

Abstract
Emerging evidence demonstrates a close interplay between disturbances in mitochondrial function and ER homeostasis in the development of the metabolic syndrome. The present investigation sought to advance our understanding of the communication between mitochondrial dysfunction and ER stress in the onset of hepatic steatosis in male rodents with defective peroxisome proliferator-activated receptor-α (PPARα) signaling. Genetic depletion of PPARα or perturbation of PPARα signaling by high-fructose diet compromised the functional activity of metabolic enzymes involved in mitochondrial fatty acid β-oxidation and induced hepatic mitochondrial stress in rats and mice. Inhibition of PPARα activity further enhanced the expression of apolipoprotein B (apoB) mRNA and protein, which was associated with reduced mRNA expression of the sarco/endoplasmic reticulum calcium ATPase (SERCA), the induction of hepatic ER stress, and hepatic steatosis. Restoration of PPARα activity recovered the metabolic function of the mitochondria and ER, alleviated systemic hypertriglyceridemia, and improved hepatic steatosis. These findings unveil novel roles for PPARα in mediating stress signals between hepatic subcellular stress-responding machinery and in the onset of hepatic steatosis under conditions of metabolic stress.
Keywords: peroxisome proliferator-activated receptor-α, endoplasmic reticulum, mitochondrial and endoplasmic reticulum stress, apolipoprotein B, very-low density lipoprotein, hepatic steatosis
perturbations in lipid metabolism are involved in the pathogenesis of a cluster of chronic metabolic diseases, including fatty liver disease, insulin resistance, type 2 diabetes, and atherosclerosis. The peroxisome proliferator-activated receptor (PPAR) nuclear receptor family is a group of transcription factors that critically regulate signaling at the interface of lipid metabolism and inflammation (3). Among them, the isoform PPARα is expressed predominantly in the liver, where it promotes fatty acid β-oxidation, ketogenesis, lipid transport, and gluconeogenesis (4, 26). The majority of the genes involved in mitochondrial fatty acid β-oxidation are transcriptionally regulated by PPARα. As energy-regulating metabolic machinery, mitochondria dynamically communicate with other subcellular organelles such as the endoplasmic reticulum (ER) (10). Mitochondrial dysfunction is a contributing factor to the onset of insulin resistance; impairment of mitochondrial function in human myocytes increases predisposition to intramyocellular lipid accumulation and insulin resistance (17, 28).
Research over the past decade has established a compelling connection between lipid-induced ER stress and the metabolic syndrome (18, 23). The onset of obesity and hepatic steatosis are usually accompanied by increased secretion of hepatic very-low-density lipoprotein-triglyceride (VLDL-TG) and VLDL-apolipoprotein B (apoB) (5). Biosynthesis of apoB (a key structural component of VLDL) is modulated by both insulin and ER stress (21, 24, 29). Modest ER stress induced by free fatty acids increases apoB secretion, whereas greater lipid loading results in severe ER stress and inhibits apoB production in McA-RH777 cells (21). Furthermore, constant overproduction of apoB in hepatocytes stimulated by an atherogenic diet has been reported to directly provoke hepatic ER stress and may contribute to the onset of hepatic insulin resistance (30).
In the present study, we report that genetic depletion of PPARα (PPARα−/−) or suppression of PPARα signaling by high-fructose feeding compromises mitochondrial integrity and the function of metabolic enzymes involved in fatty acid β-oxidation. Impairment of PPARα signaling upregulated hepatic apoB expression at both the transcriptional and translational levels, which in turn suppressed expression of the sarco/endoplasmic reticulum calcium ATPase (SERCA) and induced hepatic ER stress, leading to systemic dyslipidemia and hepatic steatosis. Restoration of PPARα activity relieved mitochondrial and ER stress, alleviated systemic hypertriglyceridemia, and improved hepatic steatosis.
MATERIALS AND METHODS
Cell culture and treatments.
Primary mouse and rat hepatocytes and a rat hepatoma cell line, McA-RH7777 (ATCC), were maintained in DMEM containing 20% FBS at 37°C, 5% CO2. Oleic acid, fructose, PPARα antagonist MK-886, and PPARα agonist WY-14643 were used at final concentrations of 400 μM, 6 mM, 10 μM, and 40 μM, respectively.
Animal protocols.
All animal experiments were approved by the animal ethics committee of the Hospital for Sick Children and conducted according to national guidelines. Male wild-type (WT) (C57BL/6J) and PPARα-knockout mice (B6.129S4-Pparatm1Gonz/J) were purchased from The Jackson Laboratory (Bar Harbor, ME) at 12 wk of age. The PPARα-knockout mice were initially generated from a 129S4/SvJae background and then backcrossed for 10 generations on to the C57BL/6J background. Heterozygotes were then intercrossed to generate homozygotes. C57BL/6J mice were used for controls, as recommended by the vendor. Male rats (340–360 g) were purchased from Charles River (Montreal, QC, Canada) at 12 wk of age. Animals were housed on alternating 12-h light and dark cycles with free access to food and water. After 1 wk of acclimatization and baseline blood collection (fasted for 5 h), animals were placed on a control chow diet (Table 1) or a high-fructose diet (60% fructose, ID: 161.506; Dyets, Bethlehem, PA) for 3 wk. WY-14643 treatment was used to activate PPARα in a diet-induced model of insulin resistance. High-fructose-fed (Fruc) and chow-fed (Chow) rats were randomly divided into two groups (n = 6). Chow/WY and Fruc/WY were dosed with WY-14643 (6 mg/kg ip) once every 2 days throughout the 3-wk feeding period. Control groups (Chow/Veh and Fruc/Veh) received injection of the vehicle [1.5 ml/kg of 0.5% (vol/vol) DMSO ip]. Tissue collection was performed after 3 wk as follows: animals were fasted (5 h) and anesthetized using isoflurane (3% mixed with oxygen), tissues were excised and flash-frozen in liquid nitrogen, and livers were then homogenized in solubilization buffer, as described previously (32).
