

Abstract
Neurofibromatosis type I (NF1) is an autosomal dominant disease with an incidence of 1/3000, caused by mutations in the NF1 gene, which encodes the RAS/GTPase-activating protein neurofibromin. Non-bone union following fracture (pseudarthrosis) in children with NF1 remains a challenging orthopedic condition to treat. Recent progress in understanding the biology of neurofibromin suggested that NF1 pseudarthrosis stems primarily from defects in the bone mesenchymal lineage and hypersensitivity of hematopoietic cells to TGFβ. However, clinically relevant pharmacological approaches to augment bone union in these patients remain limited. In this study, we report the generation of a novel conditional mutant mouse line used to model NF1 pseudoarthrosis, in which Nf1 can be ablated in an inducible fashion in osteoprogenitors of post-natal mice, thus circumventing the dwarfism associated with previous mouse models where Nf1 is ablated in embryonic mesenchymal cell lineages. An ex vivo-based cell culture approach based on the use of Nf1flox/flox bone marrow stromal cells showed that loss of Nf1 impairs osteoprogenitor cell differentiation in a cell-autonomous manner, independent of developmental growth plate-derived or paracrine/hormonal influences. In addition, in vitro gene expression and differentiation assays indicated that chronic ERK activation in Nf1-deficient osteoprogenitors blunts the pro-osteogenic property of BMP2, based on the observation that only combination treatment with BMP2 and MEK inhibition promoted the differentiation of Nf1-deficient osteoprogenitors. The in vivo preclinical relevance of these findings was confirmed by the improved bone healing and callus strength observed in Nf1osx−/− mice receiving Trametinib (a MEK inhibitor) and BMP2 released locally at the fracture site via a novel nanoparticle and polyglycidol (PEG)-based delivery method. Collectively, these results provide novel evidence for a cell-autonomous role of neurofibromin in osteoprogenitor cells and insights about a novel targeted approach for the treatment of NF1 pseudoarthrosis.
Keywords: Neurofibromatosis, genetic animal models, preclinical studies, BMPs, stromal/mesenchymal stem cells, osteoblasts, anabolics
INTRODUCTION
Children and adults with neurofibromatosis type 1 (NF1; OMIM 162200) can present with a number of skeletal maladies. Osteopenia, idiopathic scoliosis, short stature, chest wall deformities and sphenoid wing dysplasia are amenable to acceptable clinical care (1). Contrastingly, dystrophic scoliosis and long bone dysplasia represent two skeletal conditions associated with high morbidity, costs, and burden in these patients. Dystrophic scoliosis usually presents in pre-adolescents as sharp angulation over a short segment of the spine (2), is frequently associated with paraspinal or other internal neurofibromas (3,4), and is treated with often invasive surgeries in skeletally immature and growing children. Long bone dysplasia in infants with NF1 is usually unilateral and commonly involves the tibia. Dysplastic tibiae frequently sustain fracture with minimum trauma, followed by non-union (pseudoarthrosis). Bracing techniques are used to reduce chances of fracture until children reach skeletal maturity. Invasive and repetitive surgeries make this condition a challenging one to treat and often lead to amputation. Knowledge on natural history and pathogenesis of NF1 skeletal maladies is still speculative, contributing to the lack of consensus regarding treatments.
The identification of effective drug targets to promote osteoblast differentiation and eventually bone union in individuals with NF1 pseudarthrosis will require a better understanding of the molecular basis of the differentiation defect of Nf1-deficient bone cells. TGFβ signaling inhibition has recently been shown to improve bone mass and bone healing in a model of NF1 skeletal defects (5), however it remains to be addressed whether TGFβ signaling is increased in human NF1 dysplastic bones. On the other hand, the use of rBMP2 to promote bone repair in individuals with NF1 pseudarthrosis and in related preclinical mouse models has had a limited success (6-10), leading us to test the hypothesis that Nf1 deficiency in osteoprogenitors may impair BMP2 signaling and its bone anabolic properties. In this study, we created a new mouse model characterized by Nf1 deficiency in post-natal mesenchymal progenitors to determine the potential of MEK inhibition by Trametinib, a MEK inhibitor currently in Phase III clinical studies, to increase BMP2 efficacy in promoting bone healing.
MATERIAL AND METHODS
Animals
All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Vanderbilt University Medical Center. WT and Nf1TetOff-Osx−/− mice (herein calledNf1Osx−/− mice) were generated by crossing Nf1flox/flox mice (11) and Osx-tTA, tetO-cre; Nf1flox/flox mice (12). To repress transactivation of Cre by the Osx promoter during development, 200μg/ml doxycycline was added to the drinking water of pregnant mothers and pups and refreshed every 2-3 days, until the time at which recombination/deletion of Nf1 was desired. All experimental mice were originated from the same colony to increase genetic homogeneity.
For genotyping, genomic DNA was isolated from tail snips by sodium hydroxide digestion, and PCR was performed using primers P1, P2 and P4, as described by Zhu et al (11). The Osx-Cre transgene was detected using the forward: 5′-GCG GTC TGG CAG TAA AAA CTA TC-3′ and reverse: 5′-GTG AAA CAG CAT TGC TGT CAC TT-3′ primers.
Generation of mid-diaphyseal fractures
Closed mid-diaphyseal fracture of the tibia was created by three-point bending with an Einhorn device in 2 month-old male and female mice, as previously described (13). To produce stabilized fractures, an intramedullary fixation was used by inserting a 0.25 mm metal insect pin in the tibial tuberosity through the patellar tendon, prior to the creation of the fracture. Buprenorphine was administered subcutaneously for pain control. X-rays were taken following fracture to exclude any mice with unsatisfactory fractures.
Cell culture
BMSCs were extracted from long bones by centrifugation of dissected femoral and tibial diaphyses at 2000g for 3 min. The cells were then counted, plated and grown for 7 days in αMEM supplemented with 10% FBS, 100 I.U./ml penicillin, 100μg/ml streptomycin (Cellgro, Manassas, VA, USA). At day 7, mineralization was induced by the addition of 50μg/ml of ascorbic acid and 10mM β-glycerophosphate. The media was refreshed every 2-3 days for 10 more days.
