

Abstract
CD59 is a membrane protein inhibitor of the membrane attack complex (MAC) of complement. mCd59 knockout mice reportedly exhibit hemolytic anemia and platelet activation. This phenotype is comparable to the human hemolytic anemia known as paroxysmal nocturnal hemoglobinuria (PNH), in which platelet activation and thrombosis play a critical pathogenic role. It has long been suspected but not formally demonstrated that both complement and nitric oxide (NO) contribute to PNH thrombosis. Using mCd59a and mCd59b double knockout mice (mCd59ab−/− mice) in complement sufficient (C3+/+) and deficient (C3−/−) backgrounds, we document that mCd59ab−/− platelets are sensitive to complement-mediated activation and provide evidence for possible in vivo platelet activation in mCd59ab−/− mice. Using a combination of L-NAME (a NO-synthase inhibitor) and NOC-18 or SNAP (NO-donors), we further demonstrate that NO regulates complement-mediated activation of platelets. These results indicate that the thrombotic diathesis of PNH patients could be due to a combination of increased complement-mediated platelet activation and reduced NO-bioavailability as a consequence of hemolysis.
Introduction
The complement system provides higher organisms “innate” protection against simpler pathogens such as viruses, bacteria, yeast and parasites [1–3]. Immediate recognition of the potential invader as “non-self” followed by clearing occurs through a process of “labeling and attack” that can be activated through three different biochemical pathways. The lectin and alternative pathways, which are respectively triggered or amplified by non-mammalian carbohydrate structures, label “non-self” organisms by attaching early complement activation products such as C3b to their surface. The classical pathway is activated by structural changes in the Fc region of antibodies that take place upon antibody binding (labeling) to an antigen in the surface of the potential invader [4]. Sequential activation of the complement system through either of the three pathways leads to a common “attack” phase that is mediated by formation of the cytotoxic membrane attack complex (MAC). The MAC is a macromolecular pore that inserts into the cell membrane allowing the influx of salt and water that eventually kills the target cell by colloidosmotic lysis [1,4,5].
To protect “self” cells from the potentially devastating effect of autologous complement activation, an array of plasma- and membrane-bound “complement regulatory proteins” restricts complement activation at different stages of the activation pathways [1,2,4]. Because activation/amplification of the complement pathways and formation of the MAC occur on the cell surface, proteins bound to the external surface of “self” cells, such as membrane cofactor protein (MCP), decay-accelerating factor (DAF) and CD59, operate much more effectively in restricting complement activation than “fluid phase” inhibitors. Effective restriction of complement activation on “self” cell membranes by DAF and CD59 is further optimized because both proteins are attached to the external cell surface through a glycosyl-phosphatidylinositol (GPI) anchor. This GPI anchor provides DAF and CD59 lateral mobility within the membrane lipid bilayer, and thereby, rapid access to the membrane site, where focal complement activation/amplification or MAC formation is taking place. Attachment of the GPI anchor to an encoded processing signal at the C-terminus of GPI-linked proteins occurs post translationally and is mediated by products of the PIG-A gene.
Paroxysmal nocturnal hemoglobinuria (PNH) [6] is a striking example of a human disease in which decreased restriction of the complement system uncovers the deleterious consequences of uncontrolled MAC formation on the surface of “self” cells [6]. In subjects with PNH, an acquired somatic mutation of the PIG-A gene in hematopoietic-cell precursors causes a deficiency of all GPI-linked proteins, including the key complement regulators DAF and CD59, in circulating cells. Deficiency of DAF and CD59 renders red blood cells (RBC) and platelets highly susceptible to complement-mediated lysis and activation ex vivo [7], and is likely to contribute to both the hemolytic anemia and thrombotic diathesis that constitute the main clinical features of the disease [8].
Experimental and clinical evidence indicate that complement is the main effector of the hemolytic anemia characteristic of PNH [6]. In contrast, the mechanism mediating increased platelet activation in PNH is not clearly understood. Although PNH-platelets are highly sensitive to MAC-mediated activation ex vivo [9,10] it has been proposed that a nitric oxide (NO)-scavenger effect of free-plasma hemoglobin may contribute to abnormal platelet activation and thrombosis in PNH patients [11].
The complement regulatory protein CD59 is a specific inhibitor of MAC formation and mCd59 knockout mice exhibit a PNH-like phenotype characterized by hemolytic anemia with increased reticulocytes [12–14]; this phenotype is complement-mediated because it is rescued by the deficiency of C3 [14,15]. mCd59 KO mice also exhibit evidence of increased platelet activation with formation of platelet-derived micro-particles (PMP) but the mechanism underlying this phenotypic expression has not been formally established in vivo [13].
Mice have two Cd59 genes (termed mCd59a and mCd59b) [16,17]. mCd59a is universally expressed in mouse tissues and is a primary complement regulator in mice; mCd59b is expressed in testis and at a lower level in homeopathic cells including erythrocytes and platelets [14,15,18]. Here, we report the use of mCd59a and mCd59b double knockout mice in complement-sufficient (mCd59ab−/−/C3+/+) and complement-deficient (mCd59ab−/−/C3−/−) backgrounds to establish the role of complement in platelet activation in vivo, and describe a novel homeostatic effect of NO balancing complement-mediated activation of platelets.
