

Abstract
The TSH receptor (TSHR) A-subunit is more effective than the holoreceptor in inducing thyroid-stimulating antibodies (TSAb) that cause Graves' disease. A puzzling phenomenon is that 2 recombinant, eukaryotic forms of A-subunits (residues 22–289), termed active and inactive, are recognized mutually exclusively by pathogenic TSAb and mouse monoclonal antibody 3BD10, respectively. Understanding the structural difference between these TSHR A-subunit forms could provide insight into Graves' disease pathogenesis. The 3-dimensional structure of the active A-subunit (in complex with a human TSAb Fab, M22) is known, but the structural difference with inactive A-subunits is unknown. We solved the 3BD10 Fab 3-dimensional crystal structure. Guided by prior knowledge of a portion of its epitope, 3BD10 docked in silico with the known active TSHR-289 monomeric structure. Because both TSAb and 3BD10 recognize the active TSHR A-subunit monomer, this form of the molecule can be excluded as the basis for the active-inactive dichotomy, suggesting, instead a role for A-subunit quaternary structure. Indeed, in silico analysis revealed that M22, but not 3BD10, bound to a TSHR-289 trimer. In contrast, 3BD10, but not M22, bound to a TSHR-289 dimer. The validity of these models is supported experimentally by the temperature-dependent balance between active and inactive TSHR-289. In summary, we provide evidence for a structural basis to explain the conformational heterogeneity of TSHR A-subunits (TSHR-289). The pathophysiologic importance of these findings is that affinity maturation of pathogenic TSAb in Graves' disease is likely to involve a trimer of the shed TSHR A-subunit.
Graves' disease is one of the most common organ-specific autoimmune diseases affecting humans, with a prevalence in the female population of ∼2% (reviewed in Ref. 1). Thyroid-stimulating autoantibodies (TSAb) mimic the action of TSH on the TSH receptor (TSHR) and are the direct cause of hyperthyroidism in this disease (2–4). These ligands bind to the very large TSHR extracellular domain (ECD; amino acid residues 22–410 after signal peptide removal) and lead to G protein activation by a conformational change in the heptahelical transmembrane domain (TMD) (reviewed in Ref. 5). Despite the central role for the TSH holoreceptor in increasing thyroid hormone synthesis and secretion after ligand binding, there is strong evidence that it is not the TSH holoreceptor, or even the entire ECD, but a shed component of the ECD that is the primary immunogen in the induction and affinity maturation of pathologic TSAb (6, 7). Therefore, aside from the functional importance of the TSH holoreceptor in Graves' disease, insight into the structure of the TSHR ECD shed component will contribute to understanding the pathogenesis of this disease.
The TSHR ECD comprises an N-terminal leucine-rich repeat domain (LRD) linked to the TMD by a hinge region that is approximately 50 amino acid residues longer than in the other glycoprotein hormone receptors (GPHR) (residues 317–366) (8, 9). Posttranslational intramolecular cleavage within the TSHR hinge region excises a C-peptide region with poorly defined boundaries, including and extending slightly beyond, these 50 amino acid residues (10, 11), resulting in an N-terminal A-subunit linked by disulfide bonds to a B-subunit (C-terminal portion of the hinge and the TMD) (reviewed in Ref. 12) (Figure 1). Dissolution of the disulfide bonds either by disulfide isomerase (13) or by continued proteolytic digestion (14) leads to shedding of the A-subunit (LRD and N-terminal portion of the hinge region). Although the crystal structure of the major portion of the shed A-subunit (amino acid residues 22–260) in complex with a human monoclonal TSAb fragment, antigen binding (Fab) (15), as well as with a human TSH blocking antibody (16), has been solved, important structural and functional questions remain unanswered. In particular, this crystal structure does not provide information on a puzzling phenomenon involving TSHR A-subunit structural heterogeneity, described below.
Figure 1.
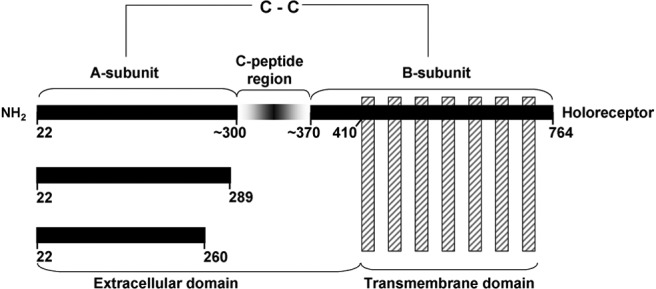
Schematic representation of TSHR components. Intramolecular cleavage of the TSH holoreceptor on the cell surface results in A- and B-subunits linked by disulfide bonds (C-C). This process is associated with deletion of an intervening C-peptide region with indistinct borders, but approximating amino acid residue 300 at the C terminus of the A-subunit and amino acid residue 370 at the N terminus of the B-subunit (10–12). Residue 22 represents the N terminus of the TSHR after signal peptide removal. The purified, recombinant TSHR A-subunit that is the subject of the present report extends to residue 289, the latter chosen because the Arg and Lys cluster in this region was a potential cleavage site and because TSHR-289 was secreted by transfected mammalian cells more effectively than a slightly longer A-subunit. The crystal structure of the A-subunit truncated further upstream at residue 260 has been solved (structure data bank PDB IDs: 3GO4 and 2XWT).
