

Abstract
Objective
Fibroblast-like synoviocytes (FLS) participate in joint inflammation and damage during rheumatoid arthritis (RA) and its animal models. The purpose of this study was to define the importance of KCa1.1 (BK, Maxi-K, Slo1, KCNMA1) channel expression and function in FLS and to establish these channels as potential new targets for RA therapy.
Methods
We compared KCa1.1 expression levels in FLS from rats with the pristane-induced arthritis (PIA) model of RA and in FLS from healthy rats. We then used ex vivo functional assays combined with siRNA-induced knock-down, over-expression, and functional modulation of KCa1.1 in PIA-FLS. Finally, we determined the effectiveness of modulating KCa1.1 in two rat models of RA, moderate PIA and severe complete Freund’s adjuvant collagen-induced arthritis (CFA-CIA).
Results
We found that PIA-FLS express the KCa1.1 channel as their major potassium channel, as do FLS from patients with RA. In contrast, FLS from healthy rats expressed fewer of these channels. Inhibiting the function or expression of KCa1.1 ex vivo reduced the proliferation, production of proteases, and invasive properties of PIA-FLS whereas opening native KCa1.1 or over-expressing the channel enhanced the invasiveness of both PIA-FLS and FLS isolated from healthy rats. Treatment with a KCa1.1 channel blocker starting at onset of clinical signs stopped disease progression in both PIA and CFA-CIA, reduced joint and bone damage, and inhibited FLS invasiveness and proliferation.
Conclusion
Our results demonstrate a critical role for KCa1.1 channels in the regulation of FLS invasiveness and suggest they represent a potential therapeutic target for RA.
Rheumatoid arthritis (RA) is a chronic and systemic inflammatory disease that preferentially targets diarthrodial joints (1, 2). It is characterized by extensive synovial hyperplasia and cartilage and bone damage, leading to disability. While the etiology of RA is not fully understood, it involves the activation of endothelial and synovial cells, as well as the activation and recruitment of immune cells to the synovium.
Fibroblast-like synoviocytes (FLS) are prominent in the RA pannus where they secrete proteases that degrade collagen, cytokines and chemokines that induce the accumulation and activation of inflammatory cells, and growth factors that induce angiogenesis (3, 4). Importantly, FLS from patients with RA (RA-FLS) are highly invasive and can migrate from affected to healthy joints (5). Their ex vivo invasive properties tightly correlate with histological and radiographic damage in RA and its experimental models (6, 7); this damage itself being correlated with disease severity and an increased risk of disability, deformities, and premature death (8). Thus, reducing the pathogenic properties of RA-FLS represents an attractive target for the treatment of RA, particularly since no RA therapies have been developed to specifically target these cells.
We have previously identified the KCa1.1 channel (BK, maxi-K, Slo1, KCNMA1) as the major potassium channel expressed by RA-FLS (9). Inhibiting the function of this channel in RA-FLS through blocking its pore-forming α subunit ex vivo perturbs the calcium homeostasis of the cells and inhibits their proliferation, migration, and invasiveness, as well as their production of proteases, chemokines, and growth factors (9). These results suggest KCa1.1 channels as important regulators of the destructive phenotype of RA-FLS and as therapeutic target for RA by attenuating these pathogenic functions. We tested this possibility in the current study, using experimental arthritis in rats. We first demonstrated that functional KCa1.1 are the major potassium channels at the plasma membrane of FLS from rats with the pristane-induced arthritis (PIA) model of RA and are expressed in larger numbers by PIA-FLS when compared to FLS from healthy animals. Blocking KCa1.1 inhibited the proliferation of PIA-FLS and reduced their ability to produce the matrix metalloproteinase (MMP) pro-MMP-2. Importantly, blocking KCa1.1 or reducing its expression reduced the invasiveness of PIA-FLS. In contrast, opening native KCa1.1 or over-expression of the channel enhanced the invasiveness of PIA-FLS and of healthy rat FLS. Treatment of rats at onset of clinical signs in two models of RA with a KCa1.1-specific blocker reduced disease severity, synovial inflammation, cartilage and bone damage, and inhibited the ex vivo invasiveness of FLS.
Materials and Methods
Animals and cells
Experiments involving rats were conducted after IACUC approval. Female Dark Agouti (DA) rats, 8-11 weeks old (Harlan-Sprague-Dawley), and Lewis rats, 8-11 weeks old (Charles River), were provided food and water ad libitum. FLS were isolated from synovial tissues and maintained in culture as described (10). All ex vivo assays were performed with FLS after passage 3 ex vivo (> 95% purity).
Manipulation of ion channel expression and function
We used two well-characterized small molecule blockers of KCa1.1, paxilline (Fermentek) and tetraethyl ammonium chloride (TEA; Sigma-Aldrich), and the selective peptide blocker of KCa1.1 iberiotoxin (Peptides International) (11). As an agonist of KCa1.1, we used phloretin (Sigma-Aldrich) (12). The KCa3.1 blocker TRAM-34 and the Kv1.3 blocker PAP-1 (11) were gifts from Dr. Wulff (Department of Pharmacology, University of California, Davis). The KCa2.x blocker apamin (11) and the Kv1.3 blocker ShK-186 (13) were from CS Bio. SMARTpool siRNA directed to KCa1.1 (target sequences: GACCUGAUCUUCUGCUUAA, GAUCCAAGAAGGUACUUUA, GAAUUUACCGGCUGAGAGA, UCGAAUAUCAUGAGAGUAA) was purchased from Thermo Scientific and transfected into FLS following manufacturer's instructions for analyzed 48 hrs later. KCa1.1 and GFP were overexpressed in FLS using the Bacmam baculovirus system. KCa1.1 and GFP were subcloned into a pFastbac vector (Invitrogen) modified by replacing the insect polyhedron promoter with a mammalian cytomegalovirus promoter (14). This donor plasmid was recombined into the baculovirus genome using the Bac-to-bac system (Invitrogen) and transfected into SF9 insect cells for virus production. FLS were transduced with the virus at a multiplicity of infection of 10 and analyzed 6 hrs later.
Immunocytochemistry
Cells were stained to detect the α subunit of KCa1.1, as described (9). Wheat germ agglutinin was used to detect the plasma membrane and DAPI to visualize the nucleus (Invitrogen). Photos were taken on a Zeiss LSM 510 inverted Laser Scanning Microscope with a 40x-oil Fluor objective.