Results
Complement mediates platelet activation in mCd59ab−/−/C3+/+ mice
Bleeding time is reduced in mCd59ab−/−/C3+/+ mice
Experimental evidence indicates that complement activation and MAC formation induces platelet activation [9]. The bleeding time measured by venostasis is a test routinely used in clinical settings to assess platelet function including platelet reactivity, one of the critical variable affecting the test results [19]. We hypothesized that if deficiency of CD59 increases spontaneous MAC-mediated platelet reactivity in vivo, this abnormality should be reflected in the bleeding time of mCd59ab−/−/C3+/+ mice. Figure 1A shows that mCd59ab−/−/C3+/+ mice exhibit a significantly shorter bleeding time than mCd59ab+/+/C3+/+; this shorter bleeding time is not associated with different platelet counts (see Fig. 2) and is normalized by the deficiency of C3 in mCd59ab−/−/C3−/− mice. These results suggest that in mCd59ab−/−/C3+/+ mice there is an increased platelet reactivity that is complement-mediated because it is rescued by the simultaneous deficiency of C3 (Fig. 1A). Increased complement-mediated platelet activation in mCd59ab−/−/C3+/+ mice was confirmed by measuring the release of sP-selectin [20] induced ex vivo by CVF, an activator of the alternative complement pathway (Fig. 1B). Of note, we did not find any histological evidence of thrombosis after autopsying mCd59ab−/−/C3+/+ mice, nor was bleeding the tail from these animals more difficult than bleeding the tails of mCd59ab+/+/C3+/+ mice.
Figure 1.
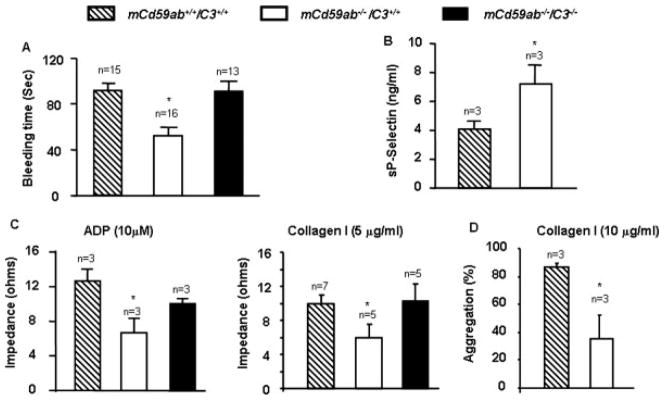
Complement-mediated platelet activation of mCd59ab−/−/C3+/+. (A) Bleeding time. *P < 0.001 vs. mCd59ab+/+/C3+/+ and **P < 0.01 vs. mCd59ab−/−/C3−/− (B) Release of sP-selectin ex vivo triggered by CVF-mediated C activation and 10% of rat serum. *P < 0.05 vs. mCd59ab+/+/C3+/+. (C) ADP and collagen-induced platelet aggregation in whole blood. *P < 0.05 vs. mCd59ab+/+/C3+/+. No other pair-wise comparisons of results vs. mCd59ab+/+/C3+/+ were statistically significant. (D) Collagen-induced platelet aggregation in PRP. *P < 0.05 vs. mCd59ab+/+/C3+/+.
Figure 2.

Increased platelet consumption in mCd59ab−/− mice blood. Left panel: platelet count at 0 min. Middle panel: platelet count at 1 min after blood extraction. *P< 0.05 vs. mCd59ab+/+/C3+/+ or mCd59ab−/−/C3−/−. Inset: relative (%) platelet consumption 1 min after blood extraction. *P < 0.05 vs. mCd59ab+/+/C3+/+ or mCd59ab−/−/C3−/−. All other pair-wise comparisons vs. mCd59ab+/+/C3+/+ were not statistically significant.
Platelet aggregation appears to be decreased in mCd59ab−/−/C3+/+ mice
Platelet aggregation in response to agonists such as adenosine diphosphate (ADP) and collagen is an established ex vivo method to assess platelet function. ADP stimulates platelet aggregation by the exposure of specific and saturable binding sites for fibrinogen at the platelet surface [21], whereas collagen’s agonist effect is mediated by the adhesion of platelets followed by ADP release from collagen-attached platelets [22]. ADP and collagen-induced platelet aggregation in whole blood appeared to be decreased in mCd59ab−/−/C3+/+ mice as compared to both mCd59ab+/+/C3+/+ and mCd59ab−/−/C3−/− mice (Fig. 1C). Specific platelet aggregation assessed in platelet rich plasma (PRP) using collagen stimulation was also reduced in mCd59ab−/−/C3+/+ as compared with mCd59ab+/+/C3+/+ mice (Fig. 1D). The normalization of ex vivo ADP and collagen-induced aggregation in mCd59ab−/−/C3−/− mice implies that the reduced platelet aggregation observed in mCd59ab−/−/C3+/+ is complement dependent and not due to an intrinsic defect of the mCd59ab−/−/C3+/+ platelets.