For many years before and consequent to the report on the atomic structure of TSHR 22–260 (15), we have studied a TSHR ECD component (amino acid residues 22–289) that more closely represents the shed A-subunit. We focused on TSHR-289 because the full TSHR ECD was retained incorrectly folded within transfected eukaryotic cells, whereas inserting stop codons at potential TSHR intramolecular cleavage sites permitted secretion of the TSHR A-subunit (17). However, affinity purification of TSHR-289 using mouse monoclonal antibody (mAb) 3BD10 generated to this protein (partially purified) led to a major surprise. Despite its capture by 3BD10, the TSHR-289 eluted from the column was not recognized by pathogenic TSAb in Graves' patients' sera. This material remained in the flow-through from the column (18). Consequently, we could separate and purify 2 types of TSHR-289 (or A-subunits), one recognized by TSAb (termed active), the other recognized by 3BD10 (termed inactive) (19). These two A-subunit forms were secreted by transfected CHO cells in varying, but typically equimolar, amounts. Although reciprocally recognizing active and inactive A-subunits, TSAb and 3BD10 have similar properties, including small epitopic components at the N terminus of the A-subunit (15, 20), as well as partial steric hindrance in binding to the TSH holoreceptor on the cell surface (18, 21). However, the full 3BD10 epitope, necessary to determine the orientation of the antibody relative to the TSHR, remains unknown.
In the present study, we determined the crystal structure of the 3BD10 Fab in our effort to understand the structural basis for the difference between active and inactive TSHR A-subunit conformers. We provide evidence that this difference resides in the degree of A-subunit multimerization (trimer vs dimer). These findings suggest that the shed A-subunit trimer is a critical immunogen in Graves' disease.
Materials and Methods
3BD10 Fab preparation
Mouse mAb 3BD10 (18) secreted by SP2/0 hybridoma cells was purified by a HiTrap Protein G HP column (17-0405-01; GE Healthcare) and Fab prepared by immobilized ficin digestion (Mouse IgG1 Fab Preparation Kit; Thermo Scientific). 3BD10 Fab fragments were concentrated to 7.5 mg/mL. The amino acid sequences of the 3BD10 heavy (H) and light (L) variable regions (Figure 2) determined by nucleotide sequencing of cDNA prepared from the SP2/0 hybridoma cells (GenBank accession numbers KM393208 and KM393209, respectively) were confirmed by the 3BD10 Fab crystal structure, as described below.
Figure 2.
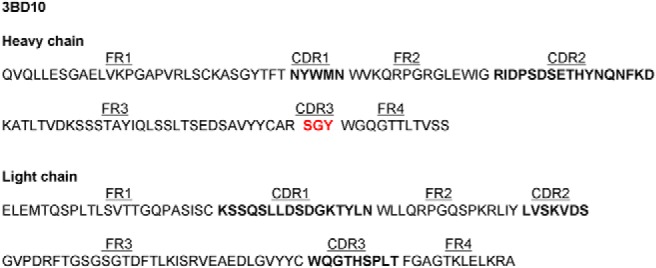
Primary amino acid sequence of the 3BD10 H-chain and L-chain variable regions. The CDRs are in bold. The unusual very short H-chain CDR3 (SGY) is shown in red. Abbreviation: FR, framework region.
Structure determination of 3BD10 Fab
Fab fragments from 3BD10 were crystallized from solution containing 7.5 mg/mL Fab, 20mM Tris-HCl (pH 7.4) and 11% PEG4K at room temperature by the hanging drop method. Crystals grew over 8 weeks as plates with the largest dimension of 0.3 mm.
Diffraction data were collected in-house on a Rigaku MicroMax 007 HF x-ray generator equipped with a Rigaku R-AXIS IV++ image plate detector, with the crystal cooled to 100°K. To minimize overlap of reflections while maintaining high-resolution completeness, 2 datasets were collected, one at a crystal to detector distance of 145 mm and the other at 190 mm. Each dataset was indexed separately using XDS (22) and then combined and scaled using AIMLESS (23, 24), with statistics shown in Table 1. Initial phases were calculated using the molecular replacement program PHASER (25) with both heavy chains (H-chains) and light chains (L-chains) of PBD 4K23 (26) used as the search model. Two Fab complexes were found in the asymmetric unit giving a final Z-score of 9.0. Initial electron density maps were improved by jelly body refinement of the molecular replacement solution using REFMAC5 (27). These partially refined electron density maps were of sufficient quality to allow manual rebuilding using COOT (28), followed by alternating rounds of reciprocal space refinement and manual adjustment until RFree convergence was achieved. The refined model was validated using MOLPROBITY (29), with final refinement and model statistics are shown in Table 1. The molecular structure data of the 3BD10 Fab has been deposited as PDB code 4QT5.
Table 1.
Crystallographic Data
| Data | |
|---|---|
| Resolution, Å | 19.81–2.50 |
| Space group | P212121 |
| Unit cell dimensions | |
| Å | 44.0, 137.0, 149.1 |
| Degrees | 90.0, 90.0, 90.0 |
| Rmerge, % | 11.0 (62.6)a |
| Total number of observations | 285 221 (32 620) |
| Total number of unique observations | 32 117 (3591) |
| I/s (I) | 7.6 (1.7) |
| Completeness, % | 99.8 (100.0) |
| Average mosaicity, ° | 0.36 |
| *Rwork/Rfree, % | 23.5/27.9 |
| Ramachandran plot (favored/allowed/outliers), % | 97.4/2.4/0.2 |
| Number of protein residues modeled | 846 |
| Number of protein atoms modeled | 6478 |
| Number of water molecules modeled | 105 |
| Average B factor of protein atoms, Å2 | 39.4 |
| Average B factor of water molecules, Å2 | 26.2 |
| Root mean square bond lengths, Å | 0.011 |
| Root mean square bond angles, ° | 1.491 |
Values in parentheses correspond to the highest-resolution shell.
TSH receptor A-subunit protein (TSHR-289)
Aliquots (1–2 μg) of affinity-purified active or inactive recombinant TSHR-289 protein (19) were subjected to 12% polyacrylamide gel electrophoresis before or after reduction (100mM dithiothreitol for 45 minutes at 50°C) followed by Coomassie blue staining.
