

Abstract
Double minute chromosomes (DMs) are extrachromosomal cytogenetic structures found in tumour cells. As hallmarks of gene amplification, DMs often carry oncogenes and drug-resistance genes and play important roles in malignant tumour progression and drug resistance. The mitogen-activated protein kinase (MAPK) signalling pathway is frequently dysregulated in human malignant tumours, which induces genomic instability, but it remains unclear whether a close relationship exists between MAPK signalling and DMs. In the present study, we focused on three major components of MAPK signalling, ERK1/2, JNK1/2/3 and p38, to investigate the relationship between MAPK and DM production in tumour cells. We found that the constitutive phosphorylation of ERK1/2, but not JNK1/2/3 and p38, was closely associated with DMs in tumour cells. Inhibition of ERK1/2 activation in DM-containing and ERK1/2 constitutively phosphorylated tumour cells was able to markedly decrease the number of DMs, as well as the degree of amplification and expression of DM-carried genes. The mechanism was found to be an increasing tendency of DM DNA to break, become enveloped into micronuclei (MNs) and excluded from the tumour cells during the S/G2 phases of the cell cycle, events that accompanied the reversion of malignant behaviour. Our study reveals a linkage between ERK1/2 activation and DM stability in tumour cells.
Keywords: double minute chromosomes, malignant tumour, MAPK signalling pathway, ERK1/2 constitutive phosphorylation
Introduction
Gene amplification is an important molecular manifestation of genomic instability in malignant tumour cells and typically occurs via two types of abnormal chromosomal structures, homogeneously staining regions (HSRs) and double minute chromosomes (DMs) 1,2. DMs are small, paired, acentric, circular extrachromosomal elements that frequently carry oncogenes and drug-resistance genes, playing vital roles in tumourigenesis, cellular proliferation, anti-apoptosis and drug resistance 3,4. The factors that affect DM production in tumours remain unclear, yet the elimination of DMs is a feasible approach for decreasing the malignancy of cancer cells that contain oncogenic gene amplification and represents a powerful strategy for the biological therapy of harmful tumours.
Malignant tumour cells are characterized by the abnormal activation of cellular signalling pathways, including in particular the mitogen-activated protein kinase (MAPK) family, composed of a group of conserved cellular serine/threonine protein kinases 5–8. To date, six parallel MAPK signalling pathways have been identified in mammalian cells and three have been studied extensively: the extracellular-regulated protein kinase (ERK) 1/2 pathway; the c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) 1/2/3 pathway; and the p38 isomer α/β/γ/δ pathway 9–11. Dysregulated MAPK signalling is frequently related to chromosomal genomic instability, particularly gene amplification 12–14. Oncogenes involved in the activation of the MAPK signalling pathway, such as MYC, EGFR, MDM2, HER-2/NEU, C-ERBB-2, MYB, C-MET, CDK2, ETS1, FGFR2 and RAS, have been found to be amplified in the form of DMs 15–20. However, it remains unclear whether there is a direct association between dysregulation of MAPK signalling and DM production. In the present study, we aimed to elucidate the relationship between the activation of MAPK signalling pathways and DM production. We reveal a strong link between ERK1/2 activation and DM production in tumour cells, providing a target for clinical therapy by specifically eliminating DMs and halting the amplification of oncogenes from tumour cells with constitutively phosphorylated ERK1/2.
