

Significance
Intracellular Ca2+ concentration is a chemical message upstream of critical physiological processes: synaptic and endocrine signaling, muscle contraction, etc. In excitable cells, calcium influx is controlled by voltage-gated calcium (CaV) channels. The CaV protein possesses four homologous but nonidentical voltage-sensing domains (VSDs), each potentially acting as a gatekeeper for the calcium signal. However, the contribution of each VSD to calcium entry is unclear. By optically tracking the voltage-dependent movements of each human CaV1.2 VSD, we revealed the conformational rearrangements that precede and govern excitation-evoked calcium signaling: VSDs II and III primarily control CaV1.2 conduction. Global structure-based kinetic modeling also revealed the participation of VSD I, implicating principles of protein allostery in the CaV VSD–pore coupling mechanism.
Keywords: fluorometry, Ca2+ entry, allostery, gating mechanism, CaV1.2
Abstract
Excitation-evoked Ca2+ influx is the fastest and most ubiquitous chemical trigger for cellular processes, including neurotransmitter release, muscle contraction, and gene expression. The voltage dependence and timing of Ca2+ entry are thought to be functions of voltage-gated calcium (CaV) channels composed of a central pore regulated by four nonidentical voltage-sensing domains (VSDs I–IV). Currently, the individual voltage dependence and the contribution to pore opening of each VSD remain largely unknown. Using an optical approach (voltage-clamp fluorometry) to track the movement of the individual voltage sensors, we discovered that the four VSDs of CaV1.2 channels undergo voltage-evoked conformational rearrangements, each exhibiting distinct voltage- and time-dependent properties over a wide range of potentials and kinetics. The voltage dependence and fast kinetic components in the activation of VSDs II and III were compatible with the ionic current properties, suggesting that these voltage sensors are involved in CaV1.2 activation. This view is supported by an obligatory model, in which activation of VSDs II and III is necessary to open the pore. When these data were interpreted in view of an allosteric model, where pore opening is intrinsically independent but biased by VSD activation, VSDs II and III were each found to supply ∼50 meV (∼2 kT), amounting to ∼85% of the total energy, toward stabilizing the open state, with a smaller contribution from VSD I (∼16 meV). VSD IV did not appear to participate in channel opening.
Voltage-gated Ca2+ (CaV) channels respond to membrane depolarization by catalyzing Ca2+ influx. CaV-mediated elevation of intracellular [Ca2+] regulates such critical physiological functions as neurotransmitter and hormone release, axonal outgrowth, muscle contraction, and gene expression (1). Their relevance to human physiology is evident from the broad phenotypic consequences of CaV channelopathies (2). The voltage dependence of CaV-driven Ca2+ entry relies on the modular organization of the channel-forming α1 subunit (Fig. 1), which consists of four repeated motifs (I–IV), each comprising six membrane-spanning helical segments (S1–S6) (Fig. 1A). Segments S1–S4 form a voltage-sensing domain (VSD), whereas segments S5 and S6 contribute to the Ca2+-conductive pore (1). The VSDs surround the central pore (Fig. 1B). VSDs are structurally and functionally conserved modules (3–5) capable of transducing a change in the cell membrane electrical potential into a change of ion-specific permeability or enzyme activity. VSDs sense depolarization by virtue of a signature motif of positively charged Arg or Lys at every third position of helix S4 (Fig. 1D), which rearranges in response to depolarization (4, 6–10). In contrast to voltage-gated K+ (KV) channels but similar to pseudotetrameric voltage-gated Na+ (NaV) channels, the amino acid sequences encoding each VSD have evolved independently (Fig. 1D). In addition to their distinct primary structure, the four CaV VSDs may also gain distinct functional properties from the asymmetrical association of auxiliary subunits, such as β, α2δ, and calmodulin (1, 11–16) (Fig. 1C). The structural divergence among VSD-driven channels was foreseen by the classical Hodgkin–Huxley model (17), in which four independent “gating particles” control the opening of homotetrameric KV channels and only three seem sufficient to open NaV channels. An early study by Kostyuk et al. (18) suggested that only two gating particles are coupled to CaV channel opening. We recognize today that gating particles correspond to VSDs, and in NaV channels, VSDs I–III control Na+ influx, whereas VSD IV is associated with fast inactivation (19–21).
Fig. 1.

CaV membrane topology, putative structure, and S4 helix homology. (A) CaV channel-forming α1 subunits consist of four concatenated repeats, each encompassing one voltage sensor domain (VSD) and a quarter of the central pore domain (PD) (1). Stars indicate the positions of fluorophore labeling. (B) The atomic structure of an NaV channel (Protein Data Bank ID code 4EKW; top view) (56) shown as a structural representation for the CaV α1 subunit. (C) The α1 subunit asymmetrically associates with auxiliary β, α2δ, and calmodulin (CaM) subunits (11–16). (D) Sequence alignment of VSD helix S4 from each of four CaV1.2 repeats and the archetypal homotetrameric Shaker K+ channel. Conserved, positively charged Arg or Lys is in blue. Residues substituted by Cys for fluorescent labeling are marked: F231 (VSD I), L614 (VSD II), V994 (VSD II), and S1324 (VSD IV).