Modeling of TSHR-289
In comparison with the solved structure of TSHR-260 whose crystal structure has been reported (15, 16), TSHR-289 contains an additional 29 amino acid residues at its C terminus. Therefore, we chose not to use the TSHR-260 crystal structures to perform in silico analysis of potential TSHR-289 multimers. Instead, the additional residues of TSHR-289 were modeled using the Bioploymers module in Sybylx2.0 (Tripos, Centara) with the crystal structure of FSH receptor (FSHR) (PDB code: 4AY9) (30) used as a template. To obtain a stereochemically and energetically favorable model, the initial chimeric model was subjected to molecular simulation. Molecular dynamics were performed using Desmond (Desmond Molecular Dynamics System, version 2.4; D. E. Shaw Research). The model was simulated for 12 nanoseconds using the default parameters incorporated in Desmond, and structure analyses were performed using Pymol (The PyMOL Molecular Graphics System, version 1.5.0.4; Schrodinger, LLC) and CCP4mg (31). The quality of the model was assessed by analysis of the Ramachandran plot (32).
Docking of 3BD10 to TSHR-289
A portion of the 3BD10 epitope was known to be located within TSHR N-terminal residues 22 to 51 (18). Amino acid substitutions in synthetic peptides in this location further localized the 3BD10 epitope to TSHR residues Glu35, Asp36, and Phe37 (20). Nevertheless, this information was insufficient to determine the full 3BD10 epitope. In silico docking of 3BD10 to TSHR-289 was therefore performed using M-Dock (University of Massachusetts), guided by this previous experimental evidence. Contact residues between 3BD10 and TSHR-289 were calculated using CONTACT (24).
Trimeric and dimeric structures of TSHR-289 were generated using M-ZDock (33). All calculations were performed with sugars (glycosylations) identified from the crystal structures, and glycosylation played no role in either dimer or trimer formation. The best models were further subjected to rigid-body refinement using CNS (34, 35) to remove steric clashes between the protomers. The interaction energies between the 3BD10 Fab/TSHR-289 complex and the TSHR-289 protomers were calculated using FoldX (36, 37). The buried surface areas of the trimer and dimer were calculated using NACCES (Hubbard, S. J., and J. M. Thornton, 1993 NACCES computer program, Department of Biochemistry and Molecular Biology, University College London).
Results
3BD10 crystal structure
The overall structure of the 3BD10 Fab is shown in Figure 3A. Crystallographic studies revealed 2 Fab molecules per asymmetric unit related by local 2-fold noncrystallographic symmetry. The elbow angles are 131.50 and 125.50, as calculated by the method of Stanfield et al (38). For both L-chains, contiguous stretches of amino acids from Glu1 to Arg211 could be modeled in to electron density. Residues Ala129 to Asn133 of the H-chain range are missing. Complementarity determining regions (CDRs) 1 and 2 from the H- and L-chains, as well as the L-chain CDR3, adopt a canonical conformation. However, the 3BD10 H-chain CDR3 adopts a unique β-hairpin conformation (Figure 3B), being unusually short (39) and composed of only 3 amino acids (SGY) by the Kabat convention. An H-chain CDR3 loop with fewer than 4 residues is a rare occurrence.
Figure 3.
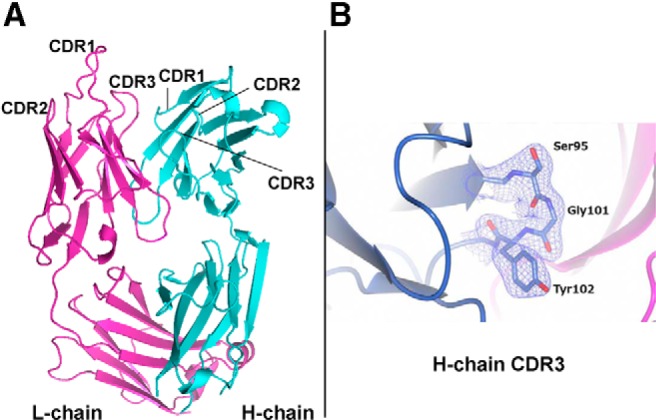
A, 3BD10 Fab 3-dimensional structure. The CDR in the H-chain (cyan) and L-chain (magenta) are indicated. B, The quality of the structure is illustrated by electron density (2fo-fc map contoured at the 1σ level) around the 3BD10 H-chain CDR3 (SGY).
Docking of 3BD10 to TSHR LRD 22–289
Two crystal structures for TSHR LRD residues 22–260 are available, one in complex with TSAb M22 (PDB ID:3G04) (15) and the other TSH blocking antibody K1–70 (PDB ID: 2XWT) (16). The LRD structures are identical except for the visualization of the extreme N terminus of cysteine-bonded loop 1 (residues 22–30) in 2XWT. Because M22 docks equally well with the 2XWT TSHR structure, we used 2XWT for in silico docking of mAb 3BD10. Because our active and inactive recombinant TSHR proteins contained amino acid residues 22 to 289, we extended the C terminus of the 2XWT structure by modeling TSHR residues 261 to 289 based on the known structure of the closely homologous FSHR (30). Docking was guided by previous experimental evidence for the location of a few amino acids of the 3BD10 epitope (see Materials and Methods).
The 3BD10 Fab docks to the N terminus of TSHR-289, on the dorsolateral convex surface of the LRD, on the side opposite to the disulfide-bonded loop 1 projection (Figure 4, A and B). As expected, because of the N-terminal location of its epitope, mAb 3BD10 docked to the same location on TSHR-260 as on TSHR-289 (data not shown). Of importance, there is no overlap between the 3BD10 epitope and that for M22, the latter binding to the ventral, concave β-sheet surface of the LRD (Figure 4C). The contact residues in the 3BD10-TSHR-289 binding surface are rich in hydrogen bonds, with no salt bridges (Table 2). The residues in the 3BD10 surface making contact with TSHR-289 are depicted in Figure 4D. The calculated energy of 3BD10 binding to TSHR-289 is −0.51 kcal/mol.
Figure 4.

