution–extrusion. The Sf value was 0.70. Purification of 4 to complete 4-DE was performed by a CherryOne automated operating system with a Tauto TBE 300B high-speed countercurrent chromatography (HSCCC) instrument. The coil volume was 300 mL, the flow rate was 2 mL/min, and the revolution speed was 800 rpm. Fraction collection was set to 2.5 min per tube. Elution was run in a head-to-tail mode for a VCM = 720 mL, followed by elution–extrusion. The Sf value was 0.82. The same procedure and conditions were used to generate purified 1, 2/3, and 4 to complete multiT-DE.
Methods for Quantitation
The chemical diversity of the original extracts and the residual complexity of the depleted extracts and target metabolites favor quantitation methods with the inherent ability to measure multiple components with a single procedure. Working without the need for identical reference materials for calibration, qHNMR fills this role very well, with the ability to quantify any component for which a fully analyzed 1H NMR spectrum is available, or for which a distinct signal is present without overlap and with reasonable assignment to a number of protons (albeit only relative molar quantitation when using the normalization/100% method). LC-MS has the ability to quantify constituents in complex mixtures provided that pure samples of the constituents are available for calibration.
Quantitative 1H NMR (qHNMR) for K-Targeted Profiling
Samples contained precisely (0.01 mg) from weighed quantities, in an analytical balance, of 0.50–5.00 mg of extracts, and were dissolved with exactly 50 μL of DMSO-d6 (D 99.98%), measured with a precision glass syringe. Samples were transferred to 1.7 mm NMR tubes, and the tubes were sealed with a propane gas torch to protect them from air moisture. 1H NMR spectra were measured at 600.13 MHz on a Bruker AVANCE-600 NMR spectrometer equipped with a 5 mm TXI cryoprobe, using standard pulse sequences. The 1D 1H NMR spectra were acquired under quantitative conditions.27,28
Quantitative 1H NMR (qHNMR) for Chemical Subtraction Experiments
Samples contained precisely (0.01 mg) weighed quantities, in an analytical balance scale, of 0.50–10.0 mg of the subtracted compounds, and 8–25 mg of the DEs weighed, into 5 mm Norell NMR tubes. Exactly 600 μL of MeOH d4 (99.8+ atom %D), measured with a precision glass syringe, were directly added to each tube. Measurements were done at 600.13 MHz on a Bruker AVANCE-600 NMR spectrometer equipped with a 5 mm TXI cryoprobe, using standard pulse sequences. The 1D 1HNMR spectra were acquired under quantitative conditions and evaluated using the 100% method.27,28 Offline 1D data processing was performed using Mnova NMR software package (v.6.0.2, MestreLab Research S.L., A Coruña, Spain), applying a Lorentzian-to-Gaussian window function (lb = −0.3 Hz, Gaussian factor = 0.05), as well as double zero filling (32 K). After manual phasing, a polynomial baseline correction was performed.
UHPLC Quantitation of Target Compounds – Depleted Extract Profiling and Quantitation
The whole hops extract, K-targeted subtracted compounds and depleted extract were prepared at 1.00–10.00 mg/mL solutions in MeOH. Samples were analyzed on a Shimadzu Nexera UHPLC equipped with a Waters Acquity UPLC BEH C18 (2.1 × 5.0 mm, 1.7 μm) column and using a UV detection mode. Wavelengths were set to 293 nm (flavanones) and 369 nm (chalcones). Solvent A = H2O with 0.1% HCO2H and solvent B = MeCN with 0.1% HCO2H. A gradient was created by pumping 5 to 57% B over 18 min, 57 to 98% B over 7 min, followed by holding B at 98% for 3 min. The flow rate was set to 0.6 mL/min. The retention times for compounds 1, 2, 3, and 4 were 12.2, 14.2, 16.0, and 16.9 min, respectively.