In this study, we used fluorometry to probe the properties of four individual VSDs in a human L-type calcium channel CaV1.2, which is a widely expressed regulator of physiological processes, such as cardiac and smooth muscle contractility (22). Although the collective transition of the CaV VSDs and the pore has been investigated in studies measuring total charge displacement (gating currents) (23, 24), the activation properties and functional roles of each VSD are unknown. Evidence for the role of each VSD in L-type CaV channel operation has been presented from charge neutralization studies, but a clear picture has yet to emerge. Work on a chimeric L-type channel suggests that VSDs I and III drive channel opening (25), whereas other studies on CaV1.2 favored the involvement of VSD II over VSD I, with the roles of VSDs III and IV remaining unclear (26, 27).
The individual optical reports of four CaV1.2 VSDs revealed that each operates with distinct biophysical parameters. We found that VSDs II and III exhibit voltage- and time-dependent characteristics compatible with channel opening and that they can be considered rate-limiting for activation. We compared the voltage and time dependence of the fluorescent signals and ionic currents with the predictions of thermodynamic models relevant to CaV domain organization. We found that CaV1.2 activation is compatible with a model of allosteric VSD–pore coupling, where VSDs II and III are the primary drivers of channel opening with a smaller contribution by VSD I. We discuss the mechanism of CaV1.2 voltage sensitivity, which exhibits similarities to but also clear differences from the related pseudotetrameric NaV1.4 channels.
Results
Optical Tracking of Individual CaV1.2 VSDs.
To optically track the conformational transitions of each VSD in human CaV1.2 channels, we used voltage-clamp fluorometry (VCF), a hybrid electrophysiological–optical approach (28–34). A Cys was introduced at the extracellular flank of S4 helices in each VSD of the CaV1.2 pore-forming subunit (α1C) and labeled with a fluorophore (Fig. 1D), which served as an optical reporter of local VSD conformational rearrangements. The voltage clamp was implemented by a cut-open oocyte Vaseline gap to provide the necessary clamp speed (35, 36). The fluorescently labeled positions included in this work resulted in well-resolved depolarization-evoked fluorescence deflections and minimally perturbed channel function.
Membrane current and fluorescence emission from Xenopus oocytes expressing human CaV1.2 channels labeled at each of four VSDs (Fig. 2A) were simultaneously acquired. Fluorescence emitted from the label attached to the VSD I S4 (Fig. 2A, blue) decreased on membrane depolarization, suggesting that this segment undergoes a voltage-dependent conformational change that results in quenching of the attached fluorophore. The emission of a fluorophore conjugated to VSD II S4 increased on depolarization (Fig. 2A, red): that is, the fluorophore is more efficiently quenched in the resting state of this domain than in the active state. Fluorescence changes were reported from VSD III (Fig. 2A, green) on depolarization to −40 mV, suggesting that this sensor exhibits distinct voltage dependence from VSD I. When the S4 of VSD IV was labeled, the fluorescence changes observed were distinctly slower than in the other VSDs (Fig. 2A, orange). The voltage-dependent fluorescence emissions reported from each of four CaV1.2 VSDs had distinct signs, voltage dependences, and kinetics, confirming that they report on the short-range transitions of their labeled domains. No voltage-evoked F was detected in oocytes expressing WT CaV1.2 channels subjected to the fluorophore-labeling protocol (Fig. 2B), confirming that label conjugation of native Cys, if present, did not contaminate voltage-dependent F in channels with introduced Cys.
Fig. 2.
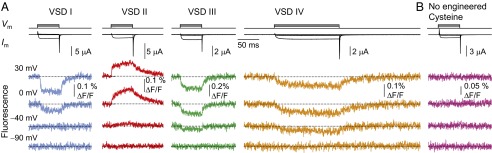
Optical tracking of the conformational rearrangements of each human CaV1.2 VSD. (A) Membrane current (Im) from oocytes expressing CaV1.2 (α1C/α2δ/β3) channels in extracellular 2 mM Ba2+. The traces acquired at pulses from −90 to −90, −40, 0, and 30 mV are shown superimposed above the simultaneously acquired fluorescence traces. The channels were labeled at helix S4 of VSDs I, III, IV (2-((5(6)-Tetramethyl-rhodamine)carboxylamino)ethyl methanethiosulfonate, MTS-TAMRA), or II (tetramethylrhodamine-5'-maleimide, TMRM). Increasing the test potential resulted in local VSD conformational rearrangements resolved as fluorescence deflections (F). (B) No voltage-dependent F is observed in labeled oocytes expressing WT CaV1.2 without engineered Cys.
Each CaV1.2 VSD Activates with Distinct Voltage Dependence.