A, Docking of the 3-dimensional crystal structures of the 3BD10 Fab and TSHR-289. The latter represented TSHR amino acid residues 22–260 (2XWT) (16) extended to TSHR residue 289 by modeling based on the homologous region of the FSHR (30). Cyan, 3BD10 H-chain; magenta, 3BD10 L-chain; green, TSHR-289. B, Rotation of molecules by 90°. C, Human monoclonal TSAb Fab M22 docked to TSHR-289 indicates no overlap with the 3BD10 epitope. Docking by Z-dock used the 3-dimensional structure of M22 (3GO4) (15) and TSHR-289. Docking was guided by including all the contact residues between M22 and TSHR-260 (15). D, 3BD10 residues at the binding surface to TSHR-289.
Table 2.
Amino Acid Contact Residues for 3BD10 Docked to TSHR-289
| TSHR-289 | 3BD10 |
|---|---|
| D36 | T28Ha |
| F37 | Y27H, T28H, Y32H |
| S49 | S56L, G57L |
| L50 | S56La |
| P52 | Y102H, D55L, R46L |
| S53 | R94H, V2H |
| Q55 | Y32H |
| H70 | S52La |
| S73 | V54La, Y49La |
| N74 | V54La, S56La, Y49L, D55La |
| L75 | Y49La |
| P76 | Y49La, R46L |
| N77 | Y32Ha, S95Ha |
| Y98 | K53La |
| N99 | K53La |
| K102 | L50L |
Hydrogen bond contacts.
Active vs inactive TSHR-289
The TSHR-289 structure used for 3BD10 docking includes the entire TSAb M22 epitope contained within the TSHR-260 crystal structure and therefore represents the active form of our recombinant eukaryotic TSHR-289 protein. Experimentally, 3BD10 binds exclusively to a recombinant TSHR-289 form that is not recognized by M22 and is therefore termed inactive. However, in silico, 3BD10 docks readily with the same monomer as M22. Therefore, the TSHR LRD monomer in the crystal structure cannot represent both the active and inactive forms that we observed experimentally. This observation suggested that a difference in TSHR-LRD quaternary structure (multimerization) could explain the active vs inactive dichotomy. Support for this possibility was the presence of higher-order forms of affinity-purified TSHR-289 to a greater extent in the inactive than in the active form (Figure 5). However, it was not possible from these data to determine the number of protomers present within each form. For example, dimers could themselves form tetramers. After reduction, the higher-order forms reverted primarily to the size expected of their monomers. From their crystal structures, the isolated FSHR LRD exists as a dimer (40) and the FSHR ECD (LRD plus hinge) exists as a trimer (30). Although the full TSHR ECD tethered to the plasma membrane by a glycosylphosphatidyl inositol anchor does form multimers (41), whether the isolated TSHR A-subunit (essentially TSHR-289) exists as a multimer is unknown.
Figure 5.
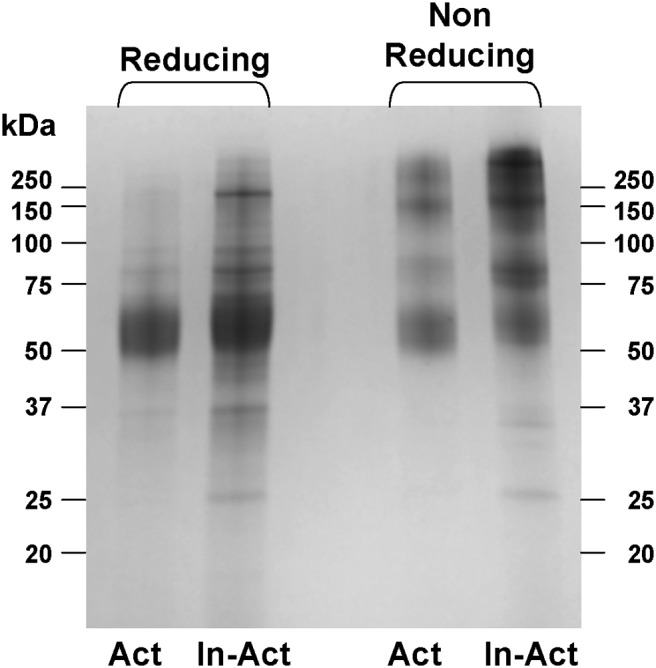
SDS-PAGE of the active and inactive forms of TSHR-289. Affinity-purified forms of TSHR-289 (19) were subjected to 12% SDS-PAGE before or after reduction (100mM dithiothreitol for 45 minutes at 50°C) followed by Coomassie blue staining. The broad bands reflect the very large degree of glycosylation. Without reduction, higher-order forms of TSHR-289 are evident, to a greater extent in inactive (In-Act) than in active (Act) molecules.
In silico analysis of TSHR-289 multimerization
To explore the potential role of TSHR-289 multimerization as an explanation for the active/inactive dichotomy, we examined the cluster of the top 5 trimer and dimer predictions generated by M-Dock for their consistency with the experimental data. For the TSHR-289 trimers, in the first prediction, both TSAb M22 and 3BD10 binding sites are exposed (as for the monomer), which therefore does not conform to the active/inactive distinction observed experimentally. In the second and third predicted trimers, neither the M22 nor 3BD10 binding sites are accessible. The fourth trimer is fully consistent with the experimental data (19); the TSAb M22 active binding site is accessible, whereas that for 3BD10 binding (see Figure 4) is obstructed (Figure 6). Turning to the cluster of TSHR-289 dimers, the first prediction is consistent with the data observed experimentally (19); namely, the 3BD10 binding site is accessible whereas that for M22 is obstructed (Figure 7). A summary of these results is provided in Table 3.
Figure 6.