Table 1.
Detailed composition of diets for the rats
| Ingredients, %mass | Chow | Fructose |
|---|---|---|
| Protein | 16.4 | 22.21 |
| Fat | 13.3 | 6.0 |
| Starch | 56.4 | Trace |
| Fructose | 0.2 | 60.0 |
| Glucose | 0.1 | Trace |
| Sucrose | 0.6 | Trace |
| Cellulose fiber | 3.5 | 7.09 |
| Salt mix | 5.9 | 3.5 |
| Vitamin mix | 3.6 | 1.0 |
| Choline bitartrate | Trace | 0.2 |
VLDL collection.
Animals were fasted for 5 h, and baseline blood was collected via tail vein bleed (100 μl). Animals were then treated with 20% poloxamer (500 mg/kg ip), and blood was collected at 1 and 2 h via tail vein bleed (100 μl), with final blood collections done at 3 h via cardiac puncture (500 μl). Plasma TG and cholesterol were assessed in all samples, as described below. VLDL fractions were separated from plasma (200 μl) via ultracentrifugation (45,000 rpm for 5 h at 4°C), and TG, cholesterol, and apoB were assessed as described below. VLDL isolation was performed only on the 3-h time point, as the other time points did not have enough volume for isolation.
Immunoblot analyses.
Immunoprecipitations and immunoblotting were performed as described previously (31). The following antibodies were used in this study: anti-human apoB, anti-apoE, and anti-albumin antibodies (Midland Bioproducts, Boone, IA); anti-KDEL monoclonal antibody (CalBiochem, San Diego, CA); anti-eukaryotic initiation factor-2α (eIF2α) and anti-phosphoserine-51 of eIF2α (Cell Signaling Technology, Danvers, MA); and anti-HSP60 polyclone antibody (Wuhan Boster). All antibodies were used at a final concentration of 0.1–1 μg/ml. After incubation with the appropriate horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG secondary antibody (1:2,000 dilution), proteins were visualized by enhanced chemiluminescence according to the manufacturer's instructions (Amersham Biosciences, Pittsburgh, PA).
Metabolic labeling, ultracentrifugation, and fractionation.
McA-RH7777 cells (1 × 106) were cultured with or without MK-866 (10 μM) for 48 h, deprived of methionine and cysteine for 1 h in the presence or absence of MK-866 (10 μM), and then pulsed with 100 μCi/ml [35S]methionine in the presence of oleate (400 μM) for 3 h. Media were collected and subjected to fractionation by ultracentrifugation. Briefly, media were collected and adjusted to 1.1 g/ml with NaBr, which was overlaid with a 1.06, 1.02, and 1.006 g/ml gradient and ultracentrifuged at 35,000 rpm for 18 h using a Beckman Optima LE-80K Ultracentrifuge. Fractions were collected, immunoprecipitated for apoB, and subjected to SDS-PAGE.
Lipid extraction and TG and cholesterol mass measurement from tissue.
Lipid extraction and analysis were performed as described previously (2). Briefly, ∼300 mg of liver tissue was added to 20 volumes of a 2:1 chloroform-methanol mixture and incubated for 24 h at room temperature. Following the incubation period, 0.2 volumes of 0.9% NaCl were added to the solvent mixture. The samples were thoroughly vortexed and then centrifuged at 2,000 rpm for 3 min. The upper aqueous phase was removed, and the solvent layer was allowed to evaporate. The dried lipid was resuspended in 1 ml of 100% ethanol, and TG and cholesterol concentrations were determined using commercially available kits from Randox (Mississauga, ON, Canada) as per the manufacturer's instructions. Lipid data are expressed in milligrams of lipid per gram of liver tissue.
RNA isolation and RT-PCR.
Total RNA was extracted from liver tissues and primary rat hepatocytes using the Qiagen (Mississauga, ON, Canada) RNeasy mini kit. cDNA was synthesized using Applied Biosystems TaqMan Reverse Transcription Reagents (Foster City, CA). RT-PCR was performed using appropriate primers to measure the mRNA levels of the indicated genes. PCR reactions were carried out using SYBR Green PCR Master Mix (Applied Biosystems, Streetsville, ON, Canada). Relative quantities of mRNA were calculated from threshold cycle (CT) values with the comparative CT method, using 18S rRNA as an internal reference.
Immunofluorescence microscopy.
McA-RH7777 cells cultured on collagen-coated coverslips in six-well microplates were fixed with 4% paraformaldehyde in 1× phosphate-buffered solution (PBS) for 15 min and then permeabilized with 0.1% Triton X-100 in 1× PBS for 5 min. Cells were incubated with anti-human apoB antibody [1:1,000 in 5% bovine serum albumin (BSA)], followed by incubation with the secondary antibody Alexa Fluor 594 donkey anti-goat IgG (Invitrogen; 1:1,000 in 5% BSA), both for 1 h at room temperature. Finally, nuclei were stained with 4,6-diamidino-2-phenylindole for 15 min at room temperature (1:1,000 in 5% BSA). Images were captured with a Quorum spinning disk confocal microscope (Quorum Technologies, Guelph, ON, Canada) and Volocity 6 software (PerkinElmer, Woodbridge, ON, Canada).
Mitochondrial JC-1 staining in primary mouse hepatocytes.
Isolated primary hepatocytes were allowed 3 h to attach on six-well plates (0.5 × 106 cells/well) and then treated for 24 h with either DMSO (0.5%), MK-886 (20 μM) or fructose (12 mM). Cells were washed twice with warm DMEM and then incubated with JC-1 staining reagent (Cayman Chemical, Ann Arbor, MI) for 15 min at 37°C (according to the manufacturer's instructions). Healthy cells (mainly JC-1 aggregates) appear red; unhealthy apoptotic cells (mainly JC-1 monomers) appear green.