LC-MS/MS of Target Compounds for Profiling and Residual Quantitation in DEs
Quantitative UHPLC-MS-MS analyses were carried out using a Shimadzu LCMS 8030 triple quadrupole mass spectrometer equipped with a Shimadzu Nexera UHPLC system and a Shimadzu XR-ODS III C18 column (2.0 × 50 mm, 1.6 μm). A gradient consisting of solvents A and B was performed at a flow rate of 500 μL/min. Solvent B was increased for 45 to 70% over 1.5 min, held at 70% for 0.1 min and then equilibrated at 45% for 0.9 min before the next injection. The total run time including equilibration was 2.5 min. Selected reaction monitoring (SRM) of two transitions (quantifier and qualifier) were used for each analyte as follows: m/z 353 to m/z 119 (quantifier) and m/z 353 to m/z 233 (qualifier) for 1 and 4; and m/z 339 to m/z 119 (quantifier) and m/z 339 to m/z 219 (qualifier) for 2 and 3. The SRM transition of m/z 341 to m/z 119 was monitored for the internal standard, 8-isopentylnaringenin. The ions of m/z 353 and m/z 339 are deprotonated molecules of isomeric 4 and 1 and of isomeric 2 and 3, respectively.50
Qualitative LC-MS analysis were carried out using Waters (Milford, MA) 2695 solvent delivery system connected to a Waters SYNAPT quadrupole/time-of-flight mass spectrometer operated in the positive ion electrospray mode. HPLC separations were carried out using a Waters XBridge C18 reversed phase column (2.0 × 50 mm, 2.5 μm) and a mobile phase consisting of solvents A and B. Compounds were separated using a linear gradient from 20–80% B over 15 min at a flow rate of 0.22 mL/min. Mass spectrometric data were acquired from m/z 150–800 at 10 000 fwhm resolution using Leu-enkephalin as the lock mass. Tandem mass spectra were taken at 15 or 25 eV using argon as collision gas.50
Acknowledgments
The authors acknowledge the long-term support provided by Mr. Harald Schwarz and Dr. Martin Biendl of Hopsteiner Inc., New York (NY)/Yakima (WA) and Mainburg (Germany), respectively. Furthermore, the authors are grateful to Samuel Pro, Warren Friedl, Tom Burdick, and Nick Novak, Wrightwood Technologies, Chicago (IL), for their kind support of countercurrent separation instrumentation used during this study. We also thank our long-term collaborators in the UIC/NIH Botanical Center for many helpful discussions of dietary supplements research. Further more, we are grateful to Dr. Benjamin Ramirez (NMR Facilities) at the UIC Center of Structural Biology (CSB) for his NMR expertise. This research was funded by ODS and NCCAM of the NIH through grants P50 AT000155, RC2 AT005899, and R44 AT004534.
Supporting Information Available
Overview of publication series on residual complexity. qNMR profiles of the total extract, DESIGNER extracts, and subtracted metabolites. LC-MS profiles of subtracted compounds. UHPLC-PDA profiles of the total extract, DESIGNER extracts, and subtracted metabolites. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Notes
⊥ This paper represents Part 24 of the series on Residual Complexity and Bioactivity (see http://go.uic.edu/residualcomplexity).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chen S.-N.; Lankin D.; Chadwick L. R.; Jaki B. U.; Pauli G. F. Planta Med. 2009, 75, 757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick L.; Pauli G. F.; Farnsworth N. R. Phytomedicine 2006, 13, 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoli P.; Zavatti M. J. Ethnopharmacol. 2008, 116, 383–396. [DOI] [PubMed] [Google Scholar]