The activation curves [F(V)] for individual CaV1.2 VSDs were obtained by measuring the F amplitude at the end of pulses at a range of potentials and normalizing as described in Materials and Methods. The F(V) curves exhibited characteristic sigmoid shapes that could be described by Boltzmann distributions (Fig. 3A and SI Appendix, Table S1). The membrane potentials at which the VSDs reached 50% probability of activation (V0.5) ranged from near −50 (VSD IV) to +5 mV (VSD I). All F(V) curves are left-shifted with respect to the conductance vs. voltage [G(V)] curve, with the exception of VSD I, which overlaps with the G(V) at negative potentials (Fig. 3A). The charge movement [Q(V)] is near the middle of four F(V) curves.
Fig. 3.

Each VSD exhibits distinct voltage dependence and kinetics. (A) Mean normalized conductance (G; black ▼) and charge movement (Q; white right-pointing triangle) from WT channels and F from VSDs I (blue ●), II (red ▲), III (green ◆), and IV (yellow ■). The curves are fits to single or (for G) the sum of two Boltzmann distributions (parameters in SI Appendix, Tables S1 and S3). Error bars indicate ± SEM. (B) Representative membrane current (gray) from WT channels for a −90- → 20-mV pulse, with superimposed F reported from VSD I (τ1 = 2.6 ms, 59%; τ2 = 8.1 ms), II (τ1 = 1.1 ms, 98%; τ2 = 20 ms), III (τ1 = 0.88 ms, 68%; τ2 = 9.2 ms), and IV (τ = 17 ms). The black dashed lines are exponential functions with the reported time constants. Mean kinetic parameters are in SI Appendix, Table S2. The sequence of activation for the CaV1.2 VSDs (half-time to maximum, t0.5) is VSD II (1.0 ms), III (1.4 ms), I (2.9 ms), and IV (11 ms). Ionic currents from Cys mutant channels superimposed to their respective fluorescence traces are in presented in SI Appendix, Fig. S2B.
The F(V) steepness, which was determined by the Boltzmann valence (z), suggests differences in the sensitivity of each VSD to changes in the membrane potential ranging from 1.2 (VSD IV) to 3.2 e0 (VSD II). The sum of all F(V) curves weighed by their respective valence reasonably approximates the gating charge voltage dependence (SI Appendix, Fig. S1). This analysis represents a rough estimate of the true VSD charge displacements, because Boltzmann analysis does not typically provide the correct valence in a multistate channel (37).
CaV1.2 Voltage Sensors Activate and Deactivate with Different Kinetics.
We characterized the time course of VSD transitions by fitting F to single or the sum of two exponential function(s) (SI Appendix, Table S2). To relate the kinetics of VSD activation to the timing of pore opening, we superimposed the ionic current from WT channels with representative F traces during a step to 20 mV (Fig. 3B). VSDs II and III are the fastest to activate and clearly precede the ionic current. VSD I is slightly slower and achieves steady state after inward current has reached its plateau. By contrast, VSD IV is an order of magnitude slower than current activation. On repolarization, VSD II exhibits the fastest deactivation component, whereas VSDs I, III, and IV deactivate at rates slower than channel closure. The fast activations of VSDs I–III are compatible with a predominant role for these VSDs in opening the CaV1.2 pore, whereas VSD IV and the lack of monoexponentiality (existence of slower kinetic components) in the other VSDs suggest that the VSDs may also be involved in slower processes, such as voltage-dependent inactivation. Alternatively, the slower fluorescence components may represent secondary transitions of the fluorescent labels.
Cysteine Substitution and Fluorescent Labeling Minimally Perturb CaV1.2 Operation.
The chosen positions for Cys substitution and labeling had minimal effect on channel voltage dependence, which is shown in SI Appendix, Fig. S2. Specifically, Cys introduction and labeling did not substantially affect the kinetics of ionic current (SI Appendix, Fig. S2A) or gating currents (SI Appendix, Fig. S2C), because representative traces of channels labeled in VSDs I (SI Appendix, Fig. S2, blue), II (SI Appendix, Fig. S2, red), and III (SI Appendix, Fig. S2, green) are shown superimposed with traces from WT channels (SI Appendix, Fig. S2, black). Mutation S1324C measurably slowed down current onset (τON = 2.7 ± 0.23 ms for −90- → 20-mV pulses; compare with WT τON = 1.0 ± 0.17 ms) (SI Appendix, Fig. S2A, orange); however, the fluorescence signal from this VSD is distinctly slower (τON = 21 ± 5.3 ms) than the current in both WT (Fig. 3B) and labeled S1324C channels (SI Appendix, Fig. S2B). Therefore, the premise that VSD IV activation is too slow to contribute significantly to current activation still holds for the S1324C mutant. The potential of 50% total charge displacement was largely unaffected by Cys introduction and labeling, whereas the macroscopic conductance curve deviated from the WT by up to 15 mV (VSD II) (SI Appendix, Fig. S2 D and E and Table S3). This analysis shows that the highly distinct voltage dependence and kinetics of the fluorescence reported from the different VSDs were caused by the conformational changes of the protein rather than effects of Cys introduction and/or fluorescent labeling.
Activation of VSDs II and III Is Rate-Limiting for CaV1.2 Channel Opening in the Context of an Obligatory Model.
