

Abstract
Hypertension causes vascular inflammation evidenced by an increase in perivascular macrophages and proinflammatory cytokines in the arterial wall. Perivascular macrophage depletion reduced tumor necrosis factor (TNF)-α expression in cerebral arteries of hypertensive rats and attenuated inward remodeling, suggesting that TNF-α might play a role in the remodeling process. We hypothesized that TNF-α inhibition would improve middle cerebral artery (MCA) structure and reduce damage after cerebral ischemia in hypertensive rats. Six-week-old male stroke-prone spontaneously hypertensive rats (SHRSP) were treated with the TNF-α inhibitor etanercept (ETN; 1.25 mg·kg−1·day−1 ip daily) or PBS (equivolume) for 6 wk. The myogenic tone generation, postischemic dilation, and passive structure of MCAs were assessed by pressure myography. Cerebral ischemia was induced by MCA occlusion (MCAO). Myogenic tone was unchanged, but MCAs from SHRSP + ETN had larger passive lumen diameter and reduced wall thickness and wall-to-lumen ratio. Cerebral infarct size was increased in SHRSP + ETN after transient MCAO, despite an improvement in dilation of nonischemic MCA. The increase in infarct size was linked to a reduction in the number of microglia in the infarct core and upregulation of markers of classical macrophage/microglia polarization. There was no difference in infarct size after permanent MCAO or when untreated SHRSP subjected to transient MCAO were given ETN at reperfusion. Our data suggests that TNF-α inhibition attenuates hypertensive MCA remodeling but exacerbates cerebral damage following ischemia/reperfusion injury likely due to inhibition of the innate immune response of the brain.
Keywords: hypertension, cerebral vascular remodeling, ischemic stroke, tumor necrosis factor-α, macrophages/microglia
cerebrovascular accidents are a major cause of adult disability in developed countries (15). In recent years much effort has been directed to developing neuroprotective strategies to improve the outcome of cerebral ischemia, with few translational achievements. Primary prevention remains an important strategy to manage individuals at risk of having a stroke. Hypertension is a primary risk factor for stroke, and hypertension-induced remodeling of cerebral arteries has been linked to larger infarcts following experimentally induced cerebral ischemia (9, 39). The mechanisms underlying hypertensive cerebral artery remodeling have not been fully elucidated, but we recently showed that macrophage-mediated vascular inflammation is involved in this process (35).
Inflammatory mediators play a role in the pathophysiology of hypertension and end-organ damage. Many circulating factors known to be important in hypertension induce vascular inflammation, including angiotensin II (28), aldosterone (3), and endothelin-I (41). These factors are also associated with hypertensive vascular remodeling (21, 38, 48). Thus, it is possible that vascular inflammation is in part responsible for the remodeling and vascular damage induced by these vasoactive mediators, particularly through proinflammatory cytokines, including TNF-α. We recently proposed that TNF-α might be involved in hypertensive remodeling of both the cerebral and mesenteric vasculatures (35). Furthermore, other studies showed that TNF-α is involved in the renal damage observed in hypertensive salt-sensitive rats (10, 11), and TNF-α inhibition reduces infarct size following cerebral ischemia in normotensive rats (45). Thus we hypothesized that TNF-α inhibition with etanercept (ETN) would attenuate remodeling of the middle cerebral artery (MCA) and mesenteric resistance arteries (MRA) in stroke-prone spontaneously hypertensive rats (SHRSP). We further hypothesized that the improvement in MCA structure would reduce infarct size after cerebral ischemia. The structure of the MCA and third order MRA was assessed by pressure myography. Pial perfusion was measured by scanning laser Doppler flowmetry, and cerebral ischemia was induced using the intraluminal suture model for MCA occlusion.
MATERIAL AND METHODS
Animals and treatment.
Six-week-old male SHRSP from the colony housed at Michigan State University were randomized into two groups: one group was treated with the TNF-α inhibitor ETN (1.25 mg/kg ip daily) or an equal volume of PBS (vehicle) ip daily. Rats were treated with ETN or PBS from 6 to 12 wk of age. We have previously shown that treatments during this period are efficacious in attenuating vascular remodeling (34–36). Rats were maintained on a 12-h:12-h light/dark cycle, with tap water and regular chow ad libitum. At 12 wk of age, rats were anesthetized with 3% isoflurane, weighed, and euthanized by decapitation after exsanguination. All organs used in this study were harvested after decapitation. The experimental protocol was approved by the Michigan State University Institutional Animal Care and Use Committee and was in accordance with the American Physiological Society's Guiding Principles in the Care and Use of Animals.
A separate group of naive, previously untreated 12-wk-old male SHRSP was used to assess the effects of acute ETN treatment on the outcome of cerebral ischemia. Rats subjected to transient middle cerebral artery occlusion (tMCAO; protocol described below) received ETN (1.25 mg/kg ip) or equivolume PBS at reperfusion and 24 h after ischemia.
Measurement of arterial pressure.
Blood pressure during the last week of treatment was measured by tail-cuff using a RTBP1001 tail-cuff blood pressure system (Kent Scientific, Torrington, CT) as described previously (34).
Pressure myography studies.
MCA function and structure were assessed by pressure myography (Danish Myo Technology, Aarhus, Denmark) as described previously (36). Briefly, we analyzed MCA spontaneous myogenic tone generation and structural and mechanical properties. MCAs were equilibrated in oxygenated (95%O2-5% CO2) physiological salt solution (PSS) containing (in mmol) 141.9 NaCl, 4.7 KCl, 1.12 KH2PO4, 1.7 MgSO4·7H2O, 2.8 CaCl2, 10 HEPES, 5 dextrose, and 0.5 EDTA (pH 7.4) until development of spontaneous myogenic tone, which was calculated using the following formula: %tone = [1 − (active lumen diameter/passive lumen diameter)] × 100. Passive structure was assessed with calcium-free PSS containing 2 mmol/l EGTA plus 10 μmol/l sodium nitroprusside (SNP), and intraluminal pressure was increased from 3 to 180 mmHg in 20-mmHg increments. The wall-to-lumen ratio and circumferential wall stress were calculated (2). Passive distensibility was calculated as described previously (7). The elastic modulus (β-coefficient) was calculated from the stress/strain curves using an exponential model (y = aeβx), where β is the slope of the curve and is directly correlated to vascular stiffness.
MRA passive structure was assessed with calcium-free PSS containing 2 mmol/l EGTA plus 10 μmol/l sodium nitroprusside, and intraluminal pressure was increased from 3 to 180 mmHg with 30-mmHg increments. All structural parameters analyzed in the MCA were analyzed in the MRA.
MCA occlusion.
Cerebral ischemia was induced using the intraluminal suture model of MCA occlusion (26). Rats were initially anesthetized with isoflurane in an induction chamber, and anesthesia was maintained with 2% isoflurane in oxygen; body temperature was maintained at 37°C. An incision was made in the top of the head to expose the skull for measurement of pial perfusion by scanning laser Doppler flowmetry and attachment of a laser Doppler flow probe to measure perfusion to the region supplied by the MCA (5 mm lateral and 1 mm posterior to the bregma). A midline incision was made to expose the carotid artery. The lingual and thyroid arteries were cauterized, and the external carotid was tied off with suture. A 3-0 nylon monofilament with a rounded end (Doccol, Redland, CA) was inserted into the common carotid artery. This monofilament was then advanced through the internal carotid artery to block blood flow to the MCA where it branches from the circle of Willis. MCA occlusion was verified by a drop in perfusion as measured by both scanning laser Doppler flowmetry and the Doppler flow probe. One set of rats was subjected to transient ischemia (tMCAO). The MCAO was performed as described above, and ischemia was maintained for 1 h. Reperfusion was achieved by retracting the monofilament until its round end was visible in the common carotid artery. Reperfusion was maintained for 47 h. Rats were then anesthetized and decapitated, and the brain was removed and sliced into 2-mm sections for subsequent staining with 2% 2,3,5-triphenyltetrazolium chloride for 20 min to assess ischemic damage. With this staining the area of viable tissue will stain pink and areas of infarcted tissue will remain white. Brain slices were fixed in 4% paraformaldehyde, and digital images were taken. The percentage of infarction was determined by the following equation: %hemisphere infarcted = ((VC - VL)/VC)*100, where VC is the volume of normal tissue in the nonischemic hemisphere and VL is the volume of normal tissue in the ischemic hemisphere (46).