Gene expression assays
Total RNA was extracted using TRIzol (Invitrogen, Grand Island, NY, USA) and cDNAs were synthesized following DNase I treatment using the high-capacity cDNA reverse-transcription kit (Applied Biosystems, USA). Quantitative PCR (qPCR) was performed by using TaqMan or SYBR green gene expression assays. The probe and primer sets for Runx2 (Mm00501578_m1); CyclinD (Mm00432359_m1); Osx (Mm00504574_m1); Tnsap (Mm00475834_m1) and the normalizer Hprt (Mm00446968_m1) were obtained from Applied Biosystems (Foster City, CA, USA). The SYBR green primers were: Nf1 (forward; GTATTGAATTGAAGCACCTTTGTTTGG, reverse; CTGCCCAAGGCTCCCCCAG); Ocn (forward; ACCCTGGCTGCGCTCTGTCTCT, reverse; GATGCGTTTGTAGGCGGTCTTCA) and Col1a1 (forward; GACATCCCTGAAGTCAGCTGC, reverse; TCCCTTGGGTCCCTCGAC). Specificity of amplification was verified by the presence of a single peak on the dissociation curve. Specific amplification conditions are available upon request. Measurements were performed in triplicate and from at least 3 independent experiments.
Western blot analyses
Whole cell lysates were separated by SDS-PAGE electrophoresis according to standard protocols. Nitrocellulose membranes were probed using standard protocols using an anti-Phospho-ERK1/2 antibody (Cell Signaling cat#9101, dilution 1:1000) and ananti-ERK1/2 antibody (Cell Signaling cat#9102, dilution 1:1000).
Cell culture staining
Tissue non-specific alkaline phosphatase (TNSALP) activity was assayed using the ALP assay kit (Sigma). Cell supernatant or lysate from adherent cells plated in 12 well plates were incubated with 20mM p-nitrophenylphosphate in reaction buffer (50mM Tris-HCl, pH 8.8, 10mM MgCl2) for 5-10 minutes at 37°C, then the reaction was stopped with 0.1N NaOH, and the absorbance was read at 405nm.
For Alizarin staining, cells in 12 well plates were washed twice in 1× PBS and stained with a solution containing 40mM Alizarin red S solution. The plates were incubated at room temperature for 20 minutes, then washed and air-dried. Bound calcium was released by adding 1ml of acetic acid and the mixture was incubated for 20 minutes. One hundred microliters of the solution was collected for OD405 measurement.
Adenovirus Infection of BMSCs
BMSCs were isolated from Nf1flox/flox mice and seeded at a density of 106 cells/well in 12 well plates. At 40% confluence, cells were incubated in complete cell culture medium (α-MEM, 10% FBS and 100 I.U./ml penicillin) containing either Ad5-CMV-GFP or Ad5-CMV-cre (Vector development lab, Baylor College of Medicine) at 2.5×109 PFUs. After 2 days of incubation, the medium in each well was replaced with complete cell culture medium. Nf1 mRNA and recombination efficiency were determined according to the protocol previously described by Wang et al (14).
Polyester nanoparticle synthesis (NP)
Poly(α-allyl-valerolactone, valerolactone) containing 4% allyl groups was epoxidized with meta-chloroperoxybenzoic acid (mcpba, 1.2 eq per alkene, Sigma) in CH2Cl2 (0.065 M alkene) for 48 hours and washed with saturated sodium bicarbonate (15). The resulting polymer was dried under reduced pressure. 2,2′-ethylenedioxy-bis(ethylamine) (7.5 μL, 0.51 mmol, Sigma) was added to a solution of poly(epoxide-valerolactone, valerolactone) (0.173 g, Mn = 3,644 Da) dissolved in CH2Cl2 (21.1 mL). The reaction mixture refluxed for 12 hours at 46° C. Residual bisamine was removed by dialyzing with Snakeskin Pleated Dialysis Tubing (MWCO = 10,000) against CH2Cl2 to yield nanoparticles (NP).
Trametinib encapsulation into nanoparticles (NP-GSK212)
A solution of nanoparticle (23.0 mg) and MEK inhibitor (Trametinib, 5.0 mg, GlaxoSmithKline) in DMSO (1.00 mL) was added drop-wise to a vortexing solution of aqueous 1% d-α-tocopherolpolyethyleneglycol (1000) succinate (Vit E-TPGS (Sigma), 14.0 mL). The resulting precipitate was washed with deionized water using three cycles of centrifugation at 7830 rpm for 20 minutes. The precipitate was freeze-dried to yield a white powder (6.4% MEK inhibitor/NP (wt/wt) determined by NanoDrop UV-Vis at 258 nm).
Polyglycidol-BMP2/NP-Trametinib dual drug delivery system
Polyglycidolhomopolymer (16) was synthesized by first stirring 3-methyl-1-butanol (0.98 mL, 1.7 M in anhydrous THF) with Sn(OTf)2 (1.28 mL, 0.037 M in anhydrous THF) in a nitrogen-purged flask. Glycidol monomer (10.0 g, 135 mmol, Sigma) was added drop-wise to the cooled reaction mixture. After the monomer addition was complete, the reaction vessel was allowed to warm up to room temperature. Once stirring was completely impeded, the crude viscous polymer product was dissolved in a minimal amount of methanol and precipitated into vigorously stirring acetone, which was then decanted to afford the pure glycidol homopolymer product as a translucent, viscous material. Recombinant human bone morphogenic protein-2 (rBMP2, Creative BioMart) was suspended in 20 mM acetic acid (10 mg/mL), added to polyglycidol, and diluted with PBS for a rBMP2 concentration of 0.50 μg/μL (6) and polyglycidol concentration of 0.46 g/mL. For Trametinibin jections and combo injections, encapsulated Trametinib was first sonicated in PBS and transferred to the polyglycidol mix to yield a Trametinib concentration of 0.188 μg/μL (17,18). Vehicle was prepared using unloaded nanoparticles and formulated similarly as described above.