Materials and methods
Cell lines, cell culture and chemical inhibitor treatment
The human ovarian cancer cell line UACC-1598, containing spontaneous DMs, was kindly provided by Dr Xin-Yuan Guan (University of Hong Kong) 21. The UACC-1598DM and UACC-1598HSR cell lines are two subclones of UACC-1598 generated by serial dilution selection. The UACC-1598DM cell line shows stable maintenance of a high number of DMs, while the UACC-1598HSR cell line contains HSRs without DMs. The human colorectal cancer cell lines NCI-H716, NCI-H508, Colo320DM, Colo320HSR, SW480 and SK-CO-1, human neuroectodermal tumour cell line SK-PN-DW, human myeloid leukaemia cell line HL-60, human gastric cancer cell line NCI-N87, human breast ductal carcinoma cell line T-47D and human embryonic kidney cell line HEK293T were all purchased from ATCC (Manassas, VA, USA). UACC-1598, UACC-1598DM, UACC-1598HSR, NCI-H716, NCI-H508, NCI-N87, Colo320DM and Colo320HSR cells were maintained in RPMI-1640 medium, SK-CO-1 and T-47D cells in minimal essential medium (MEM), SK-PN-DW and HEK293T cells in Dulbecco's modified Eagle's medium (DMEM), HL-60 cells in Iscove's modified Dubecco's medium (IMDM) and SW480 cells in Leibovitz's L-15 medium (all from Invitrogen, Grand Island, NY, USA), all supplemented with 10% fetal bovine serum (FBS). The tumour cells were treated for 2 weeks with either the MAPK-ERK1/2 inhibitor U0126 (10 µm) or PD98059 (10 µm), the MAPK-JNK inhibitor SP600125 (10 µm) or the MAPK-p38 inhibitor SB203580 (10 µm) (all from Merck KgaA, Darmstadt, Germany).
Antibodies and reagents
The anti-phospho-ERK1/2 (Thr202/Tyr204), anti-ERK1/2, anti-phospho-SAPK/JNK (Thr183/Tyr185), anti-SAPK/JNK, anti-phospho-p38 MAP kinase (Thr180/Tyr182) and anti-p38 MAP kinase antibodies were obtained from Cell Signalling Technology (Danvers, MA, USA); anti-MCL1 and anti-MYC from Santa Cruz Biotechnology (Dallas, TX, USA); anti-EIF5A2 from Sigma-Aldrich (St. Louis, MO, USA); anti-GAPDH from KangChen Bio-tech Inc. (Shanghai, China); and anti-phospho-histone H2A.X (Ser139) from Merck Millipore (Bedford, MA, USA). Anti-mouse and anti-rabbit antibodies were obtained from Rockland Immunochemicals (Gilbertsville, PA, USA). The BAC clones RP11-54A4, RP11-115 J24, RP11-89 K10, and RP11-440 N18 were purchased from the BACPAC Resources Center (Children's Hospital Oakland, Oakland, CA, USA). Cy3 was obtained from GE Healthcare (Madison, WI, USA), Green-dUTP from Enzo Life Science (New York, NY, USA), and 4,6-diamidino-2-phenylindole (DAPI) from Roche (Alameda, CA, USA). Recombinant human TNFα was purchased from BioVision (Mountain View, CA, USA).
Vector construction and generation of stable knock-down cell lines
Oligonucleotides containing siRNA sequences of the target genes were designed, and named siERK1-1, siERK1-2, siERK2-1, siERK2-2 and sicontrol (see supplementary material, Table S1). The annealed oligos were cloned into the pSUPER.retro vector (Oligoengine, Boston, MA, USA) and sequenced. The recombinant pSUPER-sh-RNA vectors were transfected into UACC-1598 cells with Lipofectamine™ 2000 transfection reagent (Invitrogen). Stable clones were selected with 1.0 µg/ml puromycin (Sigma-Aldrich) for 7 days. Knock-down efficiency was evaluated by immunoblotting.
Microarray analysis
Total RNA of UACC-1598DM and UACC-1598HSR cells were isolated using TRIzol (Invitrogen), according to the manufacturer's protocol, and then applied to the Agilent Oligo Microarray (ShanghaiBio, Shanghai, China) which contains 41093 transcripts and 30951 known genes, with three biological repeats. Data obtained from the microarray were standardized and mRNA expression values transformed using a log2 ratio. Up- and down-regulated genes were analysed separately for Gene Ontology (GO) enrichment and Pathway Express 22–24.
