

Abstract
Metabolic enzymes have been known to carry out a variety of functions besides their normal housekeeping roles known as “moonlighting functions.” These functionalities arise from structural changes induced by posttranslational modifications and/or binding of interacting proteins. Glycolysis is the sole source of energy generation for malaria parasite Plasmodium falciparum, hence a potential pathway for therapeutic intervention. Crystal structures of several P. falciparum glycolytic enzymes have been solved, revealing that they exhibit unique structural differences from the respective host enzymes, which could be exploited for their selective targeting. In addition, these enzymes carry out many parasite-specific functions, which could be of potential interest to control parasite development and transmission. This review focuses on the moonlighting functions of P. falciparum glycolytic enzymes and unique structural differences and functional features of the parasite enzymes, which could be exploited for therapeutic and transmission blocking interventions against malaria.
1. Introduction
Malaria caused by protozoan parasite Plasmodium is a major human health burden with almost half of the world population at risk [1]. Despite the availability of drugs against malaria, development of resistance against commonly used antimalarials necessitates the search for novel therapeutic targets and drugs against the disease [2]. Parasite-specific processes such as hemoglobin degradation, parasite egress from the host cell, and host cell invasion by the parasite have been the focus of drug discovery efforts against malaria [3]. In addition to these pathways, one attractive pathway for drug development is parasite's energy producing machinery. A compound targeting parasite's ATP-generating machinery could be a potential antimalarial.
2. Review
2.1. P. falciparum Glycolysis
Glycolysis is an ancient conserved metabolic energy producing machinery, which converts glucose to pyruvate and lactate under aerobic and anaerobic conditions, respectively. It has been an established fact that P. falciparum solely depends on glycolysis for energy generation and fulfills its energy needs by anaerobic metabolism of glucose to lactate as the parasite and the red blood cells (RBCs) both are devoid of functional Kreb's cycle [4]. The rate of glycolysis in P. falciparum-infected erythrocytes is 20–100 times higher than uninfected erythrocytes [5, 6]. A study by Roth Jr. et al. (1988) demonstrated that nearly all the glycolytic enzymes were upregulated in P. falciparum-infected RBCs, which was proportional to the parasitemia level. The enzymes particularly upregulated were hexokinase, aldolase, enolase, pyruvate kinase, and adenosine deaminase [7]. Several P. falciparum glycolytic enzymes, although being homologous to their human counterparts, possess distinct structural and biochemical features [8–14]. To selectively target parasite glycolytic enzymes, these differences between the host and parasite enzymes could be exploited. This review focuses on the potential of unique structural and functional properties of malarial glycolytic enzymes as targets of antimalarial drug discovery.
2.2. Moonlighting Functions of Glycolytic Enzymes and Unique Structural and Functional Properties of Malarial Glycolytic Enzymes
In prior years, it was believed that a single gene encodes a single polypeptide and was generally expected to carry out a single function [15, 16] but later it was found that a single protein could carry out multiple functions. This phenomenon was termed as “moonlighting” by Jeffery and referred to unrelated functions of glycolytic enzymes and many other metabolic enzymes exhibit within or outside the cell [17].
Moonlighting functions in proteins may arise due to structural changes occurring in the proteins due to differential binding partners and/or posttranslational modifications (PTMs). This phenomenon is involved in pathologies related to many cellular and infectious diseases; hence an improved understanding of moonlighting proteins will provide opportunity to selectively target these functions [18]. The first example of moonlighting proteins was cytokine neuroleukin, secreted by lectin-stimulated T cells that induce immunoglobulin secretion by cultured human peripheral blood mononuclear cells. This cytokine was also found to have phosphoglucose isomerase activity, which converts glucose 6-phosphate to fructose 6-phosphate during glycolysis [19–21]. In the metabolic map of glycolysis, 7 of the 10 glycolytic enzymes exhibit moonlighting activities. In addition, other metabolic enzymes closely linked to glycolysis (e.g., glycerol kinase and fructose-1,6-bisphosphatase) also possess moonlighting functions. Seven of the eight enzymes of tricarboxylic acid cycle have also been supposed to possess moonlighting functions [22]. Some of the major moonlighting functions of glycolytic enzymes are transcriptional regulation (hexokinase-2, lactate dehydrogenase-A, and enolase 1), apoptosis (hexokinase and glyceraldehyde 3-phosphate dehydrogenase), and cell motility (glucose 6-phosphate isomerase) (reviewed by Kim and Dang 2005) [23]. Examples of moonlighting functions of glycolytic enzymes in various organisms are listed in Table 1.
Table 1.
Examples of nonglycolytic functions of glycolytic enzymes in various organisms.
| Enzyme | Normal glycolytic function | Moonlighting functions |
|---|---|---|
| Hexokinase | Conversion of glucose to glucose 6-phosphate | (i) Transcription regulation [29, 30] (ii) Apoptosis [31, 33] |
| Phosphoglucoisomerase | Conversion of glucose 6-phosphate into its isomer fructose 6-phosphate | (i) Cytokine neuroleukin (a nerve growth factor) [19–21] (ii) Autocrine motility factor [40] (iii) Differentiation and maturation mediator [41] |
| Phosphofructokinase | Conversion of fructose 6-phosphate to fructose 1,6-bisphosphate | (i) Vacuolar degradation of peroxisomes (microautophagy) [43] (ii) Plasminogen binding [44] (iii) RNA processing [45] |
| Aldolase | Splitting of fructose-1,6-bisphosphate into two sugar isomers dihydroxyacetone phosphate and glyceraldehyde 3-phosphate | (i) Motility in Toxoplasma gondii [49, 50] (ii) Cell adhesion in Neisseria meningitidis [51] (iii) Regulation of WASP/Arp2/3-stimulated actin dynamics [52] |
| Triosephosphate isomerase | Interconversion of dihydroxyacetone phosphate and glyceraldehyde phosphate | (i) Defense against pathogens via contact-mediated killing by commensal organisms [58] (ii) Plasminogen binding [44] (iii) Aggregation causes neurological disorders [59] |
| Glyceraldehyde-3-phosphate dehydrogenase | Conversion of glyceraldehyde 3-phosphate to 1,3-bisphosphoglycerate | (i) Plasminogen-binding protein [61] (ii) Transcription regulation [62] (iii) Iron metabolism [63] (iv) Apoptosis [64] |
| Phosphoglycerate kinase | Conversion of 1,3-bisphosphoglycerate to 3-phosphoglycerate | (i) Plasminogen binding [67] (ii) Actin binding and streptococcal invasion of epithelial cells [68] |
| Phosphoglycerate mutase | Conversion of 3-phosphoglycerate to 2-phosphoglycerate | Plasminogen binding [44] |
| Enolase | Conversion of 2-phosphoglycerate to phosphoenol pyruvate | (i) Plasminogen receptor [69] (ii) Transcription regulation [70, 72] (iii) DNA methyltransferase DNMT2 inhibitor [71] |
| Pyruvate kinase | Conversion of phosphoenol pyruvate to pyruvate | (i) Salivary mucin binding [77] (ii) Transcriptional regulation [82] (iii) Apoptosis [83] |
| Lactate dehydrogenase | Conversion of pyruvate to lactate | Epsilon crystalin [85] |
P. falciparum glycolytic enzymes are significantly divergent from their human homologs (Table 2) and several of these enzymes possess unique structural and functional features. Sequence identity and similarity of P. falciparum glycolytic enzymes with their human homologs are listed in Table 2. Crystal structures of P. falciparum glycolytic enzymes aldolase (PfALDO) [11], triosephosphate isomerase (PfTPI) [12], glyceraldehyde 3-phosphate dehydrogenase (PfGAPDH) [13], lactate dehydrogenase (PfLDH) [14], glucose 6-phosphate isomerase (PfGPI) (Gileadi et al. 2011, details unpublished), phosphoglycerate kinase (PfPGK) [24], and phosphoglycerate mutase (PfPGM) [25] have been solved. Distinct structural differences have been identified between some of these enzymes and their human homologs, which could be exploited for their selective targeting. Generally, the active sites of enzymes or subunit interfaces in case of multimeric enzymes are targeted by inhibitors [26, 27]. Since the active site residues of parasite and host glycolytic enzymes are highly conserved, targeting the active site residues is not feasible. However, the unique structural differences and mutations at the subunit interfaces and the moonlighting functions arising from these structural differences could be specifically targeted for selective inhibition. Moonlighting functions of glycolytic enzymes and unique structural and functional properties of P. falciparum glycolytic enzymes are summarized below.
Table 2.