Another set of rats was subject to permanent MCAO (pMCAO) with the ischemia duration of 24 h, after which the rats were anesthetized and decapitated, the brain was removed, and measurement of ischemic damage was performed as described above.
Scanning laser doppler flowmetry.
Pial perfusion was measured as described previously by our laboratory (36). Briefly, under anesthesia, the rat's skull was exposed and cleaned and pial perfusion was analyzed in both cerebral hemispheres. The time points were analyzed before surgery, immediately after MCAO, before and after reperfusion (in animals subjected to tMCAO), and immediately before euthanasia. Mean perfusion in each hemisphere was measured using the LDPIwin 3.1 software (Perimed). Pial perfusion data are expressed a percentage of pre-ischemic pial perfusion in each hemisphere (for MCAO studies) or perfusion units (for basal perfusion).
Dilation of the nonischemic MCA after tMCAO.
Endothelial function in the nonischemic MCA was assessed by intraluminal perfusion of ADP in a pressure myograph. A branch-free segment of the MCA from the nonischemic hemisphere (contralateral) was isolated and mounted between two glass cannulas in a pressure myograph chamber, which was then placed in an inverted microscope (Nikon Eclipse T100; Nikon, Japan) coupled to a camera. The MCA was then bathed in PSS, pressurized at 80 mmHg, and maintained under physiological flow rate (20 dynes/cm2) to generate spontaneous myogenic tone. Increasing concentrations of ADP (0.1 nmol/l to 10 μmol/l) were then added to the intraluminal perfusate, and changes in MCA diameter were recorded using the MyoView software (Danish Myo Technology, Aarhus, Denmark).
To assess endothelium independent post-MCAO dilation, another group of MCAs from the nonischemic hemisphere were isolated, cannulated, and pressurized as described above and incubated with increasing concentrations of the nitric oxide donor endothelium-independent dilator SNP (1 nmol/l to 100 μmol/l).
Immunofluorescence.
Quantification of microglia and macrophages in the infarct core and peri-infarct region of SHRSP treated for 6 wk with ETN or PBS was performed by immunofluorescence (IF), as recently described (35). Briefly, 8 μm-thick cryosections of brain were incubated with 10% normal horse serum for 2 h, then incubated with primary antibodies against CD68 (marker of macrophages and neutrophils, mouse monoclonal anti-rat CD68; Abcam) or Iba-1 (marker of microglia, goat polyclonal anti-rat Iba-1; Abcam) for 24 h in a shaker at room temperature. Secondary antibodies conjugated to fluorescent probes were used to identify immunoreactivity. Slides without the primary antibody were negative controls for immunospecificity of the antibodies, and slides without the secondary antibody were used to control for autofluorescence of the tissue. Coverslips were mounted on the slides using the mounting media ProLong Gold anti-fade reagent with DAPI (Invitrogen Molecular Sciences, Eugene, OR), which stains nuclei in blue. A total of 10 fields from the infarct core were imaged per rat using a 40× air objective (Zeiss EC-Plan NeuFluar; numerical aperture 0.75), and five fields were imaged from the peri-infarcted area. The peri-infarct area was morphologically identified as the parenchymal tissue that maintained normal architecture immediately next to the infarct core. An investigator blinded to the experimental groups captured all images and performed the quantification. Data are shown as number of positive cells (Iba-1+ or CD68+)/infarct volume for the infarct core or as number of positive cells/total number of DAPI-nuclei in the peri-infarct area.
Quantitative real-time RT-PCR.
Markers of classical or alternative activation of macrophages/microglia (M1 and M2, respectively) in the ischemic hemisphere were assessed by quantitative RT-PCR (qRT-PCR). Total mRNA was isolated from the ischemic hemisphere of the brain using a Qiagen RNeasy Lipid Tissue Mini Kit (Qiagen Sciences) and reverse-transcribed using a qScript cDNA Synthesis Kit (Quanta Biosciences, Gaithesburg, MD). Quantitative PCR was then performed using Taqman ABI Assays on Demand probes in a 7,500 real time PCR system (Applied Biosystem, Foster City, CA). Fold changes from control were calculated using the 2−ΔΔCT method (25) with β-2-microglobulin used as endogenous control, as described previously (32).
Chemical supplies.
Unless otherwise stated, all chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Statistical analyses.
Body weight, heart:body weight, kidney:body weight, blood pressure, spontaneous myogenic tone generation, basal pial perfusion, infarct size (%HI), and IF data were analyzed by Student's t-test or a nonparametric alternative when the data did not fit a normal distribution model. Data regarding MCA dilation to ADP, MCA and MRA passive structure, and postischemic pial perfusion were analyzed by two-way ANOVA or a nonparametric test. Bonferroni's correction for multiple comparisons was performed as a post-test. All statistical analyses were carried out using the GraphPad Prism 6.0 software (GraphPad, San Diego, CA). Difference between means was considered statistically significant when P < 0.05.
RESULTS
Physiological variables.
Data regarding body and organ weight and blood pressure is summarized in Table 1. Briefly, ETN treatment did not alter final body weight, heart:body weight, and kidney:body weight in SHRSP. There was a small but insignificant (P = 0.10) decrease in blood pressure in SHRSP + ETN when compared with SHRSP + PBS.
Table 1.
Physiological variables of SHRSP treated with ETN or PBS
| SHRSP + ETN | SHRSP + PBS | |
|---|---|---|
| Final body weight, g | 246 ± 5 | 253 ± 7 |
| Body weight | ||
| Heart | 0.539 ± 0.01 | 0.534 ± 0.01 |
| Kidneys | 0.981 ± 0.01 | 0.986 ± 0.03 |
| Systolic arterial pressure, mmHg | 198 ± 3 | 208 ± 7δ |
Values are means ± SE; n = 8. Blood pressure was measure by tail-cuff at the last week of experiment. δP = 0.10, Mann-Whitney test. SHRSP, stroke-prone spontaneously hypertensive rats; ETN, etanercept.
MCA myogenic tone.
To assess if TNF-α inhibition alters the function of the MCA in SHRSP, we assessed spontaneous myogenic generation at 80 mmHg. We observed that there was no change in MCA spontaneous myogenic tone generation (Table 2).
Table 2.
