

Abstract
The vaccinia virion is a membraned, slightly flattened, barrel-shaped particle, with a complex internal structure featuring a biconcave core flanked by lateral bodies. Although the architecture of the purified mature virion has been intensely characterized by electron microscopy, the distribution of the proteins within the virion has been examined primarily using biochemical procedures. Thus, it has been shown that non-ionic and ionic detergents combined or not with a sulfhydryl reagent can be used to disrupt virions and, to a limited degree, separate the constituent proteins in different fractions. Applying a controlled degradation technique to virions adsorbed on EM grids, we were able to immuno-localize viral proteins within the virion particle. Our results show after NP40 and DTT treatment, membrane proteins are removed from the virion surface revealing proteins that are associated with the lateral bodies and the outer layer of the core wall. Combined treatment using high salt and high DTT removed lateral body proteins and exposed proteins of the internal core wall. Cores treated with proteases could be disrupted and the internal components were exposed. Cts8, a mutant in the A3 protein, produces aberrant virus that, when treated with NP-40 and DTT, release to the exterior the virus DNA associated with other internal core proteins. With these results, we are able to propose a model for the structure the vaccinia virion.
Introduction
Poxviridae comprise a family of viruses characterized by the presence of a large dsDNA genome and a complex morphology (Condit et al., 2006; Moss, 2013). Poxviruses encode a complete transcription apparatus and thus are able to replicate in the cytoplasm of infected cells. Vaccinia virus (VACV), the prototype member of this family, encodes more than 200 proteins and the role of many virus proteins during the virus replicative cycle has been determined (Goebel et al., 1990; Moss, 2013). The protein composition of purified mature virions has been determined by mass spectrometry and at least 70 virus proteins have been identified (Chung et al., 2006; Matson et al., 2014; Resch et al., 2007; Yoder et al., 2006). Although the proteomic analysis has been important for the identification of the total protein content of the mature particle, the fine localization of a significant fraction of the virion proteins is still unknown. Membrane proteins and enzymes involved in early transcription have been assigned positions in the particle, but the location of the other proteins still needs to be determined.
Electron microscopy is the preferred method for studying the morphology of the VACV particle and various electron microscopic techniques have been applied in the visualization of the virus structure (Cyrklaff et al., 2005; Dales and Siminovith, 1961; Easterbrook, 1966; Harris and Westwood, 1964; Ichihashi et al., 1984; Muller and Peters, 1963; Naginton and Horne, 1962; Peters and Muller, 1963; Westwood et al., 1964; Wilton et al., 1995). Overall, poxvirus virions have an ellipsoidal, barrel or brick shaped appearance. Analysis of VACV on a whole mount preparation using negative staining of the particles revealed the presence of a membrane that enclosed two distinct virus sub-domains: the lateral bodies and the core (Dales, 1962; Harris and Westwood, 1964; Muller and Peters, 1963; Peters and Muller, 1963; Westwood et al., 1964). The lateral bodies, which flank the core, are amorphous structures composed of proteins of unknown function. The core is comprised of a proteinaceous wall that encloses a nucleocapsid (Condit et al., 2006). Analysis of VACV by cryo-microscopy and reconstruction using electron tomography revealed pore-like structures spanning the core wall (Cyrklaff et al., 2005). No function has been determined for this structure although it could be involved in the extrusion from the core of the viral mRNA during early transcription.
Using negative staining electron microscopy, the surface of the mature virion presents two different morphological forms that are directly related to the integrity of the particle. The predominant form in a fresh virion preparation contains on its surface rodlet-like structures called surface tubule elements, creating a mulberry-like appearance (Harris and Westwood, 1964; Muller and Peters, 1963; Naginton and Horne, 1962; Westwood et al., 1964; Wilton et al., 1995). Under various conditions, the negative stain can penetrate through the virus membrane so that the surface tubule elements are no longer apparent and the virus now exhibits a capsule-like form. When virions are exposed to high pH, the lateral bodies, core wall, and the nucleocapsid can be visualized (Muller and Peters, 1963).
Analysis of VACV by atomic force microscopy has permitted a more accurate determination of the dimensions of the virus particle, because measurements are obtained with fully hydrated virions (Malkin et al., 2003). Using this approach, the virus dimensions vary between 320 and 380 nm in the major axis and 260 to 340 nm in the minor axis, similar to measurements described by other methods (Cyrklaff et al., 2005; Griffiths et al., 2001; Malkin et al., 2003; Roos et al., 1996; Sodeik and Krijnse-Locker, 2002). However, the height of the hydrated virion varies between 240 and 290 nm, a measurement significantly higher than what is observed with dried sample but similar from the cryo-electron microscopy results.
Treatment of purified virions with a non-ionic detergent and a reducing agent solubilizes the proteolipid membrane complex exposing the core and the lateral bodies (Easterbrook, 1966; Ichihashi et al., 1984). The soluble and insoluble components of the virions can be separated by centrifugation and the protein content of each fraction can be identified by various techniques (Boyd et al., 2010; Chiu and Chang, 2002; Ichihashi et al., 1984; Kato et al., 2007; Nichols et al., 2008; Resch and Moss, 2005; Unger et al., 2008). Using this approach, proteins associated with the membrane fractionate into the soluble fraction whereas the insoluble fraction contains proteins from both the lateral bodies and core. Purification of the lateral bodies away from the core has not been achieved using the current methodologies.
In this work, our goal was to develop a method to locate VACV proteins in viral subdomains. For this propose, we combined whole mount preparation of virions with immunogold labeling of the proteins. Virions attached to grids were subjected to various treatments in order to expose viral proteins located in the different sub-domains and permit their identification by immunogold labeling. Our results show after NP40 and DTT treatment, membrane proteins such as A14 and A27 are removed from the virion surface revealing proteins that are associated with the lateral bodies and the outer layer of the core wall. Combined treatment using high salt and high DTT removed lateral body proteins and exposed proteins of the internal core wall. Cores treated with proteases could be disrupted and the internal components were exposed. Cts8, a mutant in the A3 protein, produces aberrant virus that, when treated with NP-40 and DTT, release to the exterior the virus DNA associated with other internal core proteins. With these results, we are able to propose a model for the structure the vaccinia virion.
Results
Negative staining of vaccinia virus
The establishment of a standardized procedure to visualize purified virions is an important step in the understanding viral structure (De Carlo and Harris, 2011). Poxviruses were one of the first mammalian viruses to be visualized using electron microscopic techniques. The structural analyses of purified VACV in whole mount preparations revealed a unique pattern that has been confirmed by various procedures (Griffiths et al., 2001; Harris and Westwood, 1964; Heuser, 2005; Malkin et al., 2003; Muller and Peters, 1963; Westwood et al., 1964; Wilton et al., 1995). To analyze the structure of purified VACV we used five different negative staining conditions (Fig. 1). Similar to previous work (Harris and Westwood, 1964; Muller and Peters, 1963; Peters and Muller, 1963), when virions were negative-stained with phospho-tungstic acid (PTA) at pH 7.0 particles could be visualized in two different forms, the mulberry-like form and the capsule (Fig. 1A). While the mulberry form shows a rodlet-like structure surrounding the particle, the capsule form presents a defined frame that surrounds the internal structure of the virion. When the pH of the PTA solution was elevated to pH 10.5, only the capsule form was observed, presumably because the integrity of the virion membrane is compromised (Fig. 1B). Under these conditions, a tubular-like internal structure of the virion was revealed that represents the virus nucleocapsid (Muller and Peters, 1963). Virions in a whole mount preparation were also negative-stained with ammonium molybdate (AmMo), uranyl acetate (UA) and methylamine tungstate (NanoW) (Fig. 1C–E). Under all these conditions, particles with a mulberry appearance were the major form observed. When whole mount preparations of VACV were treated with the neutral detergent NP40 before negative staining, a different appearance of the particle was observed (Fig. 1F–J). In all preparations, because the lipid constituent of the virion membrane is removed allowing stain to penetrate the particles, only the capsule forms were observed and the core internal structure became apparent. It is noteworthy that after this treatment the nucleocapsid appears amorphous, different from what was observed when virions were stained with PTA at pH10.5 (Fig. 1G). The nucleocapsid is surrounded by a thick, well-defined boundary layer and the presence of the lateral bodies could be observed (Fig. 1H). In some images, this boundary layer appears to be formed by two thinner layers (Fig. 1F, H, and J). The outermost layer is composed of a protein array that comprises proteins involved in virion attachment, the entry-fusion complex and structural membrane proteins. The internal layer includes the spike-like “palisade layer” and additional core wall proteins (Condit et al., 2006; Cyrklaff et al., 2005; Roos et al., 1996). The majority of the proteins present in the outermost (membrane) layer cannot be removed from the particle by NP40 treatment alone (Chiu and Chang, 2002; Nichols et al., 2008). With this treatment, the lipid component of the membrane is extracted and virions lose their infectivity (Ichihashi and Oie, 1983; Laliberte and Moss, 2009). Remarkably, the infectivity can be restored if the lipid is replenished. After treatment of virus on the grids with NP40 and DTT, the virion membrane, including the lipid and outer protein array, is removed completely from the virion surface, so that the boundary surrounding the core is reduced in thickness or even not apparent in some of the electron micrographs (Fig. 1K–O).
