

Significance
There is immense difficulty in mapping out the complete details of an enzyme’s mechanism, especially those that catalyze an acid-base reaction, owing to the simple fact that hydrogen atom positions are rarely known with any confidence. Ultrahigh-resolution X-ray and, better still, neutron crystallography can provide this crucial layer of information. We paired these techniques to reveal the catalytic mechanism of dihydrofolate reductase (DHFR), an enzyme necessary for nucleotide biosynthesis and a classical drug target. In a complex that closely resembles the catalytically active state, DHFR stabilizes a particular substrate conformer and likely elevates the pKa of the substrate atom that is protonated. This protonation occurs directly via water, with its access to the substrate regulated by structural fluctuation of the enzyme.
Keywords: enzyme catalysis, protonation state, protein dynamics, neutron diffraction, deuterium exchange
Abstract
Dihydrofolate reductase (DHFR) catalyzes the NADPH-dependent reduction of dihydrofolate (DHF) to tetrahydrofolate (THF). An important step in the mechanism involves proton donation to the N5 atom of DHF. The inability to determine the protonation states of active site residues and substrate has led to a lack of consensus regarding the catalytic mechanism involved. To resolve this ambiguity, we conducted neutron and ultrahigh-resolution X-ray crystallographic studies of the pseudo-Michaelis ternary complex of Escherichia coli DHFR with folate and NADP+. The neutron data were collected to 2.0-Å resolution using a 3.6-mm3 crystal with the quasi-Laue technique. The structure reveals that the N3 atom of folate is protonated, whereas Asp27 is negatively charged. Previous mechanisms have proposed a keto-to-enol tautomerization of the substrate to facilitate protonation of the N5 atom. The structure supports the existence of the keto tautomer owing to protonation of the N3 atom, suggesting that tautomerization is unnecessary for catalysis. In the 1.05-Å resolution X-ray structure of the ternary complex, conformational disorder of the Met20 side chain is coupled to electron density for a partially occupied water within hydrogen-bonding distance of the N5 atom of folate; this suggests direct protonation of substrate by solvent. We propose a catalytic mechanism for DHFR that involves stabilization of the keto tautomer of the substrate, elevation of the pKa value of the N5 atom of DHF by Asp27, and protonation of N5 by water that gains access to the active site through fluctuation of the Met20 side chain even though the Met20 loop is closed.
Dihydrofolate reductase (5,6,7,8-tetrahydrofolate:NADP+ oxidoreductase) (DHFR) is a housekeeping enzyme that catalyzes the NADPH-dependent reduction of 7,8-dihydrofolate (DHF) to 5,6,7,8,-tetrahydrofolate (THF). Various redox states of THF are used in several one-carbon transfer reactions to generate thymidine, methionine, glycine, serine, and other molecules (1–3). Given its role in biosynthesis, DHFR is a target for anticancer, antimicrobial, and rheumatoid arthritis drugs, such as methotrexate (MTX) and trimethoprim (4–7).
Although the kinetics, structure, and biophysical properties of Escherichia coli DHFR (ecDHFR) have been well characterized, unresolved questions with respect to its catalytic mechanism remain (1, 3, 8–13), as evidenced by the recent controversy over whether millisecond time-scale structural fluctuations can directly affect the chemical step in catalysis (14, 15). Folate is a poor substrate for DHFR, whereas DHF is reduced more efficiently (2). In addition, unlike DHF, folate cannot be further oxidized in solution. Thus, the abortive DHFR-folate-NADP+ complex is an excellent mimic of the DHFR-DHF-NADPH Michaelis complex (3, 16), and its stability makes it well suited for structural studies.
During catalysis, a proton is donated to the N5 atom of the DHF pterin ring and a hydride equivalent is transferred from NADPH to the C6 atom of the pterin. With folate as a substrate, proton donation occurs at the N8 atom (10). The five intermediates in the catalytic cycle are E-NADPH, E-NADPH-DHF, E-NADP+-THF, E-THF, and E-NADPH-THF (3), with product release as the rate-limiting step at neutral pH. THF is released on binding of a new NADPH molecule. The enzyme displays pH dependence with a characteristic pKa value of 6.5 (8).
Previous crystallographic and NMR studies of the DHFR binary and ternary complexes have revealed the locations of the folate and nicotinamide cofactor optimal for hydride transfer and the juxtaposition of the substrate with respect to the catalytic Asp27, which forms hydrogen bonds with the N3 and NA2 atoms of folate (3, 12). The DHFR-folate-NADP+ complex structure is considered the closest mimic of the Michaelis complex and has been used as a reference model in studies of the molecular details required for proton donation and hydride transfer (2, 3, 14, 17). Although it is clear from the structure that the nicotinamide ring is optimally positioned for hydride transfer to the C6 atom of the DHF substrate, how a proton can be donated to the N5 atom is unclear, especially considering that the conserved Asp27 is almost 5 Å distant from it. Disagreement abounds as to the protonation state of the Asp27 during catalysis (10, 18, 19). The mutation of the other residues contacting the substrate diminishes but does not abrogate activity, suggesting that the enzyme is flexible and has built-in redundancies (9, 20).