ETN attenuated MCA remodeling in SHRSP
| SHRSP + PBS | SHRSP + ETN | WKY Rats (36) | |
|---|---|---|---|
| n | 7 | 8 | 4 |
| Diameter, μm | |||
| Lumen | 225 ± 6 | 253 ± 5* | 286 ± 8 |
| Outer | 278 ± 9 | 293 ± 5 | 314 ± 11 |
| Wall thickness, μm | 26 ± 1 | 20 ± 1* | 14 ± 1 |
| Wall-to-lumen ratio | 0.113 ± 0.01 | 0.079 ± 0.01* | 0.05 ± 0.004 |
| CSA, μm2 | |||
| Lumen | 40,086 ± 2,175 | 50,374 ± 2,062* | 64,398 ± 3,556 |
| Vessel | 60,422 ± 3,771 | 67,351 ± 2,426 | 77,452 ± 5,011 |
| Wall | 20,337 ± 1,683 | 16,976 ± 857* | 13,054 ± 1,607 |
| Distensibility | 41 ± 11 | 40 ± 3 | 45 ± 3 |
| Vessel stress, dynes/cm2 | 356 ± 11 | 520 ± 31* | 854 ± 74 |
| β-Coefficient | 7.12 ± 0.7 | 7.91 ± 0.7 | 8.61 ± 0.6 |
| Myogenic tone, % | 28 ± 7 | 33 ± 2 | NA |
Values are means ± SE at an intraluminal pressure of 80 mmHg. NA, nonapplicable; CSA, cross-sectional area; WKY, Wistar Kyoto.
Significantly different from SHRSP + PBS (P < 0.05). The WKY values contained in this table have been previously published (36); they are included here to provide an indication of the extent of the remodeling in SHRSP.
MCA passive structure.
To test the hypothesis that TNF-α is involved in MCA remodeling in SHRSP, we assessed MCA structure and mechanical properties by pressure myography. Treatment of SHRSP with ETN caused a small increase in the outer diameter of the MCA (Fig. 1A). Lumen diameter was significantly increased in SHRSP + ETN when compared with SHRSP + PBS at intraluminal pressures above 40 mmHg (Fig. 1B). MCA wall thickness (Fig. 1C) and wall-to-lumen ratio (Fig. 1D) were reduced by ETN treatment in SHRSP. Although no changes were observed in the MCA cross-sectional area (CSA), the lumen CSA was increased after ETN treatment, and the wall CSA was decreased (Table 2). Wall stress was higher in the MCA from SHRSP + ETN (Fig. 2A); there were no differences in distensibility (Fig. 2B) and stiffness as measured by the β-coefficient of the individual stress-strain relationships (Table 2).
Fig. 1.
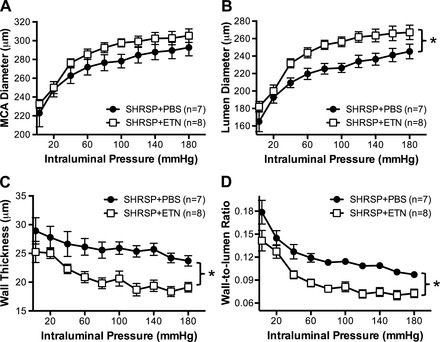
TNF-α inhibition with etanercept (ETN) improved the structure of the middle cerebral artery (MCA) in stroke-prone spontaneously hypertensive rats (SHRSP). Although there were no differences in the outer diameter of the MCA (A), ETN treatment for 6 wk caused an increase in the lumen diameter (B), suggesting attenuation in the inward remodeling process. Furthermore, there was a reduction in hypertrophic growth of the wall, observed as a decrease in the wall thickness (C). Taken together, there was an overall decrease in the wall-to-lumen ratio of the MCA (D) in SHRSP + ETN. Data are means ± SE. *P < 0.05, 2-way ANOVA.
Fig. 2.
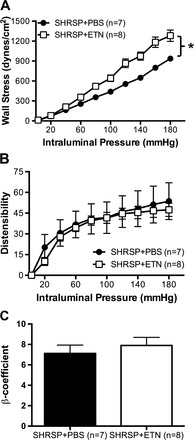
TNF-α inhibition with ETN did not change the mechanical properties of MCA. The increase in the MCA lumen diameter caused an increase in calculated wall stress (A) in SHRSP + ETN when compared with SHRSP + PBS. There were no changes in mechanical properties of the MCA, as observed by distensilibity (B), calculated as percent increase in diameter from 3 mmHg, and the β-coefficient (C) calculated from the slope of the individual stress-strain relationships. Data are means ± SE. *P < 0.01, 2-way ANOVA.
Basal pial perfusion.
To analyze if the improvement in MCA structure caused a change in basal cerebral perfusion, we assessed pial perfusion in anesthetized rats using scanning laser Doppler flowmetry. We observed a small but significant increase in basal pial perfusion in ETN-treated SHRSP (Fig. 3A).
Fig. 3.
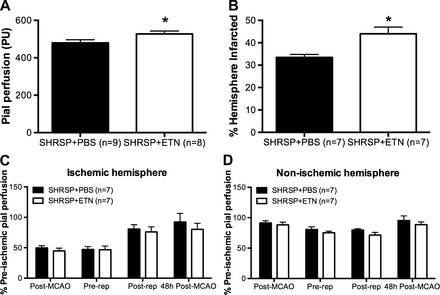
Pial perfusion and cerebral infarct after transient MCAO (tMCAO) in SHRSP treated with PBS or ETN. Basal pial perfusion, measured by scanning laser Doppler flowmetry, was increased in SHRSP + ETN when compared with SHRSP + PBS (A; *P < 0.05, Student's t-test). B: hemisphere infarcted. Cerebral infarct size after tMCAO (1 h of ischemia, 47 h of reperfusion) was larger in ETN-treated SHRSP than in PBS-treated SHRSP (*P < 0.01, Student's t-test). Pial perfusion in both the ischemic (C) and nonischemic (D) hemispheres was not different between groups. Data are means ± SE. PU, perfusion units.
Transient MCAO.
To test the hypothesis that improvement in MCA structure would reduce the cerebral infarct in SHRSP, cerebral ischemia/reperfusion was experimentally induced by tMCAO, with 1 h of ischemia and 47 h of reperfusion. Our data shows that SHRSP treated with ETN for 6 wk had significantly larger infarcts when compared with rats treated with PBS (Fig. 3B and Table 3), despite the observation that pial perfusion was not different between groups at any of the time points studied in both the ischemic (Fig. 3C) and nonischemic (Fig. 3D) hemispheres. No infarct was observed in sham-operated SHRSP (data not shown).
Table 3.
Summary of the effects of ETN treatment on the outcome of MCAO in SHRSP
| SHRSP + PBS | SHRSP + ETN | |
|---|---|---|
| Transient MCAO, %HI | 33.4 ± 1.3 | 44.3 ± 3.0* |
| Transient MCAO mortality (%) | 0/7 (0) | 0/7 (0) |
| Permanent MCAO, %HI | 65.0 ± 5.6 | 60.5 ± 2.4 |
| Permanent MCAO mortality (%) | 5/11 (45) | 0/6 (0) |
| Acute ETN transient MCAO, %HI | 37.7 ± 4.6 | 42.73 ± 9.6 |
| Acute ETN transient MCAO mortality (%) | 1/7 (14) | 0/5 (0) |
Values are means ± SE. MCAO, middle cerebral artery occlusion; %HI: percent hemisphere infarcted.
P = 0.004, Student's t-test.
Postischemic endothelium-dependent dilation of the contralateral MCA.
To evaluate if the increased infarct size observed in SHRSP + ETN was due to impaired endothelial function of cerebral arteries, we studied the dilatory response of the nonischemic MCA to ADP. Postischemic dilation of the nonischemic MCA ADP was reduced in SHRSP + PBS when compared with rats that underwent sham surgery, and ETN treatment recovered ADP-induced dilation of the MCA (Fig. 4A). Normalization of the data by the passive diameter of the MCA maintained the difference in dilation, showing that the increased dilation was not an artifact of the reduced inward remodeling (Fig. 4B). Dilation to the endothelium-independent, nitric oxide donor SNP was slightly increased in MCAs from SHRSP + ETN when compared with SHRSP + PBS (Fig. 4C). However, the difference disappeared after normalization of the data by the passive diameter of the MCA (Fig. 4D).