Percent identity and percent similarity of P. falciparum glycolytic enzymes with their major human homologs. Percent identity and percent similarity were calculated by alignment of sequences using National Center for Biotechnology Information's BLAST server (http://blast.ncbi.nlm.nih.gov/Blast.cgi/).
| P. falciparum enzyme | Human homolog | Percent identity | Percent similarity | E-value |
|---|---|---|---|---|
| Hexokinase (PfHK) | Hexokinase, isoform 2 (erythrocyte) | 32 | 50 | 2e − 64 (residues 9–458), 1e − 55 (residues 461–906) |
| Glucose 6-phosphate isomerase (PfGPI) | Glucose 6-phosphate isomerase, isoform 2 | 32 | 52 | 4e − 124 (residues 53–543), 4.5 (residues 518–543) |
| Phosphofructokinase 9 (PfPFK9) | 6-Phosphofructokinase, muscle type, isoform 1 | 26 | 39 | 3e − 08 (residues 18–232), 1e − 04 (residues 459–635), 2e − 04 (residues 377 to 574), 0.65 (residues 296 to 341) |
| Phosphofructokinase 11 (PfPFK11) | 6-Phosphofructokinase, muscle type, isoform 1 | 27 | 41 | 0.022 (residues 438–497) |
| Aldolase (PfALDO) | Aldolase A | 54 | 66 | 4e − 126 (residues 2–364) |
| Triosephosphate isomerase (PfTPI) | Triosephosphate isomerase, isoform 2 | 43 | 60 | 4e − 76 (residues 4–247) |
| Glyceraldehyde 3-phosphate dehydrogenase (PfGAPDH) | GAPDH isoform 1, liver-specific | 64 | 77 | 9e − 163 (residues 1 to 333) |
| Phosphoglycerate kinase (PfPGK) | Phosphoglycerate kinase isoform 1 | 61 | 76 | 0.0 (residues 3–416) |
| Phosphoglycerate mutase 1 (PfPGM1) | Phosphoglycerate mutase 1 | 56 | 72 | 2e − 100 (residues 1 to 253) |
| Phosphoglycerate mutase 2 (PfPGM2) | Phosphoglycerate mutase 2 | 26 | 51 | 0.019 (residues 2–66) |
| Enolase (PfENO) | Alpha enolase | 67 | 79 | 0.0 (residues 3–429) |
| Pyruvate kinase 1 (PfPK1) | Pyruvate kinase L, R | 45 | 61 | 8e − 136 (residues 75–573) |
| Pyruvate kinase 2 (PfPK2) | Pyruvate kinase L, R | 30 | 49 | 3e − 43 (residues 301–556), 6e − 26 (residues 91–302) |
| Lactate dehydrogenase (PfLDH) | LDH-A | 29 | 54 | 5e − 41 (residues 70–368) |
2.2.1. Hexokinase
In yeast, two hexokinases (HXK1 and HXK2) are found. HXK2 has been shown to exhibit nuclear localization [28]. Yeast can utilize different carbon sources, although glucose is the preferred substrate. In yeast glucose itself represses the expression of genes involved in glucose metabolism (glucose repression) through transcriptional regulation [29, 30]. In contrast, mammalian hexokinases have not been implicated in transcriptional regulation. Hexokinase 2 (HK2) localizes to the mitochondria and uses intramitochondrial ATP to phosphorylate glucose [31, 32] suggesting the efficient coupling of oxidative phosphorylation to the glycolytic pathway via mitochondrial-bound hexokinase. Type-1 and -2 hexokinases also participate in the apoptotic pathway, suggesting that mitochondrial hexokinase activity is required for growth-factor-induced cell survival. In addition, mitochondrial hexokinase activity was found to be inhibitory to apoptosis in various experimental systems [32, 33].
P. falciparum hexokinase (PfHK) is highly divergent from its vertebrate homologs and neither monoclonal nor polyclonal antibodies against PfHK cross-reacted with vertebrate hexokinases [34]. PfHK exhibits 32% sequence identity with human erythrocytic hexokinase (Table 2). Two analogues of the substrate glucose, 2-deoxy-D-glucose (2-DG) and 2-fluoro-2-deoxy-D-glucose (2-FG), showed antiplasmodial effects suggesting the potential of PfHK as a drug target [35–37]. A P. falciparum strain overexpressing PfHK was found to be less susceptible to inhibition by 2-DG, further validating the potential of PfHK as a drug target [38]. In an attempt to identify selective small molecule PfHK inhibitors, a library of small molecule hexokinase inhibitors was screened against PfHK. Isobenzothiazolinones were identified as a class of noncovalent, reversible PfHK inhibitors, some of which with 50% inhibitory concentrations (IC50s) of <1 μM [39], demonstrating the feasibility of specific targeting of PfHK.
2.2.2. Glucose 6-Phosphate Isomerase
Mammalian glucose 6-phosphate isomerase (GPI) exhibits the activity of a neurotrophic factor called neuroleukin that was purified from mouse salivary gland, molecularly cloned and expressed in monkey COS cells [19–21]. GPI also acts as an autocrine motility factor (AMF) in Gc-4 PF murine fibrosarcoma cells, which is a tumor-secreted cytokine. GPI has been independently implicated in cell motility and also used as a marker for cancer progression [40]. GPI is also known to act as differentiation and maturation mediator for human myeloid leukemia cells [41].
P. falciparum glucose 6-phosphate isomerase (PfGPI) is also highly divergent from its human homologs (32% sequence identity with human GPI isoform 2) (Table 2) and antiserum raised against recombinant PfGPI specifically inhibited parasite enzyme with no effect on host enzyme activity [42] suggesting the feasibility of its specific targeting. A high-resolution structure of PfGPI has been solved by Gileadi et al. 2011 (PDB ID: 3QKI) but its details are unpublished.
2.2.3. Phosphofructokinase
In yeast Pichia pastoris, the alpha subunit of the phosphofructokinase (PFK) enzyme complex encoded by PFK1 gene is the glucose-induced selective autophagy 1 (GSA1) protein that is responsible for selective autophagy of peroxisomes [43]. PFK was found to bind with plasminogen in clinical isolates of oral streptococci from dental plaque and inflammatory lesions [44]. In Bacillus subtilis, PFK was found to be associated with several proteins supposed to be involved in RNA processing and degradation (RNase J1, Rny, and polynucleotide phosphorylase) [45].
Two putative P. falciparum phosphofructokinase (PfPFK) genes are found on P. falciparum chromosomes 9 (PfPFK9) and 11 (PfPFK11), respectively, out of which only PfPFK9 gene possesses all the catalytic features required for enzyme activity. These enzymes also contain plant-like pyrophosphate (PPi) dependent phosphofructokinase alpha and beta subunits making them remarkably distinct from ATP-dependent erythrocytic phosphofructokinase that is a heterogeneous mixture of five isozymes [46]. However, the recombinant PFK9 and the parasite extract exhibited only ATP-dependent enzyme activity, and no activity was seen with PPi [47]. In rodent malaria parasite Plasmodium berghei, phosphofructokinase differs from human enzyme with respect to allosteric regulation. AMP known to be a potent activator of many phosphofructokinases caused only a marginal increase in enzyme activity. However, phosphoenolpyruvate, known to inhibit phosphofructokinase activity, efficiently activated the parasite enzyme, whereas no activation by ADP was observed [48].
2.2.4. Aldolase
Host cell invasion in apicomplexan parasites is dependent on the interactions between cell surface adhesins and parasite cytoskeleton. Aldolase in Toxoplasma gondii interacts with several adhesins belonging to thrombospondin-related anonymous protein (TRAP) family [49] and acts as a bridge between parasite adhesins and cytoskeleton [50]. Despite lacking an identifiable signal sequence, aldolase has been located on outer membrane of Neisseria meningitidis apart from its cytosolic location. Mutational analysis and functional complementation revealed that an aldolase-deficient mutant, although not affected in its ability to grow in vitro, showed a significant reduction in adhesion to human brain microvascular endothelial and HEp-2 cells [51]. Aldolase interacts with many cytoskeletal proteins that are known to interact with actin. It interacts both in vitro and in cells with Wiskott-Aldrich Syndrome Protein (WASP), a protein having role in controlling actin dynamics. Knockdown of aldolase resulted in a decreased cell motility and spreading that are WASP-dependent processes [52].
P. falciparum aldolase (PfALDO) shares 54% sequence identity with human aldolase A (Table 2) but possesses many distinct structural and functional properties. Immunization with PfALDO provided protective immunity to malaria in monkeys suggesting its potential as a vaccine candidate [53]. In addition to its expected cytosolic location, PfALDO also associated with P. falciparum surface via glycosylphosphatidylinositol anchor, validating its protective property [54]. PfALDO showed very high affinity in vitro binding to the cytosolic domain of the erythrocyte membrane band 3 protein. A 19-residue synthetic peptide corresponding to the binding domain sequence of band 3 protein abrogated this binding and in vitro PfALDO enzyme activity. Another 19-residue peptide with similar sequence motif from P. falciparum actin II was also an inhibitor of PfALDO in vitro enzyme activity. These binding studies suggest that PfALDO associates with the cytoskeleton of the parasite or of the host [9]. A peptide derived from P. falciparumα-tubulin also competitively inhibited PfALDO enzyme activity [55]. PfALDO has been identified a component of the apicomplexan motility machinery of the parasite known as “glideosome” and implicated in RBC invasion [56].