Fig. 1.

Negative staining of vaccinia virions. Vaccinia virions were adsorbed on a Formvar/carbon coated grid and incubated in the presence of Tris (A–E), NP-40 (F–J) or NP-40 + DTT (K–O). After 20 minutes the grids were washed and stained with PTA, pH 7.0 (A, F, K), PTA, pH 10.5 (B, G, L), AmMo (C, H, M), UA (D, I, N) or NanoW (E, J, O). The grids were examined in a TEM as described in Materials and Methods.
Electron dense structures can be observed on the top of cores stained with UA (Fig. 2A). Upon carbon-platinum shadowing of these same samples, these structures present themselves in a globular form and are presumed to be the lateral bodies (Fig. 2B). Close inspection of particles treated with NP40+DTT and stained with NanoW revealed pore-like structures with and average diameter of 11.71 (+/−1.25) on the surface of the core (Fig. 2C). The presence of pore-like structures traversing the core wall has been described, but the nature and function of these structures has yet to be determined (Cyrklaff et al., 2005).
Fig. 2.

Electron dense and pore-like structures on the surface of vaccinia core. Vaccinia cores were prepared on grids as described in Fig. 1. A) Vaccinia core stained with UA reveal an electro-dense structure in the middle of the particle. B) A globular structure on the middle of the core was enhanced when the grid was subjected to platinum-carbon shadowing. C) The presence of pore-like structures (arrows) was observed after staining cores with NanoW.
Importantly, the NP40-treated particles resemble virions in the capsule form. Furthermore, both the capsule form and NP40 treated viruses are larger than the mulberry form of the particle. Analysis of the two largest dimensions of the mulberry-like viruses (n=67) yielded 302.30 nm (+/−22.92) for the long axis and 240.90 nm (+/−22.18) for the short axis. By contrast, the dimensions of the capsule-like particles (n=65) was 319.36 nm (+/−19.65) and 251.67 nm (+/−16.38), and the NP40-treated particles (n=76) was 323.50 nm (+/−23.54) and 255.20 nm (18.14). The difference in size between the mulberry-like and the other two forms was statistically significant with p<0.01. These results are in accordance with the literature (Westwood et al., 1964). Comparing all the negative staining methods tested, the most consistent results were obtained with NanoW and UA, which we therefore used for all subsequent experiments.
Immunogold labeling of vaccinia virus after controlled degradation
Our goal in this work was to localize viral proteins within different sub-domains of the virion. Most of the current data describing the location of viral proteins in different sub-domains of the virion have been obtained using western blot analysis. In this procedure, membrane-associated proteins are removed from particles by incubating viruses with NP40/DTT, followed by the separation of the soluble fraction from the insoluble portion by centrifugation. During this procedure, the lateral bodies remain associated with the insoluble fraction and thus core and lateral body proteins are not discriminated from each other. To refine this analysis, virions attached to grids were treated using various conditions and proteins associated with the particles were identified by immunogold labeling. In parallel, virions were submitted to the same treatments in suspension, the fractions were separated by centrifugation and the proteins analyzed by western blot. A typical result of a western blot analysis (A) and an immunogold labeling of the viruses (B) is presented in Fig. 3. When viruses were incubated in Tris-HCl only, none of the assayed proteins were solubilized (Fig. 3A, Tris column). Upon treatment of the virions with NP40, virus proteins A4 and A14 were partially removed from the particle to the soluble fraction (Fig. 3A, NP column). However, when DTT was added to the NP40 treatment, the membrane-linked proteins A14 and A27 were completely removed from the virions and moved to the soluble fraction (Fig. 3A, NP+DTT column). The core proteins A4 and L4 were only partially removed from the cores. When viruses were visualized by electron microscopy after immunogold labeling of the proteins, the pattern of protein labeling was consistent with the western blot analysis. Immunolocalization of virus proteins A27 and A4 after the different treatments is presented in Fig. 3B, a–c. Upon incubation with either Tris buffer or NP40 alone, the membrane protein A27 was immuno-localized on the surface of the particle but there was no evidence for the presence of the core protein A4. When DTT was combined with NP40 during the treatment of the particles, the labelling of the A27 membrane protein disappeared from the particles and the A4 core protein was now exposed. When the assay was performed labelling proteins from the core wall (A10) and lateral bodies (F17, H1), no labeling was observed when the particles were treated with Tris buffer (Fig. 3B, d–f). Compared to Tris buffer alone, in the presence of NP40, or NP40/DTT, increased labelling of F17 was observed. The presence of the core protein A10 was observed only after treatment with NP40/DTT. When viruses attached to the grids were incubated with in Tris buffer, followed by immunogold labelling of the A14 membrane structural protein and the H1 lateral body protein, gold particles present on the virions were scarce (Fig. 3B, g–i). When NP40 was present in this treatment, the presence of both A14 and H1 became more evident. Combining DTT with the NP40 treatment, A14 was removed from the particle but H1 continued to be attached to core. The EM data are consistent with the western blot data and are consistent with the interpretation that it is necessary to remove one layer of proteins to expose the underlying layer of proteins. Specifically, in untreated virions, only the external membrane protein A27 is exposed to immunogold labeling. Treatment with NP40 alone, which removes the lipid but leaves behind the membrane protein array, retains A27 staining, and exposes the membrane structural protein, A14, and the lateral body proteins F17 and H1. Treatment with both NP40 and DTT removes the entire membrane including the membrane protein array such that staining of membrane proteins A14 and A27 is lost while staining for lateral body proteins F17 and H1 is retained, and now the core proteins A4 and A10 are exposed to antibody.
Fig. 3.


Analysis by Western blot and immunogold labeling of purified vaccinia virus after controlled degradation of virions. A) Each lane contain the equivalent of 0.06 A260nm units (~0.7 μg) of virus that was incubated in different buffers as described in Materials and Methods and labeled on the top of the figure. The presence of specific proteins in the pellet (P) or supernatant (S) fractions were determined by Western blot and labeled on each lane. B) Vaccinia virus were adsorbed on grids and incubated with Tris (A, D, G), NP-40 (B, E, H), or NP-40+DTT (C, F, I) as described in Materials and Methods. After this treatment, the proteins associated with the virions were identified by immunogold labeling using the antibodies A27 (●) A4 (•) (A, B, C), A10 (●) F17 (•) (D, E, F), A14 (●) H1 (•) (G, H, I) as described in Materials and Methods.
Disrupting the virion core
Our initial attempts to detect the core proteins A3 and L4 on intact, NP40 treated, or NP40+DTT treated virions were unsuccessful (not shown). Because A3 is part of the core wall and L4 is a DNA binding protein (Jesus et al., 2014), we hypothesize that these proteins are located in the internal region of the core and are therefore inaccessible to antibody using these procedures. We therefore experimented with various treatments intended to further strip or disrupt virion cores in order to expose internal proteins. First, cores were prepared from VACV adsorbed to grids, then further treated with various concentrations of DTT in the absence or presence of 3M NaCl. The incubation was followed with a hypotonic shock, done by incubating the grids in deionized water, before processing for negative staining (Fig. 4) or immunogold labelling (Fig. 5). The extended incubation of cores in 10 mM DTT (Fig. 4A) did not alter the morphology of the particle compared with the initial NP40+DTT treatment (Fig. 1O). Increasing the concentration of DTT to 100 mM partially disrupted the core wall however the core still maintained its overall structure (Fig. 4B). The addition of 3M of NaCl to the incubation mixture in the presence of 10 or 100 mM DTT did not disrupt the core (Fig. 4C and D), although at higher DTT and NaCl concentrations the external surface of the cores appears disordered. The fate of the proteins associated with the lateral bodies and core were analyzed after treatment with 100 mM DTT in the presence or absence of 3M NaCl by western blot and immunogold labelling of the particles (Fig. 5). The western blot shows that core proteins A10, A3, and H1 continue to be attached to the cores (P) after treatment with high DTT and NaCl (Fig. 5A). By contrast, the proteins A4 and F17 were partially removed from the core fraction to the soluble fraction (S). Immunogold labeling for electron microscopy demonstrated that after treatment with 3 M NaCl, cores were depleted of the proteins F17 and A4 from the surface, but the proteins A10 and H1 were still attached to the particle (Fig. 5B). These results are consistent with previous observations suggesting that A4 protein constitutes the palisade structure of the core wall and showing that A4 interacts with the core wall protein A10 (Cudmore et al., 1996; Risco et al., 1999). Interestingly, the NaCl/DTT treatment exposed some A3 on the surface of the core. The exposure of A3 in the immunogold labeling could be due to the partial removal of A4 from the core wall or to an increase in permeability of core wall.