Fig. 4.
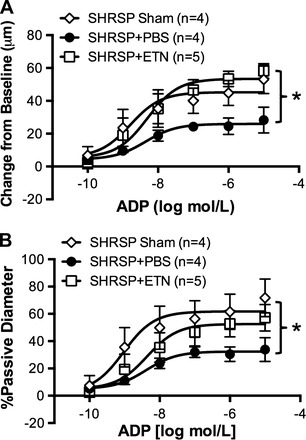
tMCAO reduces endothelium-dependent dilation of the nonischemic MCA, which is prevented by ETN treatment. The vasodilatory capacity of the nonischemic MCA (from the nonischemic hemisphere) was assessed in pressurized MCAs by intraluminal perfusion of ADP. tMCAO caused an impairment in ADP-mediated dilation of the MCA in SHRSP + PBS (●) when compared with sham-operated SHRSP (◇). This impairment seems to be mediated, at least in part, by TNF-α, since the MCA from SHRSP + ETN subjected to tMCAO showed dilation similar to that observed in sham-operated SHRSP (A). The improved dilation was not an artifact of the attenuation in inward remodeling, since normalization of the dilation data by the passive diameter maintained the difference (B). Dilation to the nitric oxide donor endothelium-independent SNP was slightly increased in the MCA from SHRSP + ETN, and normalization of the data by the passive diameter of the MCA annulled the difference. Data are means ± SE. *P < 0.01, 2-way ANOVA.
Effects of acute ETN treatment.
To assess if the increased infarct observed after TNF-α inhibition was a consequence of the chronic ETN treatment, a group of untreated 12-wk-old SHRSP were subject to tMCAO and received either ETN or PBS (1.25 mg/kg ip) at reperfusion and 24 h postischemia. Using this experimental protocol, we observed that there were no differences in infarct size (Fig. 5A and Table 3) or postischemic pial perfusion to the ischemic (Fig. 5B) and nonischemic hemispheres (Fig. 5C) between SHRSP that received PBS or ETN at reperfusion.
Fig. 5.
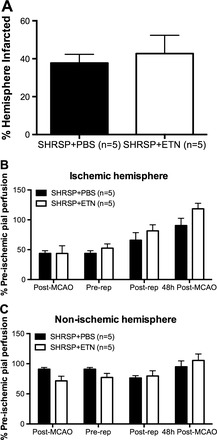
ETN treatment at reperfusion did not alter infarct size after tMCAO. To assess whether the increase in cerebral infarct size after tMCAO in SHRSP was linked to an acute effect of ETN during reperfusion injury, untreated 12-wk-old SHRSP were subjected to tMCAO and treated with ETN (1.25 mg/kg) or PBS (equivolume) at the time of reperfusion and 24 postischemia. After 47 h of reperfusion, we observed that there was no difference in infarct size between groups (A), as well as in pial perfusion in the ischemic (B) and nonischemic (C) hemispheres. Data are means ± SE.
Permanent MCAO.
To evaluate if the larger cerebral infarct observed in SHRSP + ETN after tMCAO was a consequence of the reperfusion injury, a different group of SHRSP treated with ETN for 6 wk underwent pMCAO with 24 h of ischemia. There were no differences between SHRSP + PBS and SHRSP + ETN in infarct size (Fig. 6A and Table 3) and pial perfusion to the ischemic (Fig. 6B) and nonischemic (Fig. 6C) hemispheres after pMCAO.
Fig. 6.
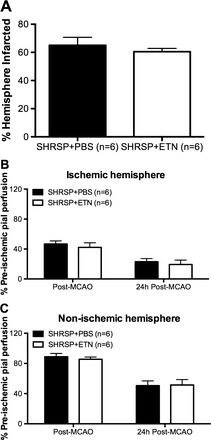
Chronic ETN treatment did not increase infarct size after permanent MCAO (pMCAO) in SHRSP. To assess if the increase in infarct size after ischemia/reperfusion was associated with the reperfusion injury, SHRSP were treated with ETN or PBS for 6 wk and subjected to pMCAO. After 24 h of ischemia, we observed that there was no difference in infarct size between groups (A). Pial perfusion in the ischemic (B) and nonischemic (C) hemispheres was also unchanged. Data are means ± SE.
Post-MCAO survival.
Data regarding post-MCAO survival is summarized in Table 3. All rats subjected to tMCAO survived for 48 h postischemia. However, five out of 11 SHRSP + PBS subjected to pMCAO did not survive for the 24 h poststroke (45% mortality), whereas six out of six SHRSP + ETN subjected to pMCAO survived for 24 h (0% mortality).
Immunofluorescence.
IF staining showed that the number of CD68+ cells (infiltrating macrophages; Fig. 7A) was not changed in both the infarct core (Fig. 7B) and the peri-infarct area (Fig. 7C) between experimental groups. However, the number of Iba-1+ cells (microglia; Fig. 8A) was reduced in the ischemic core of SHRSP + ETN when compared with SHRSP + PBS (Fig. 8B), without changes in the number of Iba-1+ cells in the peri-infarct area (Fig. 8C).
Fig. 7.
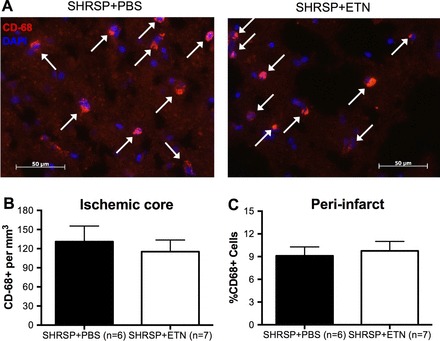
Quantification of CD68+ cells in the infarct core and peri-infarct region after tMCAO. We tested the hypothesis that chronic ETN treatment reduced macrophage infiltration to the ischemic core by quantifying the number of CD68+ cells by immunofluorescence (IF). A: representative images of CD68 labeling (red cells) colocalized with DAPI staining (blue) in the infarct core in SHRSP + PBS (left) and SHRSP + ETN (right). Note that there are no differences in the number of CD68+/DAPI cells between the groups. Quantification of the data showed that the number of CD68+/DAPI cells per volume of infarcted tissue was not different between groups (B). Similarly, the number of CD68+/DAPI cells per total number of cells in the peri-infarct area was unchanged by chronic ETN treatment (C). Data are means ± SE.
Fig. 8.
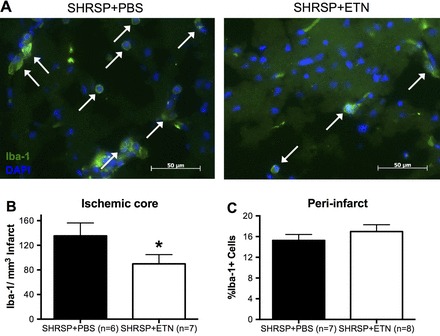
Quantification of Iba-1+ cells (microglia) in the infarct core and peri-infarct area after tMCAO. We tested the hypothesis that chronic ETN treatment reduced microglial response in the ischemic core by quantifying the number of Iba-1+ cells by IF. A: representative images of Iba-1 labeling (green cells) colocalized with DAPI staining (blue) in the infarct core in SHRSP + PBS (left) and SHRSP + ETN (right). Note that the number of Iba-1+ cells is reduced in the ischemic core of SHRSP + ETN when compared with SHRSP + PBS. Quantification of the data showed that there was a reduction in the number of Iba-1+/DAPI cells per volume of infarcted tissue in SHRSP + ETN (B; *P < 0.05, Student's t-test). However, the number of Iba-1+/DAPI cells per total number of cells in the peri-infarct area was unchanged by chronic ETN treatment (C). Data are means ± SE.
qRT-PCR.