PfALDO is a homotetramer with one 80 kDa dimer per asymmetric unit. Comparison of PfALDO crystal structure with that of human aldolase revealed the presence of a unique hydrophobic pocket on the surface of PfALDO, resulting from some amino acid changes and a single residue deletion compared to human aldolase. Another unique feature was that the overall quaternary structure of the PfALDO tetramer buried lesser surface area than human aldolase [11]. These structural differences could be exploited for specific targeting of PfALDO.
2.2.5. Triosephosphate Isomerase
It has been reported that contact-mediated killing of Cryptococcus neoformans takes place with bacterium Staphylococcus aureus [57]. The interaction between these organisms involved adhesion between triosephosphate isomerase (TPI) and α-(1A 3)-mannooligosaccharides larger than triose in S. aureus and C. neoformans, respectively [58]. TPI was one of the identified proteins that bound with plasminogen in clinical isolates of oral streptococci from dental plaque and inflammatory lesions [44]. In case of Alzheimer's disease amyloid β-peptide-induced nitro-oxidative damage promotes the nitrotyrosination of the triosephosphate isomerase in human neuroblastoma cells. Nitro-triosephosphate isomerase bound to tau monomers and induced tau aggregation to form paired helical filaments that are the characteristic of Alzheimer's disease brains [59].
P. falciparum triosephosphate isomerase (PfTPI) shows 43% sequence identity with its human homolog but also exhibits distinct structural differences. The crystal structure of PfTPI has been solved based on the structure of trypanosomal triosephosphate isomerase. Comparison of PfTPI structure with human triosephosphate isomerase (HsTPI) revealed unique differences. The conserved Glu-184 is replaced with Leu in PfTPI that is surface exposed and may be involved in binding to the erythrocyte membrane. In addition, Ser-97 near the active site of human HsTPI has been replaced with Phe-102 in PfTPI (Figures 1(a) and 1(b)) [12]. PfTPI contains four cysteine residues at positions 13, 126, 196, and 217 per subunit. Among these, Cys-13 present at the dimer interface is replaced by methionine in HsTPI. Mutation of Cys-13 in PfTPI with Asp resulted in a 7-fold reduction in the enzymatic activity [60]. These unique substitutions in PfTPI make it an attractive candidate for selective targeting.
Figure 1.

Unique structural differences between P. falciparum (PfTPI) (a) and human TPI (HsTPI) (b). Unique residues in PfTPI and their counterparts in HsTPI are labeled. PDB IDs of PfTPI and HsTPI are 1YDV and 1WYI, respectively. Structures were visualized by molecular visualization software PyMOL (DeLano Scientific).
2.2.6. Glyceraldehyde 3-Phosphate Dehydrogenase
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was found to be present on the surface and in the extracellular medium of anthrax pathogen Bacillus anthracis. Out of the three isoforms of GAPDH, isoform A was present on the surface of B. anthracis and in the extracellular medium. Human plasminogen interacted with isoform A in a lysine-dependent manner [61].
Oct-1 is a transcription factor involved in cell cycle regulation. S phase-specific cofactor(s) bind to Oct-1 and regulate the S phase-specific H2B transcription. Oct-1 coactivator OCA-S is essential for S phase-dependent histone H2B transcription. The p38 component of OCA-S which represents a nuclear form of GAPDH directly binds to Oct-1 and is essential for S phase-specific H2B transcription [62]. GAPDH is expressed on human and murine macrophage cell surface, regulated by the availability of iron in the medium. GAPDH interacts with transferrin and the GAPDH-transferrin complex is internalized into the early endosomes suggesting the transferrin uptake and regulation mechanisms in macrophages [63].
Cellular distribution of GAPDH in central nervous system (CNS) derived cells (neuroblastoma mNB41A3), in non-CNS derived cells (R6 fibroblast), and in an apoptosis-resistant Bcl2 overexpressing cell line (R6-Bcl2) was investigated. Induction of apoptosis and oxidative stress enhanced the nuclear translocation of endogenous GAPDH in all the cell types, indicating that nuclear translocation of GAPDH may play a role in apoptosis and oxidative stress [64].
P. falciparum glyceraldehyde 3-phosphate dehydrogenase (PfGAPDH) shows 63.5% sequence identity with human erythrocytic GAPDH and expressed in all developmental stages of cultured P. falciparum blood-stage parasites [65] but accumulated at the apical region of merozoites during schizogony [66]. Besides its cytosolic localization, it is also present in the parasite membrane fraction. In vitro studies using HeLa cell microsomal membranes showed that PfGAPDH was recruited to the membrane, which was dependent on the mammalian GTPase Rab2, indicating a probable role of PfGAPDH in vesicular transport and apical organelle biogenesis [66].
The crystal structure of PfGAPDH was determined based on rabbit GAPDH. PfGAPDH is a tetramer with each monomer consisting of an NAD-binding-domain containing the classical Rossman fold (residues 4–152, 320–337) and a catalytic domain (residues 153–319). The catalytic domain consists of eight beta strands connected by short loops and helices. Upon comparison of crystal structures of PfGAPDH (PDB ID: 2B4R) with the human liver GAPDH (HsGAPDH) (PDB ID: 1ZNQ), some unique differences were observed. A ridge in GAPDH structures known as S-loop separates the NAD-binding cavities of adjacent subunits. A two-residue (Lys-Gly) insert is found in this loop in PfGAPDH (residues 192–197 being Pro-Ser-Lys-Gly-Gly-Lys) compared to HsGAPDH, in which only four residues (19-Pro-Ser-Gly-Lys-194) are found (Figures 2(a) and 2(b)). The Lys-Gly in the S-loop structure creates a bulge that lines the NAD-binding cavity. This insert along with the adjacent Lys-197 creates a more constricted opening than found in HsGAPDH (Figure 2(c)). In case of HsGAPDH, Ser-192 of the insert makes a hydrogen bond with Tyr-42 (Figure 2(b)). In PfGAPDH Tyr-42 is replaced with His-42, whose side chain is unable to make hydrogen bond with Ser-193 (Figure 2(a)). Instead this histidine was found to hydrogen bond with water molecules bound to PfGAPDH, which in turn hydrogen bonded with the carbonyl oxygens of Gly-191 and Trp-199 [13]. Thus this Lys-Gly insert imparts distinct hydrogen bonding properties to PfGAPDH, which could provide differential functional properties to PfGAPDH as compared to HsGAPDH suggesting that PfGAPDH could be selectively targeted.
Figure 2.

Unique structural differences between PfGAPDH and HsGAPDH. (a) Tetrameric structure of PfGAPDH (PDB ID: 2B4R). NAD-binding domain (Rossman fold) is shown in grey and catalytic domain is shown in cyan with S-loop shown in blue (i). Zoomed image of PfGAPDH showing the residues Ser-193 and His-42 that are unable to make hydrogen bonds (ii). (b) Tetrameric structure of HsGAPDH (PDB ID: 1ZNQ). NAD-binding domain is shown in grey and catalytic domain is shown in cyan with S-loop shown in blue (i). Zoomed image of HsGAPDH showing the hydrogen bonding between residues Ser-192 and His-42 (ii). (c) Alignment of single subunits of PfGAPDH and HsGAPDH. NAD-binding domains of PfGAPDH and HsGAPDH are shown in green and cyan, respectively, and catalytic domains of PfGAPDH and HsGAPDH are shown in blue and magenta, respectively. Bound NAD is shown in orange. PfGAPDH loop containing the Lys-Gly insert (shown as sticks in blue) constricts the opening of the NAD-binding cavity more as compared to the HsGAPDH loop (shown as sticks in magenta). Structures were visualized by molecular visualization software PyMOL (DeLano Scientific).
2.2.7. Phosphoglycerate Kinase
Phosphoglycerate kinase (PGK) of group B streptococci (GBS) is expressed on the GBS cell surface. It has been shown that group B streptococcal phosphoglycerate kinase (GBS-PGK) binds to plasminogen and actin [67]. The internalization of GBS into eukaryotic cells is an actin-dependent process. In an attempt to identify host cell and/or bacterial factors involved in the process, binding of α-actinin, an actin-binding protein closely associated with cytoplasmic F-actin in the eukaryotic cell, was observed. Western blot analysis revealed that anti-α-actinin antibodies bound to a 42 kDa protein that was identified as GBS-PGK. Treatment of epithelial cells with PGK prior to performing an invasion assay inhibited GBS internalization suggesting the role of PGK in host cell invasion [68].
The crystal structure of P. falciparum phosphoglycerate kinase (PfPGK) has been solved, which did not show any unique feature other than the binding of another sulfate ion at the basic patch where the 3-phosphate of the substrate 1,3-biphosphoglycerate normally binds [24].
2.2.8. Phosphoglycerate Mutase
Phosphoglycerate mutase (PGM) was also found to be a plasminogen binding protein in clinical isolates of oral streptococci from dental plaque and inflammatory lesions [44].
In P. falciparum genome, two phosphoglycerate mutases (PfPGM1 and PfPGM2) are found. Crystal structure of PfPGM2 has been solved. Sequence and structural comparison of PfPGM2 with PfPGM1 and phosphoglycerate mutases (PGMs) from other organisms revealed a distinct conformation of the catalytic residues, not found in other known PGM structures. Characterization of PfPGM2 enzyme activity established that PfPGM2 acted as a phosphatase with a residual level of mutase activity indicating the role of PGM2 in the phosphate metabolism or regulatory functions in parasite life cycle [25].