X-rays and μCT analyses
Radiographs were obtained using a digital cabinet X-ray system (LX-60, Faxitron X-Ray, USA). μCT analyses were performed using a high-resolution benchtopScancoμ CT 40 system (Scanco Medical, Bassersdorf, Switzerland). Tomographic images were acquired at 55kVp and 145mA with an isotropic voxel size of 12μm and at an integration time of 250ms with 1000 projections collected per 360° rotation. Scans were acquired in PBS. μCT images were reconstructed, filtered (σ=0.8 and support = 1.0) and thresholded at 200. Callus trabecular bone was contoured manually on every 10 sections, and sections in between were automatically contoured with the software algorithm.
Biomechanical Testing
The fractured tibia from WT and Nf1Osx−/− littermates was cleaned of soft tissue, and stored frozen in PBS until thawed for testing. Three-point-bending was accomplished in displacement control on an InstronDynamight 4481 servo-hydraulic material testing apparatus (Instron, Norwood, MA). Hydrated samples were tested with span of 6mm at a rate of 3mm/min as per (19). Force was measured using a 100N load cell, and displacement was measured by a linear variable displacement transducer. Structural properties were extracted from force-displacement curves by custom Matlab algorithms (Mathworks, Natick, MA).
Statistical analyses
A one-way analysis of variance (ANOVA) was used to determine whether differences existed in μCT and biomechanical-derived properties among experimental groups. In the event that standard deviations were significantly different, the ANOVA was done by the non-parametric Kruskal-Wallis test. When differences existed at p<0.05, post-hoc, pair-wise comparisons were tested for significance in which the p-value was adjusted (padj<0.05) by Holm-Sidak’s method or Dunn’s method (non-parametric). Statistical analysis was performed using GraphPad PRISM (v6.0a, La Jolla, CA). Data are provided as mean +/− SD.
RESULTS
Growth plate-independent and cell-autonomous differentiation defect in Nf1-deficient osteoprogenitors
Conditional deletion of Nf1 early in the mesenchymal lineage, using the Col2a1- or Prx-cre transgenic promoters (14,20,21) triggered skeletal phenotypes in mice that phenocopy structural and cellular deficits observed in human NF1 skeletal lesions, including low bone mass, hyperosteoidosis (accumulation of non-mineralized bone matrix), increased osteoclastogenesis and delayed bone healing. However, the non-inducible nature of these promoter constructs resulted in Nf1 deficiency in growth plate chondrocytes during development, leading to pups born with a reduced stature, and thus complicating the interpretation of the cellular cause of the bone phenotypes observed, since growth plate defects impact bone development, bone accrual and osteoblast differentiation (21,22). To determine if loss of Nf1 function in osteoblast progenitors impairs their differentiation in vivo, independently of developmental growth plate osteochondroprogenitors or secreted factors, we generated a new mouse model that lacks Nf1 in post-natal Osterix (Osx)-positive osteoprogenitors (Nf1osx−/− mice). This model was generated by crossing mice with Nf1 floxed alleles (11) and mice expressing the cre-recombinase under the control of the Osterix (Osx or Sp7) promoter, which can be regulated by a doxycycline-dependent On/Off switch (Tetoff system) (12). Our breeding strategy generated 50% of Nf1flox/flox; cre-negative (WT) and 50% of Nf1flox/flox;cre-positive (Nf1osx−/−) littermates. In absence of doxycycline treatment, these mutant mice exhibited reduced stature due to Osx-cre activity in chondrocytes during development (23,24), but were born with a normal size and did not show a short stature phenotype in adults when given doxycycline (doxy) from conception to 2 weeks of age to repress cre activity and to prevent Nf1 recombination during development (Fig 1a). To assess the differentiation properties of Nf1-deficient osteoprogenitors, BMSCs were isolated from two month-old WT and Nf1osx−/− mice (treated by doxy from conception to 2 weeks of age), and the number of CFU-Ap and CFU-Ob colonies were quantified as an indirect measure of the in vivo number of bone osteoprogenitors. BMSCs from Nf1Osx−/− mice formed significantly less CFU-Ap and CFU-Ob colonies than BMSCs from WT littermates (Fig. 1b), and were characterized by a significant decrease in the expression of major osteoblast marker genes, including Alpl, Runx2, Osx, Col1a1 and Ocn compared to WT BMSCs, as quantified by qPCR analyses (Fig. 1c). These results indicate that the bones of Nf1osx−/− mice contain a reduced number of osteoprogenitors, in line with their low bone mass, cortical bone thinning and weakened mechanical properties (25), and suggest that the impaired differentiation of Nf1-deficient osteoprogenitors is growth plate-independent.
Figure1. Differentiation and gene expression changes induced by Nf1 loss of function in osteoprogenitors cells.

(a) Doxycycline (Doxy) administration corrects the growth retardation of Nf1Osx−/− mice. Doxy was given from conception to up to 10 weeks of age. (b, c) Differentiation assays. BMSCs isolated from WT and Nf1Osx−/− mice were cultured for 2 weeks in osteogenic conditions. BMSC differentiation was analyzed by ALP staining (differentiation, CFU-Ap); Alizarin red-S staining (mineralization, CFU-Ob) and crystal violet staining (cell number, CFU-F). In identical cultures, gene expression was assessed by qPCR. The results represent the mean ± SD and are representative of triplicate experiments. *:p<0.05 versus WT.