Patch-clamp electrophysiology
All experiments were conducted at room temperature in the whole-cell configuration of the patch-clamp technique using an EPC10-USB amplifier (HEKA instruments) as described (9). Cells were plated onto glass coverslips, allowed to adhere at 37°C, and placed on an inverted microscope (Olympus IX71) before the medium was exchanged for the bath solution containing (in mM): 140 K+, 20 HEPES, 2 Mg2+, 10 Cl-. Resistances of glass pipettes pulled and polished shortly before use averaged 1.8 MΩ. Pipettes were filled with solutions containing (in mM): 140 K+, 20 HEPES, 10 Cl-, 5 EGTA (0 Ca2+) or 140 K+, 20 HEPES, 10 Cl-, 5 HEDTA, and the concentration of free Ca2+, measured with a Ca2+ electrode (Orion Research), was adjusted to 5.5 μM with CaCl2. All solutions were adjusted to pH 7.2 with methanesulfonic acid and their osmolarity was 300-310 mOsm. Pipettes were brought in contact with a cell using a micromanipulator to form a high-resistance (> 1 GΩ) seal between pipette and cell. Suction (30-40 psi) was applied to the pipette to break the plasma membrane and obtain the whole-cell configuration. The pipette solution was allowed to perfuse the cell before potassium currents were recorded. The holding potential between pulses was -80 mV. Currents were elicited by 30 msec pulses from 60 mV to 180 mV in 10 mV increments. To test the effects of KCa1.1 blockers, stable potassium currents were recorded at 160 mV, followed by rapid perfusion of the modulators diluted in bath solution and recording until steady-state block was reached. A dose-response curve was generated (GraphPad Prism) to determine the IC50 of each blocker.
Flow cytometry
For KCa1.1 detection, FLS were enzymatically lifted from the culture flasks, permeabilized with saponin, and stained with antibodies against the KCa1.1 α subunit (NeuroMab) followed by fluorophore-conjugated secondary antibodies (Invitrogen). For the cytotoxicity assays, FLS staining intensity with 7-aminoactinomycin D was determined after a 48-hr incubation with vehicle, paxilline, or staurosporine, as described (9). Data were acquired on a CantoII flow cytometer with the FACSDiva software (BD Biosciences) and analyzed with FlowJo (Treestar). We used the 2-sample univariate Kolmogorov-Smirnov test with super-enhanced Dmax subtraction (SED) to compare the difference in area under the curve between two samples (15).
Western blotting
Western blots to detect KCa1.1 channel α subunits were conducted as described (9). Visualization was performed using an Odyssey Imaging System (LI-COR).
Ex vivo functional assays (invasion, proliferation, zymography)
The effects of KCa1.1 channel modulators and of KCa1.1 siRNA and over-expression on the invasion of FLS were determined using Matrigel-coated transwell systems (BD Biosciences), as described (6, 9). Proliferation assays were performed using incorporation of [3H] thymidine in the DNA of dividing cells, as described (9). We used gelatin gel zymography (Invitrogen) to measure production of pro-matrix metalloproteinase-2 (pro-MMP-2) in FLS culture supernatants, as described (9, 16).
Circulating half-life of free paxilline
Serum from healthy DA rats was spiked with known amounts of paxilline and the blocking activity on KCa1.1 channels stably expressed in HEK293 cells (gift from Dr. Wulff, University of California, Davis) was determined by patch-clamp to establish a standard dose-response curve (17). Serial serum samples from healthy DA rats were collected (18) after an intraperitoneal injection of paxilline. They were tested for KCa1.1 blocking activity by patch-clamp and free paxilline serum levels determined from the standard curve.
Experimental arthritis induction and monitoring
PIA was induced in DA rats by the subcutaneous injection of 150 μl pristane (2,6,10,14-tetramethylpentadecane; MP Biomedicals) at the base of the tail (19, 20). Complete Freund's adjuvant collagen-induced arthritis (CFA-CIA) was induced in Lewis rats by the subcutaneous injection at the base of the tail of 200 μl of a 1:1 emulsion composed of 2 mg/ml porcine type-II collagen (Chondrex) and complete Freund's adjuvant (Difco) (21). In both models, a score of 1 point was given for each swollen and red toe, and each mid-foot, digit, or knuckle, and 5 points for each swollen ankle or wrist for a maximum score of 60 per animal (20). At onset of clinical signs, rats were randomly placed in the vehicle or paxilline groups and for intraperitoneal injections every other day.
Paw X-rays, histology, and immunohistochemistry
At the end of the in vivo PIA experiments, animals were euthanized by deep anesthesia with inhaled isoflurane, followed by cardiac perfusion with saline (22). Death was ensured by decapitation. Paws were immediately collected and either used for X-ray imaging (Kodak Image Station) or fixed, decalcified, paraffin-embedded, and sectioned for staining with hematoxylin and eosin, safranin-O and fast-green (19), tartrate-resistant acid phosphatase (23), or MMP-2 (24) and scored by an investigator blinded to the treatment received by each animal, as described (25). Representative photos were taken with an Olympus BX41 microscope at 10X or 40X magnification.
Statistical analysis
Data are expressed as mean ± SEMs. Statistical analysis was performed using the non-parametric Mann-Whitney U-test for all tests but the disease severity scoring during the in vivo experiments, for which we used two-way ANOVA (GraphPad Prism). P values lower than 0.05 were considered significant in all statistical analyses.
Results
PIA-FLS express functional KCa1.1 at their plasma membrane
Since potassium channel expression varies between species in different cell types (13, 26-28) and Kv rather than KCa1.1 channels were identified in rabbit synoviocytes (29) while we have identified KCa1.1 as the major functional potassium channel in human FLS (9), we first determined whether rat FLS share the phenotype of human FLS in terms of KCa1.1 expression to validate using rat models of RA to evaluate KCa1.1 modulation. Immunocytochemistry demonstrated the expression of KCa1.1 channel pore-forming α subunits at the plasma membrane of PIA-FLS (Figure 1A), as was previously observed in RA-FLS (9). As in RA-FLS and other cell types (30), staining of α subunits of KCa1.1 was also detected in the nucleus of PIA-FLS. We used patch-clamp electrophysiology, the “gold-standard” technique to determine whether the KCa1.1 at the plasma membrane of PIA-FLS are functional (Figure 1B). Whole-cell patch-clamp recordings showed an outward potassium current whose amplitude increased with the voltage. The normalized potassium conductance was voltage-dependent and characterized by a half-activation voltage (V1/2) of 220 ± 0.9 mV in the absence of intracellular calcium. It shifted to lower voltages (V1/2 = 50 ± 0.6 mV) when 5.5 μM calcium was present in the pipette, showing that the channel is calcium-dependent. Finally, paxilline and TEA both blocked the PIA-FLS potassium currents. Since KCa1.1 is the only potassium channel to display both voltage- and calcium-dependence and to be sensitive to both paxilline and TEA (30-32), our results demonstrate that, like RA-FLS, PIA-FLS express functional KCa1.1 at their plasma membrane. In addition, more than 80% of the PIA-FLS potassium currents were blocked by paxilline and TEA, showing that KCa1.1 is the major functional potassium channel at the plasma membrane of these cells.