Fig. 4.

Morphological changes in vaccinia cores after high salt and high DTT treatment. Vaccinia cores prepared as described before were subjected to treatment with 10 mM (A, C) or 100 mM (B, D) DTT in the absence (A, B) or presence (C, D) of 3M NaCl and stained with Nano-W as described in Materials and Methods before visualization by TEM.
Fig. 5.


Analysis by Western blot and immunogold labeling of vaccinia cores after treatment with high salt. A) The equivalent of 0.06 A260nm units (~0.7 μg) of virus that was incubated in NP-40 + DTT to prepare cores as described in Materials and Methods. Cores were then incubated in buffer S (Cntl) or buffer SN (NaCl) as described in Materials and Methods and labeled on the top of the figure. The presence of specific protein in the pellet (P) or supernatant (S) fraction was determined by Western blot. B) Vaccinia virus were adsorbed on grids and incubated with NP-40+DTT to prepare cores as described in Materials and Methods. After this treatment, the grids were incubated in buffer S (A–D) or buffer SN (E–H) for 20 minutes, washed in ddH2O and proteins associated to the virions were identified by immunogold labeling using the antibodies A10 (●) A4 (•) (A, E), A10 (●) F17 (•) (B, F), A10 (●) H1 (•) (C, G), A10 (●) A3 (•) (D, H) as described in Materials and Methods.
Although some A3 was observed after treatment with DTT-NaCl, L4 was still undetectable (not shown). Because L4 is a DNA-binding protein, it may be located in the interior of the core and visualized only after rupture of the core wall.
Because the overall core integrity was relatively resistant to action of various chemical agents, we tested whether a mild treatment of the cores with protease could rupture the core and expose the interior while nevertheless preserving a recognizable core wall structure. Cores attached to the grids were incubated in the presence of pronase E or papain and analyzed by electron microscopy. As shown in Fig. 6, cores treated with pronase E (Fig. 6B) or papain (Fig. 6C) preserve their brick shape, but the core wall appears to be removed. Because the protease treatment was done after the virus was attached to the grid, the strong interaction between the particle and the carbon film on the grid could contribute to the maintenance of the shape of the virion. To determine whether lateral body and core wall proteins are removed upon protease treatment, the fate of F17 (Fig. 7A and B) and A4 (Fig. 7C and D) were analyzed by immunogold labelling of cores. As an indication of the degradation of the core wall, the release of DNA from the cores was assessed by immunogold labelling. As shown in Fig. 7, proteins F17 and A4 are associated with cores not treated with protease, and viral DNA was not labelled within these cores. After treatment with pronase E, most of the F17 and A4 proteins were digested from the cores and the exposure of viral DNA on the grids was unmistakable. Because digestion of the core wall exposed viral DNA, the presence of the A3 and L4 proteins was investigated after protease treatment. As shown in Fig. 8A and D, DNA, A3, and L4 were not detected on cores not treated with proteases. However, after incubation with pronase E (Fig. 8B) or papain (Fig. 8C) the A3 and DNA were easily seen associated with the particle or released on the grid. Interestingly, after the same treatment the presence of L4 was still not evident (Fig. 8E–F).
Fig. 6.
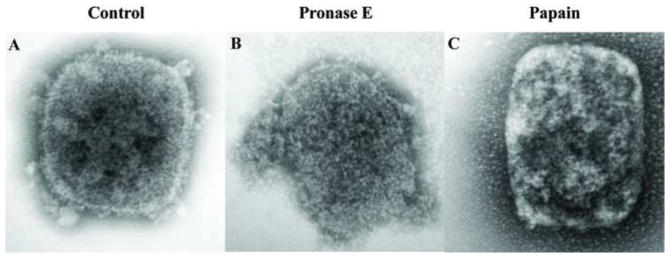
Morphological changes on vaccinia cores after protease treatment. Vaccinia cores prepared on the grids as described before were incubated in Tris (A), 2 mg/ml of Pronase E (B) or 2mg/ml Papain (C) for 120 min. After this period, the grids were negative stained with Nano-W as described in Materials and Methods before visualization by TEM.
Fig. 7.

Vaccinia DNA is exposed after Pronase E treatment. Vaccinia cores prepared on the grids as described before were incubated in Tris (A, C) or 2 mg/ml of Pronase E (B, D) for 120 min. After this treatment, the grids were washed in ddH2O and the DNA and proteins associated with the virions were identified by immunogold labeling using the antibodies F17 (●) DNA (•) (A, B), A4 (●) DNA (•) (C, D) as described in Materials and Methods.
Fig. 8.

The A3 core protein and vaccinia DNA are exposed after protease treatment. Vaccinia cores prepared on the grids as described before were incubated in Tris (A, D), or 2 mg/ml of Pronase E (B, E), or 2 mg/ml of Papain (C, F) for 120 min. After this period, the grids were washed in ddH2O and the DNA and proteins associated with the virions were identified by immunogold labeling using the antibodies A3 (●) DNA (•) (A, B, C), L4 (●) DNA (•) (D, E, F) as described in Materials and Methods.
Viruses mutant in the core wall protein A3 produce fragile particles
Vaccinia viruses with temperature sensitive mutations in the A3L gene, one of the major core wall proteins, have been previously characterized with respect to replication and morphogenesis (Kato et al., 2004). The electron microscopic analysis of the mutants reveals formation of aberrant MV. Although purified aberrant virions were indistinguishable from wt viruses in protein composition, mutant virions are inactive in a permeabilized virion transcription system. Whole mount preparation of purified A3 mutants Cts8 and Cts26 were analyzed by electron microscopy after negative staining with NanoW or uranyl acetate (Fig. 9). A3 mutant viruses incubated with buffer (Fig. 9A) present an altered morphology when compared with wt viruses (Fig. 1A). In this preparation only capsule-like particle were visualized, and virus particles have a rounded form different from the brick/barrel shape forms observed in wt viruses. In some particles, the core appears to be dislocated from the center of the particle. Surprisingly, after treatment of A3 mutant viruses with NP40+DTT the virus core wall was ruptured and the internal contents released (Figs 9B, D, and E). When cores treated with NP40+DTT were negative stained with NanoW (Fig. 9B), the internal material was released as a tangled, bead-like structure. When NP40+DTT treated cores were negative stained with UA, the material released from the core spread on the grid with a reticulated appearance while the cores seemed depleted their internal structure (Fig. 9D and E). Because the major internal component of the core is the viral genome, it is possible that the material released is DNA, either naked or associated with proteins. To analyze whether DNA was present in the released material, A3 mutants were adsorbed to grids, treated with NP40/DTT, followed by incubation with DNase. As shown Fig. 9C, in cores negative stained with NanoW the tangled structure released from the core seems relatively resistant to DNase. When cores were negative stained with UA after incubation with DNase, the reticulated material was completely removed suggesting that DNA is part of the released material. The presence of viral proteins associated with the cores and the released material were analyzed by immunogold labeling (Figs 10 and 11). As shown above, in both wt (Fig. 10A) and A3 mutant viruses (Fig. 10D) the core wall proteins A10 and A4 were easily observed associated with the particle. By contrast, the core wall protein A3 was not visible in wt virions (Fig. 10B) but exposed in the mutant virus (Fig. 10E). Similar results were observed when the presence of DNA was detected by immunogold labelling (Figs 10C and F). Specifically, the presence of DNA associated with the particle was only observed in the mutant virus and not present on the wt cores. Core wall proteins A10 (Figs 10B and E) or A4 (Figs 10C and F) were used as control for immunogold labelling and could be observed in both preparations. Although in A3 mutant viruses, internal proteins and DNA were exposed after treatment with NP40/DTT, the presence of the DNA binding protein L4 was still undetected. One possibility to be considered is that the L4 protein is wrapped in DNA in a nucleosome-like structure in a configuration that renders it unreactive to antibody. To evaluate this possibility, mutants in A3 were treated with NP40+DTT and incubated in the absence or presence of DNase (Fig. 11). In the absence of DNase, virus DNA is released from the cores and as previously observed, no L4 was detected (Fig. 11 A). When cores were incubated in the presence of DNase, the amount of immunogold labelling of DNA decreased, and the L4 protein could be unmistakably identified (Fig. 11 C). Interestingly, after incubation of the cores with DNase, the presence of A3 protein was also enhanced when compared to virus not treated with DNase (Figs 11 B and D). It is possible that both L4 and A3 play a role on the organization of the viral DNA inside the core.