Immunoblotting and quantitative polymerase chain reaction (qPCR)
For immunoblotting, cell lysates were extracted as previously described 25, subjected to 10% SDS–PAGE electrophoresis and transferred to PVDF membranes, followed by immunoblotting with primary antibodies and fluorescent labelled secondary antibodies. Images were scanned using the Odyssey Infrared Imaging System (LI-COR Biosciences, USA). For qPCR, DNA was isolated from cells using the QIAamp DNA Extraction Kit (Qiagen, Valencia, CA, USA), according to the manufacturer's protocol. DNA amplification of target genes was normalized against ACTB and each target gene was measured in triplicate. The primers were designed using Primer 3.0 software and are shown in Table S2 (see supplementary material).
Preparation of metaphase spreads, FISH analysis and micronucleus (MN) scoring
Tumour cells were harvested after demecolcine treatment (Sigma Aldrich), followed by hypotonic incubation and fixation. The cell suspension was dropped onto slides for fluorescence in situ hybridization (FISH) or Giemsa staining. The chromosomes and DMs were observed and counted by microscopy (Olympus, Japan). The BAC clones RP11-54A4, RP11-115 J24, RP11-89 K10 and RP11-440 N18 were labelled with Spectrum Cy3-dUTP or Green-dUTP, and then hybridized to interphase and metaphase spreads of UACC-1598, UACC-1598DM or Colo320DM cells, as described previously 26; the slides were counterstained with DAPI. High-quality interphase and metaphase images were captured using a Zeiss Axioskop fluorescence microscope (Oberkochen, Germany) and analysed using the MetaMorph Imaging System (Universal Imaging, West Chester, PA, USA); approximately 100 interphase cells were evaluated for each group. We also observed MNs production, and MNs with fluorescence were scored as positive (MN+), whereas MNs without fluorescence were scored as negative (MN−).
Cell proliferation, colony formation and cell invasion assays
UACC-1598 and Colo320DM cells were measured for cell viability over the course of 5–6 days, using the CellTiter 96®Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) according to the manufacturer's protocol. Colony-formation assays were performed by growing the cells for 14 days and staining with Giemsa. Three independent experiments were performed. A cell invasion assay was performed using the BD BioCoat™ Matrigel™ Invasion Chamber (BD Bioscience, Bedford, MA, USA) according to the manufacturer's protocol. Cells (5 × 104/well) were added to the upper chamber of the 24-well Transwell inserts and incubated for 72 h. Cells that had migrated to the lower surface of the membrane were fixed, stained and counted.
Results
Human ovarian cancer UACC-1598 cells and colorectal adenocarcinoma Colo320DM cells have constitutive ERK1/2 phosphorylation and contain a large number of DMs
We initially selected six human tumour cell lines known to contain DMs for the present study: UACC-1598, NCI-H716, NCI-H508, SK-PN-DW, HL-60 and NCI-N87. We found that the cell lines differed in the number of DMs, with the averages varying from < 2 DMs in NCI-N87 cells to > 40 DMs in UACC-1598, NCI-H716, NCI-H508 and SK-PN-DW cells (Figure 1A). Next, we determined the activity status of the MAPK-ERK1/2, p38 and JNK1/2/3 signalling pathways in these cell lines. We detected the phosphorylation status of ERK1/2, p38 and JNK1/2/3 by immunoblotting. As a positive control for ERK1/2, p38 and JNK1/2/3 phosphorylation, we over-expressed MEKK3, over-expressed p38 or stimulated with TNFα, respectively, in HEK293T, and showed that UACC-1598 has constitutive ERK1/2 phosphorylation, while p38 and JNK1/2/3 were not constitutively phosphorylated in any of the cell lines we tested (Figure 1B–D).
Figure 1.