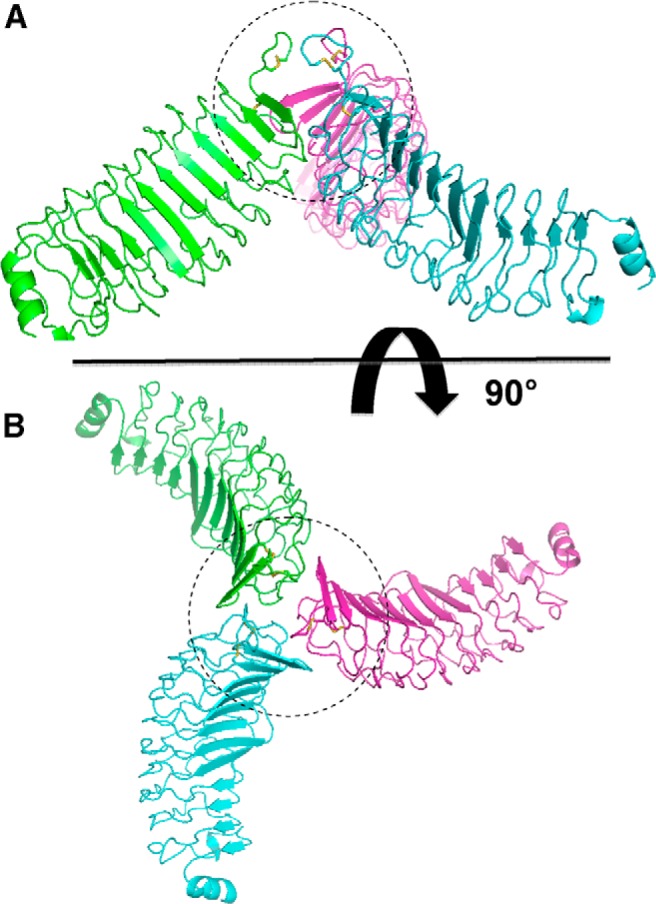
Predicted trimerization of TSHR-289 in which the M22, but not the 3BD10, binding site is accessible. This favored multimer is consistent with the experimental data for the active form of TSHR-289. The region of contact is largely at the N termini of the molecules, depicted schematically by the dashed circle. A, Lateral view. B, View from above.
Figure 7.
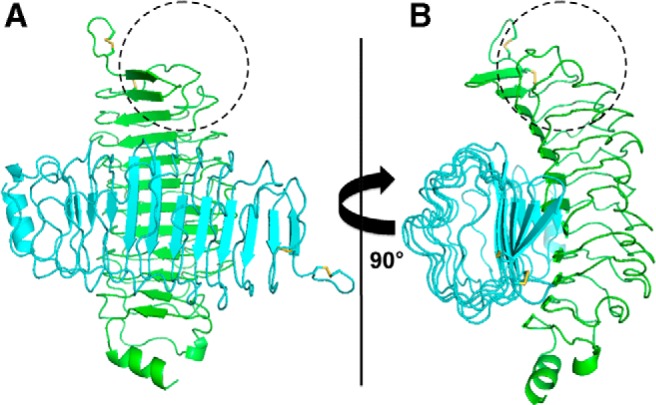
A, Predicted dimerization of TSHR-289 in which the 3BD10 epitope (depicted schematically by the dashed circle) is accessible. However, the M22 binding site is sterically hindered. This favored multimer is consistent with the experimental data for the inactive form of TSHR-289. The region of contact is largely at midzone of the concave surface of TSHR-289. B, Rotation of molecules by 90°.
Table 3.
Summary of Experimental and In Silico Dataa
| Antibody |
Antibody Properties Determined Experimentally |
Antibody Binding Site Is Accessible |
||||
|---|---|---|---|---|---|---|
| Name | Structure | Active Form of TSHR-289 | Inactive Form of TSHR-289 | TSHR-289 Monomer (Crystalb) | TSHR-289 Dimer (In Silico) | TSHR-289 Trimer (In Silico) |
| 3BD10 | Crystal | No | Yes | Yes | Yes | No |
| TSAb (M22) | Crystal | Yes | No | Yes | No | Yes |
The TSHR antigen in this study (TSHR-289, essentially the A-subunit) comprises amino acid residues 22–289 after signal peptide removal. The atomic structure of residues 22–260 has been solved (PDB IDs 3GO4 and 2XWT). To complete the structure of TSHR-289, residues 261–289 were determined by in silico modeling based on the crystal structure of the closely related FSHR ECD (PDB ID 4AY9) (see Materials and Methods).
The full epitopes for 3BD10 and M22 are present in the TSHR component (residues 22–260) whose crystal structure has been solved.
Additional important features consistent with previous experimental observations regarding the temperature-dependent balance between active and inactive TSHR-289 and the greater stability of the latter (discussed below) are 1) the TSHR-289 dimer interaction is of lower energy (ΔG −3.58 kcal/mol) than the trimer (ΔG −0.02 kcal/mol) and 2) the buried surface area of the dimer (2007 Å2) is greater than that of the trimer (477 Å2). A larger buried surface area is more stable than a small surface area (42, 43). These observations are consistent with a relationship between the TSHR-289 active/inactive dichotomy and the degree of TSHR-289 multimerization.
Discussion
Of the closely related GPHRs, only the TSHR undergoes intramolecular cleavage into subunits, followed by shedding of the N-terminal A-subunit (essentially TSHR-289), and autoantibodies only arise to the TSHR (6, 12). There is no equivalent Graves' disease of the gonads. Understanding the structural difference between the 2 forms of TSHR A-subunits recognized by TSAb (active) and mouse mAb 3BD10 (inactive) could provide insight into the pathogenesis of Graves' disease. Because epitopic components of both TSAb M22 and 3BD10 map to the TSHR N terminus (15, 44), we previously considered that a possible explanation for TSHR A-subunit active/inactive structural heterogeneity related to alternate conformations in this region. However, localization of the inactive 3BD10 epitope on the active form of TSHR-289 monomer eliminates this hypothesis. By exclusion, therefore, this structural difference can only be explained by variability in its quaternary structure.