Fig. 9.
Temperature sensitive mutant in the core wall protein A3 make fragile particle under non-permissive conditions. Purified Cts8 virions grown at the non-permissive temperature were adsorbed on a Formvar/carbon coated grid and incubated in the presence of Tris (A), or NP-40 + DTT (B–F). After 20 min, the grids were washed, and two were incubated with 40 μg/ml DNase (C, F), for 15 min. The grids were stained with Nano-W (A–C) or UA (D–F) as described in Materials and Methods.
Fig. 10.

The core wall protein A3 and DNA are released from the virions after controlled degradation of A3 mutants. Purified wt virions (A–C) and purified Cts8 virions grown at non-permissive temperature were adsorbed on Formvar/carbon coated grids and incubated in the presence NP-40 + DTT. The grids were then washed in ddH2O and the DNA and proteins associated with the virions were identified by immunogold labeling using the antibodies A10 (●) A4 (•) (A, D), A10 (●) A3 (•) (B, E), A4 (●) DNA (•) (C, F) as described in Materials and Methods.
Fig. 11.

L4 protein can be visualized only after DNase treatment. Purified Cts8 virions grown at the non-permissive temperature were adsorbed on Formvar/carbon coated grids and incubated in the presence of NP-40 + DTT. After 20 min, the grids were washed, and not treated (A, B) or treated with 40 μg/ml DNase (C, D). After this period, the grids were washed in ddH2O and the DNA and proteins associated to the virions were identified by immunogold labeling using the following antibodies L4 (●) DNA (•) (A, C), A3 (●) DNA (•) as described in Materials and Methods.
Discussion
Advances in VACV genomics, proteomics, and genetics have contributed to assigning proteins to specific genes in genome, identifying proteins as constituents of the virus particle, and understanding the role of these proteins in the replicative cycle. Electron microscopy has also played an important role in comprehending several aspects of viral morphogenesis and poxvirus virion structure (Condit et al., 2006; Cyrklaff et al., 2005; Dubochet et al., 1994; Heuser, 2005; Moss, 2013; Pedersen et al., 2000; Roos et al., 1996). Although all of these techniques have played an essential role in understanding the biology of VACV, the fine details of the virion structure have yet to be determined. The dissection of a complex viral structure such as VACV should be achieved with the aid of chemical, enzymatic, and physical implements. These tools are employed in peeling off, layer by layer, the intricate virus structure down to the nucleocapsid. Several reports have described the use of non-ionic detergent to remove the viral membrane lipid layer of VACV. When the non-ionic detergent was combined with a reducing agent, the protein array that is associated with the lipid layer was also extracted (Boyd et al., 2010; Castro et al., 2003; da Fonseca et al., 2004; Griffiths et al., 2001; Ichihashi et al., 1984; Nichols et al., 2008; Pedersen et al., 2000; Resch and Moss, 2005; Roos et al., 1996; Schmidt et al., 2013; Senkevich et al., 2008; Sood et al., 2008; Townsley et al., 2005; Unger et al., 2008; Wallengren et al., 2001; Wickramasekera and Traktman, 2010; Wilcock and Smith, 1996). Proteins dissociated from the membrane complex can be analyzed by western blot and the cores visualized by electron microscopy. With these analyses, it was possible to determine that most of the membrane-associated proteins were involved in the attachment and entry-fusion complex necessary for virus infection (Carter et al., 2005; Moss, 2006; Moss, 2012; Roberts and Smith, 2008). In this work we showed when VACV particles were treated with NP40, membranes proteins A27 and A14 became more exposed to the antibodies, and when DTT was added to the treatment these proteins are removed from the remaining cores. Because A27 is part of the attachment complex and A14 is one of the major components of the membrane complex, treating viruses with NP40 and DTT will remove the lipids and the protein array from the particle, exposing proteins from lateral bodies and core (Carter et al., 2005; Ching et al., 2009; Cudmore et al., 1996; Mercer and Traktman, 2003; Moss, 2012; Rodriguez et al., 1987; Unger et al., 2013).
One of the most intriguing features in poxvirus structure is the origin and composition of the lateral bodies. Lateral bodies are amorphous structures located between the viral membrane and the core wall, and have only be described using electron microscopy (Condit et al., 2006). Although the nature of the lateral bodies has intrigued poxvirologists for many years, the absence of adequate tools has delayed the understanding of these structures. One possible function of the proteins in the lateral bodies is to play a role in virus-host interaction, helping the virus to escape innate cellular antiviral defenses. In this case, the lateral bodies would have a role similar to the tegument in the Herpesviridae (Kalejta, 2008; Kelly et al., 2009; Tomtishen, III, 2012). In HSV, the tegument occupies the space between the capsid and the envelope and consists of at least 20 viral encoded proteins. Different properties and functions have been assigned to tegument proteins such as proteins kinases, interferon inhibitors, apoptosis and host translation regulators. Some of these proteins are conserved in the Herpesvirinae subfamily. Among the approximately 70 virion proteins associated with purified VACV MV, nearly twenty virus proteins are located in the membrane, three proteins form the core wall, and twenty two proteins constitute the transcription apparatus and nucleocapsid (Chung et al., 2006; Matson et al., 2014; Resch et al., 2007; Yoder et al., 2006). Thus the location of at least 25 proteins is still unknown. Most of these proteins could be forming the lateral bodies or could be located in another sub-domain of the virion. In this list, we include virus proteins with a known function such as: protein kinase (F10), Tyr/Ser protein phosphatase (H1), cysteine proteinase (I7); the complex of seven proteins that are involved in the formation of the MV; and proteins involved in the formation of disulfide bonds in intracellular virion membrane proteins (Schmidt et al., 2013; Senkevich et al., 2002; Szajner et al., 2004; Traktman et al., 1995; Wickramasekera and Traktman, 2010). Other proteins have been proposed to be constituent of the lateral bodies and could be involved in virus-host interactions. Among these is the essential protein F17. F17 is the second most abundant protein in the virion, it is phosphorylated by cellular kinases, and interacts with A30, one of the 7-protein complex (Wickramasekera and Traktman, 2010). Mutation of the cellular phosphorylation site in F17 does not affect virion morphogenesis but reduces the infectivity of the progeny. It has been sugested that after VACV entry F17 is degraded in a proteasome-dependent manner (Schmidt et al., 2013). Interestingly, the fact that F17 interacts with A30 suggests by association that the seven protein complex may be localized to the lateral bodies. This would be consistent with a role for the seven protein complex is forming the initial association between viroplasm and membranes during virion morphogenesis (Szajner et al., 2004). Proteins such as L3, E6, and E8 are located in the virions and have an important role in VACV replication (Boyd et al., 2010; Kato et al., 2007; Resch and Moss, 2005). These proteins contain no known motifs or other features that provide an indication of their role in the virus replicative cycle. These proteins are do not appear to be transcription factors but in their absence cores are inactive in synthetizing RNA. Finally, the VACV proteins G5 and G6 have been shown to be present in purified MV (da Fonseca et al., 2004; Senkevich et al., 2008). G5 belongs to the FEN1-like family of nucleases, and it is conserved in all poxviruses (Senkevich et al., 2009). G6 belongs to the NlpC/P60 superfamily, contributes to the virus virulence, and localizes to the lateral bodies. Interestingly, neither G5 nor G6 have been identified in any of the published proteomic studies. Because in these studies G5 and G6 were expressed under a T7 operator it is possible that under these conditions both proteins were overexpressed and passively encapsidated by the virions (Franke and Hruby, 1987; Gomez and Esteban, 2001). We showed that the treatment of cores with higher a concentration of 100 mM DTT and 3M NaCl could remove proteins from the surface of the core while preserving its structure. In our assay, F17 and A4 were partially removed after this treatment, while A10 and H1 were still attached to outside structure the cores. It is possible that proteins of the lateral bodies function not only as a connecting structure between the membrane and core wall but also as mediator of infection. Application of the techniques described here to the analysis of additional proteins of unknown location may aid in elaborating the contents of the lateral bodies.