Double minute chromosome (DM)-containing tumour cells often show constitutive activation of the ERK1/2–MAPK pathway. (A, F) The number of DMs/cell in various human malignant tumour cells (bars represent mean ± SD). (B–D, G) Phosphorylation status of ERK1/2, p38 and JNK1/2/3 in DM-containing tumour cells; phosphorylation of ERK1/2, p38 and JNK1/2/3 in MEKK3- or p38-over-expressing or TNFα-treated HEK293T cells (10 ng/ml for 30 min) are shown as positive controls. (E) Microarray analysis of UACC-1598DM compared to UACC-1598HSR cells
We established the UACC-1598DM (enriched for DMs) and UACC-1598HSR (enriched for HSRs but with no DMs) cell lines (see supplementary material, Figure S1A, B) and performed microarray analysis to evaluate the gene expression differences between the two. We found that the expression of MAPK pathway components was significantly different between the two (Figure 1E), consistent with our hypothesis that MAPK signalling might play vital roles in DM production. Furthermore, we collected five other known DM- and/or HSR-containing cell lines (Colo320DM, Colo320HSR, SW480, SK-CO-1 and T-47D) to further evaluate the abnormal pathways involved in DM production. We found that Colo320DM also carried many DMs and had constitutive ERK1/2 phosphorylation, similar to UACC-1598 (Figure 1F, G). Human ovarian cancer UACC-1598 cells and colorectal adenocarcinoma Colo320DM cells were used for the ensuing experiments.
Phosphorylated ERK1/2 mediates DM production and DM-carried gene amplification
To analyse whether a direct association exists between MAPK activation and DMs in tumour cells, we treated UACC-1598 and Colo320DM cells for 2 weeks with U0126 and PD98059 to target ERK1/2, SP600125 for JNK1/2/3 and SB203580 for p38 to inhibit the phosphorylation and activation of these kinases. By counting the number of DMs in approximately 100 cells for each group, we found that DMs were reduced by U0126 and PD98059 treatment (Figure 2A, B). We obtained the same result using UACC-1598DM cells (see supplementary material, Figure S1C). The number of DMs was not altered by SP600125 and SB203580 treatment, demonstrating that inhibiting the activation of other MAPK pathways does not affect DMs in tumour cells (Figure 2C, D). These results demonstrate that inhibiting ERK1/2 activity could decrease the number of DMs in tumour cells with constitutive ERK1/2 phosphorylation. To evaluate whether MAPK-ERK1/2 inhibitors affect DMs in DM-containing tumour cells without constitutive ERK1/2 phosphorylation, we incubated NCI-H716 and NCI-H508 cells with U0126 or PD98059 for 2 weeks. Interestingly, we did not detect an obvious variation in DM numbers with inhibitor treatment compared to DMSO treatment (Figure 2E, F).
Figure 2.

The number of double minute chromosomes (DMs) decreases after inhibition of ERK1/2 phosphorylation. (A, B) ERK1/2 inhibitors decrease the number of DMs in UACC-1598 and Colo320DM cells (mean number of DMs/cell ± SD); *p < 0.05, **p < 0.01, ***p < 0.001, by ANOVA and Dunnett's multiple comparison post-test. (C, D) The numbers of DMs in UACC-1598 or Colo320DM cells are not affected by JNK1/2/3 or p38 inhibitor treatment (SP600125 or SB203580). (E, F) The numbers of DMs in NCI-H716 and NCI-H508 cells are not affected by ERK1/2 inhibitor treatment
Based on the result that inhibiting constitutive phosphorylation of ERK1/2 is able to decrease the number of DMs, we next addressed whether inhibiting ERK1/2 phosphorylation would affect the amplification of DM-carried genes. The oncogenes MCL1, MYCN and EIF5A2 are highly amplified via DMs in UACC-1598 cells, and FAM84B and MYC are highly amplified via DMs in Colo320DM 21,27–29. After 2 weeks of U0126 and PD98059 treatment, qPCR showed that amplification levels of these genes were significantly reduced in the respective cells compared to controls (Figure 3A–E) and that their expression levels were also decreased (Figure 3F–H).