Correct interpretation of results from agonist-induced aggregation experiments using impedance aggregometry hinges upon the assumption that total platelet counts at the time of agonist addition are comparable between blood samples tested; however, this may not have been the case if mCd59ab deficiency resulted in platelet activation and “spontaneous” aggregation between blood extraction and addition of agonists, effectively reducing the number of platelets that would subsequently be counted. Given that the bleeding time of mCd59ab−/−/C3+/+ mice was clearly reduced, indicative of increased complement-mediated platelet reactivity in vivo, we reasoned that the smaller amplitude of impedance changes in blood of mCd59ab−/−/C3+/+ mice may have been due to formation of spontaneous platelet aggregates before recording began.
Platelet consumption is increased in the blood of mCd59ab−/−/C3+/+ mice
If the above interpretation is correct, we should find increased complement-mediated spontaneous platelet aggregation and/or consumption during the extraction of blood from mCd59ab−/−/C3+/+. However, spontaneous aggregation cannot be measured in complement-sufficient mCd59ab−/− because of spontaneous clot formation in the syringe during extraction. Therefore, we assessed spontaneous platelet consumption ex vivo by counting platelets in whole blood at nominal 0 time and in platelet-rich plasma (PRP) 1 min after venipuncture (1-minute PRP). Platelet counts at time 0 were not significantly different among the three mouse groups [mCd59ab+/+/C3+/+ vs. mCd59ab−/−/C3+/+ mice (P > 0.05); mCd59ab−/−/C3+/+ vs. mCd59ab−/−/C3−/− mice (P > 0.05)] (see Fig. 2). In the one-minute PRP, platelet counts in all groups were lower than at 0 time, as expected, but were significantly much lower in mCd59ab−/−/C3+/+ mice than in mCd59ab+/+/C3+/+ or mCd59ab−/−/C3−/− mice. These results suggest that during blood collection, activation of complement by a combination of injury [23], contact with the syringe’s plastic surface [24], and complement-activating factors derived from the clotting cascade [25] enhances platelet activation/consumption, to which mCd59ab−/− platelets are more sensitive owing to the absence of CD59. This interpretation is supported by the finding that one-minute PRP from mCd59ab−/−C3+/+ but not from mCd59ab+/+C3+/+ or mCd59ab−/−C3−/− contains a sub-population of platelets with increased density of membrane-bound P-selectin, which is indicative of platelet activation (Supporting Fig. 1). This subpopulation of platelets was defined by the intensity of P-selectin-specific antibody staining assessed by FACS.
In summary, the shorter bleeding time and the increased consumption of platelets in complement sufficient mCd59ab−/− mice indicate that mCd59ab−/−/C3+/+ platelets are hypersensitive to complement-mediated activation.
NO regulates complement-mediated platelet activation in mCd59ab−/− mice
NO is a physiological regulator of smooth muscle contraction and platelet reactivity [11,26,27]. It has been proposed that decreased NO (nitrite and nitrate), because of the scavenger effect of free-plasma hemoglobin, contributes to some clinical manifestations of PNH and other hemolytic anemias, including erectile dysfunction and esophageal spasms [11]; however, the role of NO in the platelet activation and thrombosis characteristic of PNH has not been studied experimentally. To assess the role of NO in vivo, we used Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME), an inhibitor of NO-synthase, to reduce the availability of NO in blood and elsewhere, and measured sP-selectin levels in plasma, an established marker of platelet activation [20]. Before L-NAME administration, plasma levels of sP-selectin were not significantly different among mCd59ab−/−/C3+/+, mCd59ab+/+/C3+/+, and mCd59ab−/−/C3−/− mice. After L-NAME administration there was a significantly higher increase in the sP-selectin level of mCd59ab−/−/C3+/+ mice compared to that of mCd59ab+/+/C3+/+ or mCd59ab−/−/C3−/− mice (Fig. 3A). It has been established that the bulk of circulating sP-selectin derives from activated platelets, and not damaged endothelium [28]. Nonetheless, the potential contribution of endothelial damage to the L-NAME-induced increase in sP-selectin observed in mCd59ab−/−/C3+/+ mice was experimentally ruled out by the absence of a parallel increase in von Willebrand factor (vWF), a well-established biomarker of endothelial dysfunction [20] (Fig. 3B). The expected effect of L-NAME on NO bioavailability in mCd59ab−/−/C3+/+ mice was confirmed by a significant decrease in NO plasma levels 4 hrs after L-NAME administration (Fig. 3C).
Figure 3.

Decreased NO availability after L-NAME injection increases complement-mediated platelet activation. (A) sP-selectin levels in plasma after L-NAME injection (0.05 mg/g body weight). *P < 0.05 vs. mCd59ab+/+/C3+/+, **P < 0.05 vs. mCd59ab−/−/C3+/+. (B) Plasma level of vWF in mCd59ab−/−/C3+/+ before and after L-NAME injection. All other pair-wise comparisons vs. mCd59ab+/+/C3+/+ were not statistically significant. (C) *P < 0.05 vs. baseline. The plasma NO levels, measured as levels of nitrites and nitrates, were determined at 4 hrs after L-NAME injection (0.05 mg/g body weight) or 4 hr after L-NAME (0.05 mg/g body weight) Plus NOC-18 injection (0.02 mg/g body weight) (15 min before L-NAME injection).
