

Abstract
Background
In clinical trials of pegloticase, a PEGylated uricase developed for treatment of gout refractory to conventional therapy, infusion-related reactions (IRs) were the second most frequent adverse event reported.
Objective
The objective of this study was to provide a detailed account of IRs with pegloticase therapy.
Methods
Data from 2 replicate, 6-month randomized trials and an open-label extension study were pooled. Infusions of pegloticase (8 mg) were administered biweekly or monthly; all patients received prophylaxis (antihistamine, acetaminophen, and corticosteroid) and were tested for urate levels prior to each infusion. An IR was defined by protocol as any otherwise unexplained adverse event or cluster of temporally related events occurring during or within 2 hours of infusion.
Results
Infusion-related reactions occurred in 94 (45%) of 208 patients receiving pegloticase; 10 patients reported IRs at first infusion and 84 during subsequent infusions. Chest discomfort (15%), flushing (12%), and dyspnea (11%) were the most common symptoms. Most IRs were rated mild or moderate; 7% were rated severe. All IRs resolved with slowing, interrupting, or stopping the infusion. No patient required blood pressure or ventilatory support. Infusion-related reactions were associated with loss of pegloticase urate-lowering efficacy: 91% of all IRs occurred in patients with preinfusion serum uric acid concentrations (sUA) greater than 6 mg/dL. For patients sustaining preinfusion sUA of less than 6 mg/dL, IRs occurred in fewer than 1 per 100 infusions.
Conclusions
Phase 3 trial data combined with post hoc analyses demonstrated that knowledge of sUA preceding each pegloticase infusion and cessation of therapy when urate-lowering efficacy is lost provide a means to optimize the safety of pegloticase in clinical practice.
Key Words: pegloticase, infusion reactions, hyperuricemia, gout, uric acid, plasma uric acid concentration
Pegloticase, a monomethoxypoly(ethyleneglycol)–conjugated mammalian recombinant uricase, was developed for the treatment of chronic gout refractory to existing therapies and is approved in the United States and the European Union.1 The tolerability and efficacy of pegloticase were demonstrated in 2 replicate 6-month, randomized placebo-controlled trials (RCTs) in patients with symptomatic gout and plasma uric acid concentration (pUA) of 8 mg/dL or greater who had failed or were intolerant of allopurinol.2 Although all patients treated with pegloticase achieved pUA of less than 6 mg/dL within 24 hours of the first infusion, not all patients fulfilled the definition of “responder” in these studies. “Responders” were patients who met the primary efficacy end point defined as pUA of less than 6.0 mg/dL for 80% of the total time or greater during the third and sixth months of treatment combined. Responder status was achieved by 42% of patients receiving biweekly pegloticase compared with 0% of those in the placebo group.2 Patients not meeting the primary end point (and all patients who did not complete the trials) were classified as “nonresponders.” Loss of response, seen as early as 2 weeks after the first infusion, was manifested by a preinfusion pUA of greater than 6 mg/dL. In the pegloticase RCTs, both serum uric acid concentration (sUA) and pUA were measured; pUA was used for the primary end point to avoid possible degradation of uric acid by circulating pegloticase. Because plasma and serum urate determinations correlated 95% of the time with respect to values greater than or less than 6 mg/dL, the more clinically accessible sUA will be described in the remainder of this article with the exception of the protocol-defined end points.
The primary safety concerns in the pegloticase RCTs were gout flares, a common finding with initiation of any urate-lowering therapy,3 and infusion-related reactions (IRs). Infusion-related reactions were the second most common adverse event and the most common reason for discontinuation in the RCTs.2 Furthermore, post hoc analyses of data from the RCTs revealed relationships between urate-lowering responses to pegloticase therapy, the development of pegloticase antibodies, and the risk for IRs. These relationships were not appreciated while the RCTs were in progress because investigators were blinded to uric acid levels and the study treatments, but they provide critical insight into factors associated with IR risk and evidence pertinent to the prevention of IRs in clinical practice. The current report presents detailed information on IRs occurring during the pegloticase trials,2,4 recommendations for managing pegloticase IRs in clinical practice, and a proposed set of pegloticase stopping rules aimed at mitigating the risk for IRs.