The VACV core wall is composed of at least three viral-encoded proteins that form a very tight structure (Condit et al., 2006; Moss, 2013). Presumably, A3 is located in the internal part of the core wall; A10 and A4 are positioned in the external part of the wall; and the molecular ratio of these proteins is 1:1:10 respectively (Chung et al., 2006; Ichihashi et al., 1984). A4 lays on the external part of the core wall, forming palisade-like structures (Cudmore et al., 1996; Roos et al., 1996). In accordance with this model are our results with the immunogold labeling of A4, where the gold particles uniformly decorate the exterior of the core wall. The partial removal of A4 from the core wall after NaCl/DTT treatment was unexpected because this protein forms a stable complex with A10 that was not removed during the procedure (Chung et al., 2006; Risco et al., 1999). Interesting, after NaCl/DTT and hypertonic shock the core wall protein A3 could be visualized by immunogold labelling, suggesting that after peeling off the A4 layer, the A3 layer became visible. The presence of a pore-like structure on the core wall that was observed in our negative staining approach has been first reported in the cryo-electron tomographic reconstruction of VACV (Cyrklaff et al., 2005). This pore-like structure seems to traverse the core wall, communicating internal and external milieu, and could be formed by one or more core wall proteins or by the insertion of another viral protein. One of the possible functions of the pore-like structure is in the extrusion of the newly transcribed early mRNA from the core to be translated by the cellular ribosomes (Gross and Shuman, 1996; Kates and Beeson, 1970). The pore-like structures could also function in the intake of nucleotides or other molecules into the core milieu.
During the course of infection, the VACV cores undergo a second uncoating process to release its genome for replication, in a mechanism that is still only partially understood (Dales, 1963; Joklik, 1964a; Joklik, 1964b; Kilcher et al., 2014; Moss, 2013). This step is dependent on virus early proteins, and cellular enzymes could be involved. In a manner similar to that which occurs with the release of proteins from the lateral bodies, a proteasome-mediated step could be engaged in the disassembling the core wall, a mechanism that has been described with other viruses (Horan et al., 2013; Lopez et al., 2011; Schmidt et al., 2013). All our attempts to break the core wall using different detergents were unsuccessful (data not shown). The core wall was resistant to different non-ionic detergents, and in the presence of ionic detergents, the cores were solubilized beyond recognition as has been shown before (Soloski et al., 1979; Soloski and Holowczak, 1981). Because chemical and physical treatment failed to disrupt the core wall, we switch to a mild protease treatment to disrupt the core wall. Under these conditions, we were able to expose the internal part of the core wall preserving the overall structure of the core. The removal of the core wall enables the labelling of A3 and the virus DNA. Interesting, the L4 protein was not consistently visualized with this approach. The identification of L4 protein was only achieved when we used A3 temperature-sensitive mutant viruses. When these mutants were grown at non-permissive temperature, aberrant virus particles were formed. Once these purified particles were treated with NP40 and DTT the core wall burst open releasing the DNA and proteins to the exterior. Using these mutants, we were able to show that L4 protein wraps DNA around itself, in a nucleosome-like structure (Smith, 1991; Turner, 2014). The packaging of the virus genome in a nucleosome-like manner could explain the tubular nucleocapsid structure observed in EM sections or with negative staining of the particle with PTA (Heuser, 2005; McFadden et al., 2012; Muller and Peters, 1963; Peters and Muller, 1963).
Combining our results with published data some considerations emerged that will help us understand the structure of the virus. In the whole mount approach, VACV can appear in both the mulberry-like or capsule-like forms (Easterbrook, 1966; Muller and Peters, 1963; Peters and Muller, 1963; Westwood et al., 1964). The difference between these two forms is related mainly to the integrity of the particle. Damage to the viral membrane permits the penetration of the stain and results in the altered the appearance of the particle (Harris and Westwood, 1964). Treatment of VACV with NP40 also damages the particle by removing the lipid layer but preserves the protein array associated with the virion. In this situation, the stain penetrates the particle and all virions have now a capsule-like form. These two forms of VACV can also be differentiated by their size, because the capsular virion has larger dimensions than the mulberry form as has been shown here and by others (Westwood et al., 1964). Our data have also shown that after NP40 treatment virions were enlarged and their dimensions were now similar to the capsule-like particles. It also important to mention that cores increase in size after being released inside the cell (Cyrklaff et al., 2007; Schmidt et al., 2013). The change in size of VACV can be viewed in a broader perspective. The first change in volume of the viral particle occurs during virion morphogenesis during the IV to MV transition. In this stage, the spherical IV particles are transformed into the brick/barrel shape of mature virions and their dimensions are significantly be reduced (Chichon et al., 2009; Condit et al., 2006). It is possible that during this contraction step, water would be excluded from the internal milieu of the cores, and consequently the internal structures of the virion would contract including nucleocapsid that would condense forming the tubular-like structure previously described (Condit et al., 2006; Heuser, 2005; McFadden et al., 2012; Peters and Muller, 1963). MV in the mulberry-like form would be impermeable to water, and after losing the membrane during the entry process rehydration of the internal milieu would occur, and an increase in the size of the particle and unfolding of the nucleocapsid would follow. We can also speculate that the rehydration of the internal component is required for virus activation. In Fig. 12, we show representations of the VACV MV in the three conditions described above. The mulberry form of the virus is represented with the intact membrane containing the lipid layer and protein array. In this situation, the virus is impermeable, the lateral bodies are compacted, and nucleocapsid is condensed. The core wall is represented by the three protein layers and the pore-like structure spanning the wall. Upon the removal of the lipid layer by NP40 treatment, there is an expansion of virion structure including the lateral bodies. In this situation, the protein array is still present but the nucleocapsid is no longer condensed. Upon incubation of virions with NP40+DTT in vitro or after virus penetration into cells in vivo both the lipid layer and the protein array are lost, lateral bodies are enlarged, and nucleocapsid expands.
Fig. 12.

A model for vaccinia virus structure. A) Last steps of morphogenesis starting with immature virion (IV), followed by the incorporation of a DNA molecule to form the immature virions with nucleoid (IVN) and the proteolysis of specific proteins to form the mature virion (MV). B) Fine detail of a sagittal section (see inset, lower left) of the MV showing the different virion sub-domains as specified on the left side of the figure. C) Same as B but the virions were treated with NP-40 and the virion lose the lipid content. D) Same as B but the virions were treated with NP-40+DTT and the virus lose the lipids and the membrane proteins.
The structure of the virion capsid is fundamental to the survival of viruses before infecting the host cell. The formation of the capsid is essential to protect the genome against changes in the environment such as pH, temperature, tonicity of the milieu or other conditions that could damage infectivity. It has been shown that in some viruses the genome is packed under high pressure that is necessary the delivery of the DNA into the infected cell (Bauer et al., 2013; Evilevitch et al., 2011; Roos et al., 2007). These viruses inject DNA directly into the cell and the viral capsid remains in the outside the cell (prokaryotes) or they inject DNA into the nucleus following transport of capsids through the cytoplasm (eukaryotes). Packing the genome at high pressure could be a way to protect the genome and deliver the DNA in an efficient manner. In VACV, the dehydration of the internal milieu may be a mechanism of protecting the genome. After virus entry, core will become permeable to water, and after hydration the nucleocapsid will acquire a relaxed form that will be now favorable to transcription. This phenomenon could be a general mechanism for viruses that contain a transcription apparatus associated with the particle and that require the removal of the membrane or capsid to be activated (Joklik, 1980; Joklik, 1981; Lopez et al., 2011; Skehel and Joklik, 1969).
Materials and Methods
Virus and cells
Wild-type vaccinia virus WR strain and temperature mutants Cts8 and Cts26 (Kato et al., 2004) were used in this work. Viruses were grown in BSC40 cells, an African green monkey cell line, and the purification procedure has been described before (Ausubel et al., 1994; Kato et al., 2004).
Preparation of vaccinia cores for high salt treatment
Virus cores were prepared by incubating 0.1 A260 nm units of virus in 100 μl of core buffer (50 mM Tris–HCl pH 8.0, 10 mM DTT, 0.05% NP40) for 10 min at room temperature. The cores were collected by a 5 min centrifugation in an Eppendorf microfuge and resuspended in 50 μl of a buffer containing 50 mM Tris-HCl, pH 8.0, 0.5% NP40, 100 mM DTT, in the absence (buffer S) or presence of 3 M NaCl (buffer SN). After a 20 min incubation on ice, 3 volumes of ddH2O and 30 ug of glycogen were added to the reaction and soluble and insoluble fractions were separated by 30 min centrifugation at 20,000 × g through a 30 μl 10% sucrose cushion in an Eppendorf microfuge. The pellet was resuspended in 100 μl SDS sample buffer. The supernatant fraction was adjusted to a final concentration of 0.1 M sodium acetate, pH 5.2. Three volumes of ethanol were added and the sample incubated for 2 h at −20°C. The sample was centrifuged for 30 min at maximum speed in an Eppendorf microfuge. The supernatant was discarded and the pellet resuspended in 100 μl SDS sample buffer. The samples were subjected to western blot analysis as described below.