Statistical analyses.
Data obtained by densitometry or fluorography were evaluated using one-way ANOVA (GraphPad Prism 5, La Jolla, CA). Posttest analysis was performed to determine the significance between groups, using unpaired two-way Student t-tests. All results are presented as means ± SE. Single and double asterisks in the figures indicate statistically significant differences of P < 0.05 or P < 0.01, respectively, compared with controls.
RESULTS
Impairment of PPARα signaling disrupts hepatic mitochondrial integrity and induces mitochondrial stress.
Mitochondrial β-oxidation of fatty acids starts with the transportation of activated long-chain fatty acids into the matrix of the mitochondria. In the liver, this process is performed mainly by the enzyme carnitine palmitoyltransferase 1α (CPT-1α), a direct target gene of PPARα. As expected, we found that in livers of PPARα−/− mice, mRNA expression of CPT-1α was significantly reduced compared with age-matched controls. Additionally, other PPARα target genes involved in fatty acid β-oxidation, including CPT-1, acetyl-CoA dehydrogenase very-long chain (ACADVL), and long-chain acyl-CoA dehydrogenase, were also decreased in livers of PPARα−/− mice (Fig. 1A). Decreased mRNA levels of metabolic enzymes involved in mitochondrial lipid metabolism were associated with reduction of fatty acid β-oxidation and altered lipid metabolism (1). We also observed that there was a significant accumulation of lipid droplets (as assessed by Oil Red O staining) and a marked elevation in lipid content, including TG and cholesterol, in the livers of PPARα−/− mice (Fig. 1B and data not shown). Further investigation of mitochondrial integrity in PPARα−/− mice showed that expression of a mitochondrial stress marker, heat shock protein 60 (HSP60), which plays a role in alleviating mitochondrial protein aggregation (11, 19, 35), was significantly increased in livers of PPARα−/− mice compared with their PPARα+/+ controls (Fig. 1C). This suggests activation of the mitochondrial stress response. The direct impact of PPARα on mitochondrial integrity was further investigated by treating primary mouse hepatocytes with a PPARα antagonist, MK-886, followed by a JC-1 staining assay (20). As shown in Fig. 1D, inhibition of PPARα signaling disturbed mitochondrial membrane integrity and altered the oxidation reduction potential of the mitochondria, which was indicated by the reduced ratio between red/green fluorescence intensity in the MK-886-treated cells (Fig. 1D, images c and d). Incubation of McA-RH7777 cells with MK-886 induced further accumulation of lipid droplets in the hepatocellular cytosol compared with the untreated cells (Fig. 1E, images c and d). These results suggest that impairment of PPARα signaling disrupts mitochondrial physiology.
Fig. 1.
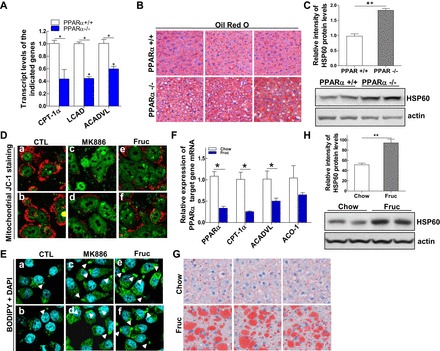
Impairment of peroxisome proliferator-activated receptor-α (PPARα) signaling disrupts hepatic mitochondrial integrity and induces mitochondrial stress. A: livers from wild-type (WT) (+/+) and PPARα-knockout (−/−) male mice at the age of 12 wk were used to prepare total RNA to detect mRNA expression of PPARα target genes carnitine palmitoyltransferase-1α (CPT-1α), long-chain acyl-CoA dehydrogenase (LCAD), and acetyl-CoA dehydrogenase very-long chain (ACADVL) by quantitative (q)RT-PCR. B: representative photographs demonstrating the appearance of livers from 12-wk-old PPARα+/+ (top) or PPARα−/− (bottom) male mice. The livers were harvested and frozen liver sections subjected to staining with the lipid-specific Oil Red O dye to positively reveal lipid droplets. C: protein mass of heat shock protein 60 (HSP60) detected by immunoblot analysis in the livers of PPARα+/+ and PPARα−/− mice. D: primary mouse hepatocytes treated for 24 h with either DMSO (0.5%), MK-886 (20 μM), or fructose (12 μM) were stained with JC-1 staining reagent for 15 min at 37°C and subjected to confocal analysis. Healthy cells (mainly JC-1 aggregates) appear red; unhealthy/apoptotic cells (mainly JC-1 monomers) appear green. E: McA cells were untreated or treated with PPARα antagonist MK-886 (10 μM) or fructose (6 mM) for 48 h and then stained with BODIPY for confocal imaging to visualize lipid droplets (green) and 4,6-diamidino-2-phenylindole (DAPI) for nuclear staining (blue). F: livers from rats after 3 wk of feeding with chow or fructose diets were used to prepare total RNA to measure relative mRNA expression of PPARα and its target genes CPT-1α, ACADVL, and acyl-CoA oxidase-1 (ACO-1) by qRT-PCR. G: livers from rats fed chow (Chow) or fructose (Fruc) for 3 wk were harvested, and frozen liver sections were subjected to staining with the lipid-specific Oil Red O dye to positively reveal lipid droplets. H: protein mass of HSP60 detected by immunoblot analysis in the livers of rats fed Chow or Fruc for 3 wk. For animal studies, results are shown as means ± SE (n = 5–6/group). For in vitro studies, data represent 3 experiments. *P < 0.05; **P < 0.01 vs. controls (CTL).