2.2.9. Enolase
Plasminogen-binding sites on nucleated cells were identified using U937 monocytoid cells as a model. Affinity chromatography was used to isolate candidate plasminogen receptors from U937 cells, which identified a major protein of Mr 54,000 that was found to be α-enolase by immunochemical and functional studies. Cell surface α-enolase was capable of binding plasminogen in ligand blotting analyses [69]. α-Enolase plays an important role in regulation of c-myc protooncogene promoter activity in the form of an alternative translation product of Myc-binding protein-1 (MBP-1) [70]. In Entamoeba histolytica, enolase exhibits both cytosolic and nuclear localization and was found to act as a cytosine-5 methyltransferase 2 (Dnmt2) inhibitor [71]. In Toxoplasma gondii two isoforms of enolase (TgENO1 and TgENO2) are found. TgENO2 is targeted to the nucleus of actively replicating parasites and regulates the transcription of a number of genes [72].
P. falciparum enolase (PfENO) shows 67% sequence identity with human α-enolase (Table 2). The unique feature of PfENO is the presence of a pentapeptide insert that is also found in other Plasmodium species and plant enolases [8]. P. falciparum exhibits diverse subcellular localization in blood stage parasites apart from its normal cytosolic location (nucleus, merozoite surface, food vacuole, and cytoskeleton) [73]. Plasmodium enolase plays a crucial role during ookinete invasion of mosquito midgut, an essential step in Plasmodium life cycle in mosquito. Enolase lines the ookinete surface and anti-enolase antibody inhibited the development of both P. falciparum and P. berghei ookinetes. Enolase captures mammalian serine protease plasminogen that assists in midgut invasion by ookinetes, suggesting that enolase may act as an invasion ligand [74]. In an attempt to identify the ookinete ligands binding to the mosquito midgut surface, a phage display screen identified a consensus peptide sequence (Pro-Tyr-Tyr-Pro) on phages that bound to P. berghei ookinete surface. The phages expressing these peptides bound actin and enolase in overlay assays of ookinete protein extracts suggesting the presence of actin and enolase on ookinete surface [75]. Immunofluorescence assay further confirmed the presence of enolase on P. falciparum ookinete surface. Plasmodium enolase recruits human plasminogen from the ingested blood via its lysine motif (277-Ala-Lys-Ser-Leu-Val-Lys-282) [74]. The peptide Ala-Lys-Ser-Leu-Val-Lys was found to be inhibitory to oocyst development [76]. These studies strongly suggest that PfENO is a promising target for transmission blocking of the parasite.
2.2.10. Pyruvate Kinase
Pyruvate kinase (PK) was identified as one of the salivary mucin-binding proteins present in the SDS extract of Streptococcus gordonii cell surface proteins [77]. Four isoforms of pyruvate kinase (M1, M2, L, and R) are expressed in mammalian cells. Mutually exclusive alternative splicing of the PKM2 pre-mRNA produces the pyruvate kinases M1 (PKM1) and M2 (PKM2) isoforms [78]. Expression level of PKM2 is upregulated in human cancer cells [79–81], where it directly regulates gene transcription by histone modification that is essential for tumorigenesis [82]. PK is converted to a less active dimer form of PKM2 isoenzyme during oncogenesis. In response to different apoptotic agents, PKM2 is translocated to the nucleus that is sufficient to induce caspase-independent cell death of tumor cells [83].
Two isoforms of pyruvate kinase (PfPK types I and II) are found in P. falciparum genome. Type I PfPK is the typical glycolytic enzyme, while type II PfPK is localized to apicoplast and involved in type II fatty acid synthesis suggesting its potential to be selectively targeted [84].
2.2.11. Lactate Dehydrogenase
Duck heart lactate dehydrogenase (LDH-B4) acts as a ε-crystallin (a member of water-soluble vertebrate lens proteins) in duck lens, [85].
P. falciparum lactate dehydrogenase (PfLDH) is essential for the anaerobic lifestyle of Plasmodium and a potential drug target [86]. PfLDH inhibitors have been designed, which reduce parasite growth in vitro in P. falciparum culture and in vivo in P. berghei [87]. Since lactate dehydrogenases (LDHs) have highly selective substrate specificities, LDH activity is also routinely used for malaria diagnosis and Plasmodium species identification based on its substrate specificity that is capable of differentiating between falciparum and nonfalciparum malaria [88]. PfLDH is divergent from human lactate dehydrogenase (HsLDH) in some unique biochemical features. In contrast to HsLDH, PfLDH is not inhibited at high concentrations of pyruvate [6] and shows a very high activity on a synthetic coenzyme 3-acetylpyridine adenine dinucleotide (APAD) at 0.5 M lactate concentration that is the basis of the parasitemia test in infected human blood. PfLDH is also highly sensitive to inhibition by gossylic nitrile diacetate (GNDA) compared to HsLDH [89].
In the crystal structure of PfLDH, a significant shift in the position of the NADH cofactor from mammalian enzymes is observed accounting for the unusual biochemical properties of this enzyme. There are two distinct regions of unique structural differences where PfLDH departs from mammalian LDHs. The antigenic loop (residues 205–221) in PfLDH adopts a different conformation than vertebrate LDHs, which may be responsible for the antigenic discrimination. Besides, the substrate specificity loop (residues 97–112) has a five-residue insert that is absent in mammalian LDHs. The insert creates a distinct cavity in the surface adjacent to the catalytic region. These structural differences could be exploited for selective targeting of the parasite enzyme [14, 90]. Overlay of PfLDH and human LDH-A subunit B highlighting the structural differences is shown in Figure 3.
Figure 3.
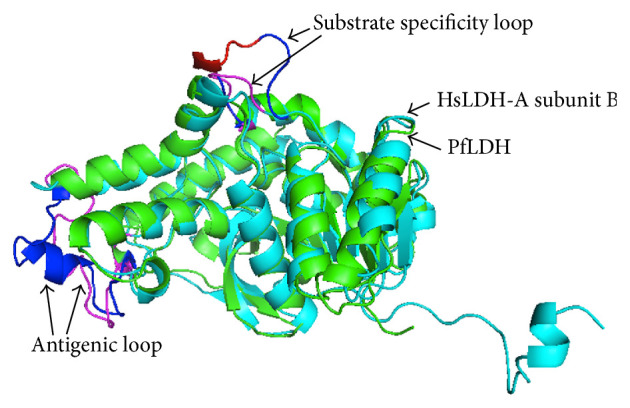
Unique structural differences between P. falciparum LDH (PfLDH) (a) and human LDH-A (HsLDH-A) (b). Aligned PfLDH (green) and HsLDH-A subunit B (cyan). Substrate specificity loop of PfLDH is shown in blue with 5-residue insert in red and that of HsLDH-A subunit B is shown in magenta. Antigenic loops of PfLDH and HsLDH-A are shown in blue and magenta, respectively.
2.3. Role of Posttranslational Modifications in Multifunctionality
Posttranslational modifications (PTMs) are a common means to impart alternative conformations and hence alternative functions to proteins [91]. PTMs change protein mass, charge, and conformation, which in turn modify protein's activity (in case of enzymes), binding affinity, and hydrophobicity [91]. Eukaryotic systems being multicompartmental, localization of proteins to different compartments is a complex process and takes place through binding partners. Localization pattern is dependent on the availability and the concentrations of the binding partners, regulated by their expression levels and PTMs [18]. For example, multifunctionality of GAPDH is dependent on PTMs like phosphorylation, nitrosylation, and acetylation [92]. Nuclear localization of GAPDH is dependent on its nitrosylation-dependent binding to Siah1, an E3 ubiquitin ligase [93]. PTMs are responsible for tremendous increase in the complexity of the proteome in the form of different variants of proteins (approximately 1.8 million protein variants from 30,000 open reading frames in humans). Proteins can undergo a single or multiple modifications at several sites, producing the variants with unique properties [94]. Role of PTMs in malaria and other diseases have been extensively reviewed by Chung et al. [95] and Karve and Cheema [96]. Some important PTMs associated with malaria and other diseases are discussed below.
Proteolytic processing of proteins to produce their variants with altered activities is a major form of PTM [95]. Proteases are a major group of Plasmodium protein, which undergo proteolytic processing by auto- or trans-proteolysis to achieve the active state [97]. Parasite-specific proteases are involved in many crucial processes necessary for parasite survival and replication (hemoglobin degradation, parasite egress, and invasion) [3]. A number of parasitophorous vacuolar and surface proteins known to have role in egress and invasion achieve their active conformation by proteolytic processing [98–100]. Proteases are activated under specific physiological conditions in a temporally regulated manner; hence therapeutic agents interfering with these conditions or targeting the enzyme activity could be potential antimalarials.