To investigate whether additional NO would compensate for the decreased NO bioavailability induced by L-NAME, we administrated two different NO donors {1-Hydroxy-2-oxo-3,3-bis(3-aminoethyl)-1-triazine (NOC-18) [29] and S-nitroso-N-acetylpenicillamine (SNAP) [30]} to the mice before L-NAME injection. The expected effect of the donors restoring NO bioavailability was confirmed in mice treated with NOC-18 before L-NAME injection (Fig. 3C). Injection of either NO donor before L-NAME brought sP-selectin values to the baseline levels seen in mCd59ab+/+/C3+/+ or mCd59ab−/−/C3+/+ mice in the absence of L-NAME (Fig. 4A). Together, these results confirm that the effects of L-NAME shown in Fig. 3A are due to reduced availability of NO, and that NO plays a regulatory role in complement-mediated platelet reactivity in vivo. Consistent with such a role, addition of the NO donor DEA/NO to the syringe used to draw blood from the mouse vena cava abrogated the increased platelet consumption observed in mCd59ab−/−/C3+/+ PRP one (Fig. 4B).
Figure 4.

NO donor abolishes complement-mediated platelet activation. (A) Normalization of sP-selectin levels in mCd59ab+/+/C3+/+ or mCd59ab−/−/C3+/+ by injection of NOC-18 (0.02 mg/g body weight) (left panel) and SNAP (10 μg/g) (right panel) 15 min before L-NAME injection. (B) NO donor DEA/NO abolished the increased platelet consumption in mCd59ab−/−/C3+/+ mice. *: percent of 1 min platelet consumption in mCd59ab−/−/C3+/+ vs. in mCd59ab+/+/C3+/+, P < 0.01. All other pair-wise comparisons vs. mCd59ab+/+/C3+/+ were not statistically significant.
Discussion
Here, we describe (1) the phenotypic characterization of mCd59ab−/− mice in complement-sufficient (C3+/+) and complement-deficient (C3−/−) conditions with emphasis on the role of complement in platelet activation in vivo; and (2) a novel homeostatic role for NO on complement-mediated platelet activation in vivo.
Abnormal platelet activation associated with thrombocytopenia, increased risk of fatal hemorrhage and thrombosis (3.7 thrombotic events per 100 patient-years) is the most serious complication of PNH and contributes significantly to its morbidity and mortality [11]. Consistent with increased platelet activation in vivo is the clinical observation that PNH patients have elevated levels of PMP [31]. We also reported that mCd59b−/−mice (in which there is total deficiency of mCd59b and lower expression of mCd59a) [32], exhibited increased platelet micro-particles [13]. Although a role of complement has long been suspected, the mechanisms underlying this increased platelet reactivity are not clearly understood. Experimentally, assembly of the MAC on the surface of purified platelets results in the initiation of a transient and reversible depolarization of the plasma membrane potential, a rise in cytosolic Ca2+, metabolic conversion of arachidonate to thromboxane, and the activation of intracellular protein kinases [33], which may be associated with platelet activation. The platelet-activating effect of the MAC is dramatically potentiated in PNH platelets lacking CD59 and in the presence of neutralizing anti-CD59 antibodies [10]. These in vitro results suggest that increased MAC deposition on unprotected platelets may contribute to the increased platelet activation with thrombocytopenia that is common in PNH patients. The shorter bleeding time associated with increased platelet consumption seen in complement-sufficient but not in complement-deficient mCd59ab−/− mice confirms the role of complement in activating CD59-defi-cient platelets in vivo. Of note, our previous demonstration of increased platelet micro-particles in mCd59b−/− [13] was possibly influenced by increased activation during blood drawing.
Activation of platelets leads to the rapid translocation of the glycoprotein P-selectin from α-granules to the membrane of platelets. For this reason, increased density of P-selectin measured by FACS analysis is an established marker of platelet activation [34]. Interestingly, it has been recently demonstrated that P-selectin in the platelet membrane binds to C3b, providing a mechanism for complement activation/amplification propagated by activated platelet [35]. It is tempting to propose that in PNH increased MAC-mediated platelet activation results from complement activation; activated platelets express increased levels of P-selectin, which binds to C3b and amplifies complement activation and MAC formation, creating a positive feedback loop for further platelet activation, consumption, thrombocytopenia induced hemorrhages and, in some instances, life threatening thrombosis. Of note, overt thrombosis in PNH patients tends to be associated with co-morbidities such as infections and dehydration. Consistent with these clinical features of PNH, the mCd59ab−/− mice described here exhibit increased platelet activation but not spontaneous thrombosis.