To determine whether the differentiation defect of Nf1−/− osteoprogenitors is indeed growth plate-independent and cell-autonomous, we used bone marrow stromal cells (BMSCs) isolated from Nf1flox/flox mice (11), infected ex vivo with GFP- or Cre-expressing adenoviruses. This approach allowed us to avoid any bias from in vivo-derived factors or conditions that could affect the early commitment, proliferation, or differentiation of Nf1-deficient osteoprogenitors, and to start the differentiation assays in cultures platted at the same density and same differentiation stage. BMSCs from long bones of Nf1flox/flox mice were isolated, cultured for 7 days in normal medium, and infected with a GFP- (Ad-CMV-GFP, control) or Cre-adenovirus (Ad-CMV-Cre). This approach led to an approximate 70% recombination efficiency, as shown by genomic PCR (Fig. 2a) and RT-PCR for Nf1 expression (Fig. 2b). Following adenoviral infection, BMSCs were induced to differentiate by supplementation of the culture medium with ascorbic acid and glycerophosphate. Differentiation CFU assays revealed that ex vivoNf1 ablation in Nf1flox/flox BMSCs induces a significant reduction in the number of CFU-Ap and CFU-Ob colonies following 14 days of differentiation (Fig. 2c, d). These results, consistent with studies using alternative models (26,27), indicate that neurofibromin, independently from growth plate, bone marrow or systemic influences, is required for normal osteoprogenitor cell differentiation and matrix mineralization.
Figure 2. Nf1 loss of function in osteoprogenitor cells impairs differentiation independently of growth plate and systemic abnormalities.
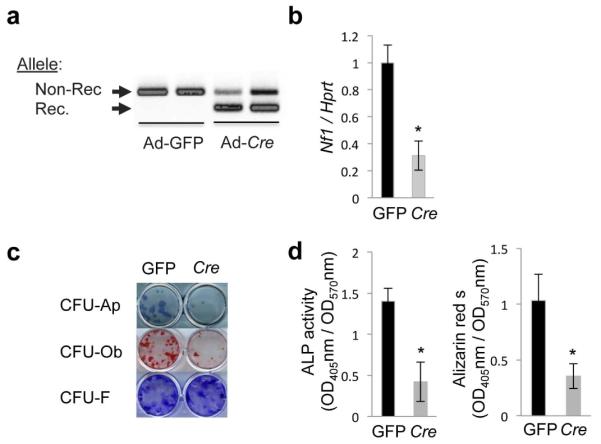
(a, b) BMSCs isolated from Nf1flox/flox mice were infected in vitro with a GFP- (control) or cre-adenovirus (Ad) and cultures were differentiated for 14 days in osteogenic conditions. (a) Non-recombined (Non-Rec.) and recombined (Rec.) Nf1 alleles detected by PCR using genomic DNA from primary cells. (b)Nf1 expression analyzed by qPCR and normalized by Hprt. (c) BMSC differentiation was analyzed by ALP staining (differentiation, CFU-Ap); Alizarin red-S staining (mineralization, CFU-Ob) and crystal violet staining (cell number, CFU-F). (d) Quantification of Alkaline Phosphatase (ALP) activity (OD405) and released Alizarin red-S (OD405), normalized by cell number (OD570) in the same cultures. The results represent the mean ± SD and are representative of triplicate experiments. *:p<0.05 versus WT.
An ERK-dependent blunted response to BMP2 contributes to the defective differentiation of Nf1−/− osteoblasts
BMPs are well known for their osteogenic properties and are clinically used in the management of skeletal conditions requiring bone anabolism. However, recombinant BMP2 failed to promote osteoblast differentiation and bone healing in Nf1+/− mice (6,7). These results suggested that deficiency in Bmp2 expression was unlikely to cause the differentiation phenotype of Nf1−/− osteoprogenitors. Because a common trait of Nf1-deficient cells, including osteoblasts, is constitutive activation of the RAS-ERK pathway, we hypothesized that chronic/uncontrolled activation of RAS-ERK signaling in Nf1−/− osteoprogenitors could antagonize the osteogenic properties of BMP2 and explain the reduced differentiation potential of these cells. To address this hypothesis, BMSCs were prepared from 2 month-old WT and Nf1Osx−/− mice (treated by doxy from conception to 2 weeks of age) and were differentiated in osteogenic conditions for 14 days in vitro, in the presence of vehicle (DMSO), rhBMP2 (100ng), the MEK inhibitor Trametinib (0.1μM), alone or in combination with BMP2. In these cultures, higher level of P-ERK was detected in BMSCs prepared from Nf1osx−/− mice compared to WT mice (Fig. 3a), and Trametinib potently abolished ERK1/2 phosphorylation, with or without BMP2 treatment. Osteogenic differentiation assays showed that rBMP2 treatment increased the number of CFU-Ob colonies in BMSC cultures from WT mice, as expected, but failed to do so in Nf1-deficient BMSCs (Fig. 3b-c). Consistent with previous data in WT osteoblasts (28,29), MEK inhibition enhanced differentiation of WT BMSCs, but had no effect on the differentiation of Nf1-deficient BMSCs either. However, combination treatment with Trametinib and rBMP2significantly increased the number of CFU-Ob colonies in Nf1-deficient BMSC cultures (Fig. 3b-c) and resulted in an increase in osteoblast marker gene expression, including Alpl, Runx2 and Ocn (Fig. 3d-f). Similar results were obtained with U0126, a commonly used MEK inhibitor (25). These results indicate that controlled RAS/ERK signaling is required for the proper response of osteoprogenitors to BMP2, and suggest that Trametinib and BMP2 treatment may have beneficial effects in the setting of NF1 skeletal maladies.
Figure3. Combination treatment with BMP2 and Trametinib corrects the differentiation defect of Nf1−/− osteoprogenitors.

(a) BMSCs isolated from WT and Nf1Osx−/− mice were serum-starved overnight and then pre-incubated with Trametinib (0.1μM) for 30 minutes. BMP2 (100ng) treatment was started 30 minutes later for 1h. Whole BMSC lysates were separated by SDS/PAGE and immunoblotted using the indicated antibodies. (b-c) BMSCs isolated from WT and Nf1Osx−/− mice were treated with vehicle (DMSO), BMP2 (100ng/ml),Trametinib (0.1μM) or both drugs for 2 weeks, and BMSC differentiation was analyzed by Alizarin red-S (CFU-Ob, b) and ALP activity (c). (d-f) Similar cultures were treated with vehicle (DMSO), BMP2 (100ng/ml), the MEK inhibitors U0126 (1μM) or Trametinib (0.1μM) or combined BMP2/U0126 or BMP2/Trametinib for 2 weeks. Expression of osteoblast marker genes was assessed by qPCR (n=3). Black bars: WT mice, grey bars: Nf1Osx−/− mice. *:p<0.05 versus WT; #:p<0.05 versus non-treated in the same genotype group.