Phosphorylation is the reversible addition of a phosphate group to an amino acid residue by a group of proteins known as protein kinases (PKs). It is one of the extensively studied PTMs in eukaryotic cells and implicated in regulation of a variety of cellular processes. Several kinase inhibitors have been found to inhibit various Plasmodium developmental stages. Inhibitor studies have suggested that protein phosphatases, enzymes, which carry out dephosphorylation of amino acids, are also essential for parasite life cycle and may be considered as potential drug targets [95]. Targeting of kinases and phosphatases is considered a potential strategy for treatment of cancer and inflammatory diseases [101, 102].
Ubiquitination is an enzymatically catalyzed covalent attachment of a small regulatory protein, ubiquitin (Ub) to proteins, which targets the ubiquitinated proteins for proteasomal degradation [103]. In addition to protein degradation, ubiquitination is implicated in regulation of a variety of cellular processes (e.g., cell signaling, cell stress-response, intracellular trafficking, etc.) [104]. A few inhibitors of ubiquitination are used as drugs against cancer and neural diseases [105]. In Plasmodium, several studies have demonstrated the essentiality of proteasomal protein turnover for cell cycle progression and proteasomal inhibitors have been shown to be inhibitory for parasite growth [106–109].
In silico analysis of PTM sites in P. falciparum and human glycolytic enzymes revealed differential PTMs in the parasite and host enzymes. Figures 4(a) and 4(b) show the differential PTMs in P. falciparum and human enolase and GAPDH enzymes. It may be speculated that these differential PTMs may account for their diverse subcellular localization and nonglycolytic functions. Study of PTMs associated with parasite-specific moonlighting functions of parasite glycolytic enzymes is an interesting area of research and needs extensive proteomic and functional characterization.
Figure 4.

Differential posttranslational modifications in P. falciparum and human enzymes, enolase (a) and GAPDH (b). PfENO, HsENO1, PfGAPDH, and HsGAPDH represent P. falciparum enolase, human enolase 1, P. falciparum GAPDH, and human GAPDH, respectively. Residues predicted to be modified are shown in boxes. Posttranslational modification sites were predicted by using online available tools. (Phosphorylation with NetPhos 2.0 (http://www.cbs.dtu.dk/services/NetPhos/), methylation with BPB-PPMS (http://www.bioinfo.bio.cuhk.edu.hk/bpbppms/intro.jsp), and ubiquitination with UbiPred (http://www.ubpred.org/), resp.)
3. Conclusion
Despite the availability of therapeutic and prophylactic drugs, malaria remains a major concern for human health because of the emergence of resistant parasites against available drugs. Keeping in mind the ability of the parasite to develop resistance, rational designing of drugs based on known targets is an urgent priority. Glycolysis, being the sole source for parasite energy requirements, presents an attractive parasite pathway to be targeted. Although it is not feasible to develop antimalarial targeting the active sites of malarial glycolytic enzymes due to strong homology with human enzymes, unique structural and functional properties of these enzymes could be exploited for antimalarial development. Unique features and feasibility of selective targeting have been described for many parasite glycolytic enzymes and reviewed in this paper.
Identification of compounds specifically targeting parasite enzymes remains an area to be extensively studied and could potentially lead to development of novel antimalarials. In this direction, attempts to identify small molecule inhibitors against PfHK have identified isobenzothiazolinones as a class of noncovalent, reversible PfHK inhibitors as lead compounds [39]. In another study, virtual screening of NADH analogs against PfLDH followed by in vitro inhibition studies has identified three lead compounds (itraconazole, atorvastatin, and posaconazole) that were effective inhibitors of P. falciparum growth in vitro [87].
In addition to development of antimalarial drugs, transmission blocking of the parasite is an effective means of controlling the spread of malaria. PfENO is an interesting transmission-blocking candidate present on the parasite ookinete surface and assists in ookinete invasion of midgut epithelium by recruiting human serine plasminogen through a lysine motif (277-Ala-Lys-Ser-Leu-Val-Lys-282) [74]. This lysine motif peptide was found to be inhibitory for oocyst development [76]. Nonpeptide stable compounds targeting the interaction between enolase and plasminogen could be potential transmission-blocking agents.
The present state of knowledge about the malarial glycolytic enzymes suggests that their unique properties could be exploited for the development of potent future antimalarials. In addition to the above-mentioned approaches, study of differential PTMs in various malarial glycolytic enzymes from the host enzymes may open novel avenues for drug discovery efforts against malaria. Although the feasibility of targeting PTMs has been demonstrated in malaria and other diseases, their role in malarial glycolytic enzymes needs to be explored.
Acknowledgments
The authors would like to acknowledge the scientists who contributed in the field of metabolic enzyme research. Asrar Alam is supported by a fellowship from Department of Atomic Energy, Government of India.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.WHO . World Malaria Report 2012. WHO; 2012. [Google Scholar]
- 2.Kell A. J. Malaria. Oxford, UK: Oxford University Press; 1991. [Google Scholar]
- 3.Rosenthal P. J. Proteases of malaria parasites: new targets for chemotherapy. Emerging Infectious Diseases. 1998;4(1):49–57. doi: 10.3201/eid0401.980107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sherman I. W. Carbohydrate metabolism of asexual stages. In: Sherman I. W., editor. Malaria: Parasite Biology, Pathogenesis and Protection. Washington, DC, USA: American Society of Microbiology; 1998. pp. 135–143. [Google Scholar]
- 5.Pfaller M. A., Krogstad D. J., Parquette A. R., Dinh P. N. Plasmodium falciparum: stage-specific lactate production in synchronized cultures. Experimental Parasitology. 1982;54(3):391–396. doi: 10.1016/0014-4894(82)90048-0. [DOI] [PubMed] [Google Scholar]
- 6.Vander Jagt D. L., Hunsaker L. A., Campos N. M., Baack B. R. d-Lactate production in erythrocytes infected with Plasmodium falciparum. Molecular and Biochemical Parasitology. 1990;42(2):277–284. doi: 10.1016/0166-6851(90)90171-H. [DOI] [PubMed] [Google Scholar]
- 7.Roth E. F., Jr., Calvin M.-C., Max-Audit I., Rosa J., Rosa R. The enzymes of the glycolytic pathway in erythrocytes infected with Plasmodium falciparum malaria parasites. Blood. 1988;72(6):1922–1925. [PubMed] [Google Scholar]
- 8.Read M., Hicks K. E., Sims P. F. G., Hyde J. E. Molecular characterisation of the enolase gene from the human malaria parasite Plasmodium falciparum—evidence for ancestry within a photosynthetic lineage. European Journal of Biochemistry. 1994;220(2):513–520. doi: 10.1111/j.1432-1033.1994.tb18650.x. [DOI] [PubMed] [Google Scholar]
- 9.Dobeli H., Trzeciak A., Gillessen D., Matile H., Srivastava I. K., Perrin L. H., Jakob P. E., Certa U. Expression, purification, biochemical characterization and inhibition of recombinant Plasmodium falciparum aldolase. Molecular and Biochemical Parasitology. 1990;41(2):259–268. doi: 10.1016/0166-6851(90)90189-S. [DOI] [PubMed] [Google Scholar]
- 10.Wanidworanun C., Nagel R. L., Shear H. L. Antisense oligonucleotides targeting malarial aldolase inhibit the asexual erythrocytic stages of Plasmodium falciparum. Molecular and Biochemical Parasitology. 1999;102(1):91–101. doi: 10.1016/S0166-6851(99)00087-0. [DOI] [PubMed] [Google Scholar]
- 11.Kim H., Certa U., Döbeli H., Jakob P., Hol W. G. J. Crystal structure of fructose-1,6-bisphosphate aldolase from the human malaria parasite plasmodium falciparum. Biochemistry. 1998;37(13):4388–4396. doi: 10.1021/bi972233h. [DOI] [PubMed] [Google Scholar]
- 12.Velanker S. S., Ray S. S., Gokhale R. S., Suma S., Balaram H., Balaram P., Murthy M. R. N. Triosephosphate isomerase from Plasmodium falciparum: the crystal structure provides insights into antimalarial drug design. Structure. 1997;5(6):751–761. doi: 10.1016/S0969-2126(97)00230-X. [DOI] [PubMed] [Google Scholar]