Figure 1.
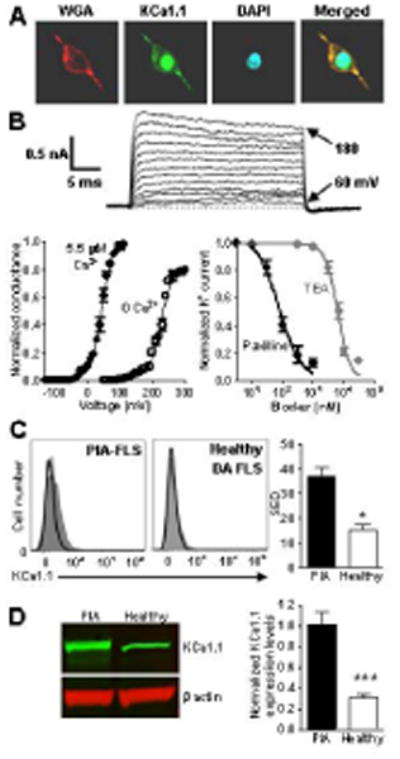
PIA-FLS express more KCa1.1 channels at their plasma membrane than FLS from healthy DA rats. A, Immunocytochemistry staining of PIA-FLS. Left, membrane staining with wheat germ agglutinin (WGA); middle left, KCa1.1 staining; middle right, nuclear staining with DAPI; right, merged. B, Electrophysiology. Top, Representative family of potassium current elicited from a PIA-FLS with a calcium-free pipette solution. Bottom-left, Conductance versus voltage relations normalized to the maximum conductance in 3 PIA-FLS with 0 Ca2+ or 5.5 μM Ca2+ in the pipette. Bottom-right, Inhibition by paxilline (black; IC50 = 61 ± 24 nM) and TEA (grey; IC50 = 426 ± 12 nM) of PIA-FLS potassium currents. Mean ± SEM; n = 3 cells. C, Left, representative flow cytometry histograms of FLS stained for KCa1.1 (shaded). Black lines = control. Right, mean ± SEM of SED value from a Kolmogorov-Smirnov comparison of staining levels in n = 3 independent experiments. D, Left, representative Western blot against the KCa1.1 α subunit on PIA-FLS and FLS from healthy DA rats. Right, KCa1.1 α subunit protein levels in PIA-FLS (black) and FLS from healthy DA rats (white) normalized to β actin levels. Mean ± SEM of 4 Western blots on different extracts. *p<0.05; ***p<0.001.
PIA-FLS express more KCa1.1 than do FLS from healthy DA rats
Flow cytometry with an antibody directed to the α subunit of KCa1.1 demonstrated channel expression in PIA-FLS (Figure 1C). In contrast, these channels were barely detectable on FLS isolated from healthy DA rats. Western blot against KCa1.1 α subunits revealed a 3-fold higher expression of this channel by PIA-FLS when compared with healthy DA rat FLS (Figure 1D), confirming a higher expression of KCa1.1 by PIA-FLS.
Blocking KCa1.1 inhibits the proliferation of PIA-FLS and their production of pro-MMP-2 without cytotoxicity
Blocking KCa1.1 channels expressed by RA-FLS with paxilline or TEA inhibits their proliferation and production of pro-MMP-2 (9). To establish whether KCa1.1 play similar regulatory functions in FLS of both species in order to validate PIA as a relevant model to assess KCa1.1 as a target for RA therapy, we measured the effect of three KCa1.1 antagonists on their proliferation and pro-MMP-2 production. Paxilline inhibited the proliferation of PIA-FLS in a dose-dependent manner with an IC50 of ≈ 15 μM (Figure 2A). Inhibition was also observed with iberiotoxin and TEA (Figure 2A, B). Antagonists of KCa2, KCa3.1, and Kv1 channels that do not target KCa1.1 did not affect PIA-FLS proliferation (Figure 2B). In toxicity assays, staurosporine induced the death of PIA-FLS whereas paxilline did not (Figure 2C); thus demonstrating that paxilline is not toxic to PIA-FLS at the concentrations used here. PIA-FLS culture supernatants contained detectable amounts of pro-MMP-2. Both paxilline and TEA induced a dose-dependent inhibition of pro-MMP-2 production by these cells with IC50s of ≈ 15 μM and ≈ 40 mM, respectively (Figure 2D).
Figure 2.
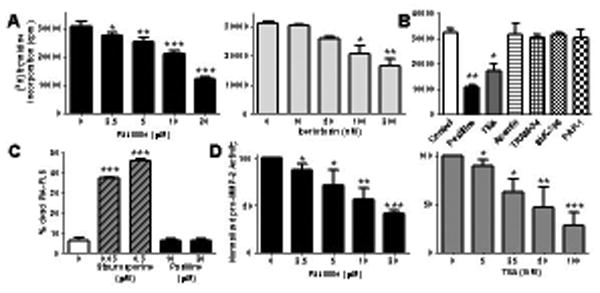
Blocking KCa1.1 channels inhibits the proliferation and pro-MMP-2 production by PIA-FLS without inducing cell death. A, Dose-dependent inhibition of PIA-FLS proliferation by paxilline (left) and iberiotoxin (right). Mean ± SEM; n = 4 independent experiment ran in triplicate. B, Effects of blockers of KCa1.1 (20 μM paxilline; 50 mM TEA), other KCa (100 nM apamin; 1 μM TRAM-34), and Kv (100 nM ShK-186; 1 μM PAP-1) channels on the proliferation of PIA-FLS. n = 3 independent experiments, each ran in triplicate and shown as mean ± SEM. C, Dose-dependent toxicity of staurosporine (striped bars) and paxilline (black) after a 48 hr incubation with PIA-FLS. Mean ± SEM; n = 3 independent experiment ran in duplicate. D, Dose-dependent inhibition of pro-MMP-2 production by paxilline (left) and TEA (right), measured by gelatin gel zymography in PIA-FLS supernatants. Mean ± SEM; n = 3 independent experiment ran in duplicate. *p<0.05; **p<0.01; ***p<0.001.