Because the onset of ionic current precedes VSD IV activation (Fig. 3B), the latter is unlikely to be rate-limiting for channel opening, which indicates that CaV1.2 channels do not operate similar to homotetrameric KV channels, which require the activation of four VSDs before channel opening (38–40). To test this hypothesis, we constructed a KV-like model of CaV1.2 activation (Fig. 4, scheme I) characterized by the obligatory gating of all four VSDs. The voltage dependence and kinetics of CaV1.2 current and VSD activations were fit simultaneously, which is shown in SI Appendix, Fig. S3. The time course of ionic current is poorly predicted by this model, because it is limited by the activation and deactivation rates of VSD IV.
Fig. 4.
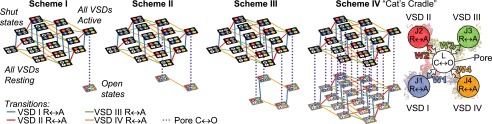
CaV structure-based kinetic models to account for optical and electrophysiological data. All schemes assume that each VSD activates independently, resulting in 16 shut states (top tiers). Resting VSDs are in white. Active VSD I is in blue, active VSD II is in red, active VSD III is in green, and active VSD IV is in orange. In scheme I, all VSDs are required to activate before channel opening. SI Appendix, Fig. S3 shows data fitting and model parameters. In scheme II, the activations of VSDs I–III are obligatory for pore opening (SI Appendix, Fig. S4). In scheme III, the activations of VSDs II and III are obligatory for pore opening (SI Appendix, Fig. S5). Scheme IV is an allosteric gating mechanism where VSD activation is not obligatory for pore opening, which in principle, allows opening transitions from any shut state. The activation of each VSD contributes energy W toward stabilizing the open state. A simplified version of scheme IV is shown to the right of scheme IV (Fig. 5 and SI Appendix, Fig. S6).
In the closely related NaV channel, which also possess a pseudotetrameric architecture (41), the activations of VSDs I–III are thought to obligatorily precede channel opening, whereas VSD IV facilitates fast inactivation (19–21, 42, 43). We tested scheme II (Fig. 4), in which open channel states are accessible only when VSDs I–III are active. Although scheme II accounted for the experimental data better than scheme I (SI Appendix, Figs. S4 and S3, respectively), it could not accurately reconcile the voltage- and time-dependent properties of VSD movement and CaV current. By contrast, scheme III (Fig. 4), in which the activations of VSDs II and III are obligatory for channel opening, fit the data very well (SI Appendix, Fig. S5). This result highlights the involvement of the activation of VSDs II and III in CaV1.2 channel opening.
CaV1.2 Activation Properties Can Be Accounted for by an Allosteric Voltage-Dependent Activation Mechanism.
An alternative to the obligatory models considered is the notion, inspired by previous work on CaV (27, 44, 45), large-conductance voltage- and Ca2+-gated (BK) channels (34, 46), and KV7.1 channels (47), that pore opening is facilitated by VSD activation through an allosteric gating mechanism: that is, each VSD contributes a weak (i.e., quantifiable) characteristic energy to the stabilization of the open state. An allosteric model of CaV1.2 activation is represented by scheme IV: “the Cat’s Cradle” (Fig. 4). Because the pore can open independently, scheme IV possesses more open states than the previous obligatory models but carries no a priori assumptions on the VSDs responsible for pore opening. The rate for each opening transition is scaled by interaction energy W, representing the energetic contribution of each active VSD toward channel opening. The values of the pore-gating charge displacement and half-activation potential parameters were fixed to values derived from linkage analysis (48, 49) on a representative G(V) (SI Appendix, Fig. S6A).
This model accurately predicted the voltage- and time-dependent properties of CaV1.2 opening and the activation of each VSD (Fig. 5). The values of VSD/pore interaction energies (W) (SI Appendix, Fig. S6B) showed that CaV1.2 opening is mostly facilitated by the activations of VSDs II and III (W2 ∼ W3 ∼ −50 meV). VSD I makes a smaller contribution toward opening the channel (W1 = −16 meV), representing ∼14% of the total allosteric energy from VSD activation stabilizing the open state. The energetic contribution of VSD IV (W4) is nearly zero, reinforcing the view that this VSD does not contribute to the voltage dependence of CaV1.2 channel opening.
Fig. 5.