- Zhao F.; Watanabe Y.; Nozawa H.; Daikonnya A.; Kondo K.; Kitanaka S. J. Nat. Prod. 2005, 48, 43–49. [DOI] [PubMed] [Google Scholar]
- de Almeida N. E. C.; do Nascimento E. S. P.; Cardoso D. R. J. Agric. Food Chem. 2012, 60, 10649–10656. [DOI] [PubMed] [Google Scholar]
- Chadwick L.; Fröhlich R.; Bolton J.; van Breemen R.; Overk C.; Burdette J.; Nikolic D.; Fong H. H. S.; Farnsworth N.; Pauli G. F. J. Nat. Prod. 2004, 67, 2024–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr G.; Gerhäuser C.; Knauft J.; Zapp J.; Becker H. J. Nat. Prod. 2005, 68, 1545–1548. [DOI] [PubMed] [Google Scholar]
- Overk C. R.; Yao P.; Chadwick L. R.; Cuendet M.; Fong H. H. S.; Pauli G. F.; Farnsworth N. R.; Bolton J. J. Agric. Food Chem. 2005, 53, 6246–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda C. L.; Stevens J. F.; Helmrich A.; Henderson M. C.; Rodriguez R. J.; Yang Y. H.; Deinzer M. L.; Barnes D. W.; Buhler D. R. Food. Chem. Toxicol. 1999, 37, 271–285. [DOI] [PubMed] [Google Scholar]
- Serwe A.; Rudolph K.; Anke T.; Erkel G. Invest. New Drugs 2012, 30, 898–915. [DOI] [PubMed] [Google Scholar]
- Yuan C. S.; Tanaka H. Curr. Drug Discovery Technol. 2011, 8, 32–41. [DOI] [PubMed] [Google Scholar]
- Fukuda N.; Tanaka H.; Shoyama Y. J. Nat. Prod. 2000, 63, 283–285. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Shoyama Y. Phytomedicine 1998, 5, 397–415. [DOI] [PubMed] [Google Scholar]
- Tanaka H.; Fukuda N.; Shoyama Y. Cytotechnology 1999, 29, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoyama Y.; Tanaka H.; Fukuda N. Cytotechnology 1999, 31, 9–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H.; Fukuda N.; Shoyama Y. J. Agric. Food Chem. 2007, 55, 3783–3787. [DOI] [PubMed] [Google Scholar]
- Uto T.; Morinaga O.; Tanaka H.; Shoyama Y. Biochem. Biophys. Res. Commun. 2012, 417, 473–478. [DOI] [PubMed] [Google Scholar]
- Haupt K. Analyst 2001, 126, 747–756. [DOI] [PubMed] [Google Scholar]
- Xie J.; Chen L.; Li C.; Xu X. J. Chromatogr. B 2003, 788, 233–242. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zhou J.-L.; Liu P.; Sun S.; Li P. J. Chromatogr. A 2010, 1217, 5239–5245. [DOI] [PubMed] [Google Scholar]
- Jin J.; Li Y.; Kipletting Tanui E.; Han L.; Jia Y.; Zhang L.; Wang Y.; Zhang X.; Zhang Y. J. Ethnopharmacol. 2013, 147, 357–365. [DOI] [PubMed] [Google Scholar]
- Chen S.-N.; Turner A.; Jaki B.; Nikolic D.; van Breemen R. B.; Friesen J. B.; Pauli G. F. J. Pharm. Biomed. Anal. 2008, 46, 692–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu F.; Friesen J. B.; McAlpine J. B.; Pauli G. F. J. Chromatogr. A 2012, 1242, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen J. B.; Pauli G. F. J. Chromatogr. A 2009, 1216, 4237–4244. [DOI] [PubMed] [Google Scholar]
- Ferreira D.; Brandt E. V.; du Volsteedt R. F.; Roux D. G. J. Chem. Soc., Perkin Trans. 1 1975, 1437–1446. [Google Scholar]