PIA-FLS invasiveness is regulated by the expression and activity of KCa1.1
We used the well-established transwell assay through Matrigel to determine the effects of modulating KCa1.1 expression and function on the invasive behavior of PIA-FLS and of FLS from healthy DA rats. Paxilline, TEA, and iberiotoxin all inhibited the invasiveness of PIA-FLS (Figure 3A), as previously shown with RA-FLS (9). Transfection of PIA-FLS with a pool of siRNA targeted to the α subunit of KCa1.1 reduced expression of the channel and the invasive capabilities of the cells (Figure 3B). The KCa1.1 channel agonist phloretin (12) increased PIA-FLS invasiveness in the absence of siRNA and in the presence of control siRNA. The invasiveness inhibited by siRNA against KCa1.1 was not rescued by the addition of phloretin, confirming that the siRNA did knock-down expression of KCa1.1 and that the phloretin-induced increase in invasiveness was due to its effects on KCa1.1. Phloretin enhanced the invasion of PIA-FLS but had no effect on the invasive properties of healthy DA rat FLS (Figure 3C). Healthy DA FLS express few KCa1.1 at their plasma membrane; opening these channels is likely insufficient for induction of cell invasiveness. We next over-expressed KCa1.1 α subunits in both PIA-FLS and healthy DA rat FLS (Figure 3D). This over-expression enhanced the invasiveness of both PIA-FLS and healthy DA FLS. In both cell types, this enhanced invasiveness was sensitive to paxilline. As a control, over-expression of GFP did not affect FLS invasiveness or sensitivity to paxilline.
Figure 3.
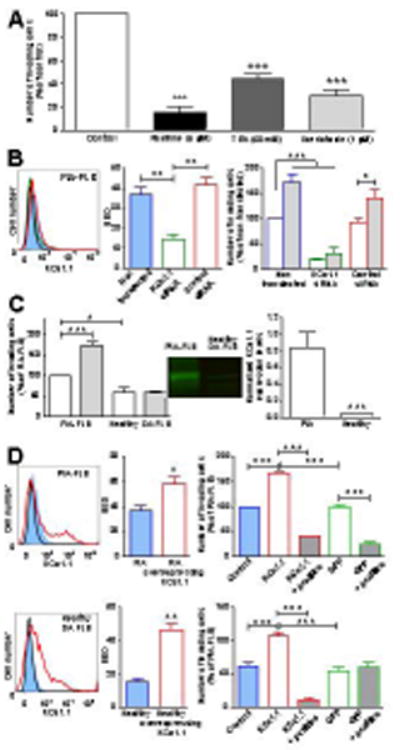
Expression of KCa1.1 regulates rat FLS invasiveness. A, Effects of KCa1.1 blockers on PIA-FLS invasiveness. Mean ± SEM, n=3. B, Left, representative flow cytometric staining of PIA-FLS for KCa1.1 (blue) after transfection with KCa1.1 siRNA (green) or control siRNA (red). Black = control. Middle, quantification of the flow cytometry. Mean ± SEM, n=3. Right, Effects of KCa1.1 siRNA (green) or control siRNA (red) in the absence (empty bars) or presence of 100 μM phloretin (grey-filled bars) on PIA-FLS invasiveness. Mean ± SEM, n=3. C, Left, Effects of phloretin (grey) and vehicle (white) on FLS invasiveness. Mean ± SEM, n=3. Middle, representative Western blot of plasma membrane fractions. Right, Western blot quantification. Mean ± SEM, n=4. D, Left, representative flow cytometric histograms of FLS stained for KCa1.1 before (blue) and after over-expression of KCa1.1 (red). Black = background control. Middle, SED values comparing staining levels against the background control; mean ± SEM, n=3. Right, FLS invasiveness. Blue = unmodified cells, red border = cells over-expressing KCa1.1, green border = cells over-expressing GFP, grey shading = cells treated with 20 μM paxilline. Mean ± SEM of 3 independent experiments. *p<0.05; **p<0.01; ***p<0.001.
Paxilline administration stops disease progression in the PIA rat model of RA and inhibits the ex vivo invasiveness and proliferation of PIA-FLS
We used patch-clamp to estimate the amount of free paxilline in the serum of DA rats following a single intraperitoneal injection (Figure 4A). Blocker activity was detectable in the serum as early as 15 min after administration and the level fell to a baseline of ≈ 400 nM over 720 min. The disappearance of free paxilline could be fit to a single exponential with a half-life of ≈ 40 min. We next induced PIA and treated the rats with either vehicle or paxilline every other day, starting at onset of clinical signs. Rats developed moderate arthritis with an average maximum score of ≈ 25. Paxilline stopped the progression of PIA and reduced overall disease severity by ≈ 50% (Figure 4B). Animals were euthanized at the end of the experiments for isolation of FLS and ex vivo invasion assays. PIA-FLS from vehicle-treated rats were significantly more invasive and proliferative than FLS from healthy DA rats. Treatment of PIA rats with paxilline reduced the ex vivo invasiveness and proliferation of PIA-FLS (Figure 4C).
Figure 4.
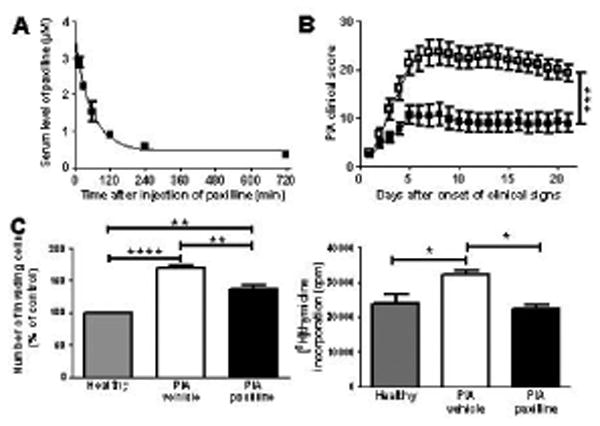
Administration of paxilline stops the progression of PIA and reduces the invasiveness and proliferation of PIA-FLS. A, Circulating half-life of free paxilline measured by patch-clamp electrophysiology against a standard curve. Data from 3 rats at each time point after a single intraperitoneal injection of 20 mg/kg paxilline were fitted to a single exponential decay. Half-life ≈ 40 min. B, Effect of paxilline (■; n=18 rats) and vehicle (□; n=18 rats), administered intraperitoneally every other day starting at onset of clinical signs, on the progression of PIA in DA rats. Mean ± SEM; 3 independent experiments. C, Ex vivo invasiveness (left) and proliferation (right) of FLS isolated from 3 vehicle-treated and 3 paxilline-treated PIA rats and from 3 healthy DA rats measured in triplicates. Mean ± SEM, n=3 independent experiments. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Paxilline administration reduces PIA-induced joint damage
As previously described (19, 33), DA rats with PIA presented with a highly abnormal joint histology including pronounced synovial hyperplasia and pannus formation with detectable MMP-2, immune infiltrates in the synovium and periosteum, angiogenesis, fibrosis, and cartilage erosions (Figure 5A-B). Treatment with paxilline starting at onset of clinical signs significantly reduced all aspects of the abnormal histology other than synovial angiogenesis and periosteum calcification and thickening. X-rays performed on hind paws from rats with PIA treated with vehicle demonstrated severe bone and joint damage that was reduced by treatment with paxilline (Figure 5C).