To corroborate this finding, we used a diet-induced animal model of insulin resistance, the high-fructose-fed rat model. We fed two groups of rats either a regular chow diet (60% of its calories from carbohydrates, predominantly from starch; Chow), or a high-fructose diet (60% of its calories from fructose; Fruc) for 3 wk. Fructose feeding resulted in about a 60% reduction of hepatic PPARα mRNA compared with control (chow-fed) rats. Reduced expression of PPARα in turn affected transcription of its target genes, with mRNA levels of CPT-1α and ACADVL decreased ∼65 and 50%, respectively, after fructose feeding (Fig. 1F). Interestingly, expression of another PPARα target gene, acyl-CoA oxidase 1, which catalyzes the initial step of peroxisomal β-oxidation, was also suppressed by the high-fructose diet, but this reduction was not statistically significant (Fig. 1F). This suggests that PPARα exerts greater regulatory effects on mitochondrial β-oxidation genes than on peroxisomal ones. The impairment of PPARα activity and reduced expression of mitochondrial β-oxidation enzymes upon high-fructose feeding was accompanied by the accumulation of lipid droplets and overexpression of HSP60 in rat livers (Fig. 1, G and H). Fructose treatment also decreased the membrane integrity and altered the oxidation reduction potential of the mitochondria (Fig. 1D, images e and f) and induced accumulation of lipid droplets in the cytoplasm of McA-RH7777 cells (Fig. 1E, images e and f). Together, these data suggest that impairment of PPARα activity compromises hepatic mitochondrial lipid metabolism and disrupts mitochondrial metabolic homeostasis.
Impairment of PPARα activity augments hepatic VLDL biogenesis.
Since dysregulation of lipid metabolism is closely related to hepatic VLDL biosynthesis and secretion, hepatic lipid accumulation of PPARα-defective rodents prompted us to investigate the secretion rate of VLDL in vivo. Hyperlipidemia has been reported previously in PPARα-defective animal models (15), and reduced lipoprotein lipase-mediated intravascular TG hydrolysis has been proposed to be one of the mechanisms for this disorder (25). To determine whether increased secretion of VLDL is also a contributing factor to the induction of hyperlipidemia, we performed an in vivo VLDL collection assay. Lipoprotein lipase activity was inhibited in both WT and PPARα-null mice by treating the mice with poloxamer (1,000 mg/kg) through ip injection. As shown in Fig. 2, A and B, despite the shutdown of lipoprotein hydrolysis in the circulating plasma, TG and cholesterol were significantly higher in the PPARα-null mice at all time points compared with WT age-matched controls. VLDL-TG and cholesterol were also increased at 3 h in PPAR−/− mice. In vitro, when determining the impact of the PPARα antagonist MK-886 on the assembly and secretion of lipoprotein particles in McA-RH7777 cells, we observed that, in lipid-rich conditions (presence of oleate), MK-886 significantly increased the secretion of VLDL particles, which was indicated by the increased secretion of newly synthesized apoB-100 in this fraction, whereas the secretion of other lipoprotein particles remained unchanged (Fig. 2C, fractions 1 and 2). Taken together, these results support a model whereby impairment of PPARα activity induces overproduction of VLDL and contributes to systemic hyperlipidemia.
Fig. 2.

Impairment of PPARα activity augments hepatic VLDL biogenesis. A and B: PPARα+/+ and PPARα−/− mice were fasted for 5 h, and blood collection was performed every hour after ip injection of poloxamer. Triglyceride (TG) and cholesterol (CHOL) levels were assessed at 0, 1, 2 and 3 h (A), and VLDL TG was assessed at 3 h (B). C: McA cells (1 × 106) were untreated or treated with MK-866 (10 μM) for 48 h, deprived of methionine and cysteine for 1 h in the presence or absence of MK-866 (10 μM), and then pulsed with 100 μCi/ml [35S]methionine in the presence of oleate (400 μM) for 3 h. Media were collected and subjected to fractionation by ultracentrifugation, fractions were collected, and 35S-labeled apolipoprotein B (apoB)-100 and 35S-labeled apolipoprotein E (apoE) were immunoprecipitated with anti-apoB and anti-apoE antibodies, respectively. For animal studies, results are shown as means ± SE (n = 5–6/group). For in vitro studies, data represent 3 experiments. *P < 0.05 vs. controls.
PPARα signaling transcriptionally regulates hepatic apoB synthesis.
The increased secretion of VLDL in the previous experiment prompted us to further investigate the regulation of apoB, a key structural protein in VLDL particles, in the PPARα-defective animal models. Protein expression of apoB-100 and apoB-48 was significantly increased in the livers of PPARα−/− mice, which was specific to apoB, since expression of another apolipoprotein, apoE, remained unchanged in the PPARα−/− mice (Fig. 3A). This phenotype was mirrored in the fructose-fed rat model; protein expression of both apoB-100 and apoB-48 was markedly elevated in the livers of rats following fructose feeding (Fig. 3B). To examine whether the increased expression of apoB-100 observed in the PPARα-defective mice was regulated by increasing de novo protein synthesis or by reducing protein degradation, we determined the mRNA levels of apoB in livers of both PPARα+/+ and PPARα−/− mice by quantitative (q)RT-PCR. As shown in Fig. 3C, compared with PPARα+/+ mice, expression of apoB mRNA was increased by ∼60% in PPARα−/− mice. The direct regulatory effect of PPARα on apoB is supported by our observation that treatment of primary rat hepatocytes with the PPARα inhibitor MK-886 also stimulated expression of apoB mRNA (Fig. 3D). Research on VLDL metabolism has suggested that apoB synthesis is prone to cotranslational and posttranslational regulation (9, 27). To investigate whether the elevated apoB mRNA contributed to the overproduction of apoB when PPARα was defective, we performed a pulse chase assay to quantify the amount of newly synthesized apoB-100 in the presence or absence of MK-886. McA-RH7777 cells were treated with MK-886 or vehicle for 48 h and then pulsed with [35S]methionine for 2 h. Labeled apoB-100 and albumin were immunoprecipitated from the cell lysates. Incubation of cells with MK-886 induced significant increases of newly synthesized apoB-100 in hepatocytes compared with the vehicle incubations at all time points (Fig. 3E, left). In contrast, no alteration in albumin protein synthesis was detected in the presence of MK-886 (Fig. 3E, right). To further demonstrate that impairment of PPARα exerts a greater impact on apoB mRNA expression than on apoB protein stabilization, we treated McA-RH7777 cells with MK-886 and immunostained the treated cells with anti-apoB antibody and BODIPY for lipid droplets. Confocal imaging showed that increased protein expression of apoB was induced by MK-886 treatment (Fig. 3F, image e). The majority of the accumulated lipid droplets induced by MK-886 treatment were colocalized with apoB in the cytoplasm (Fig. 3F, image f). It has been well accepted that proper lipidation of apoB in the ER under lipid-rich conditions prevents apoB from being retrograded to the cytoplasm and degraded by the proteasome (9), whereas apoB colocalized with lipid droplets in the cytosolic compartment is prone to proteasomal degradation (27). As such, the colocalization of apoB and lipid droplets in the MK-886-treated cells indicates that inhibition of PPARα activity is unlikely to stabilize apoB but would rather subject the protein to degradation. Hence, we conclude that accumulation of apoB-100 in the MK-886-treated cells is a consequence of increased apoB synthesis from elevated apoB mRNA transcripts.