Several catalytic mechanisms have been proposed based on X-ray and NMR structures, molecular dynamics, enzyme kinetic measurements, and Raman spectroscopy studies (1, 17, 18, 21). According to Maharaj et al. (21), the pKa of the N5 atom of DHF is 2.6 in solution. When bound in a binary complex to DHFR, its N5 pKa remains strongly acidic. However, the pKa is elevated from <4 in the binary complex to 6.5 in the catalytic mimic complex, where NADP+ is bound as well (1, 21). This value matches the pKa describing the hydride transfer step (8), suggesting that the kinetic pKa describes the level of N5 protonated substrate available. The accompanying article by Liu et al. (22) further explores the kinetic pH profile for ecDHFR and its relationship to the hydride transfer step, as well as the sequential order of the mechanism. Outstanding questions remain, including, but not limited to, the following: (i) When a catalytically competent complex is present, how does the active site environment so drastically increase the N5 pKa to promote protonation? (ii) What is the protonation state of Asp27 throughout catalysis? And (iii) what is the source and mechanism of proton donation to N5?
An oft-proposed general mechanism based on several crystallography and Raman spectroscopy studies invokes a keto-enol tautomerization of the pterin substrate, initiating at the Asp27 and triggering a proton shuttle that ultimately results in a reduction of N5 (9, 10, 23). Two versions of this mechanism have been proposed, the major differences being the protonation state of Asp27 in the ground state and the ultimate proton source for reduction of N5 (SI Materials and Methods). The caveat regarding this mechanism is that keto-enol tautomerization as a critical step in the DHFR catalytic cycle remains a major point of ambiguity (24). Blakley et al. (25) have challenged the idea that substrate undergoes tautomerization during catalysis based on the NMR finding of a persistent substrate in an N3 imino-C4 keto tautomer across a pH range.
Alternative catalytic mechanisms propose the direct involvement of water molecules in the proton transfer step. These mechanisms notably omit the necessity for a substrate tautomerization event and the requirement for protonation of Asp27 at some point in the catalytic cycle (17). Asp27 is mainly responsible for binding the substrate in a catalytically favorable conformation and maintaining a negative electrostatic field in the active site, which would be negated if its carboxylate were protonated even transiently. A recent study revealed that Met20 loop dynamics are critical for solvent access to N5, and proposed a mechanism involving direct solvent protonation of the substrate (22). There has been only one previous structural observation of a solvent molecule within hydrogen-bonding distance of the N5 atom, in a crystal structure of E. coli DHFR bound to folate and NADP+ (1RA2). It should be noted that the Met20 loop adopts an open conformation in this structure, likely because of crystal packing effects (3). A solvent molecule is typically modeled in this position in crystal structures with only substrate bound (2).
A barrier to experimentally testing most proposed enzyme mechanisms is the inability to directly visualize the positions of important catalytic protons. The initial models used for most theoretical calculations are derived from X-ray structures, and determining the location of hydrogen atoms using X-rays is difficult even at atomic resolution (1.2 Å) (26). Neutron crystallography (NC) has a proven ability to determine the positions of hydrogen atoms or ions (protons) essential for catalysis (27–30). In fact, NC defines unique positions of hydrogen atoms within ordered water molecules (31), and H3O+ molecules crucial for catalysis in xylose isomerase were recently identified (32). By virtue of the need to perform hydrogen/deuterium exchange (HDX) on crystals before data collection, NC can accurately identify hydrogen atom positions even at modest resolution. Deuterium coherently scatters neutrons with lengths similar to carbon and nitrogen, whereas hydrogen coherently scatters neutrons with negative lengths, rendering them invisible in positively contoured nuclear density maps. In the past, the determination of NC structures was hindered by the limited number of data collection facilities, low beam fluxes, and the requirement for extremely large crystals (>1 mm3 in volume). Recently, new spallation sources, enhanced deuterium labeling of samples, and improved detectors have allowed the collection of high-quality data from crystals of smaller volume, leading to a dramatic increase in the number of neutron structures deposited in the Protein Data Bank (PDB).
In a previous NC study, we resolved a question pertaining to the protonation state of the classical antifolate inhibitor MTX and Asp27 when MTX binds DHFR (28). The DHFR-MTX neutron structure demonstrates that Asp27 is negatively charged, whereas the N1 atom of MTX is protonated and thus positively charged. After our initial success with the DHFR-MTX complex, we conducted NC studies of a DHFR pseudo-Michaelis complex to identify the protonation state of Asp27 in a catalytic mimic complex, the source of protonation for N5, as well as the presence (or absence) of a substrate keto-enol tautomerization event. Here we report the neutron diffraction structure of the DHFR-folate-NADP+ complex and complementary ultrahigh-resolution X-ray structures at three different temperatures. Refinement of the neutron structure allowed determination of the positions of crucial protons on the folate substrate and the ionization state of Asp27. Furthermore, our comprehensive map of backbone HDX sheds light on the dynamically driven changes in solvent accessibility of crystalline DHFR. The ultrahigh-resolution X-ray structures provide molecular details of Met20 loop fluctuations required for the entry of solvent, identifying a water molecule possibly involved in proton donation to N5.
Results and Discussion
The structure of the E. coli DHFR-folate-NADP+ ternary complex was solved by neutron diffraction to 2.0-Å resolution at 291K and with ultrahigh-resolution X-ray diffraction at temperatures of 100 K, 277 K, and 291 K to resolutions of 0.85 Å, 1.05 Å, and 1.60 Å, respectively (Table S1) (33). The ternary complex crystallized in the P212121 space group, with one molecule in the asymmetric unit (AU). All structures were refined to satisfactory Rfree values and reasonable stereochemistry.
The Met20 loop adopts a closed conformation in all structures, consistent with the previously determined X-ray structure to 1.8-Å resolution (3). Significant nuclear density for nearly all main-chain atoms, many side-chain atoms, and most ligand atoms in the active site can be seen in the maps obtained by neutron diffraction, including many of the elusive deuterium atoms. A representative high-quality map of nuclear and electron density is provided in Fig. 1.