Figure 5.
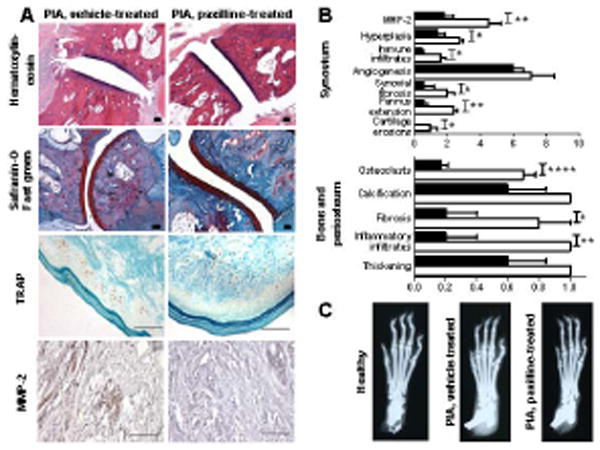
Treatment of PIA rats with paxilline reduces joint damage. A, Representative images of joints from PIA rats treated with vehicle (left) or paxilline (right) and stained with hematoxylin and eosin (10x magnification), safranin-O and fast green (10x magnification), TRAP (40x magnification), or anti-MMP-2 antibodies (40x magnification). All scale bars represent 100 μm. B, Histology scoring of PIA rats treated with vehicle (white) or paxilline (black). Mean ± SEM; n = 5 rats per group. C, Representative X-rays of paws from a healthy DA rat and from vehicle-treated and paxilline-treated PIA rats. *p<0.05; **p<0.01.
Paxilline administration stops disease progression in the CFA-CIA rat model of RA and inhibits the ex vivo invasiveness and proliferation of CFA-CIA-FLS
To determine if paxilline is beneficial in a different and even more severe model of experimental arthritis, we induced CFA-CIA in Lewis rats. In this model, all vehicle-treated animals developed severe arthritis with the maximum score of 60. As in the PIA model, treatment with paxilline every other day, starting at onset of clinical signs, stopped disease progression and reduced the overall disease severity by ≈ 30% (Figure 6A). FLS isolated from CFA-CIA rats treated with vehicle were significantly more invasive and proliferative than FLS from healthy Lewis rats; paxilline treatment of the rats significantly reduced the ex vivo invasiveness (Figure 6B) and proliferation (Figure 6C) of CFA-CIA-FLS.
Figure 6.
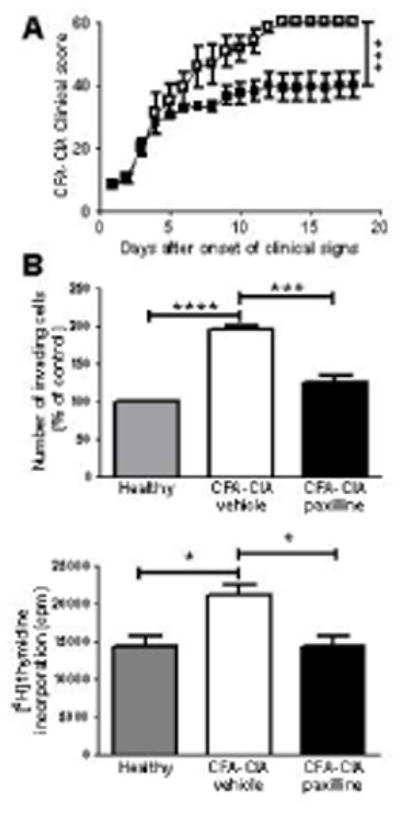
Administration of paxilline stops the progression of severe CFA-CIA and reduces the invasiveness and proliferation of CFA-CIA-FLS. A, Effect of paxilline (■) and vehicle (□), administered intraperitoneally every other day starting at onset of clinical signs, on the progression of CFA-CIA in Lewis rats. Mean ± SEM; n = 6 rats per group. B, Ex vivo invasiveness (top) and proliferation (bottom) of FLS isolated from 3 vehicle-treated and 3 paxilline-treated CFA-CIA rats and from 3 healthy Lewis rats measured in triplicates. Mean ± SEM of 3 independent experiments. ***p<0.001; ****p<0.0001.
Discussion
FLS play important roles in RA and its animal models and represent attractive targets for the treatment of RA. Here, we found that PIA-FLS express functional KCa1.1 at their plasma membrane, as do RA-FLS, validating rat models to test KCa1.1 modulators as potential therapeutics for RA. Importantly, we showed that the expression levels of these channels by PIA-FLS were higher than by FLS from healthy DA rats. Modulating the expression or function of KCa1.1 in PIA-FLS ex vivo reduced their pathogenic functions, as we had previously shown in RA-FLS (9). Finally, treatment of rats with PIA or CFA-CIA with a KCa1.1 blocker from onset of clinical signs stopped disease progression, reduced overall disease severity, FLS invasiveness and proliferation, and joint damage.
Our findings suggest that KCa1.1 is the major potassium channel at the surface of PIA-FLS because paxilline and TEA both inhibit >80% of the potassium currents in whole-cell patch-clamp. These results are in agreement with KCa1.1 being the major potassium channel in RA-FLS (9). Kv channels were described in rabbit synoviocytes (29) whereas the major potassium current in rat and human FLS is carried by KCa1.1, as demonstrated by a dependence on both changes in membrane voltage and intracellular calcium concentrations on the open probability of the channel. Such a difference in potassium channel phenotype between species has been described with other cell types (13, 26-28). Furthermore, KCa1.1 blockers and siRNA inhibited the invasiveness of PIA-FLS, suggesting that KCa1.1 plays a major role in the regulation of PIA-FLS function, regardless of the possible expression of other potassium channels. Finally, blockers of KCa2 and Kv1 channels did not affect production of pro-MMP-2 by RA-FLS (9) or the proliferation of PIA-FLS.