Figure 3.

Decreased copy number and expression level of double minute chromosome (DM)-carried genes during inhibition of ERK1/2 phosphorylation. (A–E) Copy numbers of DM-carried genes (as determined by qPCR; mean ± SD, arbitrary, normalized to control); *p < 0.05, **p < 0.01, ***p < 0.001, by ANOVA and Dunnett's multiple comparison post-test. (F–H) Immunoblotting of proteins encoded by DM-carried genes with ERK1/2 inhibitor treatments. (I, J) The level of amplification of DM-carried genes (as assayed by FISH). The classification of interphase cells into five groups according to the coverage area and distribution of fluorescent signal is shown. BAC clone RP11-54A4 (covering MCL1) and BAC clone RP11-115 J24 (covering EIF5A2), labelled in green, or Cy3, respectively, were used as FISH probes in UACC-1598 cells, and BAC clones RP11-89 K10 (covering FAM84B) and RP11-440 N18 (covering MYC), labelled in green, or Cy3, respectively, were used as FISH probes in Colo320DM cells. The chart displays the distribution of ERK1/2 inhibitor-treated interphase cells with FISH signals
We next performed cytogenetic analyses to further confirm the influence of ERK1/2 dephosphorylation on DMs and amplification of DM-carried oncogenes. We selected the BAC clones RP11-54A4 (GRCh37/hg19, Chr1:150445215–150628209) and RP11-115 J24 (Chr3:170607523–170769158), specific for the UACC-1598 DM-carried genes MCL1 (Chr1:150547032–150552066) and EIF5A2 (Chr3:170606204–170626426), labelled green or with Cy3, respectively; and BACs RP11-89 K10 (Chr8:127567520–127730253) and RP11-440 N18 (Chr8:128596756–128777986), specific for the Colo320DM DM-carried genes FAM84B (Chr8:127564683–127570711) and MYC (Chr8:128748315–128753680), as probes to perform FISH analysis of inhibitor-treated and control interphase and metaphase cells. To quantify the coverage area and distribution of fluorescence, cells were divided into five groups: dispersed 0–30% coverage; clustered 0–30% coverage; dispersed 30–60% coverage; clustered 30–60% coverage; and 60–100% coverage, indicating a dispersed or clustered distribution of the signal in the cells (Figure 3I) 29; > 100 cells for each treatment and control group were counted and the data are illustrated in Figure 3I. In control UACC-1598 cells, more cells hybridized with dispersed signals in the 0–30% and 30–60% groups (21.0% and 18.1%) than with clustered signals (12.3% and 13.8%), indicating that the amplified genes are located on DMs in a dispersed distribution. In the inhibitor-treated groups, the percentage of cells hybridizing with cluster signals in the 0–30% group (23.7% in U0126 and 33.3% in PD98059) was increased, suggesting a tendency of gene amplification to cluster when ERK1/2 phosphorylation is inhibited. Additionally, in response to inhibitor treatment, the amount of fluorescence and the percentage of cells with strongly hybridized signals were reduced (34.7% to 23.6% and 20.6%), which is statistically significant according to Ridit analysis (relative to an identified distribution unit analysis for multiple comparisons of ranked data from multiple groups; SAS9.2 software) at p < 0.01. We also found the same phenomenon in Colo320DM cells (Figure 3J) and UACC-1598DM cells (see supplementary material, Figure S1D), showing that DMs and the amplification levels of DM-carried oncogenes are reduced with MAPK–ERK1/2 inhibitor treatment, consistent with the above qPCR results. Taken together, we conclude that blocking the phosphorylation of the MAPK–ERK1/2 signalling pathway contributes to decreasing amplification levels of DM-carried oncogenes in tumour cells with constitutive ERK1/2 phosphorylation.