MATERIALS AND METHODS
Clinical Trials
Information about IRs was obtained from the replicate 6-month RCTs (studies CO405 and CO406 or GOUT 1 and 2; identifier: NCT00325195 2) and the subsequent open-label extension (OLE) study data sets (C0407; identifier NCT01356498 4). The design of the RCTs has been described previously.2 Briefly, these studies enrolled patients with refractory gout and sUA of 8.0 mg/dL or greater, who either had failed or had contraindications to allopurinol treatment, and who had at least 1 or more of the following: 3 or more self-reported gout flares over the previous 18-month period, 1 or more tophus, or gouty arthropathy. Eligible patients were randomized in a 2:2:1 ratio to receive 12 biweekly, 2-hour intravenous (IV) infusions containing either pegloticase 8 mg every 2 weeks (biweekly treatment group), pegloticase 8 mg alternating with placebo at successive infusions (monthly treatment group), or placebo. Patients were required to discontinue any pretrial urate-lowering therapies 1 week prior to the RCT treatment period. Prophylaxis against gout flares with colchicine or a nonsteroidal anti-inflammatory drug was started 1 week before the first infusion and continued throughout the RCT. All patients received IR prophylaxis prior to each infusion in the RCT and OLE studies as follows: the nonsedating H1-antihistamine fexofenadine, 60 mg on the night preceding and the morning of infusion; acetaminophen, 1000 mg the morning of infusion; and hydrocortisone, 200 mg IV given immediately prior to infusion.
Patients completing treatment in either RCT were eligible to enroll in the OLE study during which all patients received either biweekly or monthly pegloticase infusions. While still blinded to RCT treatment, the investigator and patient decided whether pegloticase would be administered biweekly or monthly at OLE study entry. Changes in dosing frequency from monthly to biweekly or vice versa were allowed once after week 25 and a second time after the data from the RCTs were unblinded. Flare prophylaxis was required for the first 3 months of the OLE study and continued at investigator discretion thereafter.
For both the RCTs and OLE study, approval of the protocol was obtained from central (IntegReview, Austin, TX) or local institutional review boards, and informed consent was obtained from all patients before any study-related procedures were performed.
The primary efficacy end point in the RCTs was the proportion of responders in each treatment group achieving pUA of less than 6.0 mg/dL for 80% of the total time or greater during months 3 and 6 combined.2 Plasma uric acid concentration determinations were completed prior to infusions and at 6 additional times during months 3 and 6. Patients who did not meet the pUA criteria, or who discontinued early, were classified as nonresponders. Safety assessments included physical examination and adverse event updates every 2 weeks and laboratory testing every month. Safety was the primary end point in the OLE; evaluation for adverse events included physical examination at each biweekly or monthly visit and laboratory testing performed every 12 weeks.
Infusion-Related Reactions
Infusion-related reactions were defined in the study protocols as any infusion-related adverse event or cluster of temporally related events that occurred during or within 2 hours after drug infusion that could not be reasonably attributed to another cause. These events prompted a standardized assessment, which included a thorough dermatologic and cardiopulmonary examination and a 12-lead electrocardiogram. Vital signs were evaluated every 30 minutes until resolution or stabilization of the event. Besides the interventions implemented by the investigator to manage the patient’s medical condition, the infusion rate could be slowed by half, interrupted and restarted at the same or slower rate, or stopped. If the event resolved, the infusion could proceed to completion at the original rate, at a reduced rate, or with increased volume of diluent. However, the infusion was to be stopped if the IR failed to resolve within 1 hour, or if the patient, in the investigator’s opinion, was at risk for anaphylaxis. In the OLE, prednisone, 20 mg on the night before the next infusion, could be added to the prophylaxis regimen if a patient had experienced an IR, and further pegloticase treatment was to be given.
RESULTS
Pooled Study Population
A total of 225 patients were randomized, and 212 received at least 1 infusion of study treatment in the RCTs.2 One hundred fifty-one (96%) of the 157 patients who completed either of the RCTs elected to enter the OLE study (149 received pegloticase, and 2 chose to be followed up by observation only). Overall, 208 patients received pegloticase in the 3 studies. A total of 6389 infusions were administered during the RCTs and the OLE study (RCT biweekly, 852; monthly, 846; placebo, 502; OLE, 4189). Eighty-five patients were allocated to biweekly pegloticase in the RCTs and received a median of 36 infusions (range, 1–63) during combined RCT and OLE participation; 84 patients were initially allocated to monthly pegloticase and received a median of 23 pegloticase infusions (range, 1–57). All 39 patients randomized to placebo and completing 1 of the RCTs started pegloticase in the OLE study; 23 received biweekly treatment with a median of 20 pegloticase infusions (range, 2–51), and 16 received monthly pegloticase with a median of 7 infusions (range, 1–26). Overall, 91 patients (44%) received more than 30 pegloticase infusions, and 40 patients (19%) received more than 50 infusions.