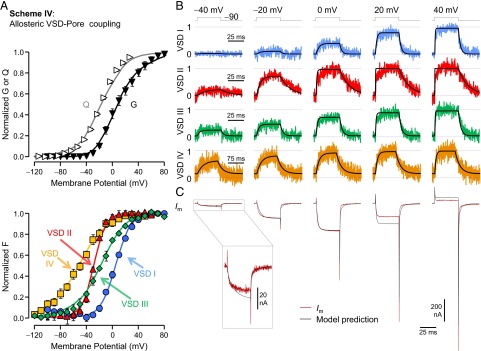
An allosteric model designates VSDs II and III as the drivers for the opening of α1C/β3/α2δ CaV1.2 channels. (A) Mean normalized total charge displacement (Q; white right-pointing triangle), ionic conductance (G; black ▼), and F reported from VSDs I (blue ●), II (red ▲), III (green ◆), and IV (yellow ■) with superimposed predictions of scheme IV (curves). (B) Fluorescence traces recorded from each VSD normalized to the steady-state probability of activation (scheme IV predictions are in black). The timescale for VSD IV traces is 75 ms. (C) Membrane current from WT channels (maroon) for the same pulses as in B with superimposed scheme IV predictions (black). Activation of VSDs II and III significantly biases the open state (W2 ∼ W3 ∼ −50 meV), and therefore, channel opening occurs at physiologically relevant potentials. VSD I makes a smaller contribution (W1 = −16 meV), whereas VSD IV is effectively not involved in CaV1.2 opening (W4 < 1 meV). Model parameters are in SI Appendix, Fig. S6B.
Discussion
By applying an optical approach (VCF) in human CaV1.2 channels, we resolved the time course of each VSD activation individually (Fig. 2A) with minimal disruption to channel function (i.e., without the use of pore blockers or charge-neutralizing mutations). An important assumption of this work is that the fluorescence traces faithfully tracked the voltage-dependent transitions of their respective VSD without missing components related to gating or reporting secondary fluorophore transitions irrelevant to channel gating. The coarse graining of VSD activation to a two-state process likely represents a simplification of the true gating mechanism, which may involve multiple charge-carrying gating transitions, such as in the Shaker VSD (50). We offer these justifications for our interpretation of the fluorescence signals and the model strategy.
-
i)
The use of relatively long, flexible fluorophores (∼12–15 Å long) should allow for the detection of conformational changes within the whole VSD. However, we cannot exclude that other gating components may have been missed.
-
ii)
The absence of a resolved delay in the fluorescence signals (Fig. 3B), which one would expect if a secondary fluorophore rearrangement followed charge movement, increases confidence in that the optical signals faithfully tracked VSD movements.
-
iii)
The global charge displacement [Q(V)] can be approximated by the charge-weighted sum of the F(V) curves (SI Appendix, Fig. S1), whereas schemes III and IV (which accounted for VSD activation and pore opening) predicted the Q(V) reasonably well, although it was not included in the fit dataset.
Schemes III and IV were less satisfactory at predicting minor slower components of the resolved fluorescence in VSDs I–III (Fig. 5B and SI Appendix, Fig. S5B). Such components may underlie slow CaV voltage-dependent processes (e.g., inactivation) or secondary fluorophore movements. In future work, longer depolarizations and expanded model schemes can be used to correlate slow VSD movements and CaV processes.
All models were less satisfactory at predicting Ig kinetics from Co2+-blocked channels. F signals from VSDs I and II acquired under Co2+ blockade were different from those from Ba2+-conducting channels, showing opposite F sign (VSD I) and faster kinetics. We cannot ascertain whether these differences are because of the CaV1.2 pore blockade or a direct effect of Co2+ on the VSDs and/or the fluorescent label. Accordingly, we focused our analysis and modeling on membrane current and fluorescence data from conducting channels.
A necessary condition for simultaneously fitting current and fluorescence by the five-particle CaV model template (one pore and four VSDs) is that only one G(V) and one set of current traces can be used to be accounted for by the activation of four VSDs. We used G(V) and current traces from WT channels. Because the current kinetics of S1324C channels (VSD IV label) were slower than WT (SI Appendix, Fig. S2), this discrepancy could have biased the model fitting to underestimate the VSD IV–pore coupling (W4). However, VSD IV movements are significantly slower than both WT and S1324C channel currents (Fig. 3B and SI Appendix, Fig. S2B, respectively); therefore, the model-independent assessment that VSD IV does not contribute to activation stands for both channels.
Which VSDs Initiate and Govern Ca2+ Entry?
The activation kinetics and voltage-dependent VSDs II and III precede ionic current in time and voltage (Fig. 3). Taken together with the modeling results, these data suggest that the voltage dependence and timing of CaV1.2 pore opening and consequently, Ca2+ influx are primarily governed by VSDs II and III. However, we cannot exclude the involvement of VSD I, which also possesses fast activation kinetics and contributes 14% of VSD energy toward channel opening. In follow-up work, to further probe the role of the CaV1.2 VSDs in gating, additional approaches (e.g., VSD immobilization) (51) should be adopted. Moreover, the auxiliary subunit composition of the CaV1.2 channel (β3 and α2δ in this work) likely plays an important role in the voltage-sensing properties and energy contribution of each VSD in α1C: considering the asymmetry encountered in both VSD–pore coupling and auxiliary subunit association with α1C (Fig. 1C), we propose that a layer of regulation for Ca2+ entry may involve the modulation of CaV VSD–pore coupling by auxiliary subunits.
Do CaV1.2 Channels Use an Allosteric Voltage-Sensing Mechanism?