NO is continuously synthesized by the endothelium and contributes to the regulation of vascular tone and platelet activation, adhesion, and aggregation [11,26,27–36]. In addition to vascular sources of NO, platelets and other blood cells have their own NO-producing machinery [37], and NO has been shown to be released during the aggregation process [26]. Under physiological conditions, sequestering of vascular-derived NO by hemoglobin is considered a critical pathway for limiting NO bioavailability. Intravascular hemolysis releases hemoglobin from RBC into the plasma medium. This cell-free hemoglobin scavenges endothelium-derived NO 600-fold faster than intra-RBC hemoglobin and disrupts NO homeostasis [38]. Extensive clinical evidence supports the notion that decreased NO levels because of scavenging by cell-free hemoglobin contributes to two characteristic clinical manifestations of intravascular hemolysis; (1) smooth muscle dystonia leading to hypertension, gastrointestinal spasmodic contractions, and erectile dysfunction; and (2) increased platelet activation and thrombosis [11]. Esophageal spasm and erectile dysfunction (suggestive of increased smooth muscle contractility) and thrombosis (suggestive of increased platelet reactivity) are common in PNH [11]. It has been recently reported that therapeutic inhibition of complement activation by administration of anti-C5 antibodies to PNH patients reduces hemolysis and free-plasma hemoglobin, and resolves clinical manifestations, such as abdominal pain, dysphasia, and erectile failure [11]. These data imply that smooth muscle constriction as a result of the NO-scavenging effect of cell-free hemoglobin may be involved in the development of the clinical phenotype of PNH. By extension, decreased levels of NO may also contribute to thrombotic diathesis in PNH patients [11], an intriguing hypothesis that would be very difficult to study in humans owing to practical and ethical considerations. Our in vivo studies in mCd59ab−/− mice demonstrate that reduced levels of NO, as a result of global inhibition of NO synthesis by L-NAME, increase complement-mediated platelet activation as determined by increased levels of sP-selectin in plasma. Moreover, increased levels of NO, produced by addition of the NO donor DEA/NO to whole blood ex vivo, abolished complement-mediated platelet consumption. These results unmask a hitherto unrecognized role for NO in balancing complement-mediated platelet activation, a phenomenon that is most likely mediated by the inhibitory effect of NO on platelet activation/aggregation; however, a direct link between NO levels and complement activity cannot be ruled out at this point.
Our previous demonstration that glycation inactivates human CD59 established a link between complement and the development of human diabetic vascular complications [39]. Patients with uncontrolled diabetes have evidence of hemolytic anemia and an increased risk of thrombosis; diabetic platelets are hyperactive [40], and plasma from patients with diabetes contains higher levels of platelet release products, including PMP and sP-selectin [41]. Our current demonstration that platelets deficient in CD59 are more sensitive to complement-mediated activation in vivo and that this effect is modulated by NO indicates that thrombotic diathesis in diabetic patients may result, in part, from glycation-inactivation of CD59 associated with decreased bioavailability of NO reported in diabetic patients.
Materials and Methods
Animal studies were reviewed and approved by the Harvard Medical School Institutional Animal Care and Use Committee (Animal Welfare Assurance no. A3431-01; International Animal Care and Use Committee approval date: Jan 10, 2005).
mCd59ab−/−/C3−/− triple-knockout mice
For the experiments described in this article, we used C3-deficient mice generated in our laboratory as described in [14].
Platelet preparation
Blood was collected by venipuncture from the mouse inferior vena cava into a syringe containing 500-μl Alsever’s solution (72.4 mM NaCl, 31 mM sodium citrate and 113.9 mM glucose) in the presence of 5 ng/ml PGI2 (Cayman chemical, Ann Arbor, Michigan) to avoid aggregation, centrifuged (200g for 7 min at room temperature), and the supernatant [platelet-rich plasma (PRP)] spun down again (2,200g for 10 min at room temperature) to collect the platelet pellet. FACS analysis with anti-CD41 antibody was used to verify the purity of this platelet population as described in [14].
Bleeding time
In preliminary experiments we optimized the method and established a reproducible bleeding time using ≈ 30 B6 wild type mice. For comparison among the different experimental groups, six- to eight-week-old mice were placed inside a restrainer and a distal tail tip (approximately 0.3 cm) was briskly cut at time 0 using a new sharp scalpel blade. The tail was immediately inserted into pre-warmed (37°C) PBS, and the time until the bleeding stopped was recorded [42].
Ex vivo measurement of platelet sensitivity to complement-mediated activation
Platelets (3 × 108 cell/ml) prepared as described above were suspended in gelatin veronal buffer (GVB++). After incubation with 5 μg/ml of cobra venom factor (CVF), a well-established complement activator [13], and 10% of rat serum (37°C, 30 min), the platelets were spun down (5000g, 10 min), and soluble P-selectin (sP-selectin) was measured in the supernatant, as described below.
Platelet Consumption
Two blood samples (150 μl each) were sequentially collected from the mouse inferior vena cava by venipuncture. After collecting the first sample into a syringe filled with 150 μl Alsever’s solution, the needle was kept in place and the syringe replaced to collect a second sample without anticoagulant. The second sample was kept in the syringe at room temperature for 1 min, then immediately mixed with 150 μl Alsever’s solution, centrifuged to collect the PRP, and the final volume adjusted to 300 μl with Alsever’s solution. Platelets in whole blood from the first sample and in the one-minute PRP collected from the second syringe were counted in the Clinical Hematology Laboratory (Boston Children’s Hospital).