Characterization of IRs
Infusion-related reactions were reported for 94 (45%) of 208 patients treated with pegloticase during the phase 3 trials. Two (5%) of 43 patients experienced at least 1 IR while receiving placebo (both during the RCTs). An IR occurred during the first pegloticase infusion in 10 (5%) of the 208 pegloticase-treated patients and in 1 (2%) of 43 patients receiving their first placebo infusion. Among the other 84 patients with at least 1 IR, these events were distributed over time as shown in Figure 1. Among patients with at least 1 IR, 93 (97%) had symptoms that arose during the infusion, and 3 (3%) had symptoms with onset in the 2-hour observation period after the infusion. Infusion-related reaction was the basis for discontinuation from study drug for 20 patients in the RCTs and for 11 patients in the OLE study. Among these 11, 6 had received placebo in the RCTs, and 5 were RCT nonresponders. No patient who was a responder in the RCTs reported an IR as the basis for discontinuation during the OLE study.
FIGURE 1.
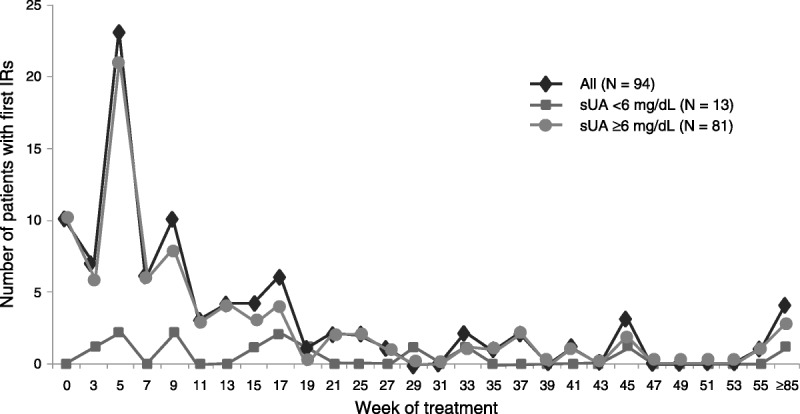
Number of first IRs with pegloticase by treatment week and sUA.
In the pooled study population from the RCTs and OLE, the most common symptoms defining IR (incidence >5% and excluding IRs associated with placebo infusions) were chest discomfort (n = 32; 15%), flushing (n = 24; 12%), dyspnea (n = 23; 11%), back pain (n = 19; 9%), hyperhidrosis (n = 18; 9%), nausea (n = 18; 9%), erythema (n = 18; 9%), urticaria (n = 17; 8%), chest pain (n = 17; 8%), pruritus (n = 16; 8%), rash (n = 13; 6%), muscle spasms (n = 13; 6%), headache (n = 12; 6%), and abdominal pain (n = 11; 5%). Other symptoms, occurring at rates of 2% to 5%, included dizziness (5%), vomiting (5%), pain (4%), chills (3%), hypertension (3%), hypotension (3%), tachycardia (3%), feeling hot (2%), musculoskeletal discomfort (2%), and wheezing (2%). Most IRs were rated mild or moderate in severity, although 12 (7%) of 169 patients who started pegloticase during the RCTs and 11 (7%) of 149 patients who received pegloticase in the OLE had IRs judged to be severe by the investigator (Table 1). Of note, in patients experiencing more than 1 pegloticase-related IR, no increase in IR severity was seen over time with treatment.
TABLE 1.
Incidence of Infusion Reactions by Severity in the RCT and OLE Populationsa

All IRs (including those classified retrospectively as anaphylaxis) in the RCTs and OLE study resolved with supportive measures, which included slowing or stopping the infusion and/or providing fluids, diphenhydramine, corticosteroids, and/or analgesics. Table 2 summarizes the adjustments to the infusions made by investigators in response to IRs during phase 3 testing. As not all reports of potential IRs were accompanied by a description of the investigator actions, the information is presented both as a percentage of all IRs (n = 381) and as a percentage of those IRs with infusion adjustments recorded (n = 264). Epinephrine was administered to 3 patients: 1 for wheezing, 1 for lip swelling, and 1 for an IR without blood pressure change. None of the patients with IRs required intubation, mechanical ventilation, vasopressors, or hospitalization within 2 hours after infusion, and there were no infusion-related deaths.