- 13.Robien M. A., Bosch J., Buckner F. S., et al. Crystal structure of glyceraldehyde-3-phosphate dehydrogenase from Plasmodium falciparum at 2.25 Å resolution reveals intriguing extra electron density in the active site. Proteins: Structure, Function, and Bioinformatics. 2006;62(3):570–577. doi: 10.1002/prot.20801. [DOI] [PubMed] [Google Scholar]
- 14.Dunn C. R., Banfield M. J., Barker J. J., Higham C. W., Moreton K. M., Turgut- Balik D., Brady R. L., Holbrook J. J. The structure of lactate dehydrogenase from Plasmodium falciparum reveals a new target for anti-malarial design. Nature Structural Biology. 1996;3(11):910–915. doi: 10.1038/nsb1196-910. [DOI] [PubMed] [Google Scholar]
- 15.Garrod A. E. Inborn Errors of Metabolism. London, UK: Hodder & Stoughton; 1923. [Google Scholar]
- 16.Beadle G. W., Tatum E. L. Genetic control of biochemical reactions in Neurospora. Proceedings of the National Academy of Sciences of the United States of America. 1941;27(11):499–506. doi: 10.1073/pnas.27.11.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeffery C. J. Moonlighting proteins. Trends in Biochemical Sciences. 1999;24(1):8–11. doi: 10.1016/S0968-0004(98)01335-8. [DOI] [PubMed] [Google Scholar]
- 18.Copley S. D. Moonlighting is mainstream: paradigm adjustment required. Bioessays. 2012;34(7):578–588. doi: 10.1002/bies.201100191. [DOI] [PubMed] [Google Scholar]
- 19.Gurney M. E., Heinrich S. P., Lee M. R., Yin H.-S. Molecular cloning and expression of neuroleukin, a neurotrophic factor for spinal and sensory neurons. Science. 1986;234(4776):566–574. doi: 10.1126/science.3764429. [DOI] [PubMed] [Google Scholar]
- 20.Chaput M., Claes V., Portetelle D., Cludts I., Cravador A., Burny A., Gras H., Tartar A. The neurotrophic factor neuroleukin is 90% homologous with phosphohexose isomerase. Nature. 1988;332(6163):454–455. doi: 10.1038/332454a0. [DOI] [PubMed] [Google Scholar]
- 21.Faik P., Walker J. I. H., Redmill A. A. M., Morgan M. J. Mouse glucose-6-phosphate isomerase and neuroleukin have identical 3' sequences. Nature. 1988;332(6163):455–456. doi: 10.1038/332455a0. [DOI] [PubMed] [Google Scholar]
- 22.Sriram G., Martinez J. A., McCabe E. R. B., Liao J. C., Dipple K. M. Single-gene disorders: what role could moonlighting enzymes play? American Journal of Human Genetics. 2005;76(6):911–924. doi: 10.1086/430799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J.-W., Dang C. V. Multifaceted roles of glycolytic enzymes. Trends in Biochemical Sciences. 2005;30(3):142–150. doi: 10.1016/j.tibs.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Smith C. D., Chattopadhyay D., Pal B. Crystal structure of Plasmodium falciparum phosphoglycerate kinase: evidence for anion binding in the basic patch. Biochemical and Biophysical Research Communications. 2011;412(2):203–206. doi: 10.1016/j.bbrc.2011.07.045. [DOI] [PubMed] [Google Scholar]
- 25.Hills T., Srivastava A., Ayi K., Wernimont A. K., Kain K., Waters A. P., Hui R., Pizarro J. C. Characterization of a new phosphatase from Plasmodium. Molecular and Biochemical Parasitology. 2011;179(2):69–79. doi: 10.1016/j.molbiopara.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 26.Welches W. R., Baldwin T. O. Active center studies on bacterial luciferase: modification of the enzyme with 2,4-dinitrofluorobenzene. Biochemistry. 1981;20(3):512–517. doi: 10.1021/bi00506a011. [DOI] [PubMed] [Google Scholar]
- 27.Shoichet B. K., Stroud R. M., Santi D. V., Kuntz I. D., Perry K. M. Structure-based discovery of inhibitors of thymidylate synthase. Science. 1993;259(5100):1445–1450. doi: 10.1126/science.8451640. [DOI] [PubMed] [Google Scholar]
- 28.Herrero P., Martínez-Campa C., Moreno F. The hexokinase 2 protein participates in regulatory DNA-protein complexes necessary for glucose repression of the SUC2 gene in Saccharomyces cerevisiae. The FEBS Letters. 1998;434(1-2):71–76. doi: 10.1016/S0014-5793(98)00872-2. [DOI] [PubMed] [Google Scholar]
- 29.Niederacher D., Entian K. D. Characterization of Hex2 protein, a negative regulatory element necessary for glucose repression in yeast. European Journal of Biochemistry. 1991;200(2):311–319. doi: 10.1111/j.1432-1033.1991.tb16187.x. [DOI] [PubMed] [Google Scholar]
- 30.Herrero P., Galindez J., Ruiz N., Martinez-Campa C., Moreno F. Transcriptional regulation of the Saccharomyces cerevisiae HXK1, HXK2 and GLK1 genes. Yeast. 1995;11(2):137–144. doi: 10.1002/yea.320110205. [DOI] [PubMed] [Google Scholar]
- 31.Bryson J. M., Coy P. E., Gottlob K., Hay N., Brooks Robey R. Increased hexokinase activity, of either ectopic or endogenous origin, protects renal epithelial cells against acute oxidant-induced cell death. Journal of Biological Chemistry. 2002;277(13):11392–11400. doi: 10.1074/jbc.M110927200. [DOI] [PubMed] [Google Scholar]
- 32.Pastorino J. G., Hoek J. B. Hexokinase II: the integration of energy metabolism and control of apoptosis. Current Medicinal Chemistry. 2003;10(16):1535–1551. doi: 10.2174/0929867033457269. [DOI] [PubMed] [Google Scholar]
- 33.Gottlob K., Majewski N., Kennedy S., Kandel E., Robey R. B., Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes and Development. 2001;15(11):1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olafsson P., Matile H., Certa U. Molecular analysis of Plasmodium falciparum hexokinase. Molecular and Biochemical Parasitology. 1992;56(1):89–101. doi: 10.1016/0166-6851(92)90157-F. [DOI] [PubMed] [Google Scholar]
- 35.Udeinya I. J., van Dyke K. 2-Deoxyglucose: inhibition of parasitemia and of glucosamine incorporation into glycosylated macromolecules, in malarial parasites (Plasmodium falciparum) Pharmacology. 1981;23(3):171–175. doi: 10.1159/000137546. [DOI] [PubMed] [Google Scholar]
- 36.De Macedo C. S., Gerold P., Jung N., Azzouz N., Kimmel J., Schwarz R. T. Inhibition of glycosyl-phosphatidylinositol biosynthesis in Plasmodium falciparum by C-2 substituted mannose analogues. European Journal of Biochemistry. 2001;268(23):6221–6228. doi: 10.1046/j.0014-2956.2001.02571.x. [DOI] [PubMed] [Google Scholar]
- 37.van Schalkwyk D. A., Priebe W., Saliba K. J. The inhibitory effect of 2-halo derivatives of D-glucose on glycolysis and on the proliferation of the human malaria parasite Plasmodium falciparum. Journal of Pharmacology and Experimental Therapeutics. 2008;327(2):511–517. doi: 10.1124/jpet.108.141929. [DOI] [PubMed] [Google Scholar]
- 38.Tjhin E. T., Staines H. M., van Schalkwyk D. A., Krishna S., Saliba K. J. Studies with the Plasmodium falciparum hexokinase reveal that PfHT limits the rate of glucose entry into glycolysis. FEBS Letters. 2013;587(19):3182–3187. doi: 10.1016/j.febslet.2013.07.052. [DOI] [PubMed] [Google Scholar]
- 39.Harris M. T., Walker D. M., Drew M. E., Mitchell W. G., Dao K., Schroeder C. E., Flaherty D. P., Weiner W. S., Golden J. E., Morris J. C. Interrogating a hexokinase-selected small-molecule library for inhibitors of Plasmodium falciparum hexokinase. Antimicrobial Agents and Chemotherapy. 2013;57(8):3731–3737. doi: 10.1128/AAC.00662-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe H., Takehana K., Date M., Shinozaki T., Raz A. Tumor cell autocrine motility factor is the neuroleukin/phosphohexose isomerase polypeptide. Cancer Research. 1996;56(13):2960–2963. [PubMed] [Google Scholar]
- 41.Xu W., Seiter K., Feldman E., Ahmed T., Chiao J. W. The differentiation and maturation mediator for human myeloid leukemia cells shares homology with neuroleukin or phosphoglucose isomerase. Blood. 1996;87(11):4502–4506. [PubMed] [Google Scholar]
- 42.Srivastava I. K., Schmidt M., Grall M., Certa U., Garcia A. M., Perrin L. H. Identification and purification of glucose phosphate isomerase of Plasmodium falciparum. Molecular and Biochemical Parasitology. 1992;54(2):153–164. doi: 10.1016/0166-6851(92)90108-V. [DOI] [PubMed] [Google Scholar]
- 43.Yuan W., Tuttle D. L., Shi Y.-J., Ralph G. S., Dunn W. A., Jr. Glucose-induced microautophagy in Pichia pastoris requires the alpha-subunit of phosphofructokinase. Journal of Cell Science. 1997;110(16):1935–1945. doi: 10.1242/jcs.110.16.1935. [DOI] [PubMed] [Google Scholar]
- 44.Kinnby B., Booth N. A., Svensäter G. Plasminogen binding by oral streptococci from dental plaque and inflammatory lesions. Microbiology. 2008;154(3):924–931. doi: 10.1099/mic.0.2007/013235-0. [DOI] [PubMed] [Google Scholar]
