

Abstract
Leptin resistance is induced by the feedback inhibitors tyrosine phosphatase-1B (PTP1B) and decreased Src homology 2 domain-containing tyrosine phosphatase-2 (SHP-2) signaling. To investigate the participation of PTP1B and SHP-2 in LPS-induced leptin resistance, we injected repeated (6-LPS) intraperitoneal LPS doses (100 μg/kg ip) for comparison with a single (1-LPS) treatment and evaluated the expression of SHP-2, PTP1B, p-ERK1/2, and p-STAT3 in the hypothalamus of male Wistar rats. The single LPS treatment increased the expression of p-STAT3 and PTP1B but not SHP-2. The repeated LPS treatment reduced SHP-2, increased PTP1B, and did not change p-STAT3. We observed that the PTP1B expression induced by the endotoxin was highly colocalized with leptin receptor cells in the hypothalamus of LepRb-IRES-Cre-tdTomato reporter mice. The single, but not the repeated, LPS treatment decreased the food intake and body weight. Leptin had no stimulatory effect on the hypophagia, body weight loss, or pSTAT3 expression in 6-LPS rats, indicating leptin unresponsiveness. Notably, the PTP1B inhibitor (3.0 nmol/rat in 5 μl icv) restored the LPS-induced hypophagia in 6-LPS rats and restored the ability of leptin to reduce food intake and body weight as well as to phosphorylate STAT3 in the arcuate, paraventricular, and ventromedial nuclei of the hypothalamus. The present data suggest that an increased PTP1B expression in the hypothalamus underlies the development of leptin resistance during repeated exposure to LPS. Our findings contribute to understanding the mechanisms involved in leptin resistance during low-grade inflammation as seen in obesity.
Keywords: leptin resistance, endotoxemia, protein tyrosine phosphatase-1B, Src homology 2 domain-containing tyrosine phosphatase-2, extracellular signal-regulated kinases
inflammation has been demonstrated to be a common underlying cause of many obesity-associated conditions, such as diabetes and atherosclerosis (13, 30, 37, 48). Rodent models of the obesity induced by a fat-enriched diet show enhancement of inflammatory cytokine secretion (37) and increased hypothalamic inflammatory signaling (44). Changes in gut microbiota are crucial factors in the development of obesity. Cani et al. (9) reported that high-fat feeding in mice augments plasma lipopolysaccharide (LPS) from gram-negative bacteria at a concentration sufficient to increase body weight and inflammation. In rats, the inflammation triggered by LPS and the subsequent activation of Toll-like receptor 4 (TLR4) has a determinant role in the development of the obese phenotype in response to high-fat/high-calorie food intake (14). Interestingly, exogenous long-term LPS infusion (300 μg·kg−1·day−1 for 4 wk) in normal-diet-fed mice caused a metabolic response that was somewhat similar to the response to high-fat feeding (9). These data suggest that LPS is an important factor linking inflammation to diet-induced obesity.
The proinflammatory adipokine leptin is also an important player connecting altered energy homeostasis and inflammation. In the arcuate (ARC) nucleus of the hypothalamus, the binding of leptin to the LepRb receptor activates the Janus kinase (JAK)-2/signal transducer and activator of transcription (STAT)3 pathway, stimulating the transcription of genes for proopiomelanocortin (POMC) and the suppressor of cytokine signaling (SOCS)3 (17, 32). In addition, the binding of leptin to LepRb recruits Src homology 2 domain-containing tyrosine phosphatase-2 (SHP-2), the major activator of extracellular signal-regulated kinase (ERK) (27, 32).
It is well established that the JAK-2/STAT3 pathway is required for the leptin regulation of appetite and energy expenditure. We previously demonstrated that the hypophagia and body weight reduction under acute inflammation, such as those observed after a single LPS administration, are also associated with the activation of STAT3 signaling in the ARC and paraventricular (PVN) nuclei, and this signaling does not increase further after leptin stimulation (6). In contrast to single LPS responses, repeated low-dose LPS exposure (100 μg·kg−1·day−1 for 6 days) induced a desensitization of the hypophagia and body weight loss. Noticeably, the repeated LPS treatment failed to change the hypothalamic pSTAT3 and SOCS3 expression induced by exogenous leptin, suggesting that the reduced capability to respond to leptin contributes to the preserved food intake and body weight gain in the endotoxin-tolerant animals (6). It has been known that SOCS3 expression inhibits the tyrosine phosphorylation of the LepRb, thus providing an important feedback mechanism for leptin signaling (17, 38). However, because we observed that SOCS3 undergoes no alteration in response to repeated LPS treatment (6), the precise mechanisms underlying the leptin resistance during endotoxin tolerance must be addressed.
Leptin signaling pathways are largely regulated by protein tyrosine phosphatases (PTP) via dephosphorylation of the phosphotyrosine residues on LepRb or other downstream signaling molecules (46). Tyrosine phosphatase-1B (PTP1B) binds to and dephosphorylates JAK-2 (25, 33, 52). Several reports demonstrated a crucial role of this protein in the development of leptin resistance (11, 47, 52). PTP1B-deficient mice show increased leptin sensitivity, reduced weight and adiposity, and increased activity and energy expenditure (1, 2, 11). Another important PTP is SHP-2, which also participates in the development of leptin resistance (16, 55). SHP-2 activates the downstream mitogen-activated protein kinase (MAPK) pathway, leading to ERK stimulation (4, 55). However, in vitro evidence demonstrates that SHP-2 can inhibit JAK-2 activation, consequently downregulating the STAT3-mediated gene transcription (10, 27). Additionally, elevated hypothalamic T cell protein tyrosine phosphatase (TCPTP) also participates in the leptin resistance in obesity (28).
Our previous observations suggest that leptin resistance in LPS-tolerant rats is not associated with increased SOCS3 expression; furthermore, the above-mentioned data show the role of the PTPs in the regulation of leptin signaling. Therefore, in this work, we assessed the expression of PTP1B and SHP-2 in rats treated with single or repeated LPS doses and evaluated the participation of these factors in the leptin resistance induced by low-grade inflammation.
MATERIALS AND METHODS
Animals.
Male Wistar rats (200–250 g, Central Animal Facility of the University of São Paulo-Campus Ribeirão Preto) were individually housed in a light- (lights on: 06:00 AM-06:00 PM) and temperature- (23 ± 1°C) controlled room with food and water available ad libitum, unless otherwise stated. The rats were adapted to the laboratory environment for at least 3 days prior to the experimental procedures. During this period, the rats were handled daily. All experimental protocols were approved by the Ethics Committee for Animal Use of the School of Medicine of Ribeirão Preto.
To investigate whether LepRb-expressing cells in the hypothalamus express PTP1B in response to the endotoxin, we performed an experiment using adult (8-wk-old) male LepRb-reporter mice. Mice expressing Cre under the control of the endogenous Lepr promoter (LepRb-IRES-Cre mice: B6.129-Leprtm2(cre)Rck/J, Jackson Laboratories) were bred with Cre-inducible tdTomato-reporter mice (B6;129S6 Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J, Jackson Laboratories), which were previously described and validated (12, 43, 50). In this mouse model, the gene reporter tdTomato is expressed selectively in the LepRb cells, and it can be visualized through its endogenous red fluorescence. Mice were housed in the Unit of Laboratory Animal Medicine (ULAM), in a light- (12 h on, 12 h off) and temperature- (21–23°C) controlled environment. They were fed a standard chow diet (Harlan Teklad Global Diet) and had ad libitum access to water. This experiment was performed in collaboration with Dr. Carol F. Elias (Department of Molecular and Integrative Physiology, University of Michigan) in accordance with the guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals as well as with those established by the University of Michigan Animal Care and Use Committee.
Animal treatment protocol.
Animals were subjected to intraperitoneal (ip) injection of sterile saline (0.15 M NaCl, 1 ml/kg) or LPS from Escherichia coli (100 μg/kg in 1 ml/kg, Serotype 026:B6, Sigma) between 04:00 PM and 04:30 PM, 2 h before lights off. The PTP1B inhibitor diluted in 1% DMSO + saline (3 nmol/rat/5 μl, Calbiochem) or vehicle (1% DMSO + saline/rat/5 μl) was intracerebroventricularly (icv) injected 90 min after the LPS or saline injection. To evaluate the sensitivity to leptin in the presence of the PTP1B inhibitor, leptin (10 μg/rat/5 μl, Calbiochem) or saline (5 μl/rat) was icv injected 2 h after the LPS or saline injections and 30 min after the icv injection of the PTP1B inhibitor or vehicle.
Measurement of PTP1B, SHP-2, and phospho- (p-)ERK1/2 expression after single or repeated LPS injections.
Rats were assigned to three different treatment groups (n = 7/group): 1) saline once daily for 6 days (6-Saline), 2) saline once daily for 5 days and an injection of LPS on the 6th day (5-Saline + 1-LPS), and 3) LPS once daily for 6 days (6-LPS). On the 6th day, 2 h after the LPS or saline injection, the animals were euthanized by decapitation, and the brains were collected and immediately stored at −80°C for later Western blotting analyses. Hypothalamic fragments were dissected out (thickness 2.7 mm) from an area 1.0 mm lateral to the midline at the anterior border of the optic chiasm and the anterior border of the mammillary bodies.
PTP1B immunostaining in LepRb reporter mice under single or repeated LPS stimulation.
To qualitatively evaluate the coexpression of the leptin receptor with PTP1B in response to endotoxin, leptin receptor Cre-tdTomato mice were assigned to three groups as described above (n = 2/group). Two hours after the last injection, the mice were anesthetized using isoflurane and transcardially perfused with saline, followed by 4% formaldehyde in 0.1 M phosphate buffer. The brain was collected, postfixed in the same fixative for 1 h, placed in PBS containing 20% sucrose, and stored at 4°C for later PTP1B immunostaining. The tdTomato red fluorescent protein is expressed only in LepRb-expressing cells, and its visualization does not require any additional staining.
Food intake and body weight measurements after single or repeated LPS injections in animals treated with the PTP1B inhibitor.
Eight days before the experiment, anesthetized rats (3:2 mixture of ketamine and xylazine in a dose of 60 mg/kg ketamine and 7.5 mg/kg xylazine at a volume of 0.1 ml/100 g) were implanted with a cannula in the lateral ventricle and divided into the three above-mentioned groups. On the 6th day, they were subdivided into six different groups (n = 10–11/group) according to the central injection of vehicle or PTP1B inhibitor: 1) 6-Saline + Vehicle, 2) 6-Saline + PTP1B inhibitor, 3) 5-Saline + 1-LPS + Vehicle, 4) 5-Saline + 1-LPS + PTP1B inhibitor, 5) 6-LPS + Vehicle, and 6) 6-LPS + PTP1B inhibitor. On the 6th day, food was withdrawn at 04:00 PM, when the rats received the last ip injection of saline or LPS, and 90 min later they received an icv injection of vehicle or PTP1B inhibitor. At 06:00 PM (lights off), the animals were refed, and food consumption was measured 2 and 14 h afterward. Body weight was determined immediately before the ip injections for all experimental periods (6 days) and 14 h after the leptin or saline injection.
Food intake and body weight measurements after leptin treatment in animals treated with repeated LPS injections and pretreated with the PTP1B inhibitor.
Rats were implanted with a cannula in the lateral ventricle 8 days before the experiment and were assigned to four groups (n = 7–8/group) as follows: 1) 6-LPS + Vehicle + Vehicle, 2) 6-LPS + Vehicle + Leptin, 3) 6-LPS + PTP1B inhibitor + Vehicle, and 4) 6-LPS + PTP1B inhibitor + Leptin. On the 6th day, food was withdrawn at 04:00 PM, when the rats received the last ip injection of LPS, and 90 min afterward they received an icv injection of vehicle or PTP1B inhibitor. Thirty minutes afterward, at 6:00 PM (lights off), the rats received an icv injection of vehicle or leptin. The rats were refed, and the food consumption was measured 2 and 14 h afterward. The body weight was determined immediately before the ip injections during all experimental periods and 14 h after the last injection.
p-STAT3 and p-ERK immunostaining after leptin administration in animals stimulated with single or repeated LPS doses and pretreated with the PTP1B inhibitor.
Rats were assigned to 12 groups (n = 6/group) as follows: 1) 6-Saline + Vehicle + Vehicle, 2) 6-Saline + Vehicle + Leptin, 3) 6-Saline + PTP1B inhibitor + Vehicle, 4) 6-Saline + PTP1B inhibitor + Leptin, 5) 5-Saline + 1-LPS + Vehicle + Vehicle, 6) 5-Saline + 1-LPS + Vehicle + Leptin, 7) 5-Saline + 1-LPS + PTP1B inhibitor + Vehicle, 8) 5-Saline + 1-LPS + PTP1B inhibitor + Leptin, 9) 6-LPS + Vehicle + Vehicle, 10) 6-LPS + Vehicle + Leptin, 11) 6-LPS + PTP1B inhibitor + Vehicle, and 12) 6-LPS + PTP1B inhibitor + Leptin. On the 6th day, at 4:00 PM, the rats received the last ip injection of saline or LPS, and 90 min afterward they received an icv injection of vehicle or PTP1B inhibitor. Thirty minutes afterward, at 6:00 PM (lights off), the rats received an icv injection of vehicle or leptin. Twenty minutes later, the animals were anesthetized using an overdose of 2,2,2-tribromoethanol (TBE, 25 mg/100 g ip; Aldrich, Milwaukee, WI); the animals were then transcardially perfused with 200 ml of saline followed by 300 ml of 4% formaldehyde in 0.1 M phosphate buffer. The brain was collected, postfixed in the same fixative for 1 h, placed in PBS containing 30% sucrose, and stored at 4°C for later immunofluorescence or immunohistochemistry procedures.
Implantation of the cannula into the lateral ventricle.
Rats were anesthetized with a mixture of ketamine (60 mg/kg) and xylazine (7.5 mg/kg) at a volume of 0.1 ml/100 g and placed in a stereotaxic instrument (Kopf, model 900). A stainless steel guide cannula (10 mm) was implanted into the right lateral ventricle of the brain. We used the following stereotaxic coordinates: AP = −0.6 mm, LL = −1.5 mm and depth = −3.6 mm from the bregma. The cannula was held in place using two stainless steel screws and dental acrylic resin in the skull. To prevent occlusion of the guide cannula, a 30-gauge metal wire filled the cannula. After surgery, the rats received a prophylactic injection of penicillin (50,000 U im). The placement of the cannula was confirmed at the end of the experiment by histological analysis.
Immunohistochemistry.
Brain coronal sections were cut at 30 μm thickness and preserved in cryoprotectant solution (ethylene glycol and glycerol) at −20°C. One of every four sections was used for pSTAT3 Tyr705 immunostaining. Briefly, the sections were rinsed with Tris, followed by 10% H2O2 in methanol for 10 min. After rinsing, nonspecific binding was prevented by immersing the sections in blocking buffer (Tris, normal goat serum, and Triton X-100) for 1 h at room temperature. The sections were incubated for 48 h at 4°C with primary antibody: rabbit anti-pSTAT Tyr705 (1:1,000, Cell Signaling no. 9145). After being rinsed, the sections were incubated for 1 h with biotinylated goat anti-rabbit secondary antibody (1:1,000, Vector, BA1000) and then processed using the Vectastain Elite avidin-biotin immunoperoxidase method (Vector Laboratories). Solutions of diaminobenzidine, nickel sulfate, and H2O2 were used to generate blue-black immunolabeling. Finally, the sections were mounted on gelatinized slides, air-dried overnight, dehydrated, cleared in xylene, and coverslipped with Ethelan. Immunopositive cells were identified in hypothalamic regions according to the coordinates from the rat brain atlas of Paxinos and Watson (35): the PVN was −0.92 mm to −2.12 mm from the bregma, and the ARC and ventromedial hypothalamic nucleus (VMH) was −2.3 mm to −3.5 mm from the bregma. Photomicrographs were captured using a Leica microscope equipped with a DC 200 digital camera attached to a contrast enhancement device. The number of immunoreactive-positive cells of all the sections in the series (8–16 sections) per rat was obtained by counting the black (nuclear) staining from a constant area of the PVN, ARC, and VMH using ImageJ software (v. 1.38, NIH).
Immunofluorescence.
To evaluate the pERK immunostaining, sections were rinsed with Tris, and nonspecific binding was prevented by immersing the sections in blocking buffer (Tris, normal donkey serum, and Triton X-100) for 1 h at room temperature. After blocking, the sections were incubated for 48 h at 4°C with rabbit anti-p-p44/42 MAPK (ERK1/2; Thr202/Tyr204) primary antibody (1:500, Cell signaling, no. 4370). After rinsing, the sections were incubated with donkey anti-rabbit conjugated with FITC secondary antibody (1:250, Jackson ImmunoResearch).
PTP1B immunolabeling was performed in sections from the leptin receptor Cre-tdTomato mice that were rinsed with 0.1 M PBS. Nonspecific binding was prevented by immersing the sections in blocking buffer (PBS, normal donkey serum, and Triton X-100) for 1 h at room temperature. After blocking, the sections were incubated for 24 h at 4°C with rabbit anti-PTP1B primary antibody (1:1,000, Abcam, ab189179). After rinsing, the sections were incubated for 2 h with donkey anti-rabbit conjugated with Alexa fluor 488 secondary antibody (1:400, Jackson ImmunoResearch).
Finally, sections were coverslipped with Fluoromont-G mounting medium (Southern Biotechnology Associates) and analyzed using a Leica confocal laser scanning microscope. The immunoreactive structures were excited using argon or helium-neon green lasers with the excitation and barrier filters set for the fluorochrome used (green), and epifluorescence was collected using a DS red filter to visualize the tdTomato protein (red). Images showing the fluorescence were obtained from sequentially acquired images of slices excited by the laser. Fluorescence images of PTP1B and LepRb were superposed to identify the presence of dual-labeled neurons.
Immunoblot analysis.
Total mediobasal hypothalamic protein was extracted using 1% Triton-X 100, 100 mM Tris·HCl (pH 7.4), 100 mM sodium pyrophosphate, 100 mM sodium fluoride, 10 mM EDTA, 10 mM sodium orthovanadate, 2 mM PMSF, 0.2 mg/ml aprotinin, and 0.2 mg/ml leupeptin at 4°C and 15,000 g for 40 min. Aliquots of the lysates containing 50 mg of protein were denatured in Laemmli buffer [6% SDS, 30% glycerol, 0.02% bromophenol blue, 200 mM Tris·HCl (pH 6.8), and 250 mM mercaptoethanol] at 95°C for 5 min. Samples were blotted onto a nitrocellulose membrane. Nonspecific binding was prevented by immersing the membranes in blocking buffer [10% BSA in Tris-buffered saline-Tween 20 (TBS-T)] for 90 min at room temperature. The membranes were then exposed overnight to the primary antibodies: rabbit anti-PTP1B (1:4,000, Abcam, ab189179), rabbit anti β-actin (1:1,000, Cell Signaling, no. 8457), rabbit anti-GAPDH (1:4000, Cell signaling, no. 5174), rabbit anti SHP-2 (1:1,000, Cell Signaling, no. 3752), rabbit anti p44/42 (ERK1/2; 1:10,000, Cell Signaling, no. 9102), or rabbit anti-p-p44/42 (p-ERK1/2; 1:1,000, Cell Signaling, no. 9101). The blots were rinsed in TBS-T and then incubated with horseradish peroxidase-conjugated goat anti-rabbit antibody (1:5,000, Cell Signaling) for 1 h at room temperature. Antibody-antigen complexes were visualized by detecting enhanced chemiluminescence using an ECL detection system (Amersham Biosciences) and digital images with Quantity One 4.5.0 software (Bio-Rad).
Statistical analysis.
Results are expressed as means ± SE and were analyzed using Statisticasoftware. One-way analysis of variance (ANOVA), followed by the Newman-Keuls post hoc test, was used to analyze the experiments with single or repeated LPS treatment. For multiple comparisons, two-way ANOVA, followed by the Newman-Keuls post hoc test, was used to analyze the experiments of food intake and body weight gain with the single or repeated LPS treatment followed by PTP1B inhibitor and leptin treatment. We used three-way ANOVA, followed by the Newman-Keuls post hoc test, to analyze the pSTAT3 and pERK expression in the animals treated with single or repeated LPS doses, PTP1B inhibitor, and leptin injection. Differences were accepted as significant at P < 0.05.
RESULTS
LPS injection increased PTP1B, reduced SHP-2, and did not change p-ERK1/2 expression in the mediobasal hypothalamus.
Figure 1 shows the relative expression (% control) of SHP-2/β-actin, p-ERK1/ERK1, p-ERK2/ERK2, p-ERK1/2/β-actin, and PTP1B/GAPDH in the mediobasal hypothalamus. SHP-2 expression decreased (P < 0.05) in the 6-LPS rats compared with the rats treated with a single dose of LPS. We did not observe changes in p-ERK1/2 expression among the groups. PTP1B expression increased (P < 0.05) in the 1-LPS- and 6-LPS-treated rats compared with the Saline group.
Fig. 1.

Percentage of Src homology 2 domain-containing tyrosine phosphatase-2(SHP-2), phospoho-extracellular signal-regulated kinase-1 (p-ERK1), p-ERK2, and protein tyrosine phosphatase-1B (PTP1B) expression in the mediobasal hypothalamus 2 h after the last injection in animals injected once a day with 6 doses of saline (6 Saline), single dose of LPS (100 μg/kg ip; 5-Saline + 1 LPS), or repeated doses of LPS (6 LPS). One-way ANOVA followed by Newman Keuls post hoc test was performed. Data are expressed as means ± SE (n = 6–7). *P < 0.05.
PTP1B expression in the ARC in response to single or repeated LPS injections colocalizes with LepRb-expressing cells.
The representative photomicrographs in Fig. 2 show the increased PTP1B immunostaining in the ARC of leptin receptor Cre-tdTomato mice under single and repeated LPS stimulation compared with the control group. PTP1B expression highly colocalized with LepRb cells in this nucleus in mice treated with single or repeated LPS injections, demonstrating the upregulation of PTP1B protein in the leptin-responsive hypothalamic neurons during low-grade inflammation.
Fig. 2.

Representative photomicrographs showing PTP1B expression (green) in the hypothalamic arcuate (ARC) nucleus 2 h after the last injection in LepRb reporter mice (red) injected once a day with 6 doses of saline, single dose of LPS (100 μg/kg ip), or repeated doses of LPS. 3V, third ventricle. Scale bar, 50 μm. Insets (A, B, and C): examples of LepRb neurons coexpressing or not PTP1B, ×40 magnification.
PTP1B inhibitor restored the ability of LPS to reduce food intake and body weight in 6-LPS-tolerant rats.
In the vehicle-treated group, a single LPS dose reduced (P < 0.05) the food intake 2 h (Fig. 3A) and 14 h (Fig. 3B) after the injection and decreased (P < 0.05) the body weight gain (Fig. 3C). Repeated LPS treatment did not change these responses compared with controls, demonstrating tolerance to LPS. Central injection of 3 nmol of the PTP1B inhibitor had no effect on the saline and 1-LPS groups. Interestingly, the 6-LPS rats showed LPS-induced hypophagia in the presence of the PTP1B inhibitor 2 h after injection but not 14 h after; these rats also showed a reduced body weight gain 14 h after injection compared with that in the rats treated with vehicle.
Fig. 3.

Cumulative food intake 2 h (A) and 14 h (B) after the last injection and body weight gain 14 h after the last injection (C) in animals injected once a day with 6 doses of saline (6 Saline), single dose of LPS (100 μg/kg ip; 5 Saline + 1 LPS), or repeated doses of LPS (6 LPS), followed by icv injection of vehicle or PTP1B inhibitor (3 nmol/rat, 5 μl). Two-way ANOVA followed by Newman-Keuls post hoc test was performed. Data are expressed as means ± SE (n = 10–11). *P < 0.05.
Leptin had no additional effect on food intake reduction but potentiated the body weight loss induced by LPS in 6-LPS-tolerant rats pretreated with the PTP1B inhibitor.
Figure 4 shows that leptin failed to reduce food intake at 2 h (4A) and 14 h (4B) and had no effect on body weight gain 14 h (4C) after treatment in the 6-LPS rats. LPS stimulation induced hypophagia and a reduction in body weight gain in the presence of the PTP1B inhibitor 2 h after injection (P < 0.05) but not after 14 h, demonstrating the acute effect of the inhibitor. Notably, in the presence of the PTP1B inhibitor, leptin had no additional effect on the food intake reduction induced by LPS 2 or 14 h after, but did have a potentiating effect on body weight loss (P < 0.05) (Fig. 4C).
Fig. 4.

Cumulative food intake 2 h (A) and 14 h (B) after the last injection and body weight gain 14 h after the last injection (C) in animals injected once a day with 6 doses of LPS (100 μg/kg ip), followed by icv injections of vehicle or PTP1B inhibitor (3 nmol/rat, 5 μl) 90 min after LPS injections, and a subsequent icv injection of vehicle or leptin (10 μg/rat) 30 min after. Two-way ANOVA followed by Newman-Keuls post hoc test was performed. Data are expressed as means ± SE (n = 7–8). *P < 0.05.
PTP1B inhibition restored the ability of leptin to phosphorylate STAT3 in the hypothalamus of 6-LPS-tolerant rats.
The single LPS stimulation increased (P < 0.05) the number of pSTAT3-positive cells in the ARC (Figs. 5 and 6), PVN (Figs. 5 and 7), and VMH (Figs. 5 and 8) compared with that in the controls. Repeated LPS stimulation did not change the STAT3 phosphorylation in these hypothalamic regions compared with that for the saline injection. Leptin increased (P < 0.05) the number of p-STAT3-positive cells in the ARC, PVN, and VMH in the animals treated with saline. In the 1-LPS-treated group, leptin had no additional effect on p-STAT3 in these regions. Noticeably, the 6-LPS rats showed no change in p-STAT3 after leptin injection compared with that for the vehicle treatment, evidencing leptin unresponsiveness. Central pretreatment with 3 nmol of the PTP1B inhibitor had no effect on p-STAT3 expression in the evaluated hypothalamic regions in any experimental group treated with vehicle. Of note, the leptin treatment in the rats pretreated with the PTP1B inhibitor increased (P < 0.05) the STAT3 phosphorylation in the ARC, PVN, and VMH of the 6-LPS rats. No additional effects of leptin on the STAT3 phosphorylation in the ARC and PVN nuclei in the saline or 1-LPS groups were observed, likely due to a ceiling effect whereby the maximal p-STAT3 expression resulted from the leptin stimulation per se in these groups. Interestingly, PTP1B inhibition potentiated the p-STAT3 expression induced by leptin in the VMH of both the saline and 1-LPS groups, similar to the result in the 6-LPS group, suggesting a different regulation of the VMH in the leptin resistance during low-grade inflammation.
Fig. 5.
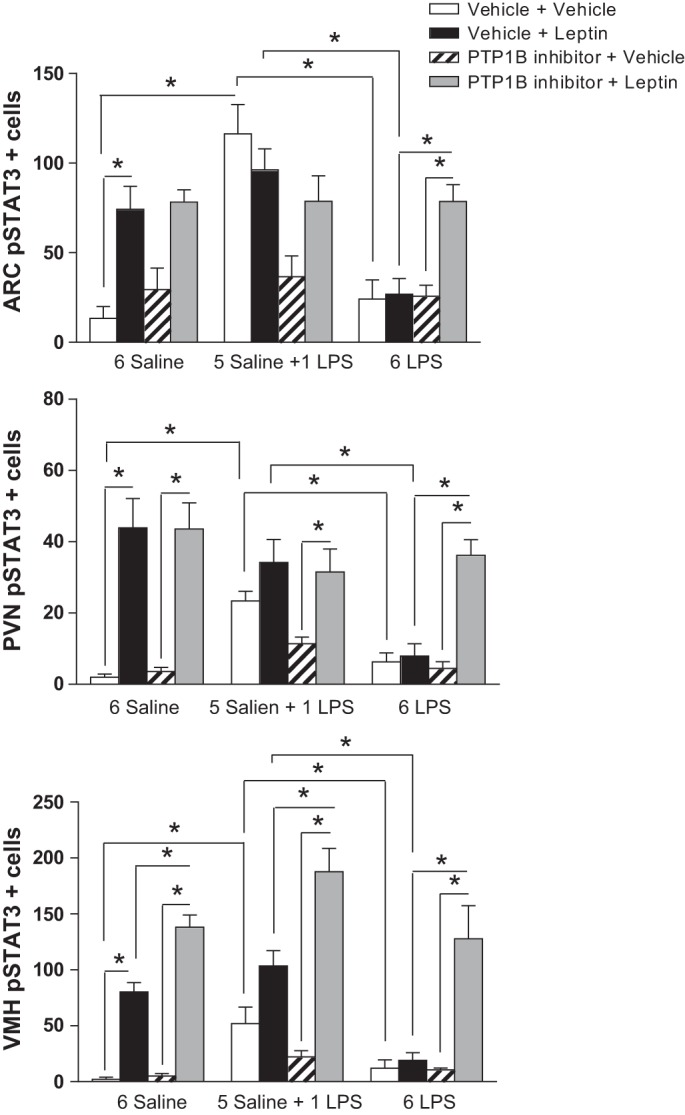
Graphs showing the number of p-STAT3-positive cells in the arcuate (ARC), paraventricular (PVN), and ventromedial (VMH) nucleus of the hypothalamus of animals injected once a day with 6 doses of saline (6 Saline), single dose of LPS (100 μg/kg ip; 5 Saline + 1 LPS), or repeated doses of LPS (6 LPS), followed by icv injections of vehicle or PTP1B inhibitor (3 nmol/rat, 5 μl) 90 min after LPS injections, and a subsequent icv injection of vehicle or leptin (10 μg/rat) 30 min after. Three-way ANOVA followed by Newman Keuls post hoc test was performed. Data are expressed as means ± SE (n = 6). *P < 0.05.
Fig. 6.

Representative photomicrographs of p-STAT3-positive cells (dark points) in the ARC (boundaries delineated with dotted line) of animals injected once a day with 6 doses of saline (6 Saline), single dose of LPS (100 μg/kg ip; 5 Saline + 1 LPS), or repeated doses of LPS (6 LPS), followed by icv injections of vehicle or PTP1B inhibitor (3 nmol/rat, 5 μl) 90 min after LPS injections, and a subsequent icv injection of vehicle or leptin (10 μg/rat) 30 min after. Scale bar, 100 μm.
Fig. 7.

Representative photomicrographs of p-STAT3-positive cells (dark points) in the PVN (boundaries delineated with dotted line) of animals injected once a day with 6 doses of saline (6 Saline), single dose of LPS (100 μg/kg ip; 5 Saline + 1 LPS), or repeated doses of LPS (6 LPS), followed by icv injections of vehicle or PTP1B inhibitor (3 nmol/rat, 5 μl) 90 min after LPS injections, and a subsequent icv injection of vehicle or leptin (10 μg/rat) 30 min after. Scale bar, 100 μm.
Fig. 8.

Representative photomicrographs of p-STAT3-positive cells (dark points) in the VMH (boundaries delineated with dotted line) of animals injected once a day with 6 doses of saline (6 Saline), single dose of LPS (100 μg/kg ip; 5 Saline + 1 LPS), or repeated doses of LPS (6 LPS), followed by icv injections of vehicle or PTP1B inhibitor (3 nmol/rat, 5 μl) 90 min after LPS injections, and a subsequent icv injection of vehicle or leptin (10 μg/rat) 30 min after. Scale bar, 100 μm.
PTP1B inhibitor had no effect on ERK phosphorylation induced by leptin in animals treated with single or repeated LPS injections.
Figure 9 shows that the number of p-ERK-expressing neurons in the PVN was not altered in any experimental group. In Fig. 10, we show representative photomicrographs of p-ERK immunolabeling in the parvocellular portion of the PVN.
Fig. 9.
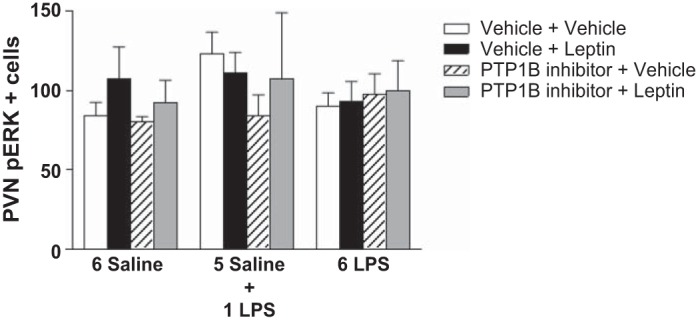
Graphs showing the number of p-ERK-positive cells in the parvocellular portion of the PVN of animals injected once a day with 6 doses of saline (6 Saline), single dose of LPS (100 μg/kg ip; 5 Saline + 1 LPS), or repeated doses of LPS (6 LPS), followed by icv injections of vehicle or PTP1B inhibitor (3 nmol/rat, 5 μl) 90 min after LPS injections, and a subsequent icv injection of vehicle or leptin (10 μg/rat) 30 min after. Three-way ANOVA followed by Newman Keuls post hoc test was performed. Data are expressed as means ± SE (n = 6). *P < 0.05.
Fig. 10.

Representative photomicrographs showing p-ERK expression (green) in the PVN of animals injected once a day with 6 doses of saline (6 Saline), single dose of LPS (100 μg/kg ip; 5 Saline + 1 LPS), or repeated doses of LPS (6 LPS), followed by icv injections of vehicle or PTP1B inhibitor (3 nmol/rat, 5 μl) 90 min after LPS injections, and a subsequent icv injection of leptin (10 μg/rat) 30 min after. Scale bar, 100 μm.
DISCUSSION
To investigate the mechanisms underlying the inability of leptin to reduce food intake and body weight as well as the inability to phosphorylate hypothalamic STAT3 protein during the development of endotoxin tolerance, we assessed the expression of the feedback inhibitor of leptin signaling PTP1B and the expression of the protein SHP-2 in the hypothalamus, which oversees the control of metabolism and energy balance. Our data reveal the essential role of PTP1B counterregulatory action in leptin resistance after repeated exposure to LPS. Additionally, we found a reduction in SHP-2 expression in the hypothalamus of rats under low-grade inflammation, indicating a likely role of this downregulation as an adjuvant in the leptin resistance in our experimental model. Nevertheless, the participation of SHP-2 in leptin resistance after endotoxemia requires further investigation.
SHP-2 is implicated in the signal transduction of a variety of cytokines, growth factors, insulin, and leptin, dephosphorylating its associated signaling molecules, thus diminishing the local signaling flow (1, 19, 39, 40). Reports have demonstrated that mice with neuronal disruption of SHP-2 are obese and resistant to leptin and insulin (26, 55). Banno et al. (1) found that POMC-Shp2−/− mice showed increased body weight after HFD or normal chow, blunted leptin response, and reduced energy expenditure compared with their littermate controls, suggesting that impairment in leptin signaling mediated by SHP-2 in POMC-Shp2−/− mice triggers an obese phenotype. Given that repeated LPS exposure leads to leptin unresponsiveness in our study, it would be interesting to evaluate whether the reduced SHP-2 expression after prolonged LPS exposure contributes to this process. Björbaek et al. (4) observed a blockade of leptin-stimulated ERK phosphorylation by LepRb in transfected cells with an inactive mutant of SHP-2. In our study, single or repeated LPS induced no changes in p-ERK expression in either the mediobasal hypothalamus extracts or in the parvocellular portion of the PVN. p-ERK can be induced in the parvocellular corticotropin-releasing factor (CRF)-expressing neurons in the PVN in response to insulin (22), whose levels may increase after infectious stimuli such as LPS (24, 42, 54). We have previously reported that a single LPS injection increased CRF mRNA and Fos-CRF expression in the parvocellular PVN (5, 41). In the present study, the LPS-induced p-ERK colocalized with CRF neurons (data not shown); however, because p-ERK expression is highly variable in the brain under several stimuli and even in basal conditions, we could not observe any significant change in p-ERK expression.
We observed that single or repeated LPS treatment increased the PTP1B expression with a high colocalization with LepRb-expressing neurons in the hypothalamus. Changes in PTP1B expression have been shown to be induced by molecules such as tumor necrosis factor (TNF)α and nuclear factor-κB (NF-κB) (20, 29, 34, 36, 53), and LPS is known to induce the production of these molecules. Picardi et al. (36) have demonstrated that central infusion of TNFα increased PTP1B protein expression and attenuated leptin sensitivity in the hypothalamus. These data support our findings suggesting that the endotoxin-induced PTP1B plays a role in leptin unresponsiveness during sustained low-grade inflammation. Overexpression of PTP1B has been reported in adipose tissue, skeletal muscle, liver, and ARC of high-fat-fed mice; this finding is coincident with increased TNFα expression (53). A high-fat diet is known to increase the circulating levels of LPS (9), whose activity is likely to account, at least partially, for the PTP1B upregulation during obesity. The low dose of LPS used in the present study is able to trigger hypothalamic inflammation without causing endotoxemic shock (5, 6, 7), thus mimicking the inflammatory environment observed in obese subjects and providing an interesting tool to investigate the low-grade inflammation and consequent leptin and insulin resistance observed during obesity.
PTP1B is known to inhibit leptin signaling by dephosphorylation of JAK-2 (25, 47). Overexpression of PTP1B in mouse hypothalamic cell lines reduced the JAK-2/STAT3 phosphorylation induced by leptin (21). PTP1B−/− mice are lean, sensitive to leptin and insulin, and resistant to the obesity induced by a fat-enriched diet (11). Considering the increased PTP1B expression in the hypothalamus of rats treated with acute or prolonged LPS, in addition to the impaired ability of leptin to reduce food intake and body weight, as well as its inability to induce p-STAT3 in 6-LPS rats, we postulated that PTP1B plays a role in leptin resistance during long-term exposure to endotoxin.
To test this hypothesis, we treated rats with the PTP1B inhibitor. As described earlier, 1-LPS rats showed hypophagia and reduced body weight gain, whereas none of these responses were observed in 6-LPS rats compared with the controls. In a previous report by our group, rats treated with the same low LPS dose exhibited increased core body temperature and oxygen consumption, contributing to body weight loss (8). Soszynski et al. in a recent report (45) demonstrated that rats treated with repeated LPS injections showed fever response after the first injection, with a complete suppression of fever after subsequent LPS injections from the second injection on. We found that the PTP1B inhibitor had no effect on food intake and body weight in Saline or 1-LPS rats, but the PTP1B inhibition acutely restored the LPS-induced hypophagia and body weight loss in 6-LPS animals. These data suggest that PTP1B lessens LPS hypophagic responses, preventing excessive anorexia and weight loss during a prolonged inflammatory challenge. PTP1B-deficient mice show increased energy expenditure due to increased leptin sensitivity (1, 2, 11). We believe that the body weight reduction in the 6-LPS rats could be due to an immediate feeding reduction in the first 2 h after PTP1B inhibition in parallel to increased leptin sensitivity to the endogenous leptin. We cannot preclude that PTP1B inhibition could restore the ability of the endotoxin to induce fever in 6-LPS-tolerant rats, contributing to the reduction of body weight gain. The role of PTP1B in LPS-induced fever is an interesting point that should be investigated in future studies.
Morrison et al. (31) demonstrated that PTP1B inhibition in aging-induced leptin-resistant rats (20 wk) promotes a reduction in food intake and increased leptin sensitivity. As previously demonstrated by our group (6), leptin had no effect on food intake and body weight in 6-LPS rats. Herein, in the presence of the PTP1B inhibitor, LPS reduced food intake and body weight gain in 6-LPS rats, but leptin had no additional effect on feeding responses. Particularly, after PTP1B inhibition leptin potentiated the body weight loss in response to LPS in the tolerant rats. This finding suggests that PTP1B may affect the leptin action on body weight irrespective of its effects on food intake. Future studies are crucial to clarify the mechanisms involved in this process.
As expected, we found that a single LPS treatment increased the number of p-STAT3-expressing neurons in the ARC, PVN, and VMH nuclei of the hypothalamus in contrast to the repeated LPS administration, which did not induce the p-STAT3 expression in these regions. Central leptin injection increased the p-STAT3 expression in the ARC, PVN, and VMH of control rats with no additional effect on p-STAT3 expression in the 1-LPS group, likely due to a ceiling effect of the acute LPS. As previously shown, the central leptin injection did not increase STAT3 phosphorylation in these hypothalamic sites in the 6-LPS rats, demonstrating the leptin resistance. Leptin resistance in the brain is known to be site specific, and the raphe pallidus and NTS in the brainstem seem to have maintained leptin signaling during diet-induced obesity (8). Therefore, the responsiveness to leptin in other brain areas after prolonged LPS exposure needs to be further studied. We observed for the first time that pretreatment with a central PTP1B inhibitor allowed leptin to phosphorylate STAT3 in the ARC, PVN, and VMH of the 6-LPS rats, restoring leptin sensitivity in these animals. In addition, pretreatment with the PTP1B inhibitor potentiated the STAT3 phosphorylation induced by leptin, particularly in the VMH, in all experimental groups. Considering the neurochemical identity of the p-STAT3-expressing neurons in the VMH, many studies have shown that the nuclear receptor steroidogenic factor 1 (SF-1), whose expression is specific in the VMH, is essential for the VMH actions on energy homeostasis (23). The role of leptin in SF-1 neurons was addressed by Dhillon et al. (15) and Bingham et al. (3) using different SF-1 Cre lines. Both groups generated mice lacking LepR in SF-1 neurons and observed that the mice were obese, indicating that LepR is required in SF-1 neurons for normal body weight homeostasis and that the SF-1 neurons in the VMH are direct targets of leptin in the control of energy balance. In view of these findings, we suggest that the p-STAT3-expressing cells in the VMH could be, at least in part, SF-1-expressing neurons.
We observed a high colocalization of PTP1B with LepRb neurons in response to LPS in the hypothalamus; however, we cannot rule out the possibility that PTP1B from other cellular populations counteracts the LPS responses. PTP1B has been demonstrated to markedly decrease the production of TNFα induced by myeloid differentiation factor 88 (MyD88), evidencing its negative regulation of Toll-like receptor signaling via suppression of the MyD88-dependent production of proinflammatory cytokines in macrophages (50). On the other hand, Grant et al. (18), using mice with selective PTP1B deletion in myeloid cells, recently demonstrated that these mice are protected against LPS-induced endotoxemia, hyperinsulinemia, macrophage infiltration into adipose tissue, and hepatic damage. These responses were associated with reduced TNFα and increased mRNA levels of the anti-inflammatory cytokine IL-10 in the macrophages, with a consequent increase in p-STAT3. Those authors revealed that the anti-inflammatory phenotype of the PTP1B-deficient mice in myeloid-cells was replicated in long-term (29 wk) HFD-fed mice, a condition that is known to chronically increase the plasma LPS concentration in a phenomenon termed metabolic endotoxemia (9). In agreement, Pike et al. (37) found an enhanced IL-10-dependent STAT3 phosphorylation in macrophages from PTP1B-null mice compared with that in wild-type mice, indicating that PTP1B inhibits STAT3 phosphorylation. We (6) have previously observed that both single and repeated LPS doses increased the IL-10 serum levels and IL-10 mRNA expression in the ARC. However, only single-LPS-treated rats showed higher p-STAT3 in this region. Both pro- and anti-inflammatory cytokines activate the JAK/STAT pathway. In our model, we believe that in response to acute LPS p-STAT3 is increased by proinflammatory cytokines. However, despite the increase in the IL-10 mRNA in the ARC, STAT3 phosphorylation is inhibited by the enhanced PTP1B expression under prolonged LPS exposure. Our data suggest that PTP1B restrains the effects of acute leptin and LPS on STAT3 phosphorylation in the hypothalamus, contributing to the prevention of excessive hypophagia. Furthermore, the PTP1B counterregulatory action is important for the development of desensitization of the LPS responses and leptin resistance during prolonged endotoxin exposure.
In conclusion, PTP1B is essential for the development of leptin resistance after low-grade endotoxemia, but the participation of the SHP-2 reduction in the mediobasal hypothalamus in this resistance requires further investigation. Our results contribute to the understanding of leptin resistance during a prolonged low-grade inflammatory challenge such as that observed in obesity. Given that the PTP1B inhibitor can act simultaneously at different central sites, future studies are required to better identify the specific sites where PTP1B acts to counteract the leptin response during long-term endotoxemia.
GRANTS
This work has been supported by FAPESP and CNPq, Brazil, and NICHD Grants HD-61539 and HD-69702, USA.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: B.d.C.B., R.C.R. and L.L.E. conception and design of research; B.d.C.B., R.C.R., E.T.U., and P.B.M. performed experiments; B.d.C.B., R.C.R., C.F.E., and L.L.E. analyzed data; B.d.C.B., R.C.R., C.F.E., J.A.-R., and L.L.E. interpreted results of experiments; B.d.C.B. and R.C.R. prepared figures; B.d.C.B., R.C.R., and L.L.E. drafted manuscript; B.d.C.B., R.C.R., E.T.U., P.B.M., C.F.E., J.A.-R., and L.L.E. edited and revised manuscript; B.d.C.B., R.C.R., E.T.U., P.B.M., C.F.E., J.A.-R., and L.L.E. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Maria Valci Aparecida dos Santos Silva, Milene Mantovani, Elizabete Rosa Milani and Lilian Eslaine Costa Mendes Silva for their technical support. We also thank Fundacao de Amparo a Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq), and National Institute of Child Health and Human Development (NICD, National Institutes of Health) for financial support.
REFERENCES
- 1.Banno R, Zimmer D, De Jonghe BC, Atienza M, Rak K, Yang W, Bence KK. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest 120: 720–734, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med 12: 917–924, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Bingham NC, Anderson KK, Reuter AL, Stallings NR, Parker KL. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology 149: 2138–2148, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bjorbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, Neel BG, Myers MG Jr, Flier JS. Divergent roles of SHP-2 in ERK activation by leptin receptors. J Biol Chem 276: 4747–4755, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Borges BC, Antunes-Rodrigues J, Castro M, Bittencourt JC, Elias CF, Elias LL. Expression of hypothalamic neuropeptides and the desensitization of pituitary-adrenal axis and hypophagia in the endotoxin tolerance. Horm Behav 52: 508–519, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Borges BC, Rorato R, Avraham Y, da Silva LE, Castro M, Vorobiav L, Berry E, Antunes-Rodrigues J, Elias LL. Leptin resistance and desensitization of hypophagia during prolonged inflammatory challenge. Am J Physiol Endocrinol Metab 300: E858–E869, 2011. [DOI] [PubMed] [Google Scholar]
- 7.Borges BC, Rorato R, Antunes-Rodrigues J, Elias LL. Glial cell activity is maintained during prolonged inflammatory challenge in rats. Braz J Med Biol Res 45: 784–791, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borges BC, Rorato R, Uchoa ET, Marangon P, da Silva GS, de Paula FJ, Branco LG, Antunes-Rodrigues J, Elias LL. High-fat diet induces site-specific unresponsiveness to LPS-stimulated STAT3 activation in the hypothalamus. Am J Physiol Regul Integr Comp Physiol 306: R34–R44, 2014. [DOI] [PubMed] [Google Scholar]
- 9.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761–1772, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Carpenter LR, Farruggella TJ, Symes A, Karow ML, Yancopoulos GD, Stahl N. Enhancing leptin response by preventing SH2-containing phosphatase 2 interaction with Ob receptor. Proc Natl Acad Sci USA 95: 6061–6066, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, Kennedy BP, Tremblay ML. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell 2: 497–503, 2002. [DOI] [PubMed] [Google Scholar]
- 12.DeFalco J, Tomishima M, Liu H, Zhao C, Cai X, Marth JD, Enquist L, Friedman JM. Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science 291(5513): 2608–2613, 2001. [DOI] [PubMed] [Google Scholar]
- 13.De Heredia FP, Gómez-Martínez S, Marcos A. Obesity, inflammation and the immune system. Proc Nutr Soc 71: 332–338, 2012. [DOI] [PubMed] [Google Scholar]
- 14.De La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol 299: G440–G448, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, Coppari R, Balthasar N, Cowley MA, Chua S Jr, Elmquist JK, Lowell BB. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49: 191–203, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Feng GS. Shp2 as a therapeutic target for leptin resistance and obesity. Expert Opin Ther Targets 10: 135–142, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Frühbeck G. Intracellular signalling pathways activated by leptin. Biochem J 393: 7–20, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grant L, Shearer KD, Czopek A, Lees EK, Owen C, Agouni A, Workman J, Martin-Granados C, Forrester JV, Wilson HM, Mody N, Delibegovic M. Myeloid-cell protein tyrosine phosphatase-1B deficiency in mice protects against high-fat diet and lipopolysaccharide-induced inflammation, hyperinsulinemia, and endotoxemia through an IL-10 STAT3-dependent mechanism. Diabetes 63: 456–470, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Hunter Signaling-2000 T, beyond. Cell 100: 113–127, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Ito Y, Banno R, Hagimoto S, Ozawa Y, Arima H, Oiso Y. TNFα increases hypothalamic PTP1B activity via the NFκB pathway in rat hypothalamic organotypic cultures. Regul Pept 174: 58–64, 2012. [DOI] [PubMed] [Google Scholar]
- 21.Kaszubska W, Falls HD, Schaefer VG, Haasch D, Frost L, Hessler P, Kroeger PE, White DW, Jirousek MR, Trevillyan JM. Protein tyrosine phosphatase 1B negatively regulates leptin signaling in a hypothalamic cell line. Mol Cell Endocrinol 195: 109–118, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Khan AM, Ponzio TA, Sanchez-Watts G, Stanley BG, Hatton GI, Watts AG. Catecholaminergic control of mitogen-activated protein kinase signaling in paraventricular neuroendocrine neurons in vivo and in vitro: a proposed role during glycemic challenges. J Neurosci 27: 7344–7360, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim KW, Sohn JW, Kohno D, Xu Y, Williams K, Elmquist JK. SF-1 in the ventral medial hypothalamic nucleus: a key regulator of homeostasis. Mol Cell Endocrinol 336: 219–223, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim YW, Kim KH, Ahn DK, Kim HS, Kim JY, Lee DC, Park SY. Time-course changes of hormones and cytokines by lipopolysaccharide and its relation with anorexia. J Physiol Sci 57: 159–165, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Koren S, Fantus IG. Inhibition of the protein tyrosine phosphatase PTP1B: potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract Res Clin Endocrinol Metab 21: 621–640, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Krajewska M, Banares S, Zhang EE, Huang X, Scadeng M, Jhala US, Feng GS, Krajewski S. Development of diabesity in mice with neuronal deletion of Shp2 tyrosine phosphatase. Am J Pathol 172: 1312–1324, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C, Friedman JM, Riedman MF. Leptin receptor activation of SH2 domain containing protein tyrosine phosphatase 2 modulates Ob receptor signal transduction. Proc Natl Acad Sci USA 96: 9677–9682, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, Simonds S, Wiede F, Reichenbach A, Hauser C, Sims NA, Bence KK, Zhang S, Zhang ZY, Kahn BB, Neel BG, Andrews ZB, Cowley MA, Tiganis T. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab 14: 684–699, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorenzo M, Fernández-Veledo S, Vila-Bedmar R, Garcia-Guerra L, De Alvaro C, Nieto-Vazquez I. Insulin resistance induced by tumor necrosis factor-alpha in myocytes and brown adipocytes. J Anim Sci 86 (14Suppl): E94–E104, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, Tsukumo DM, Anhe G, Amaral ME, Takahashi HK, Curi R, Oliveira HC, Carvalheira JB, Bordin S, Saad MJ, Velloso LA. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci 29: 359–370, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrison CD, White CL, Wang Z, Lee SY, Lawrence DS, Cefalu WT, Zhang ZY, Gettys TW. Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology 48: 433–440, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myers MG., Jr.Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog Horm Res 59: 287–304, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien JP, Salmeen A, Barford D, Tonks NK. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J Biol Chem 276: 47771–47774, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Nieto-Vazquez I, Fernández-Veledo S, de Alvaro C, Rondinone CM, Valverde AM, Lorenzo M. Protein-tyrosine phosphatase 1B-deficient myocytes show increased insulin sensitivity and protection against tumor necrosis factor-alpha-induced insulin resistance. Diabetes 56(2):404–13, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. NY: Academic, 1997. [DOI] [PubMed] [Google Scholar]
- 36.Picardi PK, Caricilli AM, de Abreu LL, Carvalheira JB, Velloso LA, Saad MJ. Modulation of hypothalamic PTP1B in the TNF-alpha-induced insulin and leptin resistance. FEBS Lett 584: 3179–3184, 2010. [DOI] [PubMed] [Google Scholar]
- 37.Pike KA, Hutchins AP, Vinette V, Théberge JF, Sabbagh L, Tremblay ML, Miranda-Saavedra D. Protein tyrosine phosphatase 1B is a regulator of the interleukin-10-induced transcriptional program in macrophages. Sci Signal 7(324): ra43, 2014. [DOI] [PubMed] [Google Scholar]
- 38.Pohl J, Woodside B, Luheshi GN. Changes in hypothalamically mediated acute-phase inflammatory responses to lipopolysaccharide in diet-induced obese rats. Endocrinology 150: 4901–4910, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Qin H, Roberts KL, Niyongere SA, Gong Y, Elson CO, Benveniste EN. Molecular mechanism of lipopolysaccharide-induced SOCS-3 gene expression in macrophages and microglia. J Immunol 179: 5966–5976, 2007. [DOI] [PubMed] [Google Scholar]
- 40.Qu CK. The SHP-2 tyrosine phosphatase: signaling mechanisms and biological functions. Cell Res 10: 279–288, 2000. [DOI] [PubMed] [Google Scholar]
- 41.Qu CK. Role of the SHP-2 tyrosine phosphatase in cytokine-induced signaling and cellular response. Biochim Biophys Acta 1592: 297–301, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Rorato R, Reis WL, Antunes-Rodrigues J, Elias LL. Cholecystokinin and hypothalamic corticotrophin-releasing factor participate in endotoxin-induced hypophagia. Exp Physiol 96: 439–450, 2011. [DOI] [PubMed] [Google Scholar]
- 43.Sato T, Laviano A, Meguid MM, Chen C, Rossi-Fanelli F, Hatakeyama K. Involvement of plasma leptin, insulin and free tryptophan in cytokine-induced anorexia. Clin Nutr 22: 139–146, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol 514: 518–532, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soszynski D, Daniluk M, Galazka M, Dmitruk K. Blockade of nitric oxide formation in the rat brain does not disturb development of endotoxin tolerance. J Physiol Pharmacol 64: 779–788, 2013. [PubMed] [Google Scholar]
- 46.Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, Nguyen HT, Fischer JD, Matsen ME, Wisse BE, Morton GJ, Horvath TL, Baskin DG, Tschöp MH, Schwartz MW. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122: 153–162, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsou RC, Bence KK. Central regulation of metabolism by protein tyrosine phosphatases. Front Neurosci 6 (192): 1–11, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tups A. Physiological models of leptin resistance. J Neuroendocrinol 21: 961–971, 2009. [DOI] [PubMed] [Google Scholar]
- 49.Velloso LA, Araujo EP, de Souza CT. Diet-induced inflammation of the hypothalamus in obesity. Neuroimmunomodulation 15: 189–193, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Xu H, An H, Hou J, Han C, Wang P, Yu Y, Cao X. Phosphatase PTP1B negatively regulates MyD88- and TRIF-dependent proinflammatory cytokine and type I interferon production in TLR-triggered macrophages. Mol Immunol 45: 3545–3552, 2008. [DOI] [PubMed] [Google Scholar]
- 51.Williams KW, Sohn JW, Donato J, Lee CE, Zhao JJ, Elmquist JK, Elias CF. The acute effects of leptin require PI3K signaling in the hypothalamic ventral premammillary nucleus. J Neurosci 31: 13147–13156, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, Neel BG. PTP1B regulates leptin signal transduction in vivo. Dev Cell 2: 489–495, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem 23;283: 14230–14241, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zawalich WS, Zawalich KC. Interleukin 1 is a potent stimulator of islet insulin secretion and phosphoinositide hydrolysis. Am J Physiol Endocrinol Metab 256: E19–E24, 1989. [DOI] [PubMed] [Google Scholar]
- 55.Zhang EE, Chapeau E, Hagihara K, Feng GS. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci USA 101: 16064–16069, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]