Paxilline inhibited KCa1.1 currents measured by patch-clamp in PIA-FLS with an IC50≈60 nM whereas concentrations in the low μM range are necessary for the inhibition of cell function. This apparent discrepancy was observed with these and other potassium channel blockers in other cells (13, 28, 34). A number of factors likely participate in this difference in responses. First, it may be necessary to block >90% of the channels for a sustained period of time to alter cell function. Second, the solutions used during patch-clamp are well-defined salt solutions whereas the tissue culture media used during functional assays are more complex and ion channel modulators are known to bind to medium components such as serum proteins, thereby decreasing the concentration of free compound to block the channel. Finally, patch-clamp provides a direct and rapid readout of the effects of the modulators on the channels whereas functional assays likely involve multiple cell signaling pathways with varying dependence on KCa1.1 channel activity.
We have chosen phloretin as an agonist of KCa1.1 because it opens the channels regardless of its activation state prior to addition of the drug (12). It should be noted that phloretin, while widely used to open KCa1.1, is not selective for this channel. For example, it also inhibits the GLUT2 glucose transporter (35). However, our data suggest that the increase in PIA-FLS invasiveness induced by phloretin is mediated through opening KCa1.1 as these effects are absent following transfection of PIA-FLS with KCa1.1 siRNA and on healthy DA FLS that express minimal amounts of KCa1.1 at their plasma membrane.
We found that the expression of functional KCa1.1 is necessary to regulate PIA-FLS invasiveness. However, the molecular mechanisms of this regulation remain to be established and are likely to involve multiple signaling pathways. We have previously shown that blocking KCa1.1 induces a calcium transient in RA-FLS (9), suggesting that this channel plays a role in FLS calcium homeostasis, an important factor in cell migration and invasion. In addition, the C-terminus of KCa1.1 contains a binding site for cortactin (36) and may therefore directly affect actin rearrangements during cell migration. Furthermore, the C-terminus of KCa1.1 contains an immunoreceptor tyrosine-based activation motif (ITAM) that can recruit the non-receptor spleen tyrosine kinase Syk (37), itself over-expressed by RA-FLS and involved in MMP production (38).
We used patch-clamp to estimate the pharmacokinetics of paxilline. This technique only allows the detection of free paxilline and is therefore likely to underestimate the total amount of circulating paxilline as some of this lipophilic compound is probably in intracellular compartments and in tissues.
FLS isolated from rats with PIA or CFA-CIA and treated with paxilline remained less invasive and proliferative ex vivo than the cells isolated from rats with PIA or CFA-CIA treated with vehicle. The invasion assays were conducted after 3 passages and multiple changes in culture media between passages. It is unlikely that sufficient amounts of paxilline from the in vivo administration would remain in the cultures at the time of the invasion assays to explain the results. In addition, we have found that PIA-FLS incubated with paxilline regained their invasive properties upon paxilline wash off (data not shown). Taken together, these results suggest that the reduction in immune infiltrates observed after treatment with paxilline decreased the overall inflammatory environment of the joints and thereby prevented activation and acquisition of invasive properties by FLS that were not yet activated at the time treatment began.
Although important in disease pathogenesis, FLS are not the only players in RA and its animal models. Infiltrating immune cells and other resident cells play a significant role in this disease. Current treatments for RA consist of disease-modifying anti-rheumatic drugs and drugs that target TNFα and other components of the immune system (39). All of these therapies affect the immune system and place patients at risk of immunosuppression. Targeting KCa1.1 is unlikely to lead to immunosuppression as most immune cells do not express this channel. We and others have extensively patch-clamped human and rat T, B, and NK lymphocytes and these cells do not express KCa1.1 channels (13, 28, 30, 34, 40, 41); neither do neutrophils or eosinophils (30, 42, 43); these immune cells will therefore not be directly affected by KCa1.1 modulation. Indeed, rheumatoid factor levels in the serum of rats with PIA or CFA-CIA were not affected by paxilline (data not shown). Treatment of PIA rats with paxilline reduced joint damage and decreased immune infiltration into the synovium. We had previously shown that KCa1.1 blockers reduce the production of cytokines and chemokines by RA-FLS (9), suggesting that paxilline reduces immune infiltrates through affecting chemokine and cytokine production by PIA-FLS rather than through directly inhibiting the homing of immune cells. Human macrophages and chondrocytes from equine joints express KCa1.1 and production of cytokines stimulated by lipopolysaccharide, but not TNFα, is sensitive to paxilline (27, 30). Furthermore, mouse osteoclasts express KCa1.1 (44); our data show a decrease in osteoclast numbers after treatment of rats with paxilline. This reduction may be the result of reduced osteoclastogenesis mediated by FLS or a direct effect on osteoclasts. As these cell types may participate in RA pathogenesis (45), the beneficial effects of paxilline in animal models of RA may result in part from affecting them. However, since ion channel expression can greatly vary between species (13, 26-28), the potassium channel phenotype and function of human and rat osteoclasts and chondrocytes will have to be identified before further conclusions can be drawn. The effects of KCa1.1 modulators on chondrocyte, osteoclast, and macrophage function during RA and its animal models remain to be established.
The systemic administration of paxilline was beneficial in stopping the progression of two rat models of RA, including the very severe model of CFA-CIA. However, paxilline is a known tremorgenic compound since it can cross the blood-brain barrier and block KCa1.1 central nervous system neurons (46). Indeed, we observed reversible tremors in rats after the administration of paxilline. In addition, modulation of KCa1.1 affects blood pressure and urinary bladder function (47). Paxilline represents a useful tool for studying the role of KCa1.1 in cells and in animal models of RA and for establishing these channels as attractive therapeutic targets but, with these known side effects, it cannot be developed into a therapeutic without strategies to eliminate or significantly reduce them. An option would be to modify paxilline to deliver it specifically to inflamed joints, as has been successfully accomplished for other small molecules (48). Alternatively, novel selective KCa1.1 blockers that do not cross the blood-brain barrier or access other cell types could be developed. Finally, KCa1.1 α subunits can be associated with different regulatory β or γ subunits, each with restricted tissue distributions (30-32). KCa1.1 blockers that can discriminate between KCa1.1 in periphery and in the central nervous system have already been identified (49, 50); it is therefore conceivable that KCa1.1 blockers selective for the channels expressed by RA-FLS can be generated.
Supplementary Material
Acknowledgments
This work was supported through funding from Baylor College of Medicine (to C.B. and F.T.H.) and an Award in Experimental Therapeutics from the Alkek Foundation and the Baylor College of Medicine Institute for Clinical and Translational Research (to C.B. and F.T.H.). M.R.T. was supported by the Howard Hughes Medical Institutes Med into Grad Initiative and T32 award GM088129 from the National Institutes of Health. The Baylor College of Medicine Cytometry & Cell Sorting, Pathology & Histology, and Monoclonal Antibody/Recombinant Protein Expression cores are supported by funding from the National Institutes of Health (S10 RR024574, P30 AI036211, and P30 CA125123).