Nf1 loss of function in callus osteoprogenitor cells impairs bone healing
The inducible nature of the Nf1osx−/− mouse model allows one to ablate Nf1 in a controlled manner in periosteal and bone marrow osteoprogenitors in adult mice (hence circumventing developmental phenotypes), and at selected times during the bone healing process following fracture, by simply stopping doxy treatment. To determine the contribution of defective Nf1-deficient osteoprogenitors and their lineage-derived cells to bone healing in mice that do not exhibit the reduced stature characteristic of the Col2a1-cre and Prx-cre driven Nf1−/− mice (14,21,30), we administered doxy to pregnant mothers and their pups from conception to 2 weeks of age, to repress cre expression during growth, after which doxy treatment was stopped and a closed tibia fracture was generated at 2 months of age, as described previously (13). Doxy removal from the drinking water led to a 20 to 40% recombination efficiency of the Nf1 floxed alleles (Fig. 4a), and to a 60% reduction in Nf1 expression (Fig. 4b) in total bone lysates, which is consistent with the complex cellularity of the bone marrow and restricted cre-driven gene recombination in osteoprogenitor cells. High-resolution X-ray analyses revealed that callus formation was severely impaired in Nf1osx−/− mice compared to WT mice, as visualized by the formation of an atrophic callus 21 days post-fracture (Fig. 4c). 3D-microtomographic analyses confirmed the existence of a significantly smaller calcified callus 21 day post-fracture (callus Tissue Volume, Fig. 4d), despite normal bone shaft cross-sectional tissue area, Fig. 4e), as well as a lower callus tissue mineral density (TMD, Fig. 4f) and callus BV/TV (Fig. 4g) in Nf1osx−/− mice compared to WT mice. Most significantly, 3-point bending tests indicated that calluses from Nf1osx−/− mice were less stiff and mechanically weaker than the ones from WT mice, as measured by flexural testing (Fig. 4h, i). These results indicate that Nf1 loss of function in post-natal osteoprogenitors has significant in vivo deleterious repercussions on the bone healing process, and establish the Nf1osx−/− mouse model as a useful tool to investigate the etiology of NF1 pseudarthrosis and novel approaches to promote bone healing in NF1.
Figure4. Nf1 loss of function in osteoblasts delays bone healing in Nf1osx−/− mice.

(a) Genomic DNA was extracted from tibiae of 2 month-old mice. Nf1 genomic recombination was evaluated by PCR to detect non-recombined (Non-Rec.) and recombined (Rec.) Nf1 alleles (n=3). (b) Nf1 expression in tibiae from 2 month-old mice was measured by qPCR (n=6).(c) WT and Nf1osx−/− mice were subjected to tibia fracture and callus X-ray analyses were performed 7, 14 and 21 days post-fracture (DPF). (d-g) 3D μCT quantification of callus tissue volume (TV, d), bone cross-sectional tissue area (e), callus tissue mineral density (TMD, f) and callus bone volume over total volume (BV/TV, g) in WT and Nf1Osx−/− mice 21 DPF. (h-i) Callus strength (maximum force, h) and callus stiffness (f) measured by three-point bending tests in WT and Nf1Osx−/− 21 DPF (n=10-15 mice/group), *:p<0.05.ns: non-significant.
Local administration of Trametinib and BMP2 promotes bone healing in Nf1Osx−/− mice
Based on the beneficial effect of Trametinib and BMP2 combination treatment on Nf1−/− osteoprogenitor differentiation observed in vitro, we asked whether such treatment could promote bone healing and improve callus bone parameters in Nf1osx−/− mice. Because of the hydrophobic nature of Trametinib, we generated a drug delivery system based on polyester nanoparticles that allow progressive release of Trametinib at the fracture site. Using a 4% cross-linking density, the in vitro release of Paclitaxel (a BCS class IV hydrophobic drug like Trametinib) was continuous and reached 50% within a 10 day period at 37°C, pH 7.4 (Supplemental Fig. 1 and (31)). In addition, we used a polyglycidol (PEG)-based delivery method to limit the diffusion of BMP2 and Trametinib-loaded nanoparticles at the injection site. WT and Nf1osx−/− mice were subjected to tibia fracture following doxy withdrawal, as described above. At day 1 and 7 post fracture, mice were injected at the fracture site with 20μLvehicle (nanoparticles and PEG), 20μL rBMP2-polyglycidol (0.50μg/μL) or 20μL Trametinib nanoparticles (0.188μg/μL) or 20μL of both (Fig. 5a). In the latter group, Trametinib nanoparticles were dispersed in the polyglycidol-based delivery system containing rBMP2. Consistent with the in vitro results, rBMP2 and Trametinib treatment alone did not improve bone healing parameters in Nf1osx−/− mice (Fig. 5b-f), whereas callus TV, BV, BV/TV and TMD were significantly higher in Nf1osx−/− mice treated with the rBMP2+Trametinib combination, compared to vehicle or single drug controls (Fig. 5b-f). In addition, mice treated with BMP2+Trametinib had a callus with higher strength compared to mice treated with BMP2 or Trametinib alone, as indicated by their increased stiffness and peak force measured by 3-point bending (Fig. 5g, h). From these findings, we conclude that MEK inhibition by Trametinib potentiates BMP2 efficacy in promoting bone healing in the context of NF1 deficiency in osteoprogenitor cells.
Figure5. Combined BMP2 treatment and MEK improves bone healing in Nf1osx−/− mice.