- 45.Commichau F. M., Rothe F. M., Herzberg C., et al. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Molecular & Cellular Proteomics. 2009;8(6):1350–1360. doi: 10.1074/mcp.M800546-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vora S., Seaman C., Durham S., Piomelli S. Isozymes of human phosphofructokinase: identification and subunit structural characterization of a new system. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(1):62–66. doi: 10.1073/pnas.77.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mony B. M., Mehta M., Jarori G. K., Sharma S. Plant-like phosphofructokinase from Plasmodium falciparum belongs to a novel class of ATP-dependent enzymes. International Journal for Parasitology. 2009;39(13):1441–1453. doi: 10.1016/j.ijpara.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 48.Buckwitz D., Jacobasch G., Gerth C. Phosphofructokinase from Plasmodium berghei: a kinetic model of allosteric regulation. Molecular and Biochemical Parasitology. 1990;40:225–232. doi: 10.1016/0166-6851(90)90044-M. [DOI] [PubMed] [Google Scholar]
- 49.Jewett T. J., Sibley L. D. Aldolase forms a bridge between cell surface adhesins and the actin cytoskeleton in apicomplexan parasites. Molecular Cell. 2003;11(4):885–894. doi: 10.1016/S1097-2765(03)00113-8. [DOI] [PubMed] [Google Scholar]
- 50.Starnes G. L., Coincon M., Sygusch J., Sibley L. D. Aldolase is essential for energy production and bridging adhesin-actin cytoskeletal interactions during parasite invasion of host cells. Cell Host & Microbe. 2009;5(4):353–364. doi: 10.1016/j.chom.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tunio S. A., Oldfield N. J., Berry A., Ala'Aldeen D. A. A., Wooldridge K. G., Turner D. P. J. The moonlighting protein fructose-1, 6-bisphosphate aldolase of Neisseria meningitidis: surface localization and role in host cell adhesion. Molecular Microbiology. 2010;76(3):605–615. doi: 10.1111/j.1365-2958.2010.07098.x. [DOI] [PubMed] [Google Scholar]
- 52.Lew C. R., Tolan D. R. Aldolase sequesters WASP and affects WASP/Arp2/3-stimulated actin dynamics. Journal of Cellular Biochemistry. 2013;114(8):1928–1939. doi: 10.1002/jcb.24538. [DOI] [PubMed] [Google Scholar]
- 53.Certa U., Ghersa P., Dobeli H., Matile H., Kocher H. P., Shrivastava I. K., Shaw A. R., Perrin L. H. Aldolase activity of a Plasmodium falciparum protein with protective properties. Science. 1988;240(4855):1036–1038. doi: 10.1126/science.3285469. [DOI] [PubMed] [Google Scholar]
- 54.Knapp B., Hundt E., Kupper H. A. Plasmodium falciparum aldolase: gene structure and localization. Molecular and Biochemical Parasitology. 1990;40(1):1–12. doi: 10.1016/0166-6851(90)90074-V. [DOI] [PubMed] [Google Scholar]
- 55.Itin C., Burki Y., Certa U., Döbeli H. Selective inhibition of Plasmodium falciparum aldolase by a tubulin derived peptide and identification of the binding site. Molecular and Biochemical Parasitology. 1993;58(1):135–144. doi: 10.1016/0166-6851(93)90097-H. [DOI] [PubMed] [Google Scholar]
- 56.Baum J., Richard D., Healer J., et al. A conserved molecular motor drives cell invasion and gliding motility across malaria life cycle stages and other apicomplexan parasites. The Journal of Biological Chemistry. 2006;281(8):5197–5208. doi: 10.1074/jbc.M509807200. [DOI] [PubMed] [Google Scholar]
- 57.Saito F., Ikeda R. Killing of Cryptococcus neoformans by Staphylococcus aureus: the role of cryptococcal capsular polysaccharide in the fungal-bacteria interaction. Medical Mycology. 2005;43(7):603–612. doi: 10.1080/13693780500078417. [DOI] [PubMed] [Google Scholar]
- 58.Ikeda R., Saito F., Matsuo M., Kurokawa K., Sekimizu K., Yamaguchi M., Kawamoto S. Contribution of the mannan backbone of cryptococcal glucuronoxylomannan and a glycolytic enzyme of Staphylococcus aureus to contact-mediated killing of Cryptococcus neoformans. Journal of Bacteriology. 2007;189(13):4815–4826. doi: 10.1128/JB.00412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guix F. X., Ill-Raga G., Bravo R., Nakaya T., De Fabritiis G., Coma M., Miscione G. P., Vill-Freixa J., Suzuki T., Fernndez-Busquets X., Valverde M. A., De Strooper B., Muoz F. J. Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation. Brain. 2009;132(5):1335–1345. doi: 10.1093/brain/awp023. [DOI] [PubMed] [Google Scholar]
- 60.Maithal K., Ravindra G., Balaram H., Balaram P. Inhibition of Plasmodium falciparum triose-phosphate isomerase by chemical modification of an interface cysteine. The Journal of Biological Chemistry. 2002;277(28):25106–25114. doi: 10.1074/jbc.M202419200. [DOI] [PubMed] [Google Scholar]
- 61.Matta S. K., Agarwal S., Bhatnagar R. Surface localized and extracellular Glyceraldehyde-3-phosphate dehydrogenase of Bacillus anthracis is a plasminogen binding protein. Biochimica et Biophysica Acta: Proteins and Proteomics. 2010;1804(11):2111–2120. doi: 10.1016/j.bbapap.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 62.Zheng L., Roeder R. G., Luo Y. S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell. 2003;114(2):255–266. doi: 10.1016/S0092-8674(03)00552-X. [DOI] [PubMed] [Google Scholar]
- 63.Raje C. I., Kumar S., Harle A., Nanda J. S., Raje M. The macrophage cell surface glyceraldehyde-3-phosphate dehydrogenase is a novel transferrin receptor. Journal of Biological Chemistry. 2007;282(5):3252–3261. doi: 10.1074/jbc.M608328200. [DOI] [PubMed] [Google Scholar]
- 64.Dastoor Z., Dreyer J.-L. Potential role of nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase in apoptosis and oxidative stress. Journal of Cell Science. 2001;114(9):1643–1653. doi: 10.1242/jcs.114.9.1643. [DOI] [PubMed] [Google Scholar]
- 65.Daubenberger C. A., Pöltl-Frank F., Jiang G., Lipp J., Certa U., Pluschke G. Identification and recombinant expression of glyceraldehyde-3-phosphate dehydrogenase of Plasmodium falciparum. Gene. 2000;246(1-2):255–264. doi: 10.1016/S0378-1119(00)00069-X. [DOI] [PubMed] [Google Scholar]
- 66.Daubenberger C. A., Tisdale E. J., Curcic M., Diaz D., Silvie O., Mazier D., Eling W., Bohrmann B., Matile H., Pluschke G. The N′-terminal domain of glyceraldehyde-3-phosphate dehydrogenase of the apicomplexan Plasmodium falciparum mediates GTPase Rab2-dependent recruitment to membranes. Biological Chemistry. 2003;384(8):1227–1237. doi: 10.1515/BC.2003.135. [DOI] [PubMed] [Google Scholar]
- 67.Boone T. J., Burnham C.-A. D., Tyrrell G. J. Binding of group B streptococcal phosphoglycerate kinase to plasminogen and actin. Microbial Pathogenesis. 2011;51(4):255–261. doi: 10.1016/j.micpath.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 68.Burnham C.-A. D., Shokoples S. E., Tyrrell G. J. Phosphoglycerate kinase inhibits epithelial cell invasion by group B streptococci. Microbial Pathogenesis. 2005;38(5-6):189–200. doi: 10.1016/j.micpath.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 69.Miles L. A., Dahlberg C. M., Plescia J., Felez J., Kato K., Plow E. F. Role of cell-surface lysines in plasminogen binding to cells: identification of α-enolase as a candidate plasminogen receptor. Biochemistry. 1991;30(6):1682–1691. doi: 10.1021/bi00220a034. [DOI] [PubMed] [Google Scholar]
- 70.Subramanian A., Miller D. M. Structural analysis of alpha-enolase: mapping the functional domains involved in down-regulation of the c-myc protooncogene. Journal of Biological Chemistry. 2000;275(8):5958–5965. doi: 10.1074/jbc.275.8.5958. [DOI] [PubMed] [Google Scholar]
- 71.Tovy A., Tov R. S., Gaentzsch R., Helm M., Ankri S. A new nuclear function of the Entamoeba histolytica glycolytic enzyme enolase: the metabolic regulation of cytosine-5 methyltransferase 2 (Dnmt2) activity. PLoS Pathogens. 2010;6(2) doi: 10.1371/journal.ppat.1000775.e1000775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mouveaux T., Oria G., Werkmeister E., Slomianny C., Fox B. A., Bzik D. J., Tomavo S. Nuclear glycolytic enzyme enolase of Toxoplasma gondii functions as a transcriptional regulator. PLoS ONE. 2014;9(8) doi: 10.1371/journal.pone.0105820.e105820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bhowmick I. P., Kumar N., Sharma S., Coppens I., Jarori G. K. Plasmodium falciparum enolase: stage-specific expression and sub-cellular localization. Malaria journal. 2009;8, article 179 doi: 10.1186/1475-2875-8-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ghosh A. K., Coppens I., Gårdsvoll H., Ploug M., Jacobs-Lorena M. Plasmodium ookinetes coopt mammalian plasminogen to invade the mosquito midgut. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(41):17153–17158. doi: 10.1073/pnas.1103657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hernández-Romano J., Rodríguez M. H., Pando V., Torres-Monzón J. A., Alvarado-Delgado A., Lecona Valera A. N., Ramos R. A., Martinez-Barnetche J., Rodríguez M. C. Conserved peptide sequences bind to actin and enolase on the surface of Plasmodium berghei ookinetes. Parasitology. 2011;138(11):1341–1353. doi: 10.1017/S0031182011001296. [DOI] [PubMed] [Google Scholar]