In considering this issue in relation to previously published data on the GPHRs, it is important to distinguish between structural studies on the holoreceptors, their ECDs, or the LRD components of the ECD. First, although there is strong evidence that the TSH holoreceptor expressed on the cell surface multimerizes (45, 46), like other GPHRs (46, 47), none of these holoreceptors has, as yet, been crystallized. Second, there are no reports of any GPHR ECD or ECD component being crystallized in the absence of ligand. Thus, the molecular structures of the FSHR LRD (40), the TSHR LRD (residues 22–260) (15, 16) and the full FSHR ECD (30, 48) have all been in complex with ligand. The formation of multimers is described in some of these reports, such as a dimer for the FSH-FSHR LRD complex (40). Very recent crystallographic data on the entire FSHR ECD (LRD plus hinge region) in complex with FSH clearly indicates its trimeric quaternary structure (48). It is therefore likely that GPHR ECDs are stabilized by ligand, a logical explanation for our inability to crystallize TSHR-289 without ligand despite multiple attempts over many years. Unlike for the other GPHRs, TSH does not bind to the TSHR LRD with high enough affinity to be detected experimentally (18), most likely reflecting a greater contribution of the ECD hinge region to ligand binding. Nevertheless, of importance clinically, the autoantigen responsible for Graves' disease, TSHR A-subunits shed from the surface of thyrocytes, does not contain ligand (11, 14) and is comparable to isolated TSHR-289, the subject of the present report.
With this background, in silico evaluation of the TSHR trimer and dimer models is, at present, the only available option. However, it is possible to test these models against directly obtained experimental data. For this purpose, in the present study, we determined the crystal structure of the 3BD10 Fab. The ability of this Fab to dock with an active (crystallized in complex with M22) TSHR LRD monomer, clearly indicates that the latter cannot represent both active and inactive forms of the protein. To study potential multimerization of TSHR-289, it was necessary to augment the C terminus of TSHR-260 (amino acid residues 22–260; PDB ID: 2XWT) (16) with TSHR residues 261 to 289 modeled on the closely homologous FSHR (30). The shed A-subunit (or TSHR-289) trimer (Figure 6) and dimer (Figure 7) predicted by M-Dock are entirely consistent with the experimental data regarding selective TSAb M22 and 3BD10 binding to active and inactive forms of the molecule, respectively (19).
Further experimental evidence supporting the predicted TSHR-289 multimers is that, once purified, the active form of TSHR-289 spontaneously converts to the inactive form, a process hastened by temperature (19). Heating to 37°C for 1 hour completes the conversion from active to inactive forms. Yet approximately equal proportions of active and inactive TSHR-289 are purified from conditioned medium harvested from CHO cell monolayer cultures even after 3 days at 37°C. This information suggested a stabilizing influence of culture medium containing many additives including fetal bovine serum. Consistent with this finding, the addition of a chemical chaperone, trimethylamine N-oxide, to purified active TSHR-289 prevented its conversion to the inactive form despite heating to 37°C for 30 hours (19). Inactive TSHR-289 was not simply a denatured form of the active protein because 3BD10 recognition was lost after TSHR-289 reduction and denaturation (18). Attempts to revert inactive TSHR-289 to the active form by slow, progressing protein refolding from 8M guanidine were unsuccessful. The greater stability of the TSHR LRD dimer vs the trimer is supported by their energetics and the extent of their buried surfaces. Heating will facilitate dissociation of the trimer and formation of a more stable, lower-energy dimer to which only 3BD10 can bind, whereas M22 binding is sterically hindered.
Based on the crystal structure of the FSHR ECD in complex with FSH, an in silico model of an TSHR ECD trimer (48) has been reported that differs markedly from that of our computed ligand-free TSHR A-subunit trimer (Figure 6). However, such a difference is to be expected, for a number of reasons. First, the trimer proposed by Jiang et al (48) comprises the entire TSHR ECD extending to insertion into the plasma membrane (amino acid residue ∼410), with the C termini of the protomers being the major contact sites. In contrast, the TSHR A-subunit trimer lacks the C terminus of the ECD. Second, the binding sites for both M22 and 3BD10 are accessible in the putative TSHR ECD trimer (48), inconsistent with the experimental data of active and inactive A-subunits existing as 2 distinct molecules.
Finally, TSAb causing Graves' hyperthyroidism arise spontaneously only in humans. Remarkably, such antibodies can only be induced in animals when the TSHR is expressed in vivo by injecting TSHR-expressing cells (49), plasmid (50, 51), or viral vectors coding for the TSH holoreceptor cDNA (52, 53) or, even more effectively, the TSHR A-subunit (6, 54). Conventional immunization with the purified TSHR A-subunit protein generates nonstimulating antibodies such as 3BD10 (18, 55, 56). Indeed, injecting mice with purified TSHR A-subunit protein 1 week before adenovirus coding for the TSHR A-subunit diverts the TSHR antibody response away from TSAb toward 3BD10-like nonfunctional TSHR antibodies (57). Of note, we have been unable to detect antibodies that recognize purified inactive TSHR A-subunits in Graves' sera, even those with very high levels of functional TSHR autoantibodies (58), providing further evidence against the A-subunit monomer being the critical immunogen. Therefore, the conformation, or oligomerization, of the TSHR antigen is critical for B cell Ig gene affinity maturation leading to the development of pathogenic TSAb. Purified recombinant TSHR A-subunit protein, especially in the presence of damaging agents such as Freund's adjuvant, is likely to be in the inactive, lower-energy, dimeric form.
In summary, based on the present evidence, we propose that a TSHR A-subunit trimer is the preferential immunogen in Graves' disease. TSHR A-subunits could be shed as trimers from trimeric holoreceptors, or multimerization could occur subsequently, as may occur in lymph nodes draining the thyroid.