Western blot analysis
Virion fractions were prepared by incubating 0.1 – 0.15 A260nm units (6.8–10 μg) of wt particles in a 300 μl volume of either Tris buffer (50 mM Tris–HCl pH 8.0), NP buffer (0.5 % NP40 in 50 mM Tris–HCl pH 8.0) or NP+DTT buffer (50 mM DTT, 0.5 % NP40 in 50 mM Tris–HCl pH 8.0), for 20 minutes at 37 °C. The fractions were separated by centrifugation through 30 μl of a 10% sucrose cushion for 20 min at maximum speed in an Eppendorf microfuge. Three hundred thirty μl of supernatant fraction was removed and the pellet was resuspended in 330 μl of 50 mM Tris-HCl, pH 8.0. Sixty μl of 5x SDS-sample buffer was added to the pellet and supernatant fractions. The proteins in each fraction were separated by an 11 % SDS-PAGE, transferred to a nitrocellulose membrane and processed for Western blot analysis as described previously (Boyd et al., 2010). The membrane was blocked with a solution of TBS-T (50 mM Tris-HCl pH 7.5; 150 mM NaCl; 0.01% Tween 20) and 5% non-fat dry milk (TBS-TM) for 1 hour, and then incubated with an appropriate dilution of the primary antibody in TBS-TM overnight at 8°C. The membrane was washed 5 times for 10 minutes with TBS-T and incubated with a horseradish peroxidase conjugated secondary antibody (Santa Cruz Biotechnology) diluted in TBS-TM for 2 hours at 8°C. The membrane was washed 5 times for 10 minutes and the protein complex detected by chemiluminescence (GE Healthcare). The dilution and source of the primary antibodies were: anti-A3 (1:20,000) rabbit serum supplied by Dr. Bernard Moss (NIAID); anti-A4 (1:20,000) and anti-A10 (1:20,000) rabbit serum supplied by Dr. Mariano Esteban (Universidad Autónoma, Madrid); anti-A10 (1:2,000) monoclonal antibody supplied by Dr. Yan Xiang (University of Texas); anti-A14 (1:1,000), anti-F17 (1:20,000) and anti-H1 (1:1,000) rabbit serum supplied by Dr. Paula Traktman (Medical College of Wisconsin); anti-A27 (1:10,000) monoclonal antibody supplied by Dr. David Ulaeto (Dstl Porton Down, Salisbury); and anti-L4 (1:40,000) rabbit serum supplied by Dr. Dennis Hruby (Oregon State University); dsDNA monoclonal antibody (1: 50) clone HYB331-01, Santa Cruz Biotechnology; immunogold conjugate goat anti-rabbit and goat anti-mouse antibody (1:50) British Biocell International (BBI).
Negative staining of vaccinia virus
Five to seven μl of purified virions (1.2 × 109 particles) were adsorbed on a formvar/carbon- coated nickel grid (EMS –USA) for 10 minutes at room temperature. The grids were then washed with 10 mM Tris-HCl, pH 8.0 and incubated for 20 minutes in the presence of Tris, NP, or NP+DTT buffers described above. The grids were then washed for 5 minutes with 10 mM Tris-HCl, rinsed with ddH2O and stained for 2 minutes with one of the following solutions: 3% phosphotungstic acid (PTA) pH 7.0, 3% phosphotungstic acid (PTA) pH 10.5, 3% ammonium molybdate (AmMo) pH 7.0, 1% uranyl acetate (UA), and Nano-W (2% methylamine tungstate pH 6.8, Nano Probes). The grids were dried and the samples were viewed in a Hitachi H7000 transmission electron microscope (TEM). As described in the text, in some experiments grids were incubated with in buffers S or SN, Pronase E (Sigma-Aldrich) or Papain (Sigma Aldrich) or DNase (Sigma Aldrich) before the negative staining or immunogold labeling. For the rotatory shadowing of the sample, the dried grid was loaded into a Bal-Tec Med030 metal evaporator equipped with C-Pd E-guns. Low angle rotary shadow was carried out at 5-degree angle and 100 RPM (Hendricks, 2014). The virus was imaged on a Hitachi H-7000 transmission electron microscope, with assistance of the University of Florida ICBR Electron Microscopy Core Laboratory.
Immunogold labeling of vaccinia proteins
For the identification of virion proteins associated with the particles, virions attached to grids and treated as described above were incubated in blocking solution (3% albumin –Sigma Aldrich) for 30 minutes at room temperature. When treating virion particles previously exposed to NP40, 0.1% NP40 (USB) was added to the blocking solution to reduce the background. The grids were then incubated with antibody mixture diluted in blocking solution (see figure legend) and incubated overnight at 4°C. The grids were then washed twice for 10 minutes each in PBS-EM (10.4 mM Na2HPO4, 3.2 mM KH2PO4, and 150 mM NaCl, pH 7.4), then incubated for 10 minutes in blocking solution and then incubated with the gold conjugated antibody for 3 hours at room temperature. The grids were then washed twice for 10 minutes each in PBS-EM, fixed with McDowell Trumps fixative (EMS) (1% glutaraldehyde, 4% formaldehyde in 0.1M Na2HPO4 buffer, pH 7.2) diluted 1:4, washed in water for 5 minutes, stain for 5 minutes with Nano-van (2% methylamine vanadate (Nano Probes), and dried before observing in the TEM.
Acknowledgments
We thank Bernard Moss, Paula Traktman, David Ulaeto, Mariano Esteban and Dennis Hruby for antisera used in this work; Karen Kelley and the University of Florida ICBR Electron Microscopy Core Laboratory for excellent advice and technical assistance; Michel H Moussatche for the drawing the vaccinia model; and Desyreé Jesus, Susan D’Costa, Baron McFadden, Jessica Tate and Ken Burns for valuable suggestions during the execution of this work. This work was supported by NIH grant R01 AI055560 to RCC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York: John Wiley & Sons; 1994. [Google Scholar]
- Bauer DW, Huffman JB, Homa FL, Evilevitch A. Herpes virus genome, the pressure is on. J Am Chem Soc. 2013;135:11216–11221. doi: 10.1021/ja404008r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd O, Strahl AL, Rodeffer C, Condit RC, Moussatche N. Temperature-sensitive mutant in the vaccinia virus E6 protein produce virions that are transcriptionally inactive. Virology. 2010;399:221–230. doi: 10.1016/j.virol.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GC, Law M, Hollinshead M, Smith GL. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol. 2005;86:1279–1290. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- Castro AP, Carvalho TM, Moussatche N, Damaso CR. Redistribution of cyclophilin A to viral factories during vaccinia virus infection and its incorporation into mature particles. J Virol. 2003;77:9052–9068. doi: 10.1128/JVI.77.16.9052-9068.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chichon FJ, Rodriguez MJ, Risco C, Fraile-Ramos A, Fernandez JJ, Esteban M, Carrascosa JL. Membrane remodelling during vaccinia virus morphogenesis. Biol Cell. 2009;101:401–414. doi: 10.1042/BC20080176. [DOI] [PubMed] [Google Scholar]
- Ching YC, Chung CS, Huang CY, Hsia Y, Tang YL, Chang W. Disulfide bond formation at the C termini of vaccinia virus A26 and A27 proteins does not require viral redox enzymes and suppresses glycosaminoglycan-mediated cell fusion. J Virol. 2009;83:6464–6476. doi: 10.1128/JVI.02295-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WL, Chang W. Vaccinia virus J1R protein: a viral membrane protein that is essential for virion morphogenesis. J Virol. 2002;76:9575–9587. doi: 10.1128/JVI.76.19.9575-9587.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CS, Chen CH, Ho MY, Huang CY, Liao CL, Chang W. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J Virol. 2006;80:2127–2140. doi: 10.1128/JVI.80.5.2127-2140.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condit RC, Moussatche N, Traktman P. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res. 2006;66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- Cudmore S, Blasco R, Vincentelli R, Esteban M, Sodeik B, Griffiths G, Krijnse LJ. A vaccinia virus core protein, p39, is membrane associated. J Virol. 1996;70:6909–6921. doi: 10.1128/jvi.70.10.6909-6921.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyrklaff M, Linaroudis A, Boicu M, Chlanda P, Baumeister W, Griffiths G, Krijnse-Locker J. Whole cell cryo-electron tomography reveals distinct disassembly intermediates of vaccinia virus. PLoS ONE. 2007;2:e420. doi: 10.1371/journal.pone.0000420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyrklaff M, Risco C, Fernandez JJ, Jimenez MV, Esteban M, Baumeister W, Carrascosa JL. Cryo-electron tomography of vaccinia virus. Proc Natl Acad Sci U S A. 2005;102:2772–2777. doi: 10.1073/pnas.0409825102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Fonseca FG, Weisberg AS, Caeiro MF, Moss B. Vaccinia virus mutants with alanine substitutions in the conserved G5R gene fail to initiate morphogenesis at the nonpermissive temperature. J Virol. 