Fig. 1.

Characterization of the DHFR-folate-NADP+ interaction. The 2.0-Å resolution nuclear density map is shown for the neutron structure (A–C and E), and the 0.85-Å resolution electron density map is shown for the X-ray structure (D and F). (A) 2Fo − Fc nuclear density (1.0 σ) of the bound folate and NADP+. (B) The positive 2Fo − Fc nuclear density (light blue, 1.0 σ), the negative 2Fo − Fc nuclear density (red, −1.7 σ), and 2Fo − Fc electron density (magenta, 1.0 σ) maps for Tyr100. (C) 2Fo- Fc nuclear density map (1.8 σ) and Fo − Fc omit nuclear density map (3.0 σ) in the active site where Asp27 and folate have H-bond interactions. A deuterated water molecule (DOD18) is H-bonded with both Asp27 and folate. The omit map was created without the deuterium contribution for neutron scattering. (D) 2Fo − Fc electron density map (1.0 σ) in the active site. (E) 2Fo − Fc nuclear density map (1.8 σ) near Met20 and folate and Fo − Fc nuclear density map (3.0 σ) near the Met20 side chain. (F) 2Fo − Fc electron density map (1.0 σ) near Met20 and folate and Fo − Fc electron density map (3.0 σ) near the Met20 side chain.
Occupancy refinement of the deuterium atoms at exchangeable positions on the amide backbone and at certain side chains was performed. Deuterium atoms with occupancies refining to ≥0.7 are considered to be in positions that are fully exchanged, those with occupancies refining to values between 0.15 and 0.69 are partially exchanged, and those with occupancies refining to <0.15 are nonexchanged (27). In the latter case, the deuterium is removed from the model and replaced with hydrogen. Cancellation effects owing to the negative scattering length of hydrogen result in negative nuclear density near hydrocarbons and atoms bound to nonexchangeable hydrogens that provide another measure of quality control of the nuclear density maps.
Folate–Asp27 Interaction in the Active Site.
Because HDX of the crystals precedes neutron data collection, the crucial hydrogens at the active site can be identified by locating deuterium atoms that have been refined to occupancy values >0.7. Moreover, because deuterium is not present in the original models used for refinement, they contribute to positive peaks in the Fo − Fc maps, whereas nonexchanged hydrogen, owing to its negative scattering length, can be visualized in a negatively contoured Fo − Fc map. To locate the deuterium and hydrogen atoms in the active site around folate and Asp27, the 2Fo − Fc and the Fo − Fc unbiased omit nuclear density maps were generated and contoured at +1.8 σ and +3.0 σ, respectively. Both maps show density peaks close to the N3 atom of folate, corresponding to the presence of a deuterium atom.
These peaks are clearly associated with N3 and not with the carboxylate side chain of Asp27 (Fig. 1C), indicating that N3 is protonated in this pseudo-Michaelis complex. Thus, we modeled a deuterium atom as bonded to N3 and refined its occupancy, which was determined to be 0.98. This indicates the presence of a deuterium atom bound to the N3 atom of folate; thus, the N3 atom of folate is protonated and Asp27 remains ionized in the folate-NADP+ complex. Given the experimentally determined pKa values of N3 in folate and DHF of ∼8 and 10.8, respectively (21, 34, 35), we predict that N3 will be protonated in a DHF ternary complex. The distance between the N3 deuterium atom and the Oδ2 of Asp27 is 1.7 Å, and the angle between N3, its bound D atom, and Oδ2 is 172.5°, indicating a strong hydrogen bond between folate and the catalytic aspartate. In addition, the NA2 amide group (−ND2) of folate and the Oδ1 atom of Asp27 form a hydrogen bond (2.1 Å). A deuterated water molecule (DOD18) is hydrogen-bonded with both the Oδ2 atom of Asp27 and the O4 atom of folate, with distances of 1.8 Å and 2.5 Å, respectively (Fig. 1C). The same hydrogen-bonding configuration is observed in the 0.85-Å ultrahigh-resolution X-ray structure (Fig. 1D).
Met20 Loop Dynamics Facilitates Solvent Entry for Protonation at N5.
We observed no nuclear density in the Fo − Fc or 2Fo − Fc maps near and adjacent to the N8 atom of folate, the ultimate proton acceptor in the reaction. This suggests that this atom is not protonated in the Michaelis complex at neutral pH. Although an experimentally determined pKa for the N8 of folate is lacking, we note that the pH of the crystallization medium used (pH 7) is ∼0.5 pH units higher than the pKa value of N5 when substrate is bound to DHFR (1, 21). If the pKa values of N5 and N8 are similar (admittedly, this may not be the case), this may explain the absence of a proton at N8. In all structures, the N10 atom, which links the pteridine and the para-aminobenzoic glutamate (p-ABA-Glu) moieties of the substrate, forms a hydrogen bond with a water molecule (DOD47 in the neutron structure).
The flexible Met20 loop in DHFR undergoes conformational changes that are closely coupled to the catalytic cycle, where it adopts an open (apoenzyme), closed (Michaelis complex), or occluded (product-bound) conformation (3, 16). This flexibility results in poor nuclear density for the Met20 side chain (Fig. 1E). X-ray structures solved at 100 K, 277 K, and 291 K provide useful independent views of thermal motion and conformational disorders. Examination of the Fo − Fc maps of the 0.85-Å 100 K structure at the Met20 side chain reveals a positive electron density peak adjacent to a negative electron density peak strongly correlating with the flexible motion of the loop (Fig. 1F). Similar difference peaks are seen in the 277 K and 291 K X-ray structures.