Fig. 3.
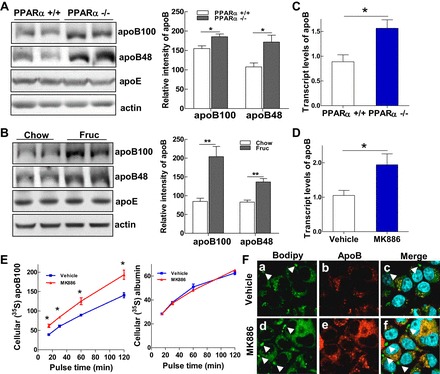
PPARα signaling transcriptionally regulates hepatic apoB synthesis. A and B: immunoblot analysis of apoB-100, apoB-48, apoE, and actin in liver homogenates of PPARα+/+ and PPARα−/− mice (A) and rats fed chow or fructose for 3 wk (B). C and D: relative mRNA expression of apoB detected by qRT-PCR in livers of PPARα+/+ and PPARα−/− mice (C) and McA cells treated with MK-886 (10 μM) for 48 h (D). E: McA cells (1 × 106) were untreated or treated with MK-866 (10 μM) for 48 h. Cells were then deprived of methionine and cysteine for 1 h in the presence or absence of MK-866 (10 μM) and pulsed with 100 μCi/ml [35S]methionine for 15, 30, 60 and 120 min. Cell lysates were prepared, and cellular 35S-labeled apoB-100 was immunoprecipitated with an anti-apoB antibody, followed by immunoprecipitation of 35S-labeled albumin, with an anti-albumin antibody as control. F: McA cells were untreated or treated with MK-886 (10 μM) for 48 h and then stained for confocal imaging with anti-apoB antibody (red), BODIPY to visualize lipid droplets (green), and DAPI for nuclear staining (blue). Colocalization of apoB with lipid droplets is shown in yellow. For animal studies, results are shown as means ± SE (n = 5–6/group). For in vitro studies, data represent 3 experiments. *P < 0.05 and **P < 0.01 vs. controls.
Accumulation of apoB in the hepatic ER induces ER stress.
SERCA is a critical enzyme involved in ER calcium metabolism. Inhibition of SERCA activity due to changes in lipid composition is a contributing factor to hepatic ER stress in obesity (8). Previously, our group had reported that accumulation of apoB-100 in hepatocytes is a causal factor in high-fat diet-induced hepatic ER stress (30). Here, we investigated whether accumulation of apoB alone was sufficient to inhibit SERCA mRNA expression. Using two McA-RH7777 cell lines that constitutively express either a COOH-terminal-truncated apoB construct (apoB-50WT) or an apoB mutant (apoB5–0N158–1496), which are more prone to protein misfolding and ER retention, we found that expression of SERCA mRNA was significantly reduced in the apoB-overexpressing cells compared with mock transfected cells (Fig. 4A). Analyzing the mRNA levels of SERCA in the livers of the experimental models, we found that either deletion of PPARα or high-fructose feeding suppressed hepatic transcription of SERCA mRNA in hepatocytes (Fig. 4, B and C). Analysis of ER stress biomarkers in the livers of PPAR−/− mice showed that deletion of PPARα significantly induced mRNA expression of C/EBP homologous protein (CHOP) and protein expression of the ER chaperones Grp94 and Grp78 compared with WT mice (Fig. 4, D and E). Phosphorylation of eIF2α was elevated without total eIF2α protein levels being changed (Fig. 4E). Increased CHOP mRNA level and phosphorylation of the eIF2α were also detected in livers of rats after 3 wk of high-fructose feeding (Fig. 4, F and G). Taken together, these data suggest that overproduction of apoB induced by defective PPARα signaling disturbs hepatic ER metabolic homeostasis.
Fig. 4.