Acknowledgments
This work was supported by National Institutes of Health Grant DK19289 and Molecular Therapeutics Core Facility, Cedars-Sinai Research Institute.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CDR
- complementarity determining region
- Fab
- fragment, antigen binding
- FSHR
- FSH receptor
- H-chain
- heavy chain
- L-chain
- light chain
- mAb
- monoclonal antibody
- LRD
- leucine-rich repeat domain
- TSAb
- thyroid-stimulating autoantibody
- TSHR
- TSH receptor
- ECD
- extracellular domain
- TMD
- transmembrane domain.
References
- 1. Boelaert K, Newby PR, Simmonds MJ, et al. Prevalence and relative risk of other autoimmune diseases in subjects with autoimmune thyroid disease. Am J Med. 2010;123:183–189. [DOI] [PubMed] [Google Scholar]
- 2. Adams DD, Purves HD. Abnormal responses in the assay of thyrotropins. Proc Univ Otago Sch Med. 1956;34:11–12. [Google Scholar]
- 3. Kriss JP, Pleshakov V, Chien JR. Isolation and identification of the long-acting thyroid stimulator and its relation to hyperthyroidism and circumscribed pretibial myxedema. J Clin Endocrinol Metab. 1964;24:1005–1028. [DOI] [PubMed] [Google Scholar]
- 4. Meek JC, Jones AE, Lewis UJ, Vanderlaan WP. Characterization of the long-acting thyroid stimulator of Graves' disease. Proc Natl Acad Sci U S A. 1964;52:342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kleinau G, Neumann S, Grüters A, Krude H, Biebermann H. Novel insights on thyroid-stimulating hormone receptor signal transduction. Endocr Rev. 2013;34:691–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen CR, Pichurin P, Nagayama Y, Latrofa F, Rapoport B, McLachlan SM. The thyrotropin receptor autoantigen in Graves disease is the culprit as well as the victim. J Clin Invest. 2003;111:1897–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mizutori Y, Chen CR, Latrofa F, McLachlan SM, Rapoport B. Evidence that shed TSH receptor A-subunits drive affinity maturation of autoantibodies causing Graves' disease. J Clin Endocrinol Metab. 2009;94:927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parmentier M, Libert F, Maenhaut C, et al. Molecular cloning of the thyrotropin receptor. Science. 1989;246:1620–1622. [DOI] [PubMed] [Google Scholar]
- 9. Nagayama Y, Kaufman KD, Seto P, Rapoport B. Molecular cloning, sequence and functional expression of the cDNA for the human thyrotropin receptor. Biochem Biophys Res Comm. 1989;165:1184–1190. [DOI] [PubMed] [Google Scholar]
- 10. Chazenbalk GD, Tanaka K, Nagayama Y, et al. Evidence that the thyrotropin receptor ectodomain contains not one, but two, cleavage sites. Endocrinology. 1997;138:2893–2899. [DOI] [PubMed] [Google Scholar]
- 11. de Bernard S, Misrahi M, Huet JC, et al. Sequential cleavage and excision of a segment of the thyrotropin receptor ectodomain. J Biol Chem. 1999;274:101–107. [DOI] [PubMed] [Google Scholar]
- 12. Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The thyrotropin (TSH) receptor: interaction with thyrotropin and autoantibodies. Endocr Rev. 1998;19:673–716. [DOI] [PubMed] [Google Scholar]
- 13. Couët J, de Bernard S, Loosfelt H, Saunier B, Milgrom E, Misrahi M. Cell surface protein disulfide-isomerase is involved in the shedding of human thyrotropin receptor ectodomain. Biochemistry. 1996;35:14800–14805. [DOI] [PubMed] [Google Scholar]
- 14. Tanaka K, Chazenbalk GD, McLachlan SM, Rapoport B. The shed thyrotropin receptor is primarily a carboxyl terminal truncated form of the A subunit, not the entire A subunit. Mol Cell Endocrinol. 1999;150:113–119. [DOI] [PubMed] [Google Scholar]
- 15. Sanders J, Chirgadze DY, Sanders P, et al. Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid. 2007;17:395–410. [DOI] [PubMed] [Google Scholar]
- 16. Sanders P, Young S, Sanders J, et al. Crystal structure of the TSH receptor (TSHR) bound to a blocking-type TSHR autoantibody. J Mol Endocrinol. 2011;46:81–99. [DOI] [PubMed] [Google Scholar]
- 17. Chazenbalk GD, Jaume JC, McLachlan SM, Rapoport B. Engineering the human thyrotropin receptor ectodomain from a non-secreted form to a secreted, highly immunoreactive glycoprotein that neutralizes autoantibodies in Graves' patients' sera. J Biol Chem. 1997;272:18959–18965. [DOI] [PubMed] [Google Scholar]
- 18. Chazenbalk GD, Wang Y, Guo J, et al. A mouse monoclonal antibody to a thyrotropin receptor ectodomain variant provides insight into the exquisite antigenic conformational requirement, epitopes and in vivo concentration of human autoantibodies. J Clin Endocrinol Metab. 1999;84:702–710. [DOI] [PubMed] [Google Scholar]
- 19. Chazenbalk GD, McLachlan SM, Pichurin P, Yan XM, Rapoport B. A prion-like shift between two conformational forms of a recombinant thyrotropin receptor A-subunit module: purification and stabilization using chemical chaperones of the form reactive with Graves' autoantibodies. J Clin Endocrinol Metab. 2001;86:1287–1293. [DOI] [PubMed] [Google Scholar]
- 20. Hamidi S, Chen CR, Murali R, McLachlan SM, Rapoport B. Probing structural variability at the N terminus of the TSH receptor with a murine monoclonal antibody that distinguishes between two receptor conformational forms. Endocrinology. 2013;154:562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chazenbalk GD, Pichurin P, Chen CR, et al. Thyroid-stimulating autoantibodies in Graves disease preferentially recognize the free A subunit, not the thyrotropin holoreceptor. J Clin Invest. 2002;110:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67:282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu X, Cai Y, Wei Y, et al. Identification of a new epitope in uPAR as a target for the cancer therapeutic monoclonal antibody ATN-658, a structural homolog of the uPAR binding integrin CD11b (alphaM). PLoS One. 2014;9:e85349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. [DOI] [PubMed] [Google Scholar]