- 76.Wang S., Ghosh A. K., Bongio N., Stebbings K. A., Lampe D. J., Jacobs-Lorena M. Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(31):12734–12739. doi: 10.1073/pnas.1204158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kesimer M., Kilic N., Mehrotra R., Thornton D. J., Sheehan J. K. Identification of salivary mucin MUC7 binding proteins from Streptococcus gordonii. BMC Microbiology. 2009;9, article 163 doi: 10.1186/1471-2180-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Noguchi T., Inoue H., Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. The Journal of Biological Chemistry. 1986;261(29):13807–13812. [PubMed] [Google Scholar]
- 79.Dombrauckas J. D., Santarsiero B. D., Mesecar A. D. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44(27):9417–9429. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- 80.Mazurek S., Boschek C. B., Hugo F., Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Seminars in Cancer Biology. 2005;15(4):300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 81.Mazurek S. Pyruvate kinase type M2: a key regulator within the tumour metabolome and a tool for metabolic profiling of tumours. Ernst Schering Foundation Symposium Proceedings. 2007;(4):99–124. doi: 10.1007/2789_2008_091. [DOI] [PubMed] [Google Scholar]
- 82.Yang W., Xia Y., Hawke D., Li X., Liang J., Xing D., Aldape K., Hunter T., Alfred Yung W. K., Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150(4):685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Steták A., Veress R., Ovádi J., Csermely P., Kéri G., Ullrich A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer Research. 2007;67(4):1602–1608. doi: 10.1158/0008-5472.CAN-06-2870. [DOI] [PubMed] [Google Scholar]
- 84.Maeda T., Saito T., Harb O. S., Roos D. S., Takeo S., Suzuki H., Tsuboi T., Takeuchi T., Asai T. Pyruvate kinase type-II isozyme in Plasmodium falciparum localizes to the apicoplast. Parasitology International. 2009;58(1):101–105. doi: 10.1016/j.parint.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hendriks W., Mulders J. W., Bibby M. A., Slingsby C., Bloemendal H., de Jong W. W. Duck lens ε-crystallin and lactate dehydrogenase B4 are identical: a single-copy gene product with two distinct functions. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(19):7114–7118. doi: 10.1073/pnas.85.19.7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Royer R. E., Deck L. M., Campos N. M., Hunsaker L. A., Vender Jagt D. L. Biologically active derivatives of gossypol: synthesis and antimalarial activities of peri-acylated gossylic nitriles. Journal of Medicinal Chemistry. 1986;29(9):1799–1801. doi: 10.1021/jm00159a043. [DOI] [PubMed] [Google Scholar]
- 87.Penna-Coutinho J., Cortopassi W. A., Oliveira A. A., França T. C. C., Krettli A. U. Antimalarial activity of potential inhibitors of Plasmodium falciparum lactate dehydrogenase enzyme selected by docking studies. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0021237.e21237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Piper R., LeBras J., Wentworth L., Hunt-Cooke A., Houzé S., Chiodini P., Makler M. Immunocapture diagnostic assays for malaria using Plasmodium lactate dehydrogenase (pLDH) American Journal of Tropical Medicine and Hygiene. 1999;60(1):109–118. doi: 10.4269/ajtmh.1999.60.109. [DOI] [PubMed] [Google Scholar]
- 89.Makler M. T., Hinrichs D. J. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. The American Journal of Tropical Medicine and Hygiene. 1993;48(2):205–210. doi: 10.4269/ajtmh.1993.48.205. [DOI] [PubMed] [Google Scholar]
- 90.Bzik D. J., Fox B. A., Gonyer K. Expression of Plasmodium falciparum lactate dehydrogenase in Escherichia coli. Molecular and Biochemical Parasitology. 1993;59(1):155–166. doi: 10.1016/0166-6851(93)90016-Q. [DOI] [PubMed] [Google Scholar]
- 91.Clark R. S. B., Bayir H., Jenkins L. W. Posttranslational protein modifications. Critical Care Medicine. 2005;33(12):S407–S409. doi: 10.1097/01.CCM.0000191712.96336.51. [DOI] [PubMed] [Google Scholar]
- 92.Sirover M. A. On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: biochemical mechanisms and regulatory control. Biochimica et Biophysica Acta: General Subjects. 2011;1810(8):741–751. doi: 10.1016/j.bbagen.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 93.Hara M. R., Agrawal N., Kim S. F., Cascio M. B., Fujimuro M., Ozeki Y., Takahashi M., Cheah J. H., Tankou S. K., Hester L. D., Ferris C. D., Hayward S. D., Snyder S. H., Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nature Cell Biology. 2005;7(7):665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 94.Jensen O. N. Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Current Opinion in Chemical Biology. 2004;8(1):33–41. doi: 10.1016/j.cbpa.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 95.Chung D.-W. D., Ponts N., Cervantes S., Le Roch K. G. Post-translational modifications in Plasmodium: more than you think! Molecular and Biochemical Parasitology. 2009;168(2):123–134. doi: 10.1016/j.molbiopara.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 96.Karve T. M., Cheema A. K. Small changes huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. Journal of Amino Acids. 2011;2011 doi: 10.4061/2011/207691.207691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhu H., Xu B.-L., Liang X., Yang Y.-R., Tang X.-F., Tang B. Molecular basis for auto- and hetero-catalytic maturation of a thermostable subtilase from thermophilic Bacillus sp. WF146. Journal of Biological Chemistry. 2013;288(48):34826–34838. doi: 10.1074/jbc.M113.498774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Koussis K., Withers-Martinez C., Yeoh S., Child M., Hackett F., Knuepfer E., Juliano L., Woehlbier U., Bujard H., Blackman M. J. A multifunctional serine protease primes the malaria parasite for red blood cell invasion. The EMBO Journal. 2009;28(6):725–735. doi: 10.1038/emboj.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Blackman M. J., Carruthers V. B. Recent insights into apicomplexan parasite egress provide new views to a kill. Current Opinion in Microbiology. 2013;16(4):459–464. doi: 10.1016/j.mib.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Alam A. Serine proteases of malaria parasite Plasmodium falciparum: potential as antimalarial drug targets. Interdisciplinary Perspectives on Infectious Diseases. 2014;2014 doi: 10.1155/2014/453186.453186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Slee E. A., Benassi B., Goldin R., Zhong S., Ratnayaka I., Blandino G., Lu X. Phosphorylation of Ser312 contributes to tumor suppression by p53 in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(45):19479–19484. doi: 10.1073/pnas.1005165107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yoboua F., Martel A., Duval A., Mukawera E., Grandvaux N. Respiratory syncytial virus-mediated NF-κB p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKKβ. Journal of Virology. 2010;84(14):7267–7277. doi: 10.1128/JVI.00142-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wilkinson K. D. Protein ubiquitination: a regulatory post-translational modification. Anti-Cancer Drug Design. 1987;2(2):211–229. [PubMed] [Google Scholar]
- 104.Hershko A., Ciechanover A. The ubiquitin system. Annual Review of Biochemistry. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 105.Colland F. The therapeutic potential of deubiquitinating enzyme inhibitors. Biochemical Society Transactions. 2010;38(1):137–143. doi: 10.1042/BST0380137. [DOI] [PubMed] [Google Scholar]
- 106.Gantt S. M., Myung J. M., Briones M. R. S., Li W. D., Corey E. J., Omura S., Nussenzweig V., Sinnis P. Proteasome inhibitors block development of Plasmodium spp. Antimicrobial Agents and Chemotherapy. 1998;42(10):2731–2738. doi: 10.1128/aac.42.10.2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lindenthal C., Weich N., Chia Y.-S., Heussler V., Klinkert M.-Q. The proteasome inhibitor MLN-273 blocks exoerythrocytic and erythrocytic development of Plasmodium parasites. Parasitology. 2005;131(1):37–44. doi: 10.1017/S003118200500747X. [DOI] [PubMed] [Google Scholar]
- 108.Reynolds J. M., El Bissati K., Brandenburg J., Günzl A., Mamoun C. B. Antimalarial activity of the anticancer and proteasome inhibitor bortezomib and its analog ZL3B. BMC Clinical Pharmacology. 2007;7, article 13 doi: 10.1186/1472-6904-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kreidenweiss A., Kremsner P. G., Mordmüller B. Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malaria Journal. 2008;7, article 187 doi: 10.1186/1475-2875-7-187. [DOI] [PMC free article] [PubMed] [Google Scholar]