In both the 100 K and 277 K structures, the Met20 side chain is modeled in two alternate conformations, whose occupancies were refined and constrained to sum to unity (Fig. 2C). As seen in all of the structures, the entry of a water molecule required to facilitate proton donation at N5 (N8 in folate) is sterically hindered by the major conformation of the Met20 side chain. Nevertheless, in the 0.85-Å resolution structure determined at 100 K, we observe a partially occupied water molecule (HOH351) within hydrogen bonding (distance, 3.1 Å) to the folate N10 atom that could possibly move to protonate N5 on a small fluctuation of the Met20 loop. This water overlaps almost perfectly with the position of the aforementioned D2O molecule (DOD47) observed in the 2.0-Å resolution neutron structure (data collected at 291 K). In the 0.85 Å X-ray structure, the water molecule hydrogen-bound with the N10 atom shows positive Fo − Fc peaks emanating toward the Met20 side chain (Fig. 1F). In addition, difference electron density maps contoured at +3.0 σ generated from the 1.05-Å resolution data (277 K) and the 1.6-Å resolution data (291 K) show similar densities, particularly for the 291 K dataset, likely owing to enhanced thermal motion, given that the data were collected at ambient temperature (Fig. 2 A and B). This finding is consistent with a previous comparative analysis of the influence of temperature on the conformational heterogeneity of the DHFR ternary complex using the 100 K and 277 K data (36). In the 1.05-Å structure determined at 277 K, both Fo − Fc and 2Fo − Fc electron densities appear for a partially occupied water molecule within generous hydrogen-bonding distance of the N5 atom (Fig. 2C). Although the distance between the water and N5 ultimately refines to 3.7 Å, the distance from the center point of a minimally biased Fo − Fc peak calculated before introduction of the water molecule to N5 is actually 3.5 Å. This partially occupied water is likely stabilized at this position by a 2.9-Å hydrogen bond with the folate O4 atom. Although this is in a similar position to the water molecule previously observed in 1RA2, a crystal structure of ecDHFR-FOL-NADP+ with the Met20 loop open (3), the key difference is that we observe this partially occupied water when the Met20 loop is closed.
Fig. 2.
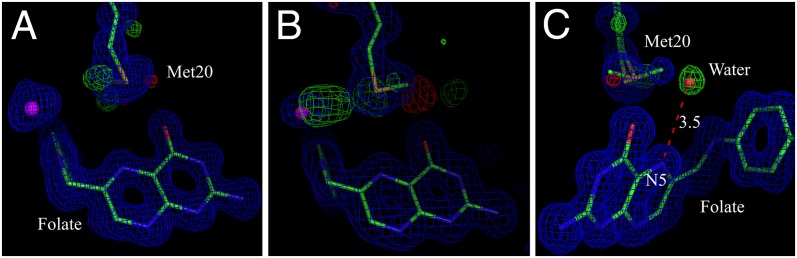
Dynamics of the Met20 side chain revealed by the high-resolution X-ray structures. (A and C) 2Fo − Fc electron density map (1.0 σ) and Fo − Fc electron density map (3.0 σ) around Met20 for the 1.05-Å resolution data collected at 277 K of the ecDHFR-folate-NADP+ complex. In C, the Met20 side chain has been modeled with alternate conformations. A water molecule is H-bonded (∼3.5 Å) to the N5 atom of folate. The image in C is rotated 180° around the y-axis compared with A. (B) 2Fo − Fc electron density map (1.0 σ) and Fo − Fc electron density map (3.0 σ) around Met20 for the 1.6-Å resolution data collected at 291 K for the ecDHFR-folate-NADP+ complex.
Solvent Structure, Amide H/D Exchange, and DHFR Dynamics.
Mobile solvent molecules and Met20 loop dynamics are important for DHFR catalysis. In contrast to X-rays, neutrons provide a means of more clearly elucidating solvent and macromolecular dynamics, because of their ability to visualize deuterium atoms (31). The analysis of solvent and protein dynamics is coupled in NC structures owing to the prominent role of protein conformational dynamics in facilitating HDX with solvent; for example, solvent forming hydrogen bonds to backbone amides or side chains that have exchangeable groups often directly assist in isotope exchange (28, 31). Thus, the level of HDX, measured by the refined occupancy of deuterium at exchangeable positions, is a measure of dynamics; that is, the greater the deuterium occupancy, the greater the dynamics. Approximately 75% of the backbone amides in the ecDHFR-folate-NADP+ structure have undergone HDX (Figs. S1 and S2), indicating at least transient interaction with deuterated solvent for three-quarters of the protein backbone. The extent of this exchange is close to the average observed in protein neutron structures available in the PDB (37); however, it is significantly higher than what was previously reported for the neutron structure of the E. coli DHFR-MTX complex (∼60%) (28).