To investigate if ETN treatment caused differential macrophage/microglia polarization in the ischemic hemisphere, mRNA expression of various polarization markers was assessed by qRT-PCR. We assessed TNF-α, inducible nitric oxide synthase (iNOS), and C-C chemokine receptor 2 (CCR2) as markers of classical (M1) activation and arginase-1, TGF-β, and macrophage mannose receptor 1 (MRC1) as markers of alternative (M2) activation. There was a small but not significant increase in TNF-α mRNA (Fig. 9A; P = 0.12), a trend toward an increase in the expression of iNOS (Fig. 9B; P = 0.06) and a twofold increase in the mRNA expression for CCR2 in the ischemic hemisphere of SHRSP + ETN when compared with SHRSP + PBS (Fig. 9C). The M2 markers arginase-1 (Fig. 9D), TGF-β (Fig. 9E), and MRC1 (Fig. 9F) were not different between SHRSP + ETN and SHRSP + PBS.
Fig. 9.
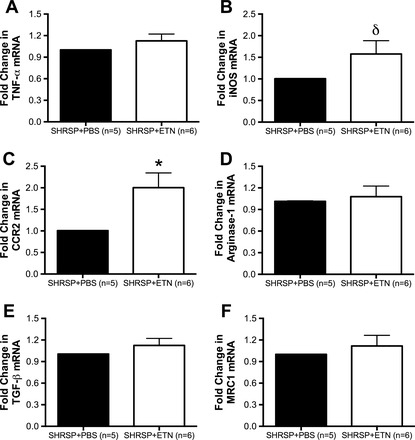
Chronic ETN treatment increased mRNA expression of marker of classical (M1) macrophage/microglia, without altering markers of alternative (M2) activation ischemic hemisphere. There was a small but not significant increase in the M1 marker TNF-α (A; P = 0.12) and a trend toward an increase in inducible nitric oxide synthase (iNOS) mRNA expression (B) in the ischemic hemisphere of SHRSP + ETN when compared with SHRSP + PBS. There was a 2-fold significant increase in the mRNA expression of C-C chemokine receptor 2 (CCR2) in the ischemic hemisphere of SHRSP + ETN (C). Expression of the M2 markers arginase-1 (D), TGF-β (E), and macrophage mannose receptor 1 (MRC1; F) was not different between groups. Data are shown as fold-change from SHRSP + PBS, and β-2-microglobulin was used as endogenous control. Total mRNA was isolated from the ischemic hemisphere of the brain. Data are means ± SE. δP = 0.06, *P < 0.05, Student's t-test.
MRA passive structure.
To assess if the improvement in vascular structure observed in the MCA after chronic ETN treatment also occurred in peripheral vascular beds, we studied the MRA. These data are summarized in Table 4 and Fig. 10. Briefly, ETN treatment attenuated MRA remodeling, as evidenced by a trend toward increase in MRA lumen diameter (Table 4; P = 0.065). The wall thickness was decreased after ETN treatment (Fig. 10A), as was the wall-to-lumen ratio (Fig. 10B). Wall stress was increased in SHRSP + ETN (Table 4), as was the MRA distensibility (Fig. 10C). MRA stiffness was reduced in SHRSP + ETN, as observed by a reduction in the β-coefficient value (Table 4 and Fig. 10D).
Table 4.
ETN attenuated remodeling of the mesenteric resistance arteries in SHRSP
| SHRSP + PBS | SHRSP + ETN | WKY Rats (36) | |
|---|---|---|---|
| n | 6 | 6 | 4 |
| Diameter, μm | |||
| Lumen | 273 ± 7 | 287 ± 9 | 280 ± 21 |
| Outer | 332 ± 8 | 334 ± 10 | 311 ± 20 |
| Wall thickness, μm | 29 ± 2 | 24 ± 1* | 16 ± 1.4 |
| Wall-to-lumen ratio | 0.11 ± 0.007 | 0.08 ± 0.005* | 0.06 ± 0.007 |
| CSA, μm2 | |||
| Lumen | 58,969 ± 3,052 | 64,776 ± 3,945 | 62,316 ± 8,843 |
| Vessel | 86,643 ± 4,179 | 88,167 ± 5,157 | 76,675 ± 9,850 |
| Wall | 27,674 ± 2,014 | 23,391 ± 1,777 | 14,386 ± 1,543 |
| Distensibility | 75 ± 5 | 91 ± 7* | 74 ± 3 |
| Vessel stress, dynes/cm2 | 433 ± 31 | 546 ± 41* | 831 ± 90 |
| β-Coefficient | 4.16 ± 0.3 | 3.34 ± 0.1* | 6.23 ± 1.02 |
Values are means ± SE at an intraluminal pressure of 90 mmHg.
Significantly different from SHRSP + PBS (P < 0.05). The WKY values contained in this table have been previously published (36); they are included here to provide an indication of the extent of the remodeling in SHRSP.
Fig. 10.
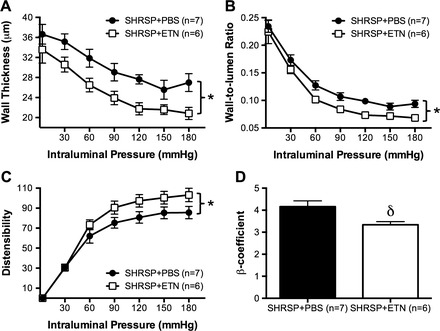
ETN treatment attenuated wall hypertrophy and improved the mechanical properties of mesenteric resistance arteries (MRA) in SHRSP. Chronic TNF-α inhibition reduced hypertrophic growth in MRA, observed as a reduction in wall thickness (A). As consequence, the wall-to-lumen ratio was smaller in SHRSP + ETN when compared with SHRSP + PBS (B). ETN treatment also improved the MRA mechanical properties, measured as an increase in distensibility (C) and in the β-coefficient (D). Data are means ± SE. *P < 0.01, 2-way ANOVA; δP < 0.05, Student's t-test.
DISCUSSION
The main findings of the present study are 1) chronic TNF-α inhibition improved the structure of the MCA, as evidenced by an increase in lumen diameter and a decrease in wall thickness and wall-to-lumen ratio; this was associated with an increase in basal pial perfusion; 2) similar to the MCA, the passive structure of the MRA was also improved, with ETN treatment causing a reduction in wall thickness and wall-to-lumen ratio; 3) despite the improvement in the MCA structure, cerebral infarct after tMCAO was increased in SHRSP + ETN, most likely a consequence of impairment in the innate immune response of the brain after chronic ETN treatment. To the best of our knowledge this is the first report to show that chronic TNF-α inhibition affects structural parameters of the vasculature independently of a reduction in blood pressure. It is also the first study to assess the effects of chronic TNF-α inhibition in the outcome of cerebral ischemia in hypertensive rodents.