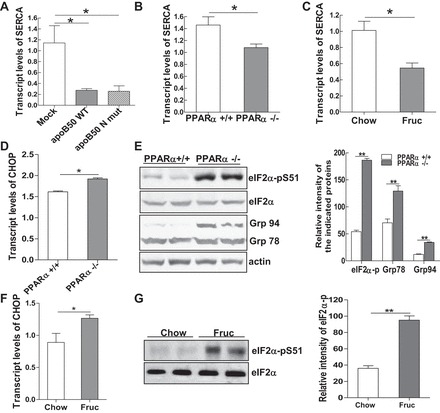
Accumulation of apoB in the hepatic endoplasmic reticulum (ER) induces ER stress. A: total mRNA was extracted from McA cells either containing a control vector or stably expressing a truncated form of apoB WT or mutant, and mRNA transcripts of the sarcoendoplasmic reticulum ATPase (SERCA) gene were measured. B and C: relative mRNA expression of SERCA detected by qRT-PCR in livers of PPARα+/+ and PPARα−/− mice (B) and rats fed Chow or Fruc diets for 3 wk (C). D: relative mRNA expression of C/EBP homologous protein (CHOP) detected by qRT-PCR in livers of PPARα+/+ and PPARα−/− mice. E: immunoblot analysis of the ER stress markers phospho-eukaryotic initiation factor-2α (eIF2α), Grp94, or Grp78 in livers of PPARα+/+ and PPARα−/− mice and quantification of their signal intensity. F: relative mRNA expression of CHOP detected by qRT-PCR in livers of rats fed Chow or Fruc diets for 3 wk. G: immunoblot analysis of phospho-eIF2α and its total protein in livers of rats fed Chow or Fruc diet for 3 wk. For animal studies, results are shown as means ± SE (n = 5–6/group). For in vitro studies, data represent 3 experiments. *P < 0.05; **P < 0.01 vs. controls.
Restoring PPARα signaling normalizes mitochondrial and ER homeostasis.
To investigate whether the restoration of PPARα activity using a PPARα agonist is able to rescue cells from both mitochondrial and ER stress, two groups of rats were fed either chow or fructose diet. Within each diet group, rats were treated with either vehicle (0.5% DMSO) or WY-14645 (6 mg/kg body wt ip) once every other day for 3 wk. At the end point, livers were isolated for analysis. Examining the mitochondrial stress marker HSP60, we noticed that expression of HSP60 protein was increased by ∼50% in the livers of fructose-fed rats compared with the chow diet group. Treating fructose-fed rats with WY-14643 reduced HSP60 expression by ∼40% (Fruc/WY vs. Fruc/Veh; Fig. 5A). Moreover, recovery of PPARα activity rescued hepatocytes from ER stress, as the expression of CHOP mRNA and phosphorylation of the c-Jun-NH2-terminal kinase (JNK) in the Fruc/WY group was reduced to a level comparable with the control group (Chow/Veh; Fig. 5, B and C). Phosphorylation of eIF2α was also significantly reduced in the WY-treated group (Fig. 5C). Ex vivo, incubation of primary rat hepatocytes with fructose (6 mM) in the presence or absence of WY-14643 for 48 h prevented the induction of GRP78 overexpression and phosphorylation of eIF2α compared with cells treated with fructose alone (Fig. 5D). These results demonstrate that recovery of PPARα activity protects hepatocytes from mitochondrial and ER stress.
Fig. 5.
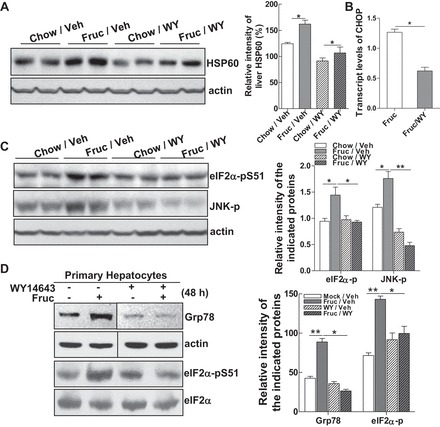
Restoring PPARα signaling normalizes mitochondrial and ER homeostasis. A and B: livers from rats fed Chow or Fruc and concomitantly treated with vehicle (Veh) or PPARα agonist WY-14643 (WY; 6 mg/kg ip) once every 2 days for 3 wk were used to prepare liver homogenants. A: protein mass of HSP60 detected by immunoblot analysis. B: relative mRNA expression of CHOP detected by qRT-PCR in livers of Fruc-fed rats untreated or treated with WY for 3 wk. C: immunoblot analysis of phospho-eIF2α and phospho-JNK levels. D: primary rat hepatocytes were untreated or treated with fructose (6 mM) in the presence or absence of WY (40 μM) for 48 h. Whole cell lysates were subjected to immunoblot analysis for Grp78 and phospho-eIF2α. Lanes were run on the same gels but were noncontiguous. For animal studies, results are shown as means ± SE (n = 5–6/group). For in vitro studies, data represent 3 experiments. *P < 0.05; **P < 0.01 vs. controls.
Restoration of mitochondrial and ER homeostasis improves hepatic steatosis and alleviates systemic hyperlipidemia.
Restoration of PPARα signaling using the PPARα agonist WY-14643 inhibited expression of apoB-100 and apoB-48 by ∼75 and 50%, respectively, in the Fruc/WY group compared with the Fruc/Veh group (Fig. 6A), whereas expression of apoE remained unchanged or even slightly increased in the WY-14643-treated groups (Fig. 6A), suggesting a specific inhibitory effect of WY-14643 on apoB biosynthesis. Measurement of the lipid content in the livers of fructose-fed rats showed that treatment with WY-14643 improved hepatic lipid metabolism, which was evident by the significant decreases in hepatic triglyceride and cholesterol. (Fig. 6B). Additionally, Oil Red O staining of liver tissue sections revealed that there were only trace levels of lipid droplets in the livers of animals treated with the PPARα agonist Fruc/WY vs. Fruc/Veh (Fig. 6C). In parallel with the changes in lipid profile of the liver, plasma TG in the Fruc/WY group rats was reduced to a level comparable with that of control rats (Chow/Veh) (Fig. 6D). Of note is that rats remained free of hepatotoxicity with short-term treatment with WY-14643, as reflected by the absence of aspartate transaminase and alanine transaminase elevation in all four groups (data not shown). These data suggest that restoration of PPARα activity protects against hepatic steatosis induced by fructose and improves systemic hyperlipidemia.
Fig. 6.