We acknowledge Dr. Michael Bloch (Department of Molecular Physiology and Biophysics, Baylor College of Medicine) for generating the KCa1.1 construct used in the Bacmam vector and Dr. Joseph M. Hyser for sharing the GFP-expressing baculovirus.
Footnotes
The authors declare no competing financial interest.
References
- 1.Demoruelle MK, Deane KD, Holers VM. When and where does inflammation begin in rheumatoid arthritis? Curr. Opin. Rheumatol. 2014;26:64–71. doi: 10.1097/BOR.0000000000000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gulko PS, Winchester RJ. Rheumatoid arthritis. In: Austen KF, Frank MM, Atkinson JP, Cantor H, editors. Samter's immunologic diseases. Baltimore: Lippincott, Williams & Wilkins; 2001. pp. 427–463. [Google Scholar]
- 3.Cooles FA, Isaacs JD. Pathophysiology of rheumatoid arthritis. Curr Opin Rheumatol. 2011;23:233–240. doi: 10.1097/BOR.0b013e32834518a3. [DOI] [PubMed] [Google Scholar]
- 4.Leech MT, Morand EF. Fibroblasts and synovial immunity. Curr Opin Pharmacol. 2013;13:565–569. doi: 10.1016/j.coph.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Lefevre S, Knedla A, Tennie C, Kampmann A, Wunrau C, Dinser R, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15:1414–1420. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laragione T, Brenner M, Mello A, Symons M, Gulko PS. The arthritis severity locus Cia5d is a novel genetic regulator of the invasive properties of synovial fibroblasts. Arthritis Rheum. 2008;58:2296–2306. doi: 10.1002/art.23610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tolboom TC, van der Helm-Van Mil AH, Nelissen RG, Breedveld FC, Toes RE, Huizinga TW. Invasiveness of fibroblast-like synoviocytes is an individual patient characteristic associated with the rate of joint destruction in patients with rheumatoid arthritis. Arthritis Rheum. 2005;52:1999–2002. doi: 10.1002/art.21118. [DOI] [PubMed] [Google Scholar]
- 8.Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233:233–255. doi: 10.1111/j.0105-2896.2009.00859.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu X, Laragione T, Sun L, Koshy S, Jones KR, Ismailov II, et al. KCa1.1 potassium channels regulate key pro-inflammatory and invasive properties of fibroblast-like synoviocytes in rheumatoid arthritis. J Biol Chem. 2012;287:4014–4022. doi: 10.1074/jbc.M111.312264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laragione T, Brenner M, Li W, Gulko PS. Cia5d regulates a new fibroblast-like synoviocyte invasion-associated gene expression signature. Arthritis Res Ther. 2008;10:R92. doi: 10.1186/ar2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wulff H, Zhorov BS. K+ channel modulators for the treatment of neurological disorders and autoimmune diseases. Chem Rev. 2008;108:1744–1773. doi: 10.1021/cr078234p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gribkoff VK, Lum-Ragan JT, Boissard CG, Post-Munson DJ, Meanwell NA, Starett JEJ, et al. Effects of channel modulators on cloned large-conductance calcium-activated potassium channels. Mol Pharmacol. 1996;50:206–217. [PubMed] [Google Scholar]
- 13.Beeton C, Pennington MW, Norton RS. Analogs of the sea anemone potassium channel blocker ShK for the treatment of autoimmune diseases. Inflamm Allergy Drug Targets. 2011;10:313–321. doi: 10.2174/187152811797200641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Condreay JP, Witherspoon SM, Clay WC, Kost TA. Transient and stable gene expression in mammalian cells transduced with a recombinant baculovirus vector. Proc Natl Acad Sci USA. 1999;96:127–132. doi: 10.1073/pnas.96.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bagwell B. A journey through flow cytometric immunofluorescence analyses - finding accurate and robust algorithms that estimate positive fraction distributions. Clin. Immunol Newsletter. 1996;16:33–37. [Google Scholar]
- 16.Hu X, Beeton C. Detection of functional matrix metalloproteinases by zymography. J Vis Exp. 2010;45:pii, 2445. doi: 10.3791/2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beeton C, Pennington MW, Wulff H, Singh S, Nugent D, Crossley G, et al. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol Pharmacol. 2005;67:1369–1381. doi: 10.1124/mol.104.008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beeton C, Chandy KG. Drawing blood from the saphenous vein of rats and by cardiac puncture. J Vis Exp. 2007;7:e266. doi: 10.3791/266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington PW, et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci U S A. 2006;103:17414–17419. doi: 10.1073/pnas.0605136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tarcha EJ, Chi V, Munoz-Elias E, Bailey D, Londono LM, Upadhyay SK, et al. Durable pharmacological responses from the peptide ShK-186, a specific Kv1.3 channel inhibitor that suppresses T cell mediators of autoimmune diseases. J Pharmacol Exp Ther. 2012;342:642–653. doi: 10.1124/jpet.112.191890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luross JA, Williams NA. The genetic and immunopathological processes underlying collagen-induced arthritis. Immunology. 2001;103:407–416. doi: 10.1046/j.1365-2567.2001.01267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beeton C, Chandy KG. Isolation of mononuclear cells from the central nervous system of rats with EAE. J Vis Exp. 2007;10:e527. doi: 10.3791/527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole AA, Walters LM. Tartrate-resistant acid phosphatase in bone and cartilage following decalcification and cold-embedding in plastic. J Histochem Cytochem. 1987;35:203–206. doi: 10.1177/35.2.3540104. [DOI] [PubMed] [Google Scholar]
- 24.Qi Q, Liu XS, Zhang G, He W, Ma R, Cong B, et al. Morphological changes of cerebral vessels and expression patterns of MMP-2 and MMP-9 on cerebrovascular wall of alcoholic rats. Int J Clin Exp Pathol. 2014;7:1880–1888. [PMC free article] [PubMed] [Google Scholar]
- 25.Brenner M, Meng HC, Yarlett NC, Griffiths MM, Remmers EF, Wilder RL, et al. The non-major histocompatibility complex quantitative trait locus Cia10 contains a major arthritis gene and regulates disease severity, pannus formation, and joint damage. Arthritis Rheum. 2005;52:322–332. doi: 10.1002/art.20782. [DOI] [PubMed] [Google Scholar]