2004;78:10238–10248. doi: 10.1128/JVI.78.19.10238-10248.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales S. The uptake and development of vaccinia virus in strain L cells followed with labeled viral deoxyribonucleic acid. J Cell Biol. 1963;18:51–72. doi: 10.1083/jcb.18.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales S. An electron microscope study of the early association between two mammalian viruses and their hosts. J Cell Biol. 1962;13:303–322. doi: 10.1083/jcb.13.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales S, Siminovith L. The development of vaccinia virus in Earle’s L strain cells as examined by electron microscopy. J Biophys Biochem Cytol. 1961;10:475–503. doi: 10.1083/jcb.10.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Carlo S, Harris JR. Negative staining and cryo-negative staining of macromolecules and viruses for TEM. Micron. 2011;42:117–131. doi: 10.1016/j.micron.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubochet J, Adrian M, Richter K, Garces J, Wittek R. Structure of intracellular mature vaccinia virus observed by cryoelectron microscopy. J Virol. 1994;68:1935–1941. doi: 10.1128/jvi.68.3.1935-1941.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easterbrook KB. Controlled degradation of vaccinia virions in vitro: an electron microscopic study. J Ultrastruct Res. 1966;14:484–496. doi: 10.1016/s0022-5320(66)80077-1. [DOI] [PubMed] [Google Scholar]
- Evilevitch A, Roos WH, Ivanovska IL, Jeembaeva M, Jonsson B, Wuite GJ. Effects of salts on internal DNA pressure and mechanical properties of phage capsids. J Mol Biol. 2011;405:18–23. doi: 10.1016/j.jmb.2010.10.039. [DOI] [PubMed] [Google Scholar]
- Franke CA, Hruby DE. Association of non-viral proteins with recombinant vaccinia virus virions. Arch Virol. 1987;94:347–351. doi: 10.1007/BF01310729. [DOI] [PubMed] [Google Scholar]
- Goebel SJ, Johnson GP, Perkus ME, Davis SW, Winslow JP, Paoletti E. The complete DNA sequence of vaccinia virus. Virology. 1990;179:247–263. doi: 10.1016/0042-6822(90)90294-2. [DOI] [PubMed] [Google Scholar]
- Gomez CE, Esteban M. Recombinant proteins produced by vaccinia virus vectors can be incorporated within the virion (IMV form) into different compartments. Arch Virol. 2001;146:875–892. doi: 10.1007/s007050170122. [DOI] [PubMed] [Google Scholar]
- Griffiths G, Wepf R, Wendt T, Locker JK, Cyrklaff M, Roos N. Structure and assembly of intracellular mature vaccinia virus: isolated-particle analysis. J Virol. 2001;75:11034–11055. doi: 10.1128/JVI.75.22.11034-11055.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross CH, Shuman S. Vaccinia virions lacking the RNA helicase nucleoside triphosphate phosphohydrolase II are defective in early transcription. J Virol. 1996;70:8549–8557. doi: 10.1128/jvi.70.12.8549-8557.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WJ, Westwood JC. Phosphotungstate Staining of vaccinia virus. J Gen Microbiol. 1964;34:491–495. doi: 10.1099/00221287-34-3-491. [DOI] [PubMed] [Google Scholar]
- Hendricks GM. Metal shadowing for electron microscopy. Methods Mol Biol. 2014;1117:73–93. doi: 10.1007/978-1-62703-776-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J. Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J Cell Biol. 2005;169:269–283. doi: 10.1083/jcb.200412169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, Ellermann-Eriksen S, Kurt-Jones E, Landolfo S, Damania B, Melchjorsen J, Bowie AG, Fitzgerald KA, Paludan SR. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol. 2013;190:2311–2319. doi: 10.4049/jimmunol.1202749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihashi Y, Oie M. The activation of vaccinia virus infectivity by the transfer of phosphatidylserine from the plasma membrane. Virology. 1983;130:306–317. doi: 10.1016/0042-6822(83)90085-5. [DOI] [PubMed] [Google Scholar]
- Ichihashi Y, Oie M, Tsuruhara T. Location of DNA-binding proteins and disulfide-linked proteins in vaccinia virus structural elements. J Virol. 1984;50:929–938. doi: 10.1128/jvi.50.3.929-938.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesus DM, Moussatche N, Condit RC. Vaccinia virus mutations in the L4R gene encoding a virion structural protein produce abnormal mature particles lacking a nucleocapsid. 2014. () [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joklik WK. The intracellular uncoating of poxvirus DNA. I The molecular basis of the uncoating process. J Mol Biol. 1964a;8:277–288. doi: 10.1016/s0022-2836(64)80137-6. [DOI] [PubMed] [Google Scholar]
- Joklik WK. The intracellular uncoating of poxvirus DNA. II The fate of radioactively-labeled rabbitpox virus. J Mol Biol. 1964b;8:263–276. doi: 10.1016/s0022-2836(64)80136-4. [DOI] [PubMed] [Google Scholar]
- Joklik WK. Virus synthesis and replication: reovirus vs. vaccinia virus Yale. J Biol Med. 1980;53:27–39. [PMC free article] [PubMed] [Google Scholar]
- Joklik WK. Structure and function of the reovirus genome. Microbiol Rev. 1981;45:483–501. doi: 10.1128/mr.45.4.483-501.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalejta RF. Tegument proteins of human cytomegalovirus. Microbiol Mol Biol Rev. 2008;72:249–65. doi: 10.1128/MMBR.00040-07. table. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates J, Beeson J. Ribonucleic acid synthesis in vaccinia virus. I The mechanism of synthesis and release of RNA in vaccinia cores. J Mol Biol. 1970;50:1–18. doi: 10.1016/0022-2836(70)90100-2. [DOI] [PubMed] [Google Scholar]
- Kato SE, Condit RC, Moussatche N. The vaccinia virus E8R gene product is required for formation of transcriptionally active virions. Virology. 2007;367:398–412. doi: 10.1016/j.virol.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato SE, Strahl AL, Moussatche N, Condit RC. Temperature-sensitive mutants in the vaccinia virus 4b virion structural protein assemble malformed, transcriptionally inactive intracellular mature virions. Virology. 2004;330:127–146. doi: 10.1016/j.virol.2004.08.038. [DOI] [PubMed] [Google Scholar]
- Kelly BJ, Fraefel C, Cunningham AL, Diefenbach RJ. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res. 2009;145:173–186. doi: 10.1016/j.virusres.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Kilcher S, Schmidt FI, Schneider C, Kopf M, Helenius A, Mercer J. siRNA screen of early poxvirus genes identifies the AAA+ ATPase D5 as the virus genome-uncoating factor. Cell Host Microbe. 2014;15:103–112. doi: 10.1016/j.chom.2013.12.008. [DOI] [PubMed] [Google Scholar]
- Laliberte JP, Moss B. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc Natl Acad Sci U S A. 2009;106:17517–17521. doi: 10.1073/pnas.0909376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez T, Silva-Ayala D, Lopez S, Arias CF. Replication of the rotavirus genome requires an active ubiquitin-proteasome system. J Virol. 2011;85:11964–11971. doi: 10.1128/JVI.05286-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin AJ, McPherson A, Gershon PD. Structure of intracellular mature vaccinia virus visualized by in situ atomic force microscopy. J Virol. 2003;77:6332–6340. doi: 10.1128/JVI.77.11.6332-6340.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson J, Chou W, Ngo T, Gershon PD. Static and dynamic protein phosphorylation in the Vaccinia virion. Virology. 2014;452–453:310–323. doi: 10.1016/j.virol.2014.01.012. [DOI] [PubMed] [Google Scholar]
- McFadden BD, Moussatche N, Kelley K, Kang BH, Condit RC. Vaccinia virions deficient in transcription enzymes lack a nucleocapsid. Virology. 2012;434:50–58. doi: 10.1016/j.virol.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J, Traktman P. Investigation of structural and functional motifs within the vaccinia virus A14 phosphoprotein, an essential component of the virion membrane. J Virol. 2003;77:8857–8871. doi: 10.1128/JVI.77.16.8857-8871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. Poxvirus entry and membrane fusion. Virology. 2006;344:48–54. doi: 10.1016/j.virol.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Moss B. Poxvirus cell entry: how many proteins does it take? Viruses. 2012;4:688–707. doi: 10.3390/v4050688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B. Poxviridae. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Wolters Kluwer-Lippincott Williams and Wilkins; 2013. pp. 2129–2159. [Google Scholar]