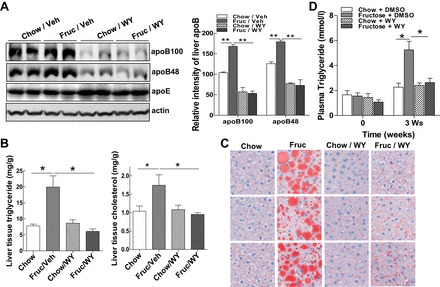
Restoration of mitochondrial and ER homeostasis improves hepatic steatosis and alleviates systemic hyperlipidemia. A: immunoblot analysis of apoB-100, apoB-48, apoE, and actin from liver homogenants of rats with conditions as shown in Fig. 5A. B: liver TG and cholesterol contents from rats in Fig. 5A were determined by procedures detailed in materials and methods. C: liver sections from rats as shown in Fig. 5A were stained with Oil Red O dye to identify neutral lipid species. D: plasma TG from rats receiving the diets and treatments as indicated for 3 wk. For animal studies, results are shown as means ± SE (n = 5–6/group). For in vitro studies, data represent 3 experiments. *P < 0.05; **P < 0.01 vs. controls.
DISCUSSION
Our goals in the present study were to characterize the role of PPARα in conveying hepatic lipid status to the mitochondria and ER as well as to determine the regulatory effect of PPARα on hepatic apoB biosynthesis during the onset of hepatic steatosis and hyperlipidemia. Over the past decade, ER stress induced by various pathogens has been well characterized as a central abnormality linking obesity, hepatic steatosis, and insulin resistance (22, 23). Here, we have demonstrated that impairment of PPARα signaling plays a role, at least partially, in the pathological metabolic stresses derived from certain lipogenic nutrients, particularly fructose.
The responses of PPARα to different dietary nutrients are distinct. Fatty acids, especially if polyunsaturated, are thought to be preferred PPARα ligands (7). Chronic consumption of a high-fat diet increases the rate of fatty acid catabolism in muscles by allowing overwhelming amounts of fatty acids to enter the mitochondria. A pharmaceutical inhibitor of CPT-1, which suppresses the hyperactive mitochondrial fatty acid β-oxidation, was able to reverse the diabetic symptoms of these animals. This study implicates mitochondrial lipid overload and increased fatty acid β-oxidation as the principal contributing factors to lipid-induced insulin resistance in skeletal muscle (14). In contrast, in the present study we found that fructose suppressed PPARα signaling, which is associated with the induction of mitochondrial and ER stress.
Fructose is a highly lipogenic sugar that has profound metabolic effects on various organs, including liver, adipose tissue, and the central nervous system. High fructose intake has been associated with many of the components of the metabolic syndrome, such as insulin resistance, elevated waist circumference, dyslipidemia, and hypertension. In contrast to glucose, consumption of fructose does not stimulate insulin secretion or require insulin for the initial steps of its hepatic metabolism. The rate of hepatic uptake of fructose from portal circulation is greater than the rate of glucose uptake, and because fructose metabolism bypasses phosphofructokinase, its metabolism is not under the regulatory control of insulin (6). The increased rate of fructose-induced de novo lipogenesis generates fatty acids that can then be incorporated into hepatic TG or other lipid species via increases in both the carbohydrate regulatory element-binding protein and sterol regulatory element-binding protein-1 activity (6). These, coupled with the lipid diverted from reduced mitochondrial fatty acid oxidation, lead to the accumulation of lipids in the livers of the PPARα−/− mice and fructose-fed rats. Increased hepatic lipid levels are associated with increased VLDL synthesis and secretion. apoB is essential for the intracellular assembly of TG into VLDL, and apoB degradation is reduced when hepatic lipid is increased, which leads to the accumulation of apoB in the hepatic ER and causes ER stress (30). Additionally, we demonstrated that impairment of PPARα signaling upregulates apoB expression at the transcriptional level. One of the potential mechanisms could be mediated by the eIF2α-activating transcription factor 4 (ATF4) pathway. Phosphorylation of eIF2α suppresses global protein synthesis by inhibiting delivery of the initiator Met-tRNAi to the initiation complex. However, phosphorylation of eIF2α also favors the translation of a selected number of mRNAs containing short upstream open reading frames (34). ATF4 is one of these proteins that plays a crucial role in adaptation to stresses by regulating the transcription of many genes (16, 33). A recent study has demonstrated that activation of ATF4 by PKR-like endoplasmic reticulum kinase upon ER stress is involved in the upregulation of hepatic VLDL receptor mRNA expression and the development of fatty liver disease (13). It is very likely that the induction of eIF2α phosphorylation in both PPARα-null mice and fructose-fed rats activates ATF4, which in turn enhances expression of apoB mRNA and protein. The potential regulatory effect of ATF4 and ATF6 on apoB biosynthesis and hepatic VLDL assembly is currently under investigation by our group.
Imbalance of ER lipid composition has been proposed to induce ER stress via inhibition of SERCA activity. Correcting the obesity-induced alteration of ER phospholipid composition or hepatic SERCA overexpression in vivo reduces chronic ER stress and improves glucose homeostasis (8). In this study, we showed that accumulation of apoB in the ER suppresses mRNA expression of SERCA, which could be one of the contributing factors to the induction of ER stress in the PPARα-defective animals. The biological activity of the ER is closely associated with the metabolic homeostasis of the mitochondria. Activation of JNK has been demonstrated to induce expression of CHOP and C/EBPβ, which in turn may stimulate transcription of the mitochondrial stress markers HSP60 and ClpP (12). Our study further demonstrated that both mitochondrial stress and ER stress are occurring in the PPARα-defective rodent models. Correction of PPARα signaling was able to prevent the induction of both mitochondrial and ER stress and reduce the biosynthesis of apoB to improve hyperlipidemia and hepatic steatosis associated with fructose consumption.