TABLE 2.
Adjustments Made to Infusions by Investigators During the RCTs and OLE

IRs as Part of Symptom Clusters
A cluster or constellation of symptoms likely to be of particular concern to clinicians was defined post hoc as the occurrence of stridor, wheezing, perioral or lingual edema, or hemodynamic instability with or without rash or urticaria. Twelve (7%) of 169 patients treated with pegloticase in the RCTs experienced such defined symptom clusters. Two patients had IRs with a constellation of defined symptoms during the first infusion, 3 during the second or third infusion, 5 during the fifth infusion, and the remaining 2 during their seventh and ninth infusions. Most IRs with a symptom constellation were moderate in severity, except for 1 rated mild and 3 rated severe by investigators. For 9 of these 12 patients (including the 2 affected during the first infusion), the IR occurred when sUA was greater than 6 mg/dL, whereas sUA data were not available at the time of these symptoms in 2 patients, and sUA was less than 6 mg/dL in the 1 remaining patient. Eighteen patients had IRs with a constellation of defined symptoms during the OLE, including 11 patients who were initially allocated to placebo in the RCT and started pegloticase in the OLE. In all instances, these IRs occurred when sUA was greater than 6 mg/dL.
Link Between IRs and Treatment Response
Pegloticase is immunogenic, and antipegloticase antibodies were detectable in 89% of patients treated in the RCTs.2 In a post hoc analysis, the formation of high-titer antibodies (titer exceeding 1:2430) was significantly associated with loss of the urate-lowering efficacy of pegloticase (P < 0.001).2 However, the majority of patients with low-titer antibodies (titers <1:2430) maintained sUA in the target range of less than 6 mg/dL. During the RCTs, most IRs occurred when sUA levels were greater than 6 mg/dL. Retrospective analyses showed that the loss of urate-lowering efficacy, as reflected by sUA of greater than 6 mg/dL, preceded a patient’s first IR, whenever it occurred, in 20 (91%) of 22 patients treated with biweekly pegloticase and 24 (71%) of 34 patients receiving monthly pegloticase.2 Infusion-related reactions occurring at the first infusion were reported for 5 patients, 1 with biweekly and 4 with the monthly pegloticase regimen. Excluding these 5 patients (whose pUA values were >6 mg/dL by definition) shows an even higher rate of patients with documented loss of response prior to their first IR (95% and 80% with biweekly and monthly dosing, respectively).
The relationship between loss of urate-lowering efficacy and increased risk of IRs was not apparent during the conduct of the phase 3 clinical trials because investigators and patients were blinded to uric acid levels. Entry into the OLE study required completion of the RCT, so that investigator’s blinding was maintained for many patients for several months into the OLE study. As noted previously, 94 patients in the pooled study population had at least 1 IR related to an infusion of pegloticase. Of these, 81 patients (86%) had sUA of greater than 6 mg/dL (including the 10 patients who experienced an IR at their first infusion), and 13 patients (14%) had sUA of less than 6 mg/dL at the time of their first IR (Fig. 1). When considered on a per-infusion rather than a per-patient basis, the rate of IRs for patients with sUA of less than 6 mg/dL was 0.5 per 100 infusions in the RCTs and 0.8 per 100 infusions in the OLE (Fig. 2). Considering all IRs during the studies (including multiple IRs for individual patients), 91% of those reported in the RCTs and 88% in the OLE study occurred when sUA was greater than 6 mg/dL.
FIGURE 2.