Stably bound solvent is found within the interior of the neutron structure of the DHFR ternary complex (Fig. 3A); namely, five water molecules—DOD47, DOD2, DOD18, DOD10, and DOD66—interacting with the substrate are seen (Fig. 3B). DOD18 forms two hydrogen bonds with the O4 atom of folate (2.3 Å) and the side chain of Asp27 (1.7 Å) (Fig. 1C). The involvement of this water molecule in a proton relay network has been proposed (9, 24); however, we believe that it mainly has a role in substrate recognition (2, 3). DOD2, another solvent molecule involved in substrate recognition, forms hydrogen bonds to Trp30 (HE1 atom), Thr113 (OG1 atom), Tyr111 (carbonyl oxygen atom), and folate (HN21 atom) (Fig. 3C). Although all five water molecules interacting with the substrate are buried (Fig. 3B), deuterium occupancy refinement indicates that they are all deuterated (refined Docc = 1), suggesting that the substrate is accessible to bulk solvent. In addition, we observe 114 water molecules on the surface of the protein (Fig. 3A); thus, 119 water molecules are modeled as bound to the E. coli DHFR-folate-NADP+ complex.
Fig. 3.
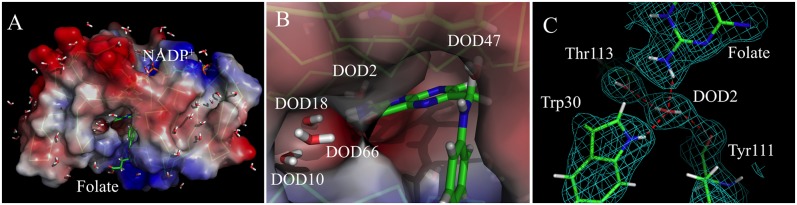
Water molecules in the ecDHFR-folate-NADP+ structure. (A) Electrostatic surface representation of the ternary complex. Negatively charged surfaces are shown in red, positively charged ones are in blue, and hydrophobic surfaces are in white. DOD molecules are shown as sticks (white for deuterium and red for oxygen). (B) Close-up view of the cavity showing the five water molecules inside. (C) H-bonding analysis between DOD2 and its nearby residues. The 2Fo − Fc nuclear density map is shown in light green.
The two most flexible regions of the DHFR molecule observed in the complex are the Met 20 loop and the αA helix (Figs. S1 and S2, Tables S2 and S3, and SI Materials and Methods). The flexibility of the Met20 loop facilitates catalytically important conformational changes during the reaction cycle (3, 12, 16). In our neutron diffraction structure, the Met20 loop is in a closed conformation and has undergone complete HDX, characteristic of an extremely dynamic region (Table S3 and Fig. S2). A small fluctuation of the loop leads to the entrance of a water molecule into the active site of the DHFR-folate-NADP+ complex, probably the solvent molecule that protonates the substrate N5 atom (Fig. 2C).
The roles of solvent access and Met20 loop dynamics were further analyzed by examining the interactions of the backbone amides with water molecules. A detailed analysis of the H-bonding network of the Met20 loop (Fig. S2) shows that residues on the N-terminal side of the Met20 loop (Val10, Asp11, Arg12, and Val13) form two hydrogen bonds with nearby water molecules, and four water molecules that could promote HDX are nearby (within 4 Å of this region). The Ile14 amide hydrogen forms a hydrogen bond with the Thr123 carbonyl oxygen, and there is no water nearby, explaining why it does not undergo HDX. The backbone amides of the apex region of the Met20 loop (Gly15, Met16, Glu17, Asn18, Ala19, and Met20) form four hydrogen bonds with nearby water molecules or a protonated NADP+ oxygen atom (the O3D atom), and there are five water molecules within 4 Å of this region, mediating HDX. In addition, there are four water molecules within 4 Å of the C-terminal side of the Met20 loop, and the loop undergoes partial or full HDX. In summary, a total of eight hydrogen bonds are formed between the amides in the Met20 loop and nearby water molecules in the ecDHFR-folate-NADP+ neutron structure. The mobility of the Met20 loop makes it accessible to interactions with solvent that promote HDX, emphasizing the roles of solvent and dynamics in DHFR catalysis.
Implications for Catalysis.
Even after many years of biochemical, biophysical, and structural studies of ecDHFR, there remain critical questions regarding the role of Asp27, its ionization state, and the source of the proton for the N5 atom on the pterin ring in the catalytic mechanism. We aimed to answer these questions with the help of neutron diffraction and ultrahigh-resolution X-ray structures of the DHFR-folate-NADP+ pseudo-Michaelis complex. In the neutron diffraction structure, the catalytic Asp27 is clearly ionized, whereas the N3 atom adjacent to it is protonated. The ionization of Asp27 is consistent with previous reports that the carboxylate does not titrate at pH 5–7 (25, 38). The protonation of the N3 atom suggests that the folate substrate is in the keto form when bound to DHFR and in the presence of the nicotinamide cofactor. Moreover, there is no nuclear density consistent with a hydrogen atom on the O4 atom to indicate the existence of the enol isomer of folate (Fig. 1C). This finding is interesting, considering that several previously proposed catalytic mechanisms include enolization of the O4 atom of the pterin ring as a key step in relaying a proton to N5 for ultimate reduction of the N5–C6 bond (2, 9, 39). In our neutron structure, Asp27 is negatively charged and stabilized by its hydrogen bonds with the N3 atom and the water molecule that bridges the catalytic aspartate via a second sphere hydrogen bond to the O4 atom (Fig. 1C). Our structures support the findings of Blakley (25), who used NMR with isotopically labeled substrate in a binary complex to rule out the possibility of enolization in the catalytic cycle of DHFR.