The identification of molecular links between PPARα and apoB and their involvement in cellular stress signaling unveils a novel role for PPARα in cellular stress response. It also reveals a novel mechanistic pathway whereby lipogenic nutrients such as fructose can induce hyperlipidemia and hepatic steatosis. Our findings suggest that pharmaceutical approaches aimed at modulating the status of PPARα may be of benefit in the treatment of the metabolic syndrome linked to excessive caloric intake.
GRANTS
This work was supported by an operating grant from the Heart and Stroke Foundation of Ontario to K. Adeli. Q. Su was supported by a Postdoctoral Research Fellowship from the Canadian Institutes of Health Research (CIHR). P. Christian is a recipient of the CIHR Strategic Training Program in Protein Folding and Interaction Dynamics Studentship from the University of Toronto.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Q.S. and K.A. conception and design of research; Q.S., C.B., P.C., M.N., X.T., K.Z., and M.S. performed experiments; Q.S. and K.A. analyzed data; Q.S. and K.A. interpreted results of experiments; Q.S. prepared figures; Q.S. drafted manuscript; Q.S., C.B., P.C., M.N., and K.A. edited and revised manuscript; Q.S., K.Z., and K.A. approved final version of manuscript.
REFERENCES
- 1.Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem 273: 5678–5684, 1998. [DOI] [PubMed] [Google Scholar]
- 2.Basciano H, Miller AE, Naples M, Baker C, Kohen R, Xu E, Su Q, Allister EM, Wheeler MB, Adeli K. Metabolic effects of dietary cholesterol in an animal model of insulin resistance and hepatic steatosis. Am J Physiol Endocrinol Metab 297: E462–E473, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 454: 470–477, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Bernal-Mizrachi C, Weng S, Feng C, Finck BN, Knutsen RH, Leone TC, Coleman T, Mecham RP, Kelly DP, Semenkovich CF. Dexamethasone induction of hypertension and diabetes is PPAR-alpha dependent in LDL receptor-null mice. Nat Med 9: 1069–1075, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab 22: 353–363, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dekker MJ, Su Q, Baker C, Rutledge AC, Adeli K. Fructose: a highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am J Physiol Endocrinol Metab 299: E685–E694, 2010. [DOI] [PubMed] [Google Scholar]
- 7.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci USA 94: 4312–4317, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473: 528–531, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ginsberg HN, Fisher EA. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J Lipid Res 50 Suppl: S162–S166, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol 19: 81–88, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci 123: 3849–3855, 2010. [DOI] [PubMed] [Google Scholar]
- 12.Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C elegans. Mol Cell 37: 529–540, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jo H, Choe SS, Shin KC, Jang H, Lee JH, Seong JK, Back SH, Kim JB. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 57: 1366–1377, 2013. [DOI] [PubMed] [Google Scholar]
- 14.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab 7: 45–56, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Linden D, Alsterholm M, Wennbo H, Oscarsson J. PPARalpha deficiency increases secretion and serum levels of apolipoprotein B-containing lipoproteins. J Lipid Res 42: 1831–1840, 2001. [PubMed] [Google Scholar]
- 16.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol 167: 27–33, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes 55, Suppl 2: S9–S15, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muoio DM, Newgard CB. Obesity-related derangements in metabolic regulation. Annu Rev Biochem 75: 367–401, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem 76: 723–749, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Nury T, Samadi M, Varin A, Lopez T, Zarrouk A, Boumhras M, Riedinger JM, Masson D, Vejux A, Lizard G. Biological activities of the LXRα and β agonist, 4β-hydroxycholesterol, and of its isomer, 4α-hydroxycholesterol, on oligodendrocytes: effects on cell growth and viability, oxidative and inflammatory status. Biochimie 95: 518–530, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Ota T, Gayet C, Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Invest 118: 316–332, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozawa K, Miyazaki M, Matsuhisa M, Takano K, Nakatani Y, Hatazaki M, Tamatani T, Yamagata K, Miyagawa J, Kitao Y, Hori O, Yamasaki Y, Ogawa S. The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes 54: 657–663, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Qiu W, Su Q, Rutledge AC, Zhang J, Adeli K. Glucosamine-induced endoplasmic reticulum stress attenuates apolipoprotein B100 synthesis via PERK signaling. J Lipid Res 50: 1814–1823, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qu S, Su D, Altomonte J, Kamagate A, He J, Perdomo G, Tse T, Jiang Y, Dong HH. PPARα mediates the hypolipidemic action of fibrates by antagonizing FoxO1. Am J Physiol Endocrinol Metab 292: E421–E434, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr 21: 193–230, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Roglans N, Vila L, Farre M, Alegret M, Sanchez RM, Vazquez-Carrera M, Laguna JC. Impairment of hepatic Stat-3 activation and reduction of PPARalpha activity in fructose-fed rats. Hepatology 45: 778–788, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Schrauwen P, Hesselink MK. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes 53: 1412–1417, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Sparks JD, Dong HH. FoxO1 and hepatic lipid metabolism. Curr Opin Lipidol 20: 217–226, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Su Q, Tsai J, Xu E, Qiu W, Bereczki E, Santha M, Adeli K. Apolipoprotein B100 acts as a molecular link between lipid-induced endoplasmic reticulum stress and hepatic insulin resistance. Hepatology 50: 77–84, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Su Q, Wang S, Baltzis D, Qu LK, Wong AH, Koromilas AE. Tyrosine phosphorylation acts as a molecular switch to full-scale activation of the eIF2alpha RNA-dependent protein kinase. Proc Natl Acad Sci USA 103: 63–68, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai J, Zhang R, Qiu W, Su Q, Naples M, Adeli K. Inflammatory NF-κB activation promotes hepatic apolipoprotein B100 secretion: evidence for a link between hepatic inflammation and lipoprotein production. Am J Physiol Gastrointest Liver Physiol 296: G1287–G1298, 2009. [DOI] [PubMed] [Google Scholar]
- 33.Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA 101: 11269–11274, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal 9: 2357–2371, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J 21: 4411–4419, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]