Schemes I–III (Fig. 4), which allow channel opening only after the obligatory movement of a defined set of VSDs that is analogous to the opening of a bicycle lock for a specific combination, can be viewed as the strong coupling limit of the allosteric model (scheme IV), in which the interaction energies between the VSDs and the pore (W) are too large to be resolved. Obligatory coupling as the limit of strong allosteric coupling is a reasonable conjecture given the modular construction of ion channels and the widespread use of allosteric control among regulatory proteins in general (52). The ability of this model to satisfactorily fit the data (Fig. 5) showed that an allosteric gating mechanism is compatible with CaV1.2 channel activation. Interestingly, the allosteric model fitting assigned a negligible (<1 meV) energetic contribution of VSD IV to pore opening. It should be noted that this result does not exclude physical coupling between VSD IV and the pore (which are, indeed, parts of the same protein) (Fig. 1 A and B) but only that this VSD, in its resting or active state, contributes the same energy toward pore opening. Consequently, a mutation in a specific VSD can still affect channel properties, regardless of whether the activation transition of that VSD facilitates channel opening.
A CaV Paradigm for Voltage Sensing?
Because the activation of another pseudotetrameric channel, NaV1.4, has been studied by VCF (20, 42, 43, 53), we may compare the voltage-dependent operations of the NaV and CaV voltage sensors. The NaV1.4 VSDs activate within a more restricted voltage range (V0.5 = −76 mV for VSD III to −58 mV for VSD II) (43) than those of CaV1.2 (VSD IV, −52 mV; VSD I, 5 mV). Overall, the CaV1.2 VSDs are slower than their NaV1.4 counterparts (43) (SI Appendix, Table S2). However, in both channels, VSD IV is the slowest, a finding consistent with the theory that VSD IV evolution diverged from that of VSDs I–III in pseudotetrameric channels (5). NaV1.4 VSD IV activation exhibits a measurable delay, suggesting that it moves later in the activation sequence of this channel (43), a feature not resolved in any CaV1.2 VSDs (Fig. 3B). Finally, an obligatory model consistent with NaV channel gating (Fig. 4, scheme II), which is partly based on the scheme in ref. 43, could not account for CaV1.2 activation (SI Appendix, Fig. S4). The differences between NaV1.4 and CaV1.2 VSDs support the view that, although the two channel types are related and share a repeated domain structure, they may not use their voltage sensors in the same way to open the pore.
The functional heterogeneity of CaV1.2 VSDs reported in this work likely arises from the CaV channel complex structural asymmetry, because (i) the VSDs possess nonidentical amino acid sequences and charge distributions along S4 helices (Fig. 1D) and (ii) auxiliary subunits α2δ and β3 associate with channel forming-α1C in 1:1:1 stoichiometry (Fig. 1C) and likely modulate each VSD differently, fine-tuning the magnitude and timing of voltage-dependent Ca2+ influx. In conclusion, we found evidence that VSDs II and III govern CaV1.2 conductance, with a smaller contribution by VSD I. It will be of prime interest to explore the roles of each VSD in additional voltage-dependent functions, such as voltage-dependent inactivation and facilitation, and their modulation by CaV auxiliary subunits.
Materials and Methods
The human L-type Ca2+ channel α1C77 (gb CAA84346) (54) was used with β3- (Uniprot P54286) and α2δ-1 (Uniprot P13806) subunits. The voltage-dependent rearrangements of each VSD were optically tracked by a cut-open oocyte Vaseline gap voltage clamp complemented with fluorometry (35, 36, 55) as previously described (30, 32–34). Complete details are in SI Appendix.
Supplementary Material
Acknowledgments
The α1C77 (CaV1.2) clone was a gift from Nicolaj Soldatov. This work was supported in part by National Institutes of Health/National Heart, Lung, and Blood Institute Grant P01HL078931 and General Medicine Grant R01GM110276; American Heart Association Beginning Grant-in-Aid 12BGIA10560007 and Scientist Development Grant 14SDG20300018 (to A.P.), and Postdoctoral Fellowship 14POST18780018 (to N.S.); and Chilean Government Grants FONDECYT1120864 and ACT1104 (to A.N.). The Centro Interdisciplinario de Neurociencia de Valparaíso is a Millennium Institute supported by the Millennium Scientific Initiative of the Chilean Ministry of Economy.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1411127112/-/DCSupplemental.