- Muller G, Peters D. Substructures of vaccinia virus, demonstrated by negative contrast. Arch Gesamte Virusforsch. 1963;13:435–451. [PubMed] [Google Scholar]
- Naginton J, Horne RW. Morphological studies of orf and vaccinia viruses. Virology. 1962;16:248–260. doi: 10.1016/0042-6822(62)90245-3. [DOI] [PubMed] [Google Scholar]
- Nichols RJ, Stanitsa E, Unger B, Traktman P. The vaccinia virus gene I2L encodes a membrane protein with an essential role in virion entry. J Virol. 2008;82:10247–10261. doi: 10.1128/JVI.01035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen K, Snijder EJ, Schleich S, Roos N, Griffiths G, Locker JK. Characterization of vaccinia virus intracellular cores: implications for viral uncoating and core structure. J Virol. 2000;74:3525–3536. doi: 10.1128/jvi.74.8.3525-3536.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters D, Muller G. The fine structure of the DNA-containing core of vaccinia virus. Virology. 1963;21:267–269. doi: 10.1016/0042-6822(63)90267-8. [DOI] [PubMed] [Google Scholar]
- Resch W, Hixson KK, Moore RJ, Lipton MS, Moss B. Protein composition of the vaccinia virus mature virion. Virology. 2007;358:233–247. doi: 10.1016/j.virol.2006.08.025. [DOI] [PubMed] [Google Scholar]
- Resch W, Moss B. The conserved poxvirus L3 virion protein is required for transcription of vaccinia virus early genes. Journal of Virology. 2005;79:14719–14729. doi: 10.1128/JVI.79.23.14719-14729.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risco C, Rodriguez JR, Demkowicz W, Heljasvaara R, Carrascosa JL, Esteban M, Rodriguez D. The vaccinia virus 39-kDa protein forms a stable complex with the p4a/4a major core protein early in morphogenesis. Virology. 1999;265:375–386. doi: 10.1006/viro.1999.0046. [DOI] [PubMed] [Google Scholar]
- Roberts KL, Smith GL. Vaccinia virus morphogenesis and dissemination. Trends Microbiol. 2008;16:472–479. doi: 10.1016/j.tim.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Rodriguez JF, Paez E, Esteban M. A 14,000-Mr envelope protein of vaccinia virus is involved in cell fusion and forms covalently linked trimers. J Virol. 1987;61:395–404. doi: 10.1128/jvi.61.2.395-404.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos N, Cyrklaff M, Cudmore S, Blasco R, Krijnse-Locker J, Griffiths G. A novel immunogold cryoelectron microscopic approach to investigate the structure of the intracellular and extracellular forms of vaccinia virus. EMBO J. 1996;15:2343–2355. [PMC free article] [PubMed] [Google Scholar]
- Roos WH, Ivanovska IL, Evilevitch A, Wuite GJ. Viral capsids: mechanical characteristics, genome packaging and delivery mechanisms. Cell Mol Life Sci. 2007;64:1484–1497. doi: 10.1007/s00018-007-6451-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt FI, Bleck CK, Reh L, Novy K, Wollscheid B, Helenius A, Stahlberg H, Mercer J. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep. 2013;4:464–476. doi: 10.1016/j.celrep.2013.06.028. [DOI] [PubMed] [Google Scholar]
- Senkevich TG, Koonin EV, Moss B. Predicted poxvirus FEN1-like nuclease required for homologous recombination, double-strand break repair and full-size genome formation. Proc Natl Acad Sci U S A. 2009;106:17921–17926. doi: 10.1073/pnas.0909529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich TG, White CL, Koonin EV, Moss B. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc Natl Acad Sci U S A. 2002;99:6667–6672. doi: 10.1073/pnas.062163799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich TG, Wyatt LS, Weisberg AS, Koonin EV, Moss B. A conserved poxvirus NlpC/P60 superfamily protein contributes to vaccinia virus virulence in mice but not to replication in cell culture. Virology. 2008;374:506–514. doi: 10.1016/j.virol.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel JJ, Joklik WK. Studies on the in vitro transcription of reovirus RNA catalyzed by reovirus cores. Virology. 1969;39:822–831. doi: 10.1016/0042-6822(69)90019-1. [DOI] [PubMed] [Google Scholar]
- Smith MM. Histone structure and function. Curr Opin Cell Biol. 1991;3:429–437. doi: 10.1016/0955-0674(91)90070-f. [DOI] [PubMed] [Google Scholar]
- Sodeik B, Krijnse-Locker J. Assembly of vaccinia virus revisited: de novo membrane synthesis or acquisition from the host? Trends Microbiol. 2002;10:15–24. doi: 10.1016/s0966-842x(01)02256-9. [DOI] [PubMed] [Google Scholar]
- Soloski MJ, Cabrera CV, Esteban M, Holowczak JA. Studies concerning the structure and organization of the vaccinia virus nucleoid. I Isolation and characterization of subviral particles prepared by treating virions with guanidine-HCL, nonidet-P40, and 2-mercaptoethanol. Virology. 1979;99:209–217. doi: 10.1016/0042-6822(79)90001-1. [DOI] [PubMed] [Google Scholar]
- Soloski MJ, Holowczak JA. Characterization of supercoiled nucleoprotein complexes released from detergent-treated vaccinia virions. J Virol. 1981;37:770–783. doi: 10.1128/jvi.37.2.770-783.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood CL, Ward JM, Moss B. Vaccinia virus encodes I5, a small hydrophobic virion membrane protein that enhances replication and virulence in mice. J Virol. 2008;82:10071–10078. doi: 10.1128/JVI.01355-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szajner P, Jaffe H, Weisberg AS, Moss B. A complex of seven vaccinia virus proteins conserved in all chordopoxviruses is required for the association of membranes and viroplasm to form immature virions. Virology. 2004;330:447–459. doi: 10.1016/j.virol.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Tomtishen JP., III Human cytomegalovirus tegument proteins (pp65, pp71, pp150, pp28) Virol J. 2012;9:22. doi: 10.1186/1743-422X-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley AC, Senkevich TG, Moss B. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J Virol. 2005;79:9458–9469. doi: 10.1128/JVI.79.15.9458-9469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traktman P, Caligiuri A, Jesty SA, Liu K, Sankar U. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of virion morphogenesis. J Virol. 1995;69:6581–6587. doi: 10.1128/jvi.69.10.6581-6587.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BM. Nucleosome signalling; An evolving concept. Biochim Biophys Acta. 2014;1839:623–626. doi: 10.1016/j.bbagrm.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Unger B, Mercer J, Boyle KA, Traktman P. Biogenesis of the vaccinia virus membrane: genetic and ultrastructural analysis of the contributions of the A14 and A17 proteins. J Virol. 2013;87:1083–1097. doi: 10.1128/JVI.02529-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger B, Nichols RJ, Stanitsa ES, Traktman P. Functional characterization of the vaccinia virus I5 protein. Virol J. 2008;5:148. doi: 10.1186/1743-422X-5-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallengren K, Risco C, Krijnse-Locker J, Esteban M, Rodriguez D. The A17L gene product of vaccinia virus is exposed on the surface of IMV. Virology. 2001;290:143–152. doi: 10.1006/viro.2001.1131. [DOI] [PubMed] [Google Scholar]
- Westwood JC, Harris WJ, Zwartouw HT, Titmuss DH, Appleyard G. Studies on the structure of vaccinia virus. J Gen Microbiol. 1964;34:67–78. doi: 10.1099/00221287-34-1-67. [DOI] [PubMed] [Google Scholar]
- Wickramasekera NT, Traktman P. Structure/Function analysis of the vaccinia virus F18 phosphoprotein, an abundant core component required for virion maturation and infectivity. J Virol. 2010;84:6846–6860. doi: 10.1128/JVI.00399-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock D, Smith GL. Vaccinia virions lacking core protein VP8 are deficient in early transcription. Journal of Virology. 1996;70:934–943. doi: 10.1128/jvi.70.2.934-943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton S, Mohandas AR, Dales S. Organization of the vaccinia envelope and relationship to the structure of intracellular mature virions. Virology. 1995;214:503–511. doi: 10.1006/viro.1995.0061. [DOI] [PubMed] [Google Scholar]
- Yoder JD, Chen TS, Gagnier CR, Vemulapalli S, Maier CS, Hruby DE. Pox proteomics: mass spectrometry analysis and identification of Vaccinia virion proteins. Virol J. 2006;3:10. doi: 10.1186/1743-422X-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]