- Schinkovitz A.; Pro S. M.; Main M.; Chen S. N.; Jaki B. U.; Lankin D. C.; Pauli G. F. F. J. Nat. Prod. 2008, 71, 1604–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli G. F.; Jaki B. U.; Gödecke T.; Lankin D. C. J. Nat. Prod. 2012, 75, 834–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli G. F.; Jaki B. U.; Lankin D. C. J. Nat. Prod. 2005, 68, 133–149. [DOI] [PubMed] [Google Scholar]
- Ramos Alvarenga R.; Nikolic D.; van Breemen R. B.; Chen S.-N.; Pauli G. F. Planta Med. 2013, 79, PP5. [Google Scholar]
- Ramos Alvarenga R.; Nikolic D.; van Breemen R. B.; Chen S.-N.; Pauli G. F. Planta Med. 2012, 78, PJ108. [Google Scholar]
- Kueger S.; Steinhauser D.; Willmitzer L.; Giavalisco P. Plant J. 2012, 70, 39–50. [DOI] [PubMed] [Google Scholar]
- Shyur L. F.; Yang N. S. Curr. Opin. Chem. Biol. 2008, 12, 66–71. [DOI] [PubMed] [Google Scholar]
- Hegeman A. D. Briefings Funct. Genomics 2010, 9, 139–148. [DOI] [PubMed] [Google Scholar]
- Simmler C.; Napolitano J. G.; McAlpine J. B.; Chen S.-N.; Pauli G. F. Curr. Opin. Biotechnol. 2014, 25, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano J. G.; Lankin D. C.; Chen S.-N.; Pauli G. F. Magn. Reson. Chem. 2012, 50, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Chen S.-N.; McAlpine J. B.; Klein L. L.; Friesen J. B.; Lankin D. C.; Pauli G. F. J. Nat. Prod. 2014, 77, 611–617. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Qiu X.; Nikolić D.; Chen S.-N.; Huang K.; Li G.; Pauli G. F.; van Breemen R. B. Eur. J. Pharm. Sci. 2014, 53, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano J. G.; Gödecke T.; Rodriguez Brasco M. F.; Jaki B. U.; Chen S.-N.; Lankin D. C.; Pauli G. F. J. Nat. Prod. 2012, 75, 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano J. G.; Lankin D. C.; McAlpine J. B.; Niemitz M.; Korhonen S.-P.; Chen S.-N.; Pauli G. F. J. Org. Chem. 2013, 78, 9963–9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IUPAC. Compendium of Chemical Terminology, Gold Book, Online Version 2.3.2.; International Union of Pure and Applied Chemistry: Research Triangle Park, NC, 2012; pp 512–513. http://goldbook.iupac.org/. [Google Scholar]
- Ramallo I. A.; Salazar M. O.; Mendez L.; Furlan R. L. Acc. Chem. Res. 2011, 44, 241–250. [DOI] [PubMed] [Google Scholar]
- López S. N.; Ramallo I. A.; Sierra M. G.; Zacchino S. A.; Furlan R. L. E. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 441–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmler C.; Hajirahimkhan A.; Lankin D. C.; Bolton J.; Jones T.; Soejarto D. D.; Chen S.-N.; Pauli G. F. J. Agric. Food Chem. 2013, 61, 2146–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli G. F.; Pro S.; Friesen J. B. J. Nat. Prod. 2008, 71, 1489–1508. [DOI] [PubMed] [Google Scholar]
- Ito Y. J. Chromatogr. A 2005, 1065, 145–168. [DOI] [PubMed] [Google Scholar]
- Friesen B.; Pauli G. F. J. Chromatogr. A 2009, 1216, 4225–4231. [DOI] [PubMed] [Google Scholar]
- Friesen J. B.; Pauli G. F. J. Chromatogr. A 2007, 1151, 51–59. [DOI] [PubMed] [Google Scholar]
- Friesen J. B.; Pauli G. F. J. Liq. Chromatogr. Relat. Technol. 2005, 28, 2877–2806. [Google Scholar]
- Friesen J. B.; Pauli G. F. Anal. Chem. 2007, 79, 2320–2324. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Qiu X.; Nikolic D.; Dahl J. H.; van Breemen R. B. J. AOAC Int. 2012, 95, 1744–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.