(a-g) Following tibia fracture, WT an Nf1osx−/− mice were treated (Tx) with vehicle (nanoparticles and polyglycidol) or rBMP2-polyglycidol or Trametinib nanoparticles or both, 1 and 7 days post fracture. (a) Treatment scheme. (b-f) 3D μCT representative reconstruction images (b), quantification of callus bone volume (BV, c); callus tissue volume (TV, d); callus bone volume over total volume (BV/TV, e) and callus tissue mineral density (TMD, f) in WT and Nf1Osx−/− mice 21 days after fracture. (g-h) Callus strength (maximum peak force, f) and callus stiffness (h) measured by three-point bending tests were significantly lower in Nf1Osx−/− mice compared to WT mice,21 DPF. *:p<0.05 versus WT, #:p<0.05 versus single treatment in theNf1Osx−/− mouse group (n=10-15 mice/group).
DISCUSSION
The clinical management of NF1 pseudarthrosis is challenging. This reflects the current poor understanding of the underlying pathophysiology of this condition and its variability among individuals with NF1. In this study, we show that lack of Nf1 in osteoprogenitors, in a cell-autonomous fashion and independently from growth plate developmental alterations, is sufficient to impair their differentiation into mature osteoblasts, and that the response of osteoprogenitors to BMP2 requires regulation of RAS/ERK signaling by neurofibromin. We also show that BMP2 can only promote bone healing in mice lacking Nf1 in adult osteoprogenitors when the constitutive activation of RAS/ERK signaling typical of Nf1-deficient cells is suppressed.
These results differ from the ones from a previous report by Sharma et al, which describes a beneficial effect the MEK inhibitor PD98059 in a mouse model of NF1 impaired healing, without the need of combined BMP2 treatment (32). Apart from dosing considerations, the main difference between the two studies is the differentiation stage at which Nf1 is ablated to generate Nf1-deficient bone cells. The use of the 2.3kb Col1a1-cre mice to prepare Nf1-deficient BMSC cultures in the first study leads to Nf1 deletion very specifically in osteoblasts, but in committed mature osteoblasts, whereas the Osx-cre and ex vivo adeno-cre approaches used in the current study lead to Nf1 deletion in proliferative osteoprogenitor cells, which more closely reflects what occurs in individuals with NF1 pseudarthrosis. It is thus possible that the response of undifferentiated BMSCs to MEK inhibition and BMP2 is distinct from the one of committed Col1a1-positive osteoblasts, and that the effects seen in the model using the 2.3kb Col1a1-cre mice are in fact caused by the rescue of other defects associated with Nf1 loss of function and ERK activation, such as altered mineralization, collagen synthesis and osteoclastogenesis, rather than differentiation (33,34). In any case, our study indicates that Nf1-deficient osteoprogenitors display a blunted response to the osteogenic property of BMP2, which may directly contribute to the bone repair defect observed in Nf1osx−/− mice. The beneficial effect of BMP2 combined with MEK inhibition in this model supports such a contribution, that may coexist with the previously reported increase in hyperactive TGFβ signaling (5). The relative contribution of these two mechanisms to the delay in bone healing observed in the NF1 setting will require further investigations, as both can independently modulate in vivo bone repair (35-37).
Despite its well-recognized osteogenic properties, BMP2/7 was inefficient, on its own, in promoting bone union in patients with NF1 pseudarthrosis and in a mouse model of NF1 pseudarthrosis. Only a combination treatment with BMP2 and bisphosphonates was associated with better outcomes in mice and in a study assessing fracture union in seven patients with NF1 pseudarthrosis (6,7,38). The current study shows that BMP2 and MEK inhibition are both required to improve bone healing parameters in mice lacking Nf1 in osteoprogenitors. Although it is difficult to compare treatment efficacy in independent studies using different mouse models and drugs, these results point to the necessity of a dual treatment to significantly improve bone healing in the context of NF1pseudarthrosis, reflecting the multitude and complexity of molecular and cellular processes affected by Nf1 loss of function.
In summary, this study revealed a blunted BMP2 response as a cause of the defective differentiation of Nf1-deficient osteoprogenitors, and led to preclinical data supporting the use of a novel targeted combination therapy based on MEK inhibition (Trametinib) and rhBMP2 to promote bone healing in individuals with NF1 pseudarthrosis. Trametinib is FDA-approved for the treatment of patients with unresectable or metastatic melanoma with BRAF mutations. Hence, its use in the NF1 setting could have an advantage over the development of other drugs.
Supplementary Material
Supplementary Figure 1. (a) Cumulative release of Paclitaxel (0.15mM) in vitro at 37°C, pH 7.4 over 21 days from 4 and 10% cross-linking density polyester nanoparticles. (b) Schematic of the degradation and diffusion of the polyester nanoparticles and polyglycidol polymer to allow progressive release of Trametinib and BMP2.
Acknowledgments
This work was funded by a Young Investigator Award (2012-01-028) from the Children’s Tumor Foundation (JN) and by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the National Institutes of Health, under Award Number 5R01 AR055966 (FE). Support for the micro computed tomography equipment was provided by the National Institutes of Health, Award Number S10 RR027631 (DSP)
Footnotes
Disclosures:
The authors have no conflict of interest to declare.
Authors’ roles: Study design: JDLCN, FE. Study conduct: JDLCN, FE. Data collection: JDLCN, DMS, GV, SU. Data analysis: JDLCN, JSN, DSP, FE. Data interpretation: JDLCN, FE. Drafting manuscript: JDLCN, XY, FE. Approving final version of manuscript: JDLCN, DMS, GV, SU, DSP, JSN, XY, EH, FE. FE takes responsibility for the integrity of the data analysis.