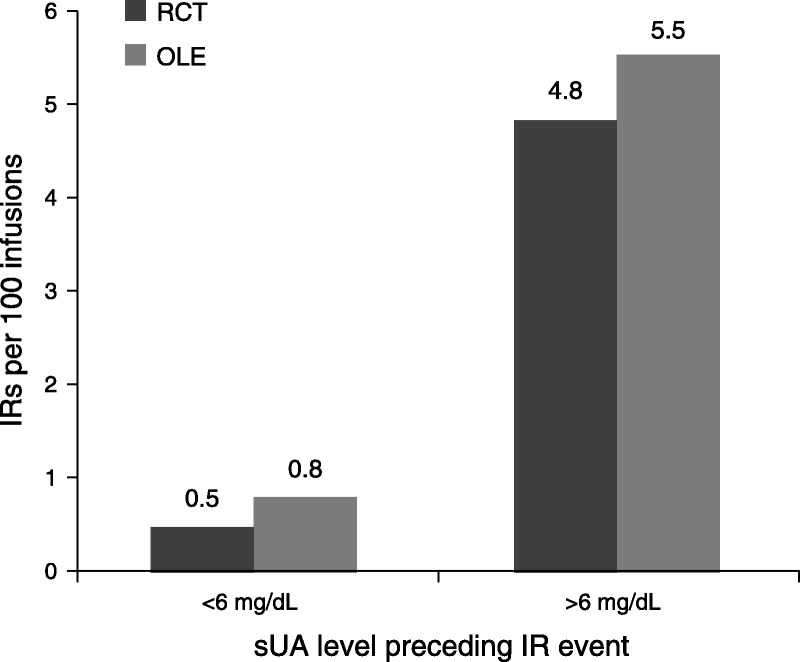
Incidence of IRs per 100 infusions of pegloticase according to sUA.
Discontinuation of pegloticase therapy in patients who lose urate-lowering response offers a valuable approach for mitigating the risk of IR. Using data from the RCTs as an example, 36 (42%) of 85 patients treated with biweekly pegloticase achieved the primary urate-lowering end point, and 22 (26%) of 85 patients in this dosing group had at least 1 IR.2 These results occurred in the absence of specific rules for stopping pegloticase treatment. A post hoc analysis of the biweekly pegloticase dosing group in the RCTs illustrated the impact of several possible stopping rules based on sUA. As shown in Table 3, a stopping rule based on 2 consecutive uric acid measurements of greater than 6 mg/dL reduced the risk of IR by nearly half (from 26% to 14%) while having little effect on the treatment response rate (from 42% to 41%). Use of a stopping rule based on a single sUA measurement of greater than 6 mg/dL would have resulted in a much greater reduction in IR risk (to 8%), but fewer patients would have remained in the study to achieve the primary end point (36%). Comparable changes in IR risk and response rates were seen for stopping rules based on 1 or 2 consecutive sUA measurements of greater than 7 mg/dL.
TABLE 3.
Stopping Rules to Evaluate Benefit (Treatment Response) Versus Risk (IRs) With Biweekly Pegloticase Based on Data Pooled From the RCTs

Serious IRs
Serious adverse events are defined by the US Food and Drug Administration (FDA) as those that are life threatening, require initial or prolonged hospitalization, lead to disability or permanent damage, cause death, or jeopardize the patient such that medical or surgical intervention is needed to prevent 1 of these outcomes.5 In the pegloticase-treated pooled population, 22 (11%) of the 208 patients had a serious IR, including 6 (7%) of 85 patients who received biweekly pegloticase in the RCT, 9 (11%) of 84 patients who received monthly pegloticase, and 7 (18%) of 39 patients who were initially allocated to placebo and then started pegloticase in the OLE study. Serious IRs occurred more frequently at early time points, with 2 episodes at the first infusion of pegloticase, 14 episodes through 6 months, and the remaining 6 episodes occurring after 6 months. As noted above, no serious IR occurred in the placebo groups of the RCTs. All serious IRs resolved with supportive measures.
Anaphylaxis
There was no prespecified definition of anaphylaxis in the pegloticase study protocols, and there were no reports of anaphylaxis in the database from investigators who participated in the RCTs. However, a post hoc analysis was conducted during the FDA review process, in which the following diagnostic criteria for anaphylaxis were used: skin or tissue mucosal involvement and either airway compromise and/or reduced blood pressure with or without associated symptoms, which showed a temporal relationship to the pegloticase or placebo infusion with no other identifiable cause. Using this definition, the FDA identified 14 cases (5%) of anaphylaxis or potential anaphylaxis among the 273 patients tested in the complete phases 2 and 3 clinical development program. Among patients treated with biweekly pegloticase in all clinical trials irrespective of dose, 8 (7%) of 123 met the FDA’s clinical criteria for anaphylaxis. Four of these patients were identified by the FDA as receiving biweekly pegloticase in the RCTs and are presented in Table 4.