References
- 1.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3(8):a003947. doi: 10.1101/cshperspect.a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lorenzon NM, Beam KG. Disease causing mutations of calcium channels. Channels (Austin) 2008;2(3):163–179. doi: 10.4161/chan.2.3.5950. [DOI] [PubMed] [Google Scholar]
- 3.Yu FH, Catterall WA. The VGL-chanome: A protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE. 2004;2004(253):re15. doi: 10.1126/stke.2532004re15. [DOI] [PubMed] [Google Scholar]
- 4.Chanda B, Bezanilla F. A common pathway for charge transport through voltage-sensing domains. Neuron. 2008;57(3):345–351. doi: 10.1016/j.neuron.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 5.Palovcak E, Delemotte L, Klein ML, Carnevale V. Evolutionary imprint of activation: The design principles of VSDs. J Gen Physiol. 2014;143(2):145–156. doi: 10.1085/jgp.201311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tombola F, Pathak MM, Isacoff EY. How does voltage open an ion channel? Annu Rev Cell Dev Biol. 2006;22:23–52. doi: 10.1146/annurev.cellbio.21.020404.145837. [DOI] [PubMed] [Google Scholar]
- 7.Swartz KJ. Sensing voltage across lipid membranes. Nature. 2008;456(7224):891–897. doi: 10.1038/nature07620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catterall WA. Ion channel voltage sensors: Structure, function, and pathophysiology. Neuron. 2010;67(6):915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vardanyan V, Pongs O. Coupling of voltage-sensors to the channel pore: A comparative view. Front Pharmacol. 2012;3:145. doi: 10.3389/fphar.2012.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blunck R, Batulan Z. Mechanism of electromechanical coupling in voltage-gated potassium channels. Front Pharmacol. 2012;3:166. doi: 10.3389/fphar.2012.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hidalgo P, Neely A. Multiplicity of protein interactions and functions of the voltage-gated calcium channel β-subunit. Cell Calcium. 2007;42(4-5):389–396. doi: 10.1016/j.ceca.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Minor DL, Jr, Findeisen F. Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels (Austin) 2010;4(6):459–474. doi: 10.4161/chan.4.6.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dolphin AC. The α2δ subunits of voltage-gated calcium channels. Biochim Biophys Acta. 2013;1828(7):1541–1549. doi: 10.1016/j.bbamem.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 14.Buraei Z, Yang J. Structure and function of the β subunit of voltage-gated Ca2+ channels. Biochim Biophys Acta. 2013;1828(7):1530–1540. doi: 10.1016/j.bbamem.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ben-Johny M, Yue DT. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J Gen Physiol. 2014;143(6):679–692. doi: 10.1085/jgp.201311153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neely A, Hidalgo P. Structure-function of proteins interacting with the α1 pore-forming subunit of high-voltage-activated calcium channels. Front Physiol. 2014;5:209. doi: 10.3389/fphys.2014.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117(4):500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kostyuk PG, Krishtal OA, Shakhovalov YA. Separation of sodium and calcium currents in the somatic membrane of mollusc neurones. J Physiol. 1977;270(3):545–568. doi: 10.1113/jphysiol.1977.sp011968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen LQ, Santarelli V, Horn R, Kallen RG. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J Gen Physiol. 1996;108(6):549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldschen-Ohm MP, Capes DL, Oelstrom KM, Chanda B. Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel. Nat Commun. 2013;4:1350. doi: 10.1038/ncomms2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Capes DL, Goldschen-Ohm MP, Arcisio-Miranda M, Bezanilla F, Chanda B. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J Gen Physiol. 2013;142(2):101–112. doi: 10.1085/jgp.201310998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw RM, Colecraft HM. L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc Res. 2013;98(2):177–186. doi: 10.1093/cvr/cvt021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shirokov R, Levis R, Shirokova N, Ríos E. Two classes of gating current from L-type Ca channels in guinea pig ventricular myocytes. J Gen Physiol. 1992;99(6):863–895. doi: 10.1085/jgp.99.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neely A, Wei X, Olcese R, Birnbaumer L, Stefani E. Potentiation by the β subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 1993;262(5133):575–578. doi: 10.1126/science.8211185. [DOI] [PubMed] [Google Scholar]
- 25.García J, Nakai J, Imoto K, Beam KG. Role of S4 segments and the leucine heptad motif in the activation of an L-type calcium channel. Biophys J. 1997;72(6):2515–2523. doi: 10.1016/S0006-3495(97)78896-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beyl S, et al. Neutralisation of a single voltage sensor affects gating determinants in all four pore-forming S6 segments of Ca(V)1.2: A cooperative gating model. Pflugers Arch. 2012;464(4):391–401. doi: 10.1007/s00424-012-1144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beyl S, et al. Methods for quantification of pore-voltage sensor interaction in Ca(V)1.2. Pflugers Arch. 2014;466(2):265–274. doi: 10.1007/s00424-013-1319-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mannuzzu LM, Moronne MM, Isacoff EY. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271(5246):213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- 29.Cha A, Bezanilla F. Characterizing voltage-dependent conformational changes in the Shaker K+ channel with fluorescence. Neuron. 1997;19(5):1127–1140. doi: 10.1016/s0896-6273(00)80403-1. [DOI] [PubMed] [Google Scholar]