REFERENCES
- 1.Stevenson DA, Little D, Armstrong L, Crawford AH, Eastwood D, Friedman JM, Greggi T, Gutierrez G, Hunter-Schaedle K, Kendler DL, Kolanczyk M, Monsell F, Oetgen M, Richards BS, Schindeler A, Schorry EK, Wilkes D, Viskochil DH, Yang FC, Elefteriou F. Approaches to Treating NF1 Tibial Pseudarthrosis: Consensus From the Children’s Tumor Foundation NF1 Bone Abnormalities Consortium. J Pediatr Orthop. 2013;33(3):269–275. doi: 10.1097/BPO.0b013e31828121b8. [DOI] [PubMed] [Google Scholar]
- 2.Crawford AH, Herrera-Soto J. Scoliosis associated with neurofibromatosis. Orthop Clin North Am. 2007;38(4):553–62. vii. doi: 10.1016/j.ocl.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Khong PL, Goh WH, Wong VC, Fung CW, Ooi GC. MR imaging of spinal tumors in children with neurofibromatosis 1. AJR Am J Roentgenol. 2003;180(2):413–7. doi: 10.2214/ajr.180.2.1800413. [DOI] [PubMed] [Google Scholar]
- 4.Ramachandran M, Tsirikos AI, Lee J, Saifuddin A. Whole-spine magnetic resonance imaging in patients with neurofibromatosis type 1 and spinal deformity. J Spinal Disord Tech. 2004;17(6):483–91. doi: 10.1097/01.bsd.0000133466.97241.50. [DOI] [PubMed] [Google Scholar]
- 5.Rhodes SD, Wu X, He Y, Chen S, Yang H, Staser KW, Wang J, Zhang P, Jiang C, Yokota H, Dong R, Peng X, Yang X, Murthy S, Azhar M, Mohammad KS, Xu M, Guise TA, Yang FC. Hyperactive transforming growth factor-beta1 signaling potentiates skeletal defects in a neurofibromatosis type 1 mouse model. J Bone Miner Res. 2013 doi: 10.1002/jbmr.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schindeler A, Ramachandran M, Godfrey C, Morse A, McDonald M, Mikulec K, Little DG. Modeling bone morphogenetic protein and bisphosphonate combination therapy in wild-type and Nf1 haploinsufficient mice. J Orthop Res. 2008;26(1):65–74. doi: 10.1002/jor.20481. [DOI] [PubMed] [Google Scholar]
- 7.Schindeler A, Birke O, Yu NY, Morse A, Ruys A, Baldock PA, Little DG. Distal tibial fracture repair in a neurofibromatosis type 1-deficient mouse treated with recombinant bone morphogenetic protein and a bisphosphonate. J Bone Joint Surg Br. 2011;93(8):1134–9. doi: 10.1302/0301-620X.93B8.25940. [DOI] [PubMed] [Google Scholar]
- 8.Anticevic D, Jelic M, Vukicevic S. Treatment of a congenital pseudarthrosis of the tibia by osteogenic protein-1 (bone morphogenetic protein-7): a case report. J Pediatr Orthop B. 2006;15(3):220–1. doi: 10.1097/01.bpb.0000194439.75378.ac. [DOI] [PubMed] [Google Scholar]
- 9.Lee FY, Sinicropi SM, Lee FS, Vitale MG, Roye DP, Jr., Choi IH. Treatment of congenital pseudarthrosis of the tibia with recombinant human bone morphogenetic protein-7 (rhBMP-7). A report of five cases. J Bone Joint Surg Am. 2006;88(3):627–33. doi: 10.2106/JBJS.D.02201. [DOI] [PubMed] [Google Scholar]
- 10.Fabeck L, Ghafil D, Gerroudj M, Baillon R, Delince P. Bone morphogenetic protein 7 in the treatment of congenital pseudarthrosis of the tibia. J Bone Joint Surg Br. 2006;88(1):116–8. doi: 10.1302/0301-620X.88B1.16619. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15(7):859–76. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133(16):3231–44. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 13.Wang W, Nyman JS, Moss HE, Gutierrez G, Mundy GR, Yang X, Elefteriou F. Local low-dose lovastatin delivery improves the bone-healing defect caused by Nf1 loss of function in osteoblasts. J Bone Miner Res. 2010;25(7):1658–67. doi: 10.1002/jbmr.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Nyman JS, Ono K, Stevenson DA, Yang X, Elefteriou F. Mice lacking Nf1 in osteochondroprogenitor cells display skeletal dysplasia similar to patients with neurofibromatosis type I. Hum Mol Genet. 2011;20(20):3910–24. doi: 10.1093/hmg/ddr310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stevens DM, Watson HA, LeBlanc MA, Wang RY, Chou J, Bauer WS, Harth E. Practical polymerization of functionalized lactones and carbonates with Sn(OTf)(2) in metal catalysed ring-opening polymerization methods. Polymer Chemistry. 2013;4(8):2470–2474. [Google Scholar]
- 16.Spears BR, Waksal J, McQuade C, Lanier L, Harth E. Controlled branching of polyglycidol and formation of protein-glycidol bioconjugates via a graft-from approach with “PEG-like” arms. Chem Commun (Camb) 2013;49(24):2394–6. doi: 10.1039/c3cc38369e. [DOI] [PubMed] [Google Scholar]
- 17.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, Long B, Liu J, Dinunzio E, Windsor W, Zhang R, Zhao S, Angagaw MH, Pinheiro EM, Desai J, Xiao L, Shipps G, Hruza A, Wang J, Kelly J, Paliwal S, Gao X, Babu BS, Zhu L, Daublain P, Zhang L, Lutterbach BA, Pelletier MR, Philippar U, Siliphaivanh P, Witter D, Kirschmeier P, Bishop WR, Hicklin D, Gilliland DG, Jayaraman L, Zawel L, Fawell S, Samatar AA. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3(7):742–50. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 18.Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011;39(1):23–31. doi: 10.3892/ijo.2011.1015. [DOI] [PubMed] [Google Scholar]
- 19.Tan AM, Tan CL, Phua KB, Joseph VT. Chemotherapy for hepatoblastoma in children. Ann Acad Med Singapore. 1990;19(2):286–9. [PubMed] [Google Scholar]