- 28. Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 29. Chen VB, Arendall WB, 3rd, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang X, Liu H, Chen X, et al. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc Natl Acad Sci U S A. 2012;109:12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McNicholas S, Potterton E, Wilson KS, Noble ME. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67:386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963;7:95–99. [DOI] [PubMed] [Google Scholar]
- 33. Pierce B, Tong W, Weng Z. M-ZDOCK: a grid-based approach for Cn symmetric multimer docking. Bioinformatics. 2005;21:1472–1478. [DOI] [PubMed] [Google Scholar]
- 34. Brünger AT, Adams PD, Clore GM, et al. Crystallography, NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. [DOI] [PubMed] [Google Scholar]
- 35. Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc. 2007;2:2728–2733. [DOI] [PubMed] [Google Scholar]
- 36. Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. [DOI] [PubMed] [Google Scholar]
- 37. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res 2005; 33(Web Server issue):W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stanfield RL, Zemla A, Wilson IA, Rupp B. Antibody elbow angles are influenced by their light chain class. J Mol Biol. 2006;357:1566–1574. [DOI] [PubMed] [Google Scholar]
- 39. Zemlin M, Klinger M, Link J, et al. Expressed murine and human CDR-H3 intervals of equal length exhibit distinct repertoires that differ in their amino acid composition and predicted range of structures. J Mol Biol. 2003;334:733–749. [DOI] [PubMed] [Google Scholar]
- 40. Fan QR, Hendrickson WA. Structure of human follicle-stimulating hormone in complex with its receptor. Nature. 2005;433:269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Latif R, Michalek K, Morshed SA, Davies TF. A tyrosine residue on the TSH receptor stabilizes multimer formation. PLoS One. 2010;5:e9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chothia C, Janin J. Principles of protein-protein recognition. Nature. 1975;256:705–708. [DOI] [PubMed] [Google Scholar]
- 43. Horton N, Lewis M. Calculation of the free energy of association for protein complexes. Protein Sci. 1992;1:169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hamidi S, Chen CR, McLachlan SM, Rapoport B. Insight into thyroid-stimulating autoantibody interaction with the thyrotropin receptor N-terminus based on mutagenesis and re-evaluation of ambiguity in this region of the receptor crystal structure. Thyroid. 2011;21:1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Latif R, Graves P, Davies TF. Oligomerization of the human thyrotropin receptor: fluorescent protein-tagged hTSHR reveals post-translational complexes. J Biol Chem. 2001;276:45217–45224. [DOI] [PubMed] [Google Scholar]
- 46. Urizar E, Montanelli L, Loy T, et al. Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J. 2005;24:1954–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guan R, Feng X, Wu X, et al. Bioluminescence resonance energy transfer studies reveal constitutive dimerization of the human lutropin receptor and a lack of correlation between receptor activation and the propensity for dimerization. J Biol Chem. 2009;284:7483–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiang X, Fischer D, Chen X, et al. Evidence for follicle-stimulating hormone receptor as a functional trimer. J Biol Chem. 2014;289:14273–14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shimojo N, Kohno Y, Yamaguchi K, et al. Induction of Graves-like disease in mice by immunization with fibroblasts transfected with the thyrotropin receptor and a class II molecule. Proc Natl Acad Sci U S A. 1996;93:11074–11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Costagliola S, Many MC, Denef JF, Pohlenz J, Refetoff S, Vassart G. Genetic immunization of outbred mice with thyrotropin receptor cDNA provides a model of Graves' disease. J Clin Invest. 2000;105:803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaneda T, Honda A, Hakozaki A, Fuse T, Muto A, Yoshida T. An improved Graves' disease model established by using in vivo electroporation exhibited long-term immunity to hyperthyroidism in BALB/c mice. Endocrinology. 2007;148:2335–2344. [DOI] [PubMed] [Google Scholar]
- 52. Nagayama Y, Kita-Furuyama M, Ando T, et al. A novel murine model of Graves' hyperthyroidism with intramuscular injection of adenovirus expressing the thyrotropin receptor. J Immunol. 2002;168:2789–2794. [DOI] [PubMed] [Google Scholar]
- 53. Ando T, Imaizumi M, Graves P, Unger P, Davies TF. Induction of thyroid-stimulating hormone receptor autoimmunity in hamsters. Endocrinology. 2003;144:671–680. [DOI] [PubMed] [Google Scholar]
- 54. Gilbert JA, Gianoukakis AG, Salehi S, et al. Monoclonal pathogenic antibodies to the thyroid-stimulating hormone receptor in Graves' disease with potent thyroid stimulating activity but differential blocking activity activate multiple signaling pathways. J Immunol. 2006;176:5084–5092. [DOI] [PubMed] [Google Scholar]
- 55. Schwarz-Lauer L, Pichurin PN, Chen CR, et al. The cysteine-rich amino terminus of the thyrotropin receptor is the immunodominant linear antibody epitope in mice immunized using naked deoxyribonucleic acid or adenovirus vectors. Endocrinology. 2003;144:1718–1725. [DOI] [PubMed] [Google Scholar]
- 56. McLachlan SM, Nagayama Y, Rapoport B. Insight into Graves' hyperthyroidism from animal models. Endocr Rev. 2005;26:800–832. [DOI] [PubMed] [Google Scholar]
- 57. Misharin AV, Nagayama Y, Aliesky HA, Mizutori Y, Rapoport B, McLachlan SM. Attenuation of induced hyperthyroidism in mice by pretreatment with thyrotropin receptor protein: deviation of thyroid-stimulating antibody to nonfunctional antibodies. Endocrinology. 2009;150:3944–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chazenbalk GD, Pichurin P, McLachlan SM, Rapoport B. A direct binding assay for thyrotropin receptor autoantibodies. Thyroid. 1999;9:1057–1061. [DOI] [PubMed] [Google Scholar]