To assess the effect of the NO-donor 2-(N,N-diethylamino)-diazenolate-2-oxide (DEA/NO, kindly provided by Dr. David Wink, NCI, Bethesda, MD) [43], we used a similar protocol, except that 100 μl of blood was collected into 200 μl of Alsever’s solution (first syringe) and another 100-μl blood into the second syringe containing 50 μl of 5 × 10−6 M DEA/NO in PBS (diluted from an alkaline 10−4 M DEA/NO stock solution immediately before use).
Ex vivo platelet aggregation
Platelet reactivity was assessed by whole blood impedance aggregometry in venous blood from mCd59ab+/+/C3+/+, mCd59ab−/−/C3+/+, and mCd59ab−/−/C3−/− mice.
ADP-induced whole blood platelet aggregation
Blood was obtained from the inferior vena cava and collected into heparinized saline (1:4 dilution), incubated for 1 min at 37°C under stirring (1,200 rpm), and aggregation was induced by addition of 10 μM ADP (Helena Laboratories, U.S.A.). Maximal changes in impedance, typically occurring at 4–5 min, were recorded using a dual-channel whole blood aggregometer (Chronolog-Log Corporation, Model 590; Haverton, PA) [44].
Collagen-induced whole blood platelet aggregation
Blood was collected into 3.8% sodium citrate (9:1 v/v). Blood (250 uL) was then diluted with 650 μl of 0.9% saline and preincubated at 37°C with constant stirring (1,200 rpm) for 1 min. before inserting an electrode to monitor impedance. Upon reaching a stable baseline, human collagen (5 μg/ml) (Chrono-Log Corporation) was added to the blood mixture and aggregation monitored for 10 min.
Collagen-induced platelet-rich plasma aggregation
Platelet aggregation in platelet-rich plasma (PRP) was analyzed using a PAP-4 platelet aggregometer (Bio/Data Corporation, Horsham, PA). Blood collected in sodium citrate was centrifuged at 400g for 7 min. to separate PRP and the remaining fraction further centrifuged at 2,200 g for 10 min to obtain platelet-poor plasma (PPP). PPP was used to equalize platelet counts between samples and as a blank reference for the aggregometer. Agonist-stimulated aggregation was assessed by adding collagen (10 μg/ml) to stirred PRP and percent aggregation was 10 min later [45].
Soluble P-selectin
Blood samples from 10 to 12-week old animals were collected by venipuncture from the inferior vena cava into a syringe containing 10-mM EDTA and PGI2 (5 ng/ml), and centrifuged (2,000g, 20 min). Plasma was further centrifuged (10,000g, 10 min) and sP-selectin in the platelet-free plasma was assayed by ELISA (R&D Systems. Minneapolis, MN) as described in [20]. NOC-18 (EMD Biosciences. San Diego, CA), SNAP (EMD Biosciences), L-NAME (Sigma-Aldrich, St. Louis, MO) or vehicle PBS were administered by tail vein injection.
Plasma vWF measurement
Mouse plasma vWF was measured by ELISA [20].
FACS analysis
Platelets were fixed with 3.7% paraformaldehyde, incubated with anti-P selectin (CHEMICON International. Temecula, CA) antibodies for 30 min at room temperature, washed three times with PBS/3% BSA buffer, and then incubated for 30 min with an appropriate FITC-conjugated secondary antibody. The cells were washed in PBS three times before analyzing the fluorescence intensity using a FACScan (Becton Dickinson, Franklin Lakes, NJ). The result of P-selectin staining on platelets is shown in Supporting Fig. 1.
Measurement of nitric oxide products, nitrite, and nitrate
It is difficult to directly measure NO because of its very short half-life and rapid oxidization to nitrite and nitrate under physiological conditions [20]. For this reason, we measured the concentration of the NO products nitrite and nitrate with a Nitric Oxide Quantitation Kit (Active Motif), as described in [20].
Statistical analysis
Comparisons of results between two groups (in Figs. 1B,C, 2 and 4), were conducted by 2-tailed Student t test for unpaired data. Differences among multiple groups in sP-selectin levels in Fig. 2 were analyzed by two-way ANOVA using the SigmaStat program. Data are presented as mean ± SEM. Level of significance for all comparisons was established at 0.05.
Supplementary Material
Acknowledgments
Contract grant sponsor: American Heart Association; Contract grant number: 0435483N; Contract grant sponsor: NIH; Contract grant numbers: RO1 DK060979, RO1052855, and RO1 AI061174.
We are grateful to Dr. B. P. Morgan for providing anti-mCd59a and mCd59b monoclonal antibodies, to Dr. Dr. E. Medof for providing anti-mDAF antibody, to Dr. Jane E. Freedman for analyzing platelet aggregation, and to the BWH Editorial Service for helpful editorial assistance.