The involvement of TNF-α in the development of hypertension has been elucidated in the recent years. Anti-TNF-α therapy caused a modest reduction in both systolic and diastolic blood pressures in humans with rheumatoid arthritis (40). In the angiotensin II-salt model of hypertension in rodents, ETN treatment delayed the development of hypertension (11), although after the delay the blood pressure was similar between control and ETN-treated groups. Deletion of TNF-α in mice prevented the rise in mean arterial pressure after angiotensin II infusion, and this effect was lost after administration of exogenous TNF-α, suggesting a direct link between TNF-α and angiotensin II-induced hypertension (44). In our study we observed a small but insignificant (P = 0.10) reduction in systolic blood pressure in the genetically hypertensive SHRSP after ETN treatment. One caveat of the present study is that blood pressure was assessed by tail cuff instead of radiotelemetry. However, we showed previously that blood pressure measurements in SHRSP using tail cuff are similar to those obtained by radiotelemetry (39). Independently of the method used, it is unlikely that a 10-mmHg reduction in blood pressure will be physiologically relevant in SHRSP.
The main finding of this study is that ETN treatment during the exponential rise in blood pressure in SHRSP attenuates MCA remodeling. The inward remodeling process is thought to be initially a protective process to increase vascular resistance and reduce intraluminal pressure in downstream arterioles and capillaries, thus preventing ruptures and hemorrhages (16). However, in the long-term this process becomes maladaptive and is linked to increased risk of end-organ damage (31), because it can have deleterious effects on local regulation of blood flow. We observed an increase in lumen diameter and a reduction in wall thickness and wall-to-lumen ratio in the MCA of SHRSP after 6 wk of ETN treatment. This finding is not without precedence, since TNF-α is a known proliferative stimulus for vascular smooth muscle cells (20, 24, 37). TNF-α also induces the expression and activity of matrix metalloproteinases (20). Vascular smooth muscle cell proliferation and matrix metalloproteinases activity are involved in hypertensive remodeling (36). The source of TNF-α in cerebral arteries seems to be perivascular macrophages, since we recently showed that reducing the number of these cells in the cerebral vasculature reduces TNF-α mRNA expression and attenuates MCA remodeling (35). We also observed a small increase in basal pial perfusion in SHRSP after ETN treatment. This increase could be due to increased perfusion through the large arteries of the Circle of Willis that feed into the pial circulation, an increase in the lumen diameter of pial arteries and arterioles, or a reduction in basal myogenic tone of cerebral arteries. Although we did not observe reduced myogenic tone in the MCA of SHRSP, a recent study showed that TNF-α mediates increased myogenic tone in the posterior cerebral artery in gerbils with heart failure (50).
Despite the improvement in the structure of the MCA, SHRSP treated with ETN for 6 wk had larger infarcts after tMCAO. This observation is in disagreement with a recent report for normotensive animals (45). We hypothesized that the increased infarct could be a consequence of reduced blood flow due to impaired postreperfusion dilation of cerebral arteries or an exacerbation of the reperfusion injury due to the chronic immunosuppression. We will discuss these two hypotheses separately.
To assess if ETN impairs postreperfusion vasodilation, SHRSP were treated with ETN or PBS for 6 wk before tMCAO, and we isolated the MCA from the nonischemic hemisphere to study endothelium-dependent and independent dilation. We observed that MCA from SHRSP subjected to tMCAO showed reduced dilation to intraluminal perfusion of ADP when compared with MCAs from sham-operated rats and that ETN treatment restored dilation to levels similar to sham-operated rats. Dilation to the endothelium-independent, nitric oxide donor SNP was slightly increased the nonischemic MCA from SHRSP + ETN and disappeared after normalization of the data by the passive diameter. However, the small increase in dilation does not entirely explain the improved dilation observed after exposure to ADP, suggesting a combination of improved endothelial function and increased sensitivity of the media layer to nitric oxide. Impaired endothelium-dependent dilation of cerebral arteries postischemia occurs in the basilar artery (8) and in peripheral vascular beds, including the mesenteric circulation (30). Our data suggest that part of the endothelial dysfunction is mediated by TNF-α. In fact, patients with rheumatoid arthritis treated with anti-TNF-α therapies show improved endothelium-dependent dilation of brachial arteries (5, 12). In the MCA, ADP elicits dilation by endothelial production of nitric oxide and endothelium-derived hyperpolarizing factor (EDHF) (52). TNF-α was shown to impair dilations mediated by nitric oxide in coronary arteries (33) and EDHF in human omental arteries (14). Importantly, EDHF-mediated dilation is prevalent in the postischemic MCA (29). It is possible that the prolonged ETN treatment either enhanced the EDHF-mediated dilation or preserved nitric oxide-dependent dilation, leading to a dilatory response similar to that observed in sham-operated SHRSP.
To test the hypothesis that the increased damage is a consequence of exacerbated reperfusion injury due to chronic immunosuppression, we designed two studies. In the first we subjected SHRSP treated with ETN for 6 wk to pMCAO; in the second we subjected untreated SHRSP to tMCAO and administered ETN at the time of reperfusion and 24 h postischemia. In both studies we observed that there were no differences in infarct size or pial perfusion in ETN-treated SHRSP. Together, these data suggest that the increase in infarct following tMCAO is likely due to a deleterious effect of the prolonged ETN treatment on the outcome of reperfusion injury. Thus we investigated the innate immune response in the brain after reperfusion by IF.
A reduction in macrophage infiltration to the ischemic hemisphere could explain the increase in infarct size, since macrophages are important to remove cytotoxic cell debris (27). Macrophage infiltration peaks at 2 days after ischemia/reperfusion injury in rodents (19), and this event is dependent on the expression of adhesion molecules by endothelial cells (51). TNF-α is a potent inducer of adhesion molecules (18, 43); thus it is possible that the prolonged inhibition reduced the levels of adhesion molecules, consequently reducing leukocyte infiltration and exacerbating the damage. However, our data do not support this possibility. The number of CD68+ cells, a marker of macrophages, monocytes, and neutrophils, in the ischemic core and peri-infarct area was not different between SHRSP treated with ETN or PBS. This suggests that there is an effect of ETN directly in the postischemic neurons or brain resident immune cells, such as microglia.
The effects of TNF-α and its inhibition on the outcome of cerebral ischemia are controversial. Some studies show that acute TNF-α generation is involved in neuronal death and infarct development following MCAO in SHR (1) and that TNF-α inhibition reduces infarct development after cerebral ischemia/reperfusion (45). In addition, TNF-α increases blood-brain-barrier permeability in the early hours following ischemia/reperfusion, increasing secondary ischemic damage (49). However, others show that knock-out of TNF-α receptors increases damage after MCAO (4, 13). It seems that TNF-α has a dual role in the evolution of the infarct following cerebral ischemia/reperfusion injury: on the one hand TNF-α induces neuronal apoptosis (32), thus leading to increased neuronal death and larger infarcts; conversely, TNF-α stimulates activation of microglia/macrophages (4), which are important for neuronal survival in the early stages following ischemia, particularly alternatively polarized microglia (17). Our data show that the number of Iba-1+ microglia/macrophages is reduced in the infarct core of SHRSP + ETN, suggesting that the innate cerebral immune response was impaired in those rats. Microglia are also important for eliminating cytotoxic molecules generated after cell death in the ischemic hemisphere (42). A reduction in the number of Iba-1+ microglia could lead to an accumulation of cytotoxic molecules, increasing cell death and expansion of the infarct core. In addition, there seems to be an upregulation of mRNA for markers of classical microglia/macrophage activation (M1), such as iNOS and CCR2, without changes in markers for M2 polarization. M1 polarization was shown to exacerbate ischemic damage, leading to increased cell death (16). Thus, it is possible that the chronic ETN treatment primed macrophage/microglia towards a M1 phenotype, exacerbating ischemic neuronal death and leading to larger infarcts.