- 20.Kolanczyk M, Kuhnisch J, Kossler N, Osswald M, Stumpp S, Thurisch B, Kornak U, Mundlos S. Modelling neurofibromatosis type 1 tibial dysplasia and its treatment with lovastatin. Bmc Medicine. 2008;6 doi: 10.1186/1741-7015-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ono K, Karolak MR, Ndong JD, Wang W, Yang X, Elefteriou F. The Ras-GTPase activity of neurofibromin restrains ERK-dependent FGFR signaling during endochondral bone formation. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang W, Lian N, Ma Y, Li L, Gallant RC, Elefteriou F, Yang X. Chondrocytic Atf4 regulates osteoblast differentiation and function via Ihh. Development. 2012;139(3):601–11. doi: 10.1242/dev.069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Shi Y, Regan J, Karuppaiah K, Ornitz DM, Long F. Osx-Cre targets multiple cell types besides osteoblast lineage in postnatal mice. PLoS One. 2014;9(1):e85161. doi: 10.1371/journal.pone.0085161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, Carmeliet G, Kronenberg HM. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell. 2010;19(2):329–44. doi: 10.1016/j.devcel.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ndong J, Makowski AJ, Uppuganti S, Vignaux G, Ono K, Perrien DS, Joubert S, Baglio SR, Granchi D, Stevenson DA, Rios JJ, Nyman JS, Elefteriou F. Asfotase-alphaimproves bone growth, mineralization and strength in mouse models of neurofibromatosis type-1. Nature Medicine. 2014 doi: 10.1038/nm.3583. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma R, Wu X, Rhodes SD, Chen S, He Y, Yuan J, Li J, Yang X, Li X, Jiang L, Kim ET, Stevenson DA, Viskochil D, Xu M, Yang FC. Hyperactive Ras/MAPK signaling is critical for tibial nonunion fracture in neurofibromin-deficient mice. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leskela HV, Kuorilehto T, Risteli J, Koivunen J, Nissinen M, Peltonen S, Kinnunen P, Messiaen L, Lehenkari P, Peltonen J. Congenital pseudarthrosis of neurofibromatosis type 1: impaired osteoblast differentiation and function and altered NF1 gene expression. Bone. 2009;44(2):243–50. doi: 10.1016/j.bone.2008.10.050. [DOI] [PubMed] [Google Scholar]
- 28.Doan TKP, Park KS, Kim HK, Park DS, Kim JH, Yoon TR. Inhibition of JNK and ERK Pathways by SP600125-and U0126-Enhanced Osteogenic Differentiation of Bone Marrow Stromal Cells. Tissue Engineering and Regenerative Medicine. 2012;9(6):283–294. [Google Scholar]
- 29.Higuchi C, Myoui A, Hashimoto N, Kuriyama K, Yoshioka K, Yoshikawa H, Itoh K. Continuous inhibition of MAPK signaling promotes the early osteoblastic differentiation and mineralization of the extracellular matrix. J Bone Miner Res. 2002;17(10):1785–94. doi: 10.1359/jbmr.2002.17.10.1785. [DOI] [PubMed] [Google Scholar]
- 30.Kolanczyk M, Kossler N, Kuhnisch J, Lavitas L, Stricker S, Wilkening U, Manjubala I, Fratzl P, Sporle R, Herrmann BG, Parada LF, Kornak U, Mundlos S. Multiple roles for neurofibromin in skeletal development and growth. Hum Mol Genet. 2007;16(8):874–86. doi: 10.1093/hmg/ddm032. [DOI] [PubMed] [Google Scholar]
- 31.Stevens DM, Gilmore KA, Harth E. An assessment of nanosponges for intravenous and oral drug delivery of BCS class IV drugs: Drug delivery kinetics and solubilization. Polymer Chemistry. 2014 In press. [Google Scholar]
- 32.Sharma R, Wu X, Rhodes SD, Chen S, He Y, Yuan J, Li J, Yang X, Li X, Jiang L, Kim ET, Stevenson DA, Viskochil D, Xu M, Yang FC. Hyperactive Ras/MAPK signaling is critical for tibial nonunion fracture in neurofibromin-deficient mice. Hum Mol Genet. 2013;22(23):4818–28. doi: 10.1093/hmg/ddt333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, Parada LF, Karsenty G. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab. 2006;4(6):441–51. doi: 10.1016/j.cmet.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ndong J, Makowski AJ, Uppuganti S, Vignaux G, Ono K, Perrien DS, Joubert S, Baglio SR, Granchi D, Stevenson DA, Rios JJ, Nyman JS, Elefteriou F. Asfotase-alpha improves bone growth, mineralization and strength in mouse models of neurofibromatosis type-1. Nature Medicine. 2014 doi: 10.1038/nm.3583. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsuji K, Cox K, Bandyopadhyay A, Harfe BD, Tabin CJ, Rosen V. BMP4 is dispensable for skeletogenesis and fracture-healing in the limb. J Bone Joint Surg Am. 2008;90(Suppl 1):14–8. doi: 10.2106/JBJS.G.01109. [DOI] [PubMed] [Google Scholar]
- 36.Alam N, St-Arnaud R, Lauzier D, Rosen V, Hamdy RC. Are endogenous BMPs necessary for bone healing during distraction osteogenesis? Clin Orthop Relat Res. 2009;467(12):3190–8. doi: 10.1007/s11999-009-1065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, Zhao L, Nagy TR, Peng X, Hu J, Feng X, Van Hul W, Wan M, Cao X. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. 2009;15(7):757–65. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Birke O, Schindeler A, Ramachandran M, Cowell CT, Munns CF, Bellemore M, Little DG. Preliminary experience with the combined use of recombinant bone morphogenetic protein and bisphosphonates in the treatment of congenital pseudarthrosis of the tibia. J Child Orthop. 2010;4(6):507–17. doi: 10.1007/s11832-010-0293-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. (a) Cumulative release of Paclitaxel (0.15mM) in vitro at 37°C, pH 7.4 over 21 days from 4 and 10% cross-linking density polyester nanoparticles. (b) Schematic of the degradation and diffusion of the polyester nanoparticles and polyglycidol polymer to allow progressive release of Trametinib and BMP2.