Before the availability of the structures of DHFR in a binary complex with substrate (39), the role of Asp27 was associated with protonation of the N5 atom of the pterin ring and the kinetically determined pKa of 6.5 (8). The crystal structures clearly reveal that Asp27 lies at least 5 Å away from the substrate’s N5 atom and cannot directly facilitate its protonation; however, its importance is demonstrated by a 3,600-fold decrease in catalytic efficiency (kcat/KM DHF) associated with an Asp27-to-Ser mutation (20, 40). Furthermore, the pKa of Asp27 is <6.5, thus ruling out a role as the titrating group in pH profiles (25, 38). Our structure supports previous suggestions that the ionized Asp27 contributes to a negative electrostatic environment that shifts the solution pKa of the substrate N5 atom from 2.4 to 6.5 when bound in a ternary complex (1, 21). In addition, Asp27 interacts with structurally relevant waters at the active site that are important for substrate binding; for example, one of the water molecules (DOD18 in the neutron structure) that forms a hydrogen bond to Asp27 also interacts with the O4 atom of the pterin ring.
After substrate and cofactor binding, the next step in the catalytic mechanism is protonation of the substrate N5 atom that may precede or occur concomitantly with hydride transfer. Some previously proposed catalytic mechanisms suggest that the enol tautomer leads to protonation of the N5 atom, with proton transfer directly from the O4 (9, 10); however, this step is considered unfavorable owing to poor geometry (2). Other alternative catalytic mechanisms invoke the direct involvement of a water molecule protonating the N5 atom (1, 2, 18, 19). Such a water molecule has been observed in multiple DHFR-folate binary complexes and chicken DHFR complexes (2, 3); however, a water in this location has been observed in a DHFR-folate-NADP+ pseudo-Michaelis complex in only one structure, 1RA2 (3). A caveat of this finding is that it was observed in an open Met20 loop conformation and was likely stabilized by crystal packing. Based on molecular dynamics simulation studies, it has been proposed that a small fluctuation (0.79 Å rmsd) of the Met20 loop permits the entrance of a water molecule to be in position for protonation of the N5 atom (18).
Owing to the mobility of the Met20 side chain, no clear nuclear density defines it in our neutron diffraction structure determined at 2.0-Å resolution. Evidence of considerable loop mobility is present in our ultrahigh-resolution X-ray structures determined at 100 K and 277 K. In both the 100 K and 277 K X-ray structures, the Met20 side chain is modeled in two alternate conformations and may be more extensively disordered. A water molecule located near both Met20 and the N10 atom of the folate also shows evidence of disorder that may be correlated with the Met20 displacement. Importantly, the 277 K crystal structure features electron density for an additional partially occupied water molecule that is near the Cε atom of one conformation of Met20 and the N5 atom of folate (distance, 3.5–3.7 Å). It is likely stabilized in this near-catalytic position by the conformation of the Met20 side chain and its hydrogen bond with the folate O4 (distance, 2.9 Å). This partially occupied water is an attractive candidate for Met20-gated entry into the active site and potential protonation of the N5 atom (Fig. 2C). This is the first direct experimental observation of a water molecule in position to directly protonate the N5 atom in a closed loop structure.
Conclusions
Based on the foregoing data, Fig. 4 B–E illustrates our proposed catalytic mechanism for E. coli DHFR. In brief, we propose that the substrate in the DHFR-folate-NADP+ complex, a pseudotransition state assembly, exists in the keto form (Fig. 1C). The conserved aspartate is negatively charged and, along with the low dielectric environment of the active site, contributes to the elevation of pKa of N5 from 2.4 to 6.5 in the complex (1, 21). Consistent with the role proposed for the conformational cycle of the Met20 loop throughout the reaction coordinate, a small fluctuation of the Met20 side chain leads to the entry of a water molecule to be in position for direct protonation of the N5 atom. After protonating N5, the resultant hydroxide ion is likely stabilized by neighboring water molecules. These findings illustrate that enolization likely is not required for the proton donation step. Protonation may precede or occur concomitantly with hydride transfer between the C4 atom of NADPH and the C6 atom of pterin, although we are unable to be definitive about the order of these two steps. The accompanying paper by Liu et al. (22) sheds light on the chronology of events in the DHFR mechanism.
Fig. 4.

Interactions between folate and surrounding residues and waters (A) and the proposed reaction mechanism (B–E). Mechanistic details are provided in the text. Water molecules are numbered according to the D2O molecules modeled in the neutron structure (i.e., W18 = DOD18).
Materials and Methods
For neutron diffraction, a single 3.6-mm3 cocrystal of the ecDHFR-folate-NADP+ complex was obtained by seeding. A 2.0-Å resolution dataset was collected at 291 K using the quasi-Laue technique on IMAGINE at the High Flux Isotope Reactor at Oak Ridge National Laboratory. A corresponding 291 K X-ray dataset was collected on a smaller crystal. The neutron structure was refined in Phenix using joint X-ray/neutron refinement (41). For ultrahigh-resolution X-ray diffraction, 100 K and 277 K datasets were collected at beamline 11-1 at the Stanford Synchrotron Radiation Laboratory. The respective structures were refined in SHELX-97 (42). More details are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Greg Petsko and Dagmar Ringe (Brandeis University) for their generous contributions to this project, Pavel Afonine for his help with the joint X-ray/neutron refinement, and Drs. Krzysztof Palczewski and Mark Yeager for supporting neutron diffraction. Research conducted at Oak Ridge National Laboratory’s High-Flux Isotope Reactor was sponsored by the US Department of Energy’s (DOE) Office of Basic Energy Sciences, Scientific User Facilities Division. The IMAGINE Project was partially supported by the National Science Foundation (Grant 0922719). Use of the Stanford Synchrotron Radiation Light Source is supported by the US DOE’s Office of Basic Energy Sciences (Contract DE-AC02-76SF00515). The Stanford Synchrotron Radiation Light Source Structural Molecular Biology Program is supported by the US DOE’s Office of Biological and Environmental Research and by the National Institutes of Health (NIH), National Institute of General Medical Sciences (NIGMS) (including Grant P41GM103393). C.D. was supported by NIH Grant 1R01 GM100887-01. M.A.W. was supported by NIH Grant GM092999. A.K. and P.L. were supported by the US DOE’s Office of Basic Energy Sciences. P.L. was partially supported by NIH NIGMS Grant R01 GM071939-01 to develop computational tools for neutron protein crystallography. Q.W. is sponsored by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB), www.pdb.org (PDB ID codes 4PDJ, 4PSY, and 4RGC).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1415856111/-/DCSupplemental.