Finally, the lack of a protective effect of ETN in the outcome of pMCAO could be explained by survival bias. We observed a 45% mortality (5 out of 11) in SHRSP treated with PBS, whereas in the SHRSP + ETN group there was no mortality. It is possible that the rats treated with vehicle that survived for 24 h had smaller infarcts; and the rats with larger infarcts did not survive until the time point established for euthanasia and analysis of infarct size. Because we did not evaluate infarct size in the rats that died before 24 h post-pMCAO, we might have skewed our results towards smaller infarcts in vehicle-treated SHRSP subjected to pMCAO.
Chronic ETN treatment attenuated hypertrophic remodeling of MRA in SHRSP. This finding is at odds with our previous report showing that macrophage depletion did not alter MRA remodeling in SHRSP (35). Macrophage depletion in SHRSP caused an increase in TNF-α mRNA expression in perivascular adipose tissue, suggesting that these cells might produce TNF-α, which appears to be an important mediator of hypertensive remodeling in the mesenteric vasculature. The findings in this study support that hypothesis, since ETN improved MRA structure in the SHRSP. Interestingly, MRA showed an improvement in mechanical properties, including reduced stiffness and increased distensibility, which was not observed in the MCA. TNF-α was shown to increase collagen deposition in the rodent heart (22); thus it is possible that ETN decreased collagen deposition in the MRA, increasing distensibility and reducing stiffness. Moreover, TNF-α was shown to increase expression and activity of matrix metalloproteases-2 and -9 in cultured human aortic smooth muscle cells (23). We (36) and others (6) have shown that matrix metalloproteases are important mediators of hypertensive artery remodeling. It is possible that TNF-α acts upstream of matrix metalloproteases, inducing their expression and activity, thus worsening remodeling in the peripheral vasculature.
In summary, we show that TNF-α inhibition with ETN improves the structure of resistance arteries in both the cerebral and mesenteric vasculature in chronic hypertension. Despite the improvement in cerebral vascular structure, infarct size following cerebral ischemia/reperfusion injury was larger in SHRSP + ETN. This effect seems to be a consequence of reduced microglia in the ischemic core after tMCAO, coupled to an increase in M1 polarization, which could lead to exacerbation in neuronal ischemic death.
Limitations of the study.
The present study has a few limitations and caveats that need to be mentioned. First, we did not perform quantitative measurements of cerebral blood flow. The scanning laser Doppler flowmetry measures perfusion in arbitrary units (perfusion units), although the calibration of the laser detector is not changed between subjects. Therefore, it still allows for comparative analyses between experimental groups. The laser has a penetration depth of ∼1 mm into the tissue. The skull of a 12-wk-old SHRSP is no thicker than 0.5 mm; thus the equipment measures perfusion in surface pial circulation and the outermost layer of the underlying cerebral cortex (36). Second, all perfusion measurements were performed with the rat under deep general anesthesia induced by 3% isoflurane in pure oxygen. Isoflurane is a known dilator of cerebral arteries (47), and the increase in basal pial perfusion could be linked to the increased lumen diameter of fully dilated cerebral arteries and arterioles. Regarding the MCAO surgical procedure, the rats were not mechanically ventilated, which could impact cerebral perfusion. Combined with the effects of isoflurane, the increased infarct in ETN-treated SHRSP could have been caused by a reduction in cerebral blood flow that was below the detection limits of the technology used in the present study. However, we propose that a shift in macrophage/microglia polarization rather than alterations in cerebral perfusion is responsible for the effects of ETN on the outcome of cerebral ischemia. Finally, the conclusions of this work are supported by associative evidence rather than direct links. Due to the terminal nature of the measurements performed, it is difficult to elucidate direct cause-and-effect links between the treatment and end-points measured.
GRANTS
The present study was funded by the American Heart Association (0840122N:AMD and 12PRE8960019:PWP).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: P.W.P. and A.M.D. conception and design of research; P.W.P., S.S.G., G.M., and J.L.M. performed experiments; P.W.P. and S.S.G. analyzed data; P.W.P. and A.M.D. interpreted results of experiments; P.W.P. prepared figures; P.W.P. drafted manuscript; P.W.P. and A.M.D. edited and revised manuscript; P.W.P. and A.M.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Nusrat Matin and Naiomy Rios for the critical evaluation of the manuscript.
REFERENCES
- 1.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 28: 1233–1244, 1997. [DOI] [PubMed] [Google Scholar]
- 2.Baumbach GL, Hajdu MA. Mechanics and composition of cerebral arterioles in renal and spontaneously hypertensive rats. Hypertension 21: 816–826, 1993. [DOI] [PubMed] [Google Scholar]
- 3.Briet M, Schiffrin EL. Vascular actions of aldosterone. J Vasc Res 50: 89–99, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med 2: 788–794, 1996. [DOI] [PubMed] [Google Scholar]
- 5.Capria A, De Nardo D, Baffetti FR, Barbini U, Violo A, Tondo T, Fontana L. Long-term anti-TNF-alpha treatments reverse the endothelial dysfunction in rheumatoid arthritis: the biological coherence between synovial and endothelial inflammation. Int J Immunopathol Pharmacol 23: 255–262, 2010. [DOI] [PubMed] [Google Scholar]
- 6.Castro MM, Rizzi E, Prado CM, Rossi MA, Tanus-Santos JE, Gerlach RF. Imbalance between matrix metalloproteinases and tissue inhibitor of metalloproteinases in hypertensive vascular remodeling. Matrix Biol 29: 194–201, 2010. [DOI] [PubMed] [Google Scholar]
- 7.Chan SL, Chapman AC, Sweet JG, Gokina NI, Cipolla MJ. Effect of PPARgamma inhibition during pregnancy on posterior cerebral artery function and structure. Front Physiol 1: 130, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coucha M, Li W, Ergul A. The effect of endothelin receptor A antagonism on basilar artery endothelium-dependent relaxation after ischemic stroke. Life Sci 91: 676–680, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorrance AM, Osborn HL, Grekin R, Webb RC. Spironolactone reduces cerebral infarct size and EGF-receptor mRNA in stroke-prone rats. Am J Physiol Regul Integr Comp Physiol 281: R944–R950, 2001. [DOI] [PubMed] [Google Scholar]
- 10.Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM. TNF-α inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol 294: R76–R83, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elmarakby AA, Quigley JE, Pollock DM, Imig JD. Tumor necrosis factor alpha blockade increases renal Cyp2c23 expression and slows the progression of renal damage in salt-sensitive hypertension. Hypertension 47: 557–562, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Galarraga B, Belch JJ, Pullar T, Ogston S, Khan F. Clinical improvement in rheumatoid arthritis is associated with healthier microvascular function in patients who respond to antirheumatic therapy. J Rheumatol 37: 521–528, 2010. [DOI] [PubMed] [Google Scholar]
- 13.Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab 18: 1283–1287, 1998. [DOI] [PubMed] [Google Scholar]
- 14.Gillham JC, Myers JE, Baker PN, Taggart MJ. TNF-alpha alters nitric oxide- and endothelium-derived hyperpolarizing factor-mediated vasodilatation in human omental arteries. Hypertens Pregnancy 27: 29–38, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 127: e6–e245, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi K, Naiki T. Adaptation and remodeling of vascular wall; biomechanical response to hypertension. J Mech Behav Biomed Mater 2: 3–19, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43: 3063–3070, 2012. [DOI] [PubMed] [Google Scholar]