Footnotes
Conflict of interest: Nothing to report.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Morgan BP, Harris CL. Complement Regulatory Proteins. London: Academic Press; 1999. [Google Scholar]
- 2.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 3.Walport MJ. Complement. Second of two parts. N Engl J Med. 2001;344:1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 4.Zhou X, Hu W, Qin X. The role of complement in the mechanism of action of rituximab for B-cell lymphoma: implications for therapy. Oncologist. 2008;13:954–966. doi: 10.1634/theoncologist.2008-0089. [DOI] [PubMed] [Google Scholar]
- 5.Acosta J, Qin X, Halperin J. Complement and complement regulatory proteins as potential molecular targets for vascular diseases. Curr Pharm Des. 2004;10:203–211. doi: 10.2174/1381612043453441. [DOI] [PubMed] [Google Scholar]
- 6.Hillmen P, Richards SJ. Implications of recent insights into the pathophysiology of paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2000;108:470–479. doi: 10.1046/j.1365-2141.2000.01802.x. [DOI] [PubMed] [Google Scholar]
- 7.Luzzatto L, Bessler M. The dual pathogenesis of paroxysmal nocturnal hemoglobinuria. Curr Opin Hematol. 1996;3:101–110. doi: 10.1097/00062752-199603020-00001. [DOI] [PubMed] [Google Scholar]
- 8.Takeda J, Miyata T, Kawagoe K, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-t. [DOI] [PubMed] [Google Scholar]
- 9.Sims PJ, Faioni EM, Wiedmer T, Shattil SJ. Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express pro-thrombinase activity. J Biol Chem. 1988;263:18205–18212. [PubMed] [Google Scholar]
- 10.Wiedmer T, Hall SE, Ortel TL, et al. Complement-induced vesiculation and exposure of membrane prothrombinase sites in platelets of paroxysmal nocturnal hemoglobinuria. Blood. 1993;82:1192–1196. [PubMed] [Google Scholar]
- 11.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293:1653–1662. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 12.Holt DS, Botto M, Bygrave AE, et al. Targeted deletion of the CD59 gene causes spontaneous intravascular hemolysis and hemoglobinuria. Blood. 2001;98:442–449. doi: 10.1182/blood.v98.2.442. [DOI] [PubMed] [Google Scholar]
- 13.Qin X, Krumrei N, Grubissich L, et al. Deficiency of the mouse complement regulatory protein mCd59b results in spontaneous hemolytic anemia with platelet activation and progressive male infertility. Immunity. 2003;18:217–227. doi: 10.1016/s1074-7613(03)00022-0. [DOI] [PubMed] [Google Scholar]
- 14.Qin X, Hu W, Song W, et al. Generation and phenotyping of mCd59a and mCd59b double-knockout mice. Am J Hematol. 2008;84:65–70. doi: 10.1002/ajh.21319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin X, Dobarro M, Bedford SJ, et al. Further characterization of reproductive abnormalities in mCd59b knockout mice: a potential new function of mCd59 in male reproduction. J Immunol. 2005;175:6294–6302. doi: 10.4049/jimmunol.175.10.6294. [DOI] [PubMed] [Google Scholar]
- 16.Qian YM, Qin X, Miwa T, et al. Identification and functional characterization of a new gene encoding the mouse terminal complement inhibitor CD59. J Immunol. 2000;165:2528–2534. doi: 10.4049/jimmunol.165.5.2528. [DOI] [PubMed] [Google Scholar]
- 17.Qin X, Miwa T, Aktas H, et al. Genomic structure, functional comparison, and tissue distribution of mouse Cd59a and Cd59b. Mamm Genome. 2001;12:582–589. doi: 10.1007/s00335-001-2060-8. [DOI] [PubMed] [Google Scholar]
- 18.Baalasubramanian S, Harris CL, Donev RM, et al. CD59a is the primary regulator of membrane attack complex assembly in the mouse. J Immunol. 2004;173:3684–3692. doi: 10.4049/jimmunol.173.6.3684. [DOI] [PubMed] [Google Scholar]
- 19.Harker LA, Slichter SJ. The bleeding time as a screening test for evaluation of platelet function. N Engl J Med. 1972;287:155–159. doi: 10.1056/NEJM197207272870401. [DOI] [PubMed] [Google Scholar]
- 20.Hu W, Ferris SP, Tweten RK, et al. Rapid conditional targeted ablation of cells expressing human CD59 in transgenic mice by intermedilysin. Nat Med. 2008;14:98–103. doi: 10.1038/nm1674. [DOI] [PubMed] [Google Scholar]
- 21.Mustard JF, Packham MA, Kinlough-Rathbone RL, et al. Fibrinogen and ADP-induced platelet aggregation. Blood. 1978;52:453–466. [PubMed] [Google Scholar]
- 22.Nossel HL, Wilner GD, Drillings M. Inhibition of collagen-induced platelet aggregation by normal plasma. J Clin Invest. 1971;50:2168–2175. doi: 10.1172/JCI106711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bellander BM, Singhrao SK, Ohlsson M, et al. Complement activation in the human brain after traumatic head injury. J Neurotrauma. 2001;18:1295–1311. doi: 10.1089/08977150152725605. [DOI] [PubMed] [Google Scholar]
- 24.Mollnes TE, Garred P, Bergseth G. Effect of time, temperature and anticoagulants on in vitro complement activation: consequences for collection and preservation of samples to be examined for complement activation. Clin Exp Immunol. 1988;73:484–488. [PMC free article] [PubMed] [Google Scholar]