References
- 1.Chen YQ, Kraut J, Blakley RL, Callender R. Determination by Raman spectroscopy of the pKa of N5 of dihydrofolate bound to dihydrofolate reductase: Mechanistic implications. Biochemistry. 1994;33(23):7021–7026. doi: 10.1021/bi00189a001. [DOI] [PubMed] [Google Scholar]
- 2.Reyes VM, Sawaya MR, Brown KA, Kraut J. Isomorphous crystal structures of Escherichia coli dihydrofolate reductase complexed with folate, 5-deazafolate, and 5,10-dideazatetrahydrofolate: Mechanistic implications. Biochemistry. 1995;34(8):2710–2723. doi: 10.1021/bi00008a039. [DOI] [PubMed] [Google Scholar]
- 3.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry. 1997;36(3):586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 4.Chan DC, Anderson AC. Towards species-specific antifolates. Curr Med Chem. 2006;13(4):377–398. doi: 10.2174/092986706775527938. [DOI] [PubMed] [Google Scholar]
- 5.Bertino JR. Cancer research: From folate antagonism to molecular targets. Best Pract Res Clin Haematol. 2009;22(4):577–582. doi: 10.1016/j.beha.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Volpato JP, Pelletier JN. Mutational “hot-spots” in mammalian, bacterial and protozoal dihydrofolate reductases associated with antifolate resistance: Sequence and structural comparison. Drug Resist Updat. 2009;12(1-2):28–41. doi: 10.1016/j.drup.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Hashkes PJ, et al. Methotrexate: New uses for an old drug. J Pediatr. 2014;164(2):231–236. doi: 10.1016/j.jpeds.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 8.Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry. 1987;26(13):4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 9.Miller GP, Benkovic SJ. Stretching exercises—flexibility in dihydrofolate reductase catalysis. Chem Biol. 1998;5(5):R105–R113. doi: 10.1016/s1074-5521(98)90616-0. [DOI] [PubMed] [Google Scholar]
- 10.Cummins PL, Gready JE. Energetically most likely substrate and active-site protonation sites and pathways in the catalytic mechanism of dihydrofolate reductase. J Am Chem Soc. 2001;123(15):3418–3428. doi: 10.1021/ja0038474. [DOI] [PubMed] [Google Scholar]
- 11.Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc Natl Acad Sci USA. 2002;99(5):2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schnell JR, Dyson HJ, Wright PE. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu Rev Biophys Biomol Struct. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- 13.Loveridge EJ, Allemann RK. Effect of pH on hydride transfer by Escherichia coli dihydrofolate reductase. ChemBioChem. 2011;12(8):1258–1262. doi: 10.1002/cbic.201000794. [DOI] [PubMed] [Google Scholar]
- 14.Bhabha G, et al. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science. 2011;332(6026):234–238. doi: 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loveridge EJ, Behiry EM, Guo J, Allemann RK. Evidence that a “dynamic knockout” in Escherichia coli dihydrofolate reductase does not affect the chemical step of catalysis. Nat Chem. 2012;4(4):292–297. doi: 10.1038/nchem.1296. [DOI] [PubMed] [Google Scholar]
- 16.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313(5793):1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 17.Falzone CJ, Wright PE, Benkovic SJ. Dynamics of a flexible loop in dihydrofolate reductase from Escherichia coli and its implication for catalysis. Biochemistry. 1994;33(2):439–442. doi: 10.1021/bi00168a007. [DOI] [PubMed] [Google Scholar]
- 18.Shrimpton P, Allemann RK. Role of water in the catalytic cycle of E. coli dihydrofolate reductase. Protein Sci. 2002;11(6):1442–1451. doi: 10.1110/ps.5060102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khavrutskii IV, Price DJ, Lee J, Brooks CL., 3rd Conformational change of the methionine 20 loop of Escherichia coli dihydrofolate reductase modulates pKa of the bound dihydrofolate. Protein Sci. 2007;16(6):1087–1100. doi: 10.1110/ps.062724307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howell EE, Villafranca JE, Warren MS, Oatley SJ, Kraut J. Functional role of aspartic acid-27 in dihydrofolate reductase revealed by mutagenesis. Science. 1986;231(4742):1123–1128. doi: 10.1126/science.3511529. [DOI] [PubMed] [Google Scholar]
- 21.Maharaj G, et al. Dissociation constants for dihydrofolic acid and dihydrobiopterin and implications for mechanistic models for dihydrofolate reductase. Biochemistry. 1990;29(19):4554–4560. doi: 10.1021/bi00471a008. [DOI] [PubMed] [Google Scholar]