TABLE 4.
Summary of 4 IRs Listed in Pegloticase Label

An additional and independent post hoc analysis was conducted using the National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network criteria for anaphylaxis with an unknown allergen.6 These criteria mandate acute onset of a reaction over minutes to several hours with involvement of the skin, mucosal tissues, or both (eg, hives, pruritus, flushing, or swollen lips, tongue, or uvula) and either respiratory compromise (eg, dyspnea, wheezing, stridor, or hypoxemia) and/or reduced blood pressure or associated symptoms of end-organ dysfunction (eg, hypotonia, syncope, or incontinence). Applying these criteria, 3 (4%) of 85 patients treated with biweekly pegloticase met the National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network clinical criteria for anaphylaxis.2
DISCUSSION
These data from the RCTs and OLE study show that the risk of IR with pegloticase is associated with loss of urate-lowering efficacy as reflected by sUA of greater than 6 mg/dL. For patients who maintain therapeutic response to pegloticase with sUA of less than 6 mg/dL, the risk of IRs is relatively low (<1 event per 100 infusions). In comparison, the rate increases to approximately 5 events per 100 infusions in patients with sUA of greater than 6 mg/dL. Similarly, the analyses of various subsets of IRs, including those characterized by a constellation of symptoms, those meeting the FDA definition for serious adverse events, and those suggestive of anaphylaxis, indicated that the great majority of events occurred in patients with preinfusion sUA measurement exceeding 6 mg/dL.
The loss of urate-lowering efficacy has been mechanistically associated with the production of high-titer (>1:2430) antipegloticase antibodies. In a post hoc analysis, only 2% of patients with antibody titers exceeding 1:2430 maintained a urate-lowering response to pegloticase compared with 63% of patients who were treated for at least 2 months without developing high-titer antibodies (P < 0.001).2,7 These findings imply that the risk of IRs may be associated with antibody titer. Indeed, the incidence of IRs was higher among patients who developed high-titer antibodies compared with those who had titers that did not exceed 1:2430 (60% vs 19%; P < 0.001).2 It is, however, important to note that antibody titers at the time of a first IR (versus the final highest titer) were not a reliable predictor of urate response and IR risk.
The mechanism(s) underlying the development of IRs with pegloticase remain unclear. Pegloticase induces production of antibodies of the immunoglobulin M and immunoglobulin G isotypes, which appear to target the polyethylene glycol moiety rather than the uricase.8,9 In the RCTs, these antibodies did not neutralize pegloticase activity in vitro except in 1 patient.7
From a practical perspective, the risk of IR and anaphylaxis with pegloticase therapy is likely to be reducible through routine monitoring of sUA levels prior to each pegloticase infusion, and then discontinuing pegloticase therapy if sUA exceeds 6 mg/dL, particularly when 2 consecutive measurements show sUA greater than 6 mg/dL or an IR occurs. In fact, early postapproval safety surveillance in the United States has demonstrated a 69% reduction in the rate of IR compared with the IR rate recorded during the RCTs.10 The stopping rules exercise shown in Table 3 provides a framework for appreciating the shift in risk-benefit that is possible with preinfusion uric acid–guided treatment within the RCT population and should be augmented in real-world practice with clinical experience and individualized care.
All patients treated in the phase 3 studies were premedicated with H1-antihistamines and corticosteroids and then monitored closely for signs and symptoms of IRs during the 2-hour infusion of pegloticase. Monitoring is recommended even if no adverse events were observed during or after previous infusions, and sUA testing should be done as near as is practical to each successive infusion.
In the event of an IR, besides any interventions implemented to manage the patient’s medical condition, the infusion rate could be slowed by half or stopped. If the reaction resolved, then the infusion can proceed to completion, either at the original rate, at a reduced rate, or using an increased volume of diluent. In no instance during the clinical trials did a second IR occur upon restarting the infusion on the same day. However, the infusion should be stopped and not restarted if the IR fails to resolve within 1 hour, or if the patient is believed to be at risk for an anaphylactic reaction. If a severe or serious IR occurs, the infusion should be stopped, and supportive treatment with antihistamines, fluids, corticosteroids, analgesics, and/or epinephrine provided. Physician discretion should guide further treatment if a patient experiences an IR and their preinfusion sUA is less than 6 mg/dL. In summary, monitoring sUA levels, stopping pegloticase if urate-lowering treatment response is lost, and monitoring patients during infusions are effective steps that should be taken to minimize the risk of IRs in patients treated with pegloticase.