- 30.Savalli N, Kondratiev A, Toro L, Olcese R. Voltage-dependent conformational changes in human Ca(2+)- and voltage-activated K(+) channel, revealed by voltage-clamp fluorometry. Proc Natl Acad Sci USA. 2006;103(33):12619–12624. doi: 10.1073/pnas.0601176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horne AJ, Fedida D. Use of voltage clamp fluorimetry in understanding potassium channel gating: A review of Shaker fluorescence data. Can J Physiol Pharmacol. 2009;87(6):411–418. doi: 10.1139/y09-024. [DOI] [PubMed] [Google Scholar]
- 32.Pantazis A, Gudzenko V, Savalli N, Sigg D, Olcese R. Operation of the voltage sensor of a human voltage- and Ca2+-activated K+ channel. Proc Natl Acad Sci USA. 2010;107(9):4459–4464. doi: 10.1073/pnas.0911959107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pantazis A, Olcese R. Relative transmembrane segment rearrangements during BK channel activation resolved by structurally assigned fluorophore-quencher pairing. J Gen Physiol. 2012;140(2):207–218. doi: 10.1085/jgp.201210807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Savalli N, Pantazis A, Yusifov T, Sigg D, Olcese R. The contribution of RCK domains to human BK channel allosteric activation. J Biol Chem. 2012;287(26):21741–21750. doi: 10.1074/jbc.M112.346171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gandhi CS, Olcese R. The voltage-clamp fluorometry technique. Methods Mol Biol. 2008;491:213–231. doi: 10.1007/978-1-59745-526-8_17. [DOI] [PubMed] [Google Scholar]
- 36.Pantazis A, Olcese R. Cut-open oocyte voltage clamp technique. In: Roberts GCK, editor. Encyclopedia of Biophysics. Springer; Berlin: 2013. pp. 406–413. [Google Scholar]
- 37.Bezanilla F, Villalba-Galea CA. The gating charge should not be estimated by fitting a two-state model to a Q-V curve. J Gen Physiol. 2013;142(6):575–578. doi: 10.1085/jgp.201311056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schoppa NE, McCormack K, Tanouye MA, Sigworth FJ. The size of gating charge in wild-type and mutant Shaker potassium channels. Science. 1992;255(5052):1712–1715. doi: 10.1126/science.1553560. [DOI] [PubMed] [Google Scholar]
- 39.Bezanilla F, Perozo E, Stefani E. Gating of Shaker K+ channels: II. The components of gating currents and a model of channel activation. Biophys J. 1994;66(4):1011–1021. doi: 10.1016/S0006-3495(94)80882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zagotta WN, Hoshi T, Aldrich RW. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J Gen Physiol. 1994;103(2):321–362. doi: 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Catterall WA. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J Physiol. 2012;590(Pt 11):2577–2589. doi: 10.1113/jphysiol.2011.224204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cha A, Ruben PC, George AL, Jr, Fujimoto E, Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 1999;22(1):73–87. doi: 10.1016/s0896-6273(00)80680-7. [DOI] [PubMed] [Google Scholar]
- 43.Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol. 2002;120(5):629–645. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marks TN, Jones SW. Calcium currents in the A7r5 smooth muscle-derived cell line. An allosteric model for calcium channel activation and dihydropyridine agonist action. J Gen Physiol. 1992;99(3):367–390. doi: 10.1085/jgp.99.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beyl S, et al. Different pathways for activation and deactivation in CaV1.2: A minimal gating model. J Gen Physiol. 2009;134(3):231–241. doi: 10.1085/jgp.200910272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horrigan FT, Aldrich RW. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J Gen Physiol. 2002;120(3):267–305. doi: 10.1085/jgp.20028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osteen JD, et al. Allosteric gating mechanism underlies the flexible gating of KCNQ1 potassium channels. Proc Natl Acad Sci USA. 2012;109(18):7103–7108. doi: 10.1073/pnas.1201582109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chowdhury S, Chanda B. Deconstructing thermodynamic parameters of a coupled system from site-specific observables. Proc Natl Acad Sci USA. 2010;107(44):18856–18861. doi: 10.1073/pnas.1003609107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sigg D. A linkage analysis toolkit for studying allosteric networks in ion channels. J Gen Physiol. 2013;141(1):29–60. doi: 10.1085/jgp.201210859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sigg D, Stefani E, Bezanilla F. Gating current noise produced by elementary transitions in Shaker potassium channels. Science. 1994;264(5158):578–582. doi: 10.1126/science.8160016. [DOI] [PubMed] [Google Scholar]
- 51.DeCaen PG, Yarov-Yarovoy V, Sharp EM, Scheuer T, Catterall WA. Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc Natl Acad Sci USA. 2009;106(52):22498–22503. doi: 10.1073/pnas.0912307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Changeux JP. Allostery and the Monod-Wyman-Changeux model after 50 years. Annu Rev Biophys. 2012;41:103–133. doi: 10.1146/annurev-biophys-050511-102222. [DOI] [PubMed] [Google Scholar]
- 53.Chanda B, Asamoah OK, Bezanilla F. Coupling interactions between voltage sensors of the sodium channel as revealed by site-specific measurements. J Gen Physiol. 2004;123(3):217–230. doi: 10.1085/jgp.200308971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soldatov NM. Molecular diversity of L-type Ca2+ channel transcripts in human fibroblasts. Proc Natl Acad Sci USA. 1992;89(10):4628–4632. doi: 10.1073/pnas.89.10.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stefani E, Bezanilla F. Cut-open oocyte voltage-clamp technique. Methods Enzymol. 1998;293:300–318. doi: 10.1016/s0076-6879(98)93020-8. [DOI] [PubMed] [Google Scholar]
- 56.Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012;486(7401):135–139. doi: 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.