DM DNA double strands are prone to breakage, such that DMs cluster, collect in MNs and are lost from tumour cells
In further analysis of the FISH data, we found a number of MNs with or without fluorescent signal in UACC-1598 and Colo320DM cells (Figure 4A). MNs are abnormal structures composed of small fragments of broken chromosomes in eukaryotic cells. To clarify the mechanism for the decreasing numbers of DMs, we found that the number of MNs and MNs with fluorescent signals (MN+) were higher after inhibitor treatment compared to control (1.58- and 2.71-fold in UACC-1598 cells; 2.08- and 2.51-fold in Colo320DM cells; Tables 1 and 2). These results were consistent with that seen in UACC-1598DM cells (see supplementary material, Table S3) and indicate that DM-amplified oncogenes were lost from tumour cells after being entrapped in MNs. In metaphase cells, we also detected the induction of HSR formation with fluorescent signals in the inhibitor treatment groups (Figures 4B–D), suggesting that DMs may be converted to HSRs in these cells. Furthermore, we analysed the timing of the elimination of DMs during the cell cycle, using fluorescence-activated cell sorting (FACS). We found that the percentage of cells in G1 phase was decreased in the inhibitor treatment groups, whereas the percentage of cells in the S + G2 phases was significantly increased (Figure 4E; see also supplementary material, Figure S1E). Taken together, dispersed DMs aggregate in S/G2 with ERK1/2 dephosphorylation and participate in MN or HSR formation, with the number of DMs and the amplification levels of DM-carried oncogenes eventually decreasing.
Figure 4.
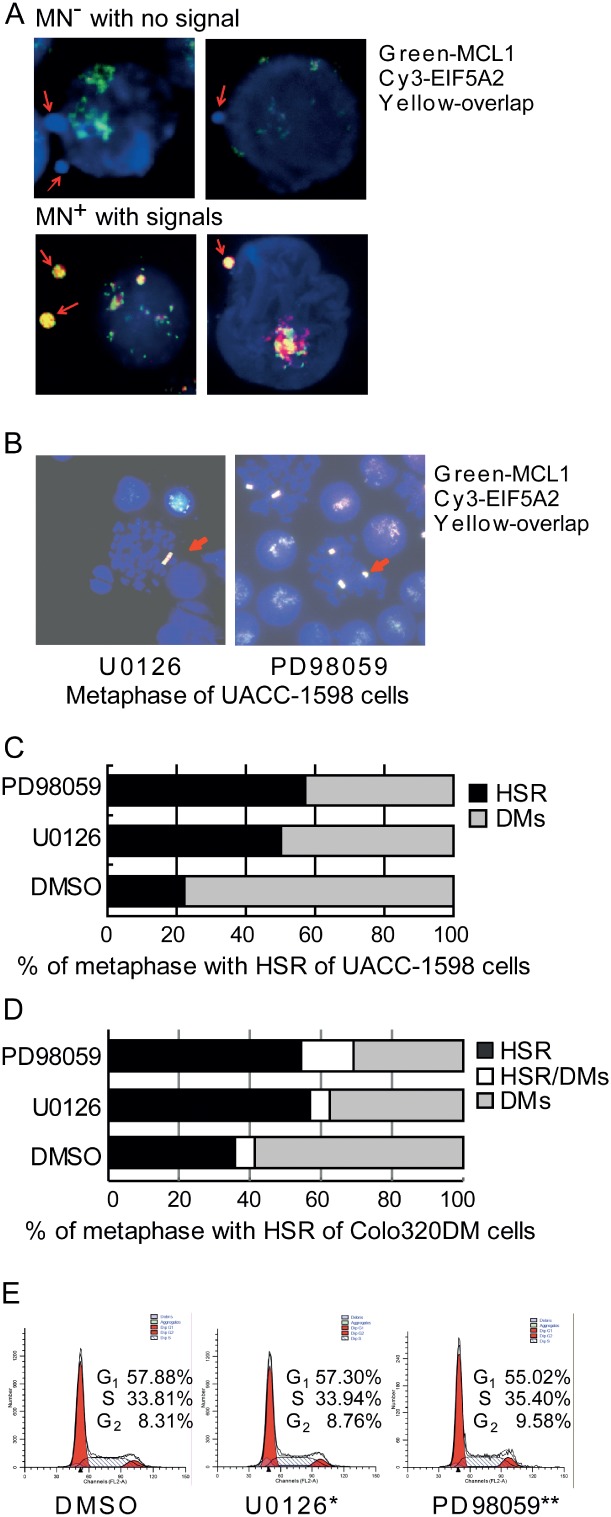
Double minute chromosome-amplified oncogenes are lost as micronuclei or transformed into homogeneously staining regions during the S/G2 phases of the cell cycle. (A) Representative micrographs indicating the two groups of miconuclei in UACC-1598 cells: those with fluorescence signals (MN+) and those without (MN−). (B) Homogeneously staining regions (HSRs) are present in metaphase UACC-1598 cells with U0126 and PD98059 treatment. (C, D) The percentage of HSRs with U0126 and PD98059 treatment. (E) The cell cycle distribution of ERK1/2 inhibitor-treated UACC-1598 cells; *p < 0.025, **p < 0.005; χ2 test compared to DMSO
Table 1.
Number of micronuclei (MN) in ERK1/2 inhibitor-treated UACC-1598 cells
|
MN-DM+ |
MN-DM− |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| UACC-1598 cells | No. of cells | No. of cells with MNs | MN frequency (×10−2) | No. of MN-DM+ | MN-DM+/ MN (%) | Frequency (×10−2) | Fold change over control | Frequency (×10−2) | Fold change over control |
| DMSO | 138 | 23 | 16.67 | 6 | 26.09 | 4.35 | 1.00 | 12.32 | 1.00 |
| U0126 | 131 | 21 | 16.03 | 9 | 42.86 | 6.87 | 1.58 | 9.16 | 0.74 |
| PD98059 | 204 | 61 | 29.90 | 24 | 39.34 | 11.76 | 2.71 | 18.14 | 1.47 |
Table 2.
Number of micronuclei (MN) in ERK1/2 inhibitor-treated Colo320DM cells
|
MN-DM+ |
MN-DM− |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Colo320DM cells | No. of cells | No. of cells with MNs | MN frequency (×10−2) | No. of MN- DM+ | MN-DM+ /MN (%) | Frequency (×10−2) | Fold change over control | Frequency (×10−2) | Fold change over control |
| DMSO | 1499 | 38 | 2.54 | 20 | 52.63 | 1.33 | 1.00 | 1.20 | 1.00 |
| U0126 | 1769 | 94 | 5.31 | 49 | 52.13 | 2.77 | 2.08 | 2.54 | 2.12 |
| PD98059 | 1552 | 91 | 5.86 | 52 | 57.14 | 3.35 | 2.51 | 2.51 | 2.09 |
DNA double-strand breakage (DSB) occurs during MN and HSR formation 30,31. γ-H2AX, a phosphorylated form of histone H2AX, is recruited to DSBs and can be detected by the formation of foci by immunostaining 32. In our study, we examined whether DSB occurred in DMs during the process of DM elimination. UACC-1598DM cells grown on coverslips were treated with U0126 or PD98059 for 24 h (with DMSO as control), followed by immunofluorescence with anti-γ-H2AX antibodies. The cells were divided into four groups, according to the area that the γ-H2AX fluorescent foci bright signal covered: no signal, 0–30% signal, 30–60% signal and 60–100% signal. Based on these data, we found that the number of cells with γ-H2AX focal signals was increased in the inhibitor treatment groups compared to controls