KEY POINTS
Monitoring preinfusion uric acid levels in patients treated with pegloticase provides critical information on the response to therapy and the appropriateness of continuing treatment.
Understanding the relationship between uric acid response and IRs seen in the pegloticase clinical trials combined with ongoing postapproval pharmacovigilance data can continue to inform patient management and minimize IR risk.
Figure.

No caption available.
Footnotes
Development of this manuscript was supported by Savient Pharmaceuticals, Inc, and Crealta Pharmaceuticals LLC. Savient Pharmaceuticals, Inc, had licensed worldwide rights to the technology related to pegloticase from Duke University and Mountain View Pharmaceuticals. Those rights are now owned by Crealta Pharmaceuticals LLC.
H.S.B.B. previously received grant/research support from Savient Pharmaceuticals; currently receives grant/research support from Takeda, Regeneron, and Ardea Biosciences; and is a consultant for Takeda and Crealta. He is a member of the speakers’ bureau for Takeda and Savient. R.A.Y. received grant/research support from Savient Pharmaceuticals, Inc, and receives support from Takeda. F.D.O. was an employee of Savient Pharmaceuticals, Inc, at the time of manuscript development and submission. J.S.S. receives research/grant and/or consulting fees from Merck, Astra Zeneca, and Eli Lilly. He previously consulted for and received research/grant support from Savient Pharmaceuticals, Inc. He has an ownership position in Academic Partners for Medical Education LLC and holds nonremunerative positions of potential influence in the Medanta Duke Research Institute. M.A.B. receives grant/research support from Takeda and is/has been a paid consultant for Takeda, Ardea Biosciences (AstraZeneca), BioCryst, Regeneron, Crealta, CymaBay, Metabolex, and Sobi. He previously consulted for and received support from Savient Pharmaceuticals, Inc.
REFERENCES
- 1. Schlesinger N, Yasothan U, Kirkpatrick P. Pegloticase. Nat Rev Drug Discov. 2011; 10: 17– 18. [DOI] [PubMed] [Google Scholar]
- 2. Sundy JS, Baraf HS, Yood RA, et al. Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: two randomized controlled trials. JAMA. 2011; 306: 711– 720. [DOI] [PubMed] [Google Scholar]
- 3. Terkeltaub R. Prophylaxis of attacks of acute gouty arthritis. In: Terkeltaub R, ed. Gout and Other Crystal Arthropathies. Philadelphia, PA: Elsevier Saunders; 2012: 187– 193. [Google Scholar]
- 4. Becker MA, Baraf HS, Yood RA, et al. Long-term safety of pegloticase in chronic gout refractory to conventional treatment. Ann Rheum Dis. 2013; 72: 1469– 1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.US Food and Drug Administration. Safety. What is a serious adverse event? [US FDA Web site]. April 18, 2013. Available at: http://www.fda.gov/safety/medwatch/howtoreport/ucm053087.htm. Accessed May 21, 2013.
- 6. Sampson HA, Muñoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report—second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. Ann Emerg Med. 2006; 47: 373– 380. [DOI] [PubMed] [Google Scholar]
- 7. Lipsky PE, Calabrese LH, Kavanaugh A, et al. Pegloticase immunogenicity: the relationship between efficacy and antibody development in patients treated for refractory chronic gout. Arthritis Res Ther. 2014; 16: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sundy JS, Ganson NJ, Kelly SJ, et al. Pharmacokinetics and pharmacodynamics of intravenous PEGylated recombinant mammalian urate oxidase in patients with refractory gout. Arthritis Rheum. 2007; 56: 1021– 1028. [DOI] [PubMed] [Google Scholar]
- 9. Ganson NJ, Kelly SJ, Scarlett E, et al. Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase. Arthritis Res Ther. 2006; 8: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malamet R, Howson T, Yeo A, et al. Real world risk of infusion reactions with pegloticase treatment: findings from post-approval US safety data. Available at: https://b-com.mci-group.com/Abstract/Statistics/AbstractStatisticsViewPage.aspx?AbstractID=148548. Accessed May 21, 2013.