- 26.Beeton C, Chandy KG. Potassium channels, memory T cells and multiple sclerosis. Neuroscientist. 2005;11:550–562. doi: 10.1177/1073858405278016. [DOI] [PubMed] [Google Scholar]
- 27.Barrett-Jolley R, Lewis R, Fallman R, Mobasheri A. The emerging chondrocyte channelome. Frontiers Physiol. 2010;1 doi: 10.3389/fphys.2010.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chi V, Pennington MW, Norton RS, Tarcha EJ, Londono LM, Sims-Fahey B, et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon. 2012;59:529–546. doi: 10.1016/j.toxicon.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Large RJ, Hollywood MA, Sergeant GP, Thornbury KD, Bourke S, Levick JR, et al. Ionic currents in intimal cultured synoviocytes from the rabbit. Am. J. Physiol. Cell Physiol. 2010;299:C1180–1194. doi: 10.1152/ajpcell.00028.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ge L, Hoa NT, Wilson Z, Arismendi-Morillo G, Kong XT, Tajhya RB, et al. Big Potassium (BK) ion channels in biology, disease and possible targets for cancer immunotherapy. Int Immunopharmacol. 2014;14:S1567. doi: 10.1016/j.intimp.2014.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Contreras GF, Castillo K, Enrique N, Carrasquel-Ursulaez W, Castillo JP, Milesi V, et al. A BK (Slo1) channel journey from molecule to physiology. Channels (Austin) 2013;7:442–458. doi: 10.4161/chan.26242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horrigan FT. Conformational coupling in BK potassium channels. J Gen Physiol. 2012;140:625–634. doi: 10.1085/jgp.201210849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brenner M, Laragione T, Shah A, Mello A, Remmers EF, Wilder RL, et al. Identification of two new arthritis severity loci that regulate levels of autoantibodies, interleukin-1β, and joint damage in pristane- and collagen-induced arthritis. Arthritis Rheum. 2012;64:1369–1378. doi: 10.1002/art.33468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chandy KG, Wulff H, Beeton C, Pennington MW, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.WuCHH YS, Tsai CY, Wang YJ, Tseng H, Wei PL, Lee CH, et al. In vitro and in vivo study of phloretin-induced apoptosis in human liver cancer cells involving inhibition of type II glucose transporter. Int J Cancer. 2009;124:2210–2219. doi: 10.1002/ijc.24189. [DOI] [PubMed] [Google Scholar]
- 36.Tian L, Chen L, McClafferty H, Sailer CA, Ruth P, Knaus HG, et al. A noncanonical SH3 domain binding motif links BK channels to the actin cytoskeleton via the SH3 adapter cortactin. FASEB J. 2006;20:2588–2590. doi: 10.1096/fj.06-6152fje. [DOI] [PubMed] [Google Scholar]
- 37.Rezzonico R, Schmid-Alliana A, Romey G, Bourget-Ponzio I, Breuil V, Breittmayer V, et al. Prostaglandin E2 induces interaction between hSlo potassium channel and Syk tyrosine kinase in osteosarcoma cells. J Bone Mineral Res. 2002;17:869–878. doi: 10.1359/jbmr.2002.17.5.869. [DOI] [PubMed] [Google Scholar]
- 38.Cha HS, Boyle DL, Inoue T, Schoot R, Tak PP, Pine P, et al. A novel spleen tyrosine kinase inhibitor blocks c-Jun N-terminal kinase-mediated gene expression in synoviocytes. J Pharmacol Exp Ther. 2006;317:571–578. doi: 10.1124/jpet.105.097436. [DOI] [PubMed] [Google Scholar]
- 39.Gibofsky A. Comparative effectiveness of current treatments for rheumatoid arthritis. Am J Manag Care. 2012;13(Suppl):S303–314. [PubMed] [Google Scholar]
- 40.Panyi G, Beeton C, Felipe A. Ion channels and anti-cancer immunity. Phil Trans R Soc B. 2014;369:20130106. doi: 10.1098/rstb.2013.0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koshy S, Wu D, Hu X, Tajhya RB, Huq R, Khan FS, et al. Blocking KCa3.1 channels increases tumor cell killing by a subset of human natural killer lymphocytes. PLoS ONE. 2013;8:e76740. doi: 10.1371/journal.pone.0076740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Essin K, Gollash M, Rolle S, Weissgerber P, Sausbier M, Bohn E, et al. BK channels in innate immune functions of neutrophils and macrophages. Blood. 2009;113:1326–1331. doi: 10.1182/blood-2008-07-166660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Femling JK, Cherny VV, Morgan D, Rada B, Davis AP, Czirjak G, et al. The antibacterial activity of human neutrophils and eosinophils requires proton channels but no BK channels. J Gen Physiol. 2006;127:659–672. doi: 10.1085/jgp.200609504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sausbier U, Dullin C, Missbach-Guentner J, Kabagema C, Flockerzie K, Kuscher GM, et al. Osteopenia due to enhanced cathepsin K release by BK channel ablation in osteoclasts. PLoS ONE. 2011;6:e21168. doi: 10.1371/journal.pone.0021168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Otero M, Goldring MB. Cells of the synovium in rheumatoid arthritis - Chondrocytes. Arthritis Res Ther. 2007;9:220. doi: 10.1186/ar2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imlach WL, Finch SC, Dunlop J, Meredith AL, Aldrich RW, Dalziel JE. The molecular mechanism of “ryegrass staggers,” a neurological disorder of K+ channels. J Pharmacol Exp Ther. 2008;327:657–664. doi: 10.1124/jpet.108.143933. [DOI] [PubMed] [Google Scholar]
- 47.Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem. 2004;279:36746–36752. doi: 10.1074/jbc.M405621200. [DOI] [PubMed] [Google Scholar]
- 48.Fiehn C, Kratz F, Sass G, Müller-Ladner U, Neumann E. Targeted drug delivery by in vivo coupling to endogenous albumin: an albumin-binding prodrug of methotrexate (MTX) is better than MTX in the treatment of murine collagen-induced arthritis. Ann Rheum Dis. 2008;67:1188–1191. doi: 10.1136/ard.2007.086843. [DOI] [PubMed] [Google Scholar]
- 49.Kaczorowski GJ, Knaus HG, Leonard RJ, McManus OB, Garcia ML. High-conductance calcium-activated potassium channels; structure, pharmacology, and function. J Bioenerg Biomembr. 1996;28(3):255–267. doi: 10.1007/BF02110699. [DOI] [PubMed] [Google Scholar]
- 50.Garcia-Valdes J, Zamudio FZ, Toro L, Possani LD. Slotoxin, αKTx1.11, a new scorpion peptide blocker of MaxiK channels that differentiates between α and α+β (β1 or β4) complexes. FEBS Lett. 2001;505:369–373. doi: 10.1016/s0014-5793(01)02791-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.