- 18.Iademarco MF, Barks JL, Dean DC. Regulation of vascular cell adhesion molecule-1 expression by IL-4 and TNF-alpha in cultured endothelial cells. J Clin Invest 95: 264–271, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87: 779–789, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee SJ, Kim WJ, Moon SK. TNF-alpha regulates vascular smooth muscle cell responses in genetic hypertension. Int Immunopharmacol 9: 837–843, 2009. [DOI] [PubMed] [Google Scholar]
- 21.Li JS, Lariviere R, Schiffrin EL. Effect of a nonselective endothelin antagonist on vascular remodeling in deoxycorticosterone acetate-salt hypertensive rats. Evidence for a role of endothelin in vascular hypertrophy. Hypertension 24: 183–188, 1994. [DOI] [PubMed] [Google Scholar]
- 22.Li YY, Feng YQ, Kadokami T, McTiernan CF, Draviam R, Watkins SC, Feldman AM. Myocardial extracellular matrix remodeling in transgenic mice overexpressing tumor necrosis factor alpha can be modulated by anti-tumor necrosis factor alpha therapy. Proc Natl Acad Sci USA 97: 12746–12751, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin CP, Huang PH, Tsai HS, Wu TC, Leu HB, Liu PL, Chen YH. Monascus purpureus-fermented rice inhibits tumor necrosis factor-alpha-induced upregulation of matrix metalloproteinase 2 and 9 in human aortic smooth muscle cells. J Pharm Pharmacol 63: 1587–1594, 2011. [DOI] [PubMed] [Google Scholar]
- 24.Lin YC, Chiang CH, Chang LT, Sun CK, Leu S, Shao PL, Hsieh MC, Tsai TH, Chua S, Chung SY, Kao YH, Yip HK. Simvastatin attenuates the additive effects of TNF-alpha and IL-18 on the connexin 43 up-regulation and over-proliferation of cultured aortic smooth muscle cells. Cytokine 62: 341–351, 2013. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20: 84–91, 1989. [DOI] [PubMed] [Google Scholar]
- 27.Mabuchi T, Kitagawa K, Ohtsuki T, Kuwabara K, Yagita Y, Yanagihara T, Hori M, Matsumoto M. Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke 31: 1735–1743, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci 29: 367–374, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Marrelli SP, Khorovets A, Johnson TD, Childres WF, Bryan RM., Jr P2 purinoceptor-mediated dilations in the rat middle cerebral artery after ischemia-reperfusion. Am J Physiol Heart Circ Physiol 276: H33–H41, 1999. [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Revelles S, Jimenez-Altayo F, Caracuel L, Perez-Asensio FJ, Planas AM, Vila E. Endothelial dysfunction in rat mesenteric resistance artery after transient middle cerebral artery occlusion. J Pharmacol Exp Ther 325: 363–369, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Mathiassen ON, Buus NH, Sihm I, Thybo NK, Morn B, Schroeder AP, Thygesen K, Aalkjaer C, Lederballe O, Mulvany MJ, Christensen KL. Small artery structure is an independent predictor of cardiovascular events in essential hypertension. J Hypertens 25: 1021–1026, 2007. [DOI] [PubMed] [Google Scholar]
- 32.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation 5: 45, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Picchi A, Gao X, Belmadani S, Potter BJ, Focardi M, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res 99: 69–77, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Pires PW, Deutsch C, McClain JL, Rogers CT, Dorrance AM. Tempol, a superoxide dismutase mimetic, prevents cerebral vessel remodeling in hypertensive rats. Microvasc Res 80: 445–452, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pires PW, Girgla SS, McClain JL, Kaminski NE, van Rooijen N, Dorrance AM. Improvement in middle cerebral artery structure and endothelial function in stroke-prone spontaneously hypertensive rats after macrophage depletion. Microcirculation 20: 650–661, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pires PW, Rogers CT, McClain JL, Garver HS, Fink GD, Dorrance AM. Doxycycline, a matrix metalloprotease inhibitor, reduces vascular remodeling and damage after cerebral ischemia in stroke-prone spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 301: H87–H97, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajesh M, Mukhopadhyay P, Hasko G, Huffman JW, Mackie K, Pacher P. CB2 cannabinoid receptor agonists attenuate TNF-alpha-induced human vascular smooth muscle cell proliferation and migration. Br J Pharmacol 153: 347–357, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rigsby CS, Ergul A, Portik Dobos V, Pollock DM, Dorrance AM. Effects of spironolactone on cerebral vessel structure in rats with sustained hypertension. Am J Hypertens 24: 708–715, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rigsby CS, Pollock DM, Dorrance AM. Spironolactone improves structure and increases tone in the cerebral vasculature of male spontaneously hypertensive stroke-prone rats. Microvasc Res 73: 198–205, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sandoo A, Panoulas VF, Toms TE, Smith JP, Stavropoulos-Kalinoglou A, Metsios GS, Gasparyan AY, Carroll D, Veldhuijzen van Zanten JJ, Kitas GD. Anti-TNFalpha therapy may lead to blood pressure reductions through improved endothelium-dependent microvascular function in patients with rheumatoid arthritis. J Hum Hypertens 25: 699–702, 2011. [DOI] [PubMed] [Google Scholar]
- 41.Schiffrin EL. Vascular endothelin in hypertension. Vascul Pharmacol 43: 19–29, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Schilling M, Besselmann M, Muller M, Strecker JK, Ringelstein EB, Kiefer R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: an investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol 196: 290–297, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76: 301–314, 1994. [DOI] [PubMed] [Google Scholar]
- 44.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51: 1345–1351, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sumbria RK, Boado RJ, Pardridge WM. Brain protection from stroke with intravenous TNFalpha decoy receptor-Trojan horse fusion protein. J Cereb Blood Flow Metab 32: 1933–1938, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab 10: 290–293, 1990. [DOI] [PubMed] [Google Scholar]
- 47.Todd MM, Weeks J. Comparative effects of propofol, pentobarbital, and isoflurane on cerebral blood flow and blood volume. J Neurosurg Anesthesiol 8: 296–303, 1996. [DOI] [PubMed] [Google Scholar]
- 48.Touyz RM. Intracellular mechanisms involved in vascular remodelling of resistance arteries in hypertension: role of angiotensin II. Exp Physiol 90: 449–455, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Yang GY, Gong C, Qin Z, Liu XH, Lorris Betz A. Tumor necrosis factor alpha expression produces increased blood-brain barrier permeability following temporary focal cerebral ischemia in mice. Brain Res Mol Brain Res 69: 135–143, 1999. [DOI] [PubMed] [Google Scholar]
- 50.Yang J, Noyan-Ashraf MH, Meissner A, Voigtlaender-Bolz J, Kroetsch JT, Foltz W, Jaffray D, Kapoor A, Momen A, Heximer SP, Zhang H, van Eede M, Henkelman RM, Matthews SG, Lidington D, Husain M, Bolz SS. Proximal cerebral arteries develop myogenic responsiveness in heart failure via tumor necrosis factor-alpha-dependent activation of sphingosine-1-phosphate signaling. Circulation 126: 196–206, 2012. [DOI] [PubMed] [Google Scholar]
- 51.Yilmaz G, Granger DN. Cell adhesion molecules and ischemic stroke. Neurol Res 30: 783–793, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.You J, Johnson TD, Childres WF, Bryan RM., Jr Endothelial-mediated dilations of rat middle cerebral arteries by ATP and ADP. Am J Physiol Heart Circ Physiol 273: H1472–H1477, 1997. [DOI] [PubMed] [Google Scholar]