- 22.Liu CT, et al. Escherichia coli dihydrofolate reductase catalyzed proton and hydride transfers: Temporal order and the roles of Asp27 and Tyr100. Proc Natl Acad Sci USA. 2014;111:18231–18236. doi: 10.1073/pnas.1415940111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bajorath J, Kraut J, Li ZQ, Kitson DH, Hagler AT. Theoretical studies on the dihydrofolate reductase mechanism: Electronic polarization of bound substrates. Proc Natl Acad Sci USA. 1991;88(15):6423–6426. doi: 10.1073/pnas.88.15.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee H, Reyes VM, Kraut J. Crystal structures of Escherichia coli dihydrofolate reductase complexed with 5-formyltetrahydrofolate (folinic acid) in two space groups: Evidence for enolization of pteridine O4. Biochemistry. 1996;35(22):7012–7020. doi: 10.1021/bi960028g. [DOI] [PubMed] [Google Scholar]
- 25.Blakley RL, Appleman JR, Freisheim JH, Jablonsky MJ. 13C and 15N nuclear magnetic resonance evidence that the active site carboxyl group of dihydrofolate reductase is not involved in the relay of a proton to substrate. Arch Biochem Biophys. 1993;306(2):501–509. doi: 10.1006/abbi.1993.1543. [DOI] [PubMed] [Google Scholar]
- 26.Fisher SJ, Blakeley MP, Cianci M, McSweeney S, Helliwell JR. Protonation-state determination in proteins using high-resolution X-ray crystallography: Effects of resolution and completeness. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 7):800–809. doi: 10.1107/S0907444912012589. [DOI] [PubMed] [Google Scholar]
- 27.Kossiakoff AA. Protein dynamics investigated by the neutron diffraction-hydrogen exchange technique. Nature. 1982;296(5859):713–721. doi: 10.1038/296713a0. [DOI] [PubMed] [Google Scholar]
- 28.Bennett BC, et al. Neutron diffraction studies of Escherichia coli dihydrofolate reductase complexed with methotrexate. Proc Natl Acad Sci USA. 2006;103(49):18493–18498. doi: 10.1073/pnas.0604977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coates L, et al. The catalytic mechanism of an aspartic proteinase explored with neutron and X-ray diffraction. J Am Chem Soc. 2008;130(23):7235–7237. doi: 10.1021/ja801269x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovalevsky AY, et al. Metal ion roles and the movement of hydrogen during reaction catalyzed by D-xylose isomerase: A joint x-ray and neutron diffraction study. Structure. 2010;18(6):688–699. doi: 10.1016/j.str.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bon C, Lehmann MS, Wilkinson C. Quasi-Laue neutron-diffraction study of the water arrangement in crystals of triclinic hen egg-white lysozyme. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 5):978–987. doi: 10.1107/s0907444998018514. [DOI] [PubMed] [Google Scholar]
- 32.Kovalevsky AY, et al. Identification of the elusive hydronium ion exchanging roles with a proton in an enzyme at lower pH values. Angew Chem Int Ed Engl. 2011;50(33):7520–7523. doi: 10.1002/anie.201101753. [DOI] [PubMed] [Google Scholar]
- 33.Wan Q, et al. Preliminary joint X-ray and neutron protein crystallographic studies of ecDHFR complexed with folate and NADP+ Acta Crystallogr F Struct Biol Commun. 2014;70(Pt 6):814–818. doi: 10.1107/S2053230X1400942X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poe M. Proton magnetic resonance studies of folate, dihydrofolate, and methotrexate: Evidence from pH and concentration studies for dimerization. J Biol Chem. 1973;248(20):7025–7032. [PubMed] [Google Scholar]
- 35.Szakács Z, Noszál B. Determination of dissociation constants of folic acid, methotrexate, and other photolabile pteridines by pressure-assisted capillary electrophoresis. Electrophoresis. 2006;27(17):3399–3409. doi: 10.1002/elps.200600128. [DOI] [PubMed] [Google Scholar]
- 36.Keedy DA, et al. Crystal cryocooling distorts conformational heterogeneity in a model Michaelis complex of DHFR. Structure. 2014;22(6):899–910. doi: 10.1016/j.str.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett BC, Gardberg AS, Blair MD, Dealwis CG. On the determinants of amide backbone exchange in proteins: A neutron crystallographic comparative study. Acta Crystallogr D Biol Crystallogr. 2008;D64(Pt 7):764–783. doi: 10.1107/S0907444908012845. [DOI] [PubMed] [Google Scholar]
- 38.Casarotto MG, Basran J, Badii R, Sze KH, Roberts GC. Direct measurement of the pKa of aspartic acid 26 in Lactobacillus casei dihydrofolate reductase: Implications for the catalytic mechanism. Biochemistry. 1999;38(25):8038–8044. doi: 10.1021/bi990301p. [DOI] [PubMed] [Google Scholar]
- 39.Bystroff C, Oatley SJ, Kraut J. Crystal structures of Escherichia coli dihydrofolate reductase: The NADP+ holoenzyme and the folate/NADP+ ternary complex. Substrate binding and a model for the transition state. Biochemistry. 1990;29(13):3263–3277. doi: 10.1021/bi00465a018. [DOI] [PubMed] [Google Scholar]
- 40.Howell EE, Booth C, Farnum M, Kraut J, Warren MS. A second-site mutation at phenylalanine-137 that increases catalytic efficiency in the mutant aspartate-27–serine Escherichia coli dihydrofolate reductase. Biochemistry. 1990;29(37):8561–8569. doi: 10.1021/bi00489a009. [DOI] [PubMed] [Google Scholar]
- 41.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheldrick GM, Schneider TR. SHELXL: High-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.