- 25.Gorbet MB, Sefton MV. Biomaterial-associated thrombosis: roles of coagulation factors, complement, platelets and leukocytes. Biomaterials. 2004;25:5681–5703. doi: 10.1016/j.biomaterials.2004.01.023. [DOI] [PubMed] [Google Scholar]
- 26.Radomski MW, Palmer RM, Moncada S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet. 1987;2:1057–1058. doi: 10.1016/s0140-6736(87)91481-4. [DOI] [PubMed] [Google Scholar]
- 27.Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res. 2001;88:756–762. doi: 10.1161/hh0801.089861. [DOI] [PubMed] [Google Scholar]
- 28.Dole VS, Bergmeier W, Mitchell HA, et al. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. 2005;106:2334–2339. doi: 10.1182/blood-2005-04-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shibuta S, Mashimoto T, Ohara A, et al. Intracerebroventricular administration of a nitric oxide-releasing compound, NOC-18, produces thermal hyperalgesia in rats. Neurosci Lett. 1995;187:103–106. doi: 10.1016/0304-3940(95)11354-1. [DOI] [PubMed] [Google Scholar]
- 30.Jansen A, Drazen J, Osborne JA, et al. The relaxant properties in guinea pig airways of S-nitrosothiols. J Pharmacol Exp Ther. 1992;261:154–160. [PubMed] [Google Scholar]
- 31.Grunewald M, Grunewald A, Schmid A, et al. The platelet function defect of paroxysmal nocturnal haemoglobinuria. Platelets. 2004;15:145–154. doi: 10.1080/09537105310001657110. [DOI] [PubMed] [Google Scholar]
- 32.Qin X, Ferris S, Hu W, et al. Analysis of the promoters and 5′-UTR of mouse Cd59 genes, and of their functional activity in erythrocytes. Genes Immun. 2006;7:287–297. doi: 10.1038/sj.gene.6364296. [DOI] [PubMed] [Google Scholar]
- 33.Wiedmer T, Sims PJ. Participation of protein kinases in complement C5b-9-induced shedding of platelet plasma membrane vesicles. Blood. 1991;78:2880–2886. [PubMed] [Google Scholar]
- 34.van der Zee PM, Biro E, et al. P-selectin- and CD63-exposing platelet micro-particles reflect platelet activation in peripheral arterial disease and myocardial infarction. Clin Chem. 2006;52:657–664. doi: 10.1373/clinchem.2005.057414. [DOI] [PubMed] [Google Scholar]
- 35.Del Conde I, Cruz MA, Zhang H, et al. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201:871–879. doi: 10.1084/jem.20041497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feelisch M, Brands F, Kelm M. Human endothelial cells bioactivate organic nitrates to nitric oxide: implications for the reinforcement of endothelial defence mechanisms. Eur J Clin Invest. 1995;25:737–745. doi: 10.1111/j.1365-2362.1995.tb01952.x. [DOI] [PubMed] [Google Scholar]
- 37.Kleinbongard P, Schulz R, Rassaf T, et al. Red blood cells express a functional endothelial nitric oxide synthase. Blood. 2006;107:2943–2951. doi: 10.1182/blood-2005-10-3992. [DOI] [PubMed] [Google Scholar]
- 38.Gladwin MT, Crawford JH, Patel RP. The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radic Biol Med. 2004;36:707–717. doi: 10.1016/j.freeradbiomed.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 39.Qin X, Goldfine A, Krumrei N, et al. Glycation inactivation of the complement regulatory protein CD59: a possible role in the pathogenesis of the vascular complications of human diabetes. Diabetes. 2004;53:2653–2661. doi: 10.2337/diabetes.53.10.2653. [DOI] [PubMed] [Google Scholar]
- 40.Taka T, Ono H, Sasaki Y, et al. Platelet reactivity in spontaneously diabetic rats is independent from blood glucose and insulin levels. Platelets. 2002;13:313–316. doi: 10.1080/0953371027000-1. [DOI] [PubMed] [Google Scholar]
- 41.Tan KT, Tayebjee MH, Lim HS, Lip GY. Clinically apparent atherosclerotic disease in diabetes is associated with an increase in platelet microparticle levels. Diabet Med. 2005;22:1657–1662. doi: 10.1111/j.1464-5491.2005.01707.x. [DOI] [PubMed] [Google Scholar]
- 42.Lee KA, Kim MS. Antiplatelet and antithrombotic activities of methanol extract of Usnea longissima. Phytother Res. 2005;19:1061–1064. doi: 10.1002/ptr.1791. [DOI] [PubMed] [Google Scholar]
- 43.Miranda KM, Katori T, Torres de Holding CL, et al. Comparison of the NO and HNO donating properties of diazeniumdiolates: primary amine adducts release HNO in Vivo. J Med Chem. 2005;48:8220–8228. doi: 10.1021/jm050151i. [DOI] [PubMed] [Google Scholar]
- 44.Sudo T, Ito H, Kimura Y. Characterization of platelet aggregation in whole blood of laboratory animals by a screen filtration pressure method. Platelets. 2003;14:239–246. doi: 10.1080/0953710031000118885. [DOI] [PubMed] [Google Scholar]
- 45.Reheman A, Yang H, Zhu G, et al. Plasma fibronectin depletion enhances platelet aggregation and thrombus formation in mice lacking fibrinogen and von Willebrand factor. Blood. 2008 doi: 10.1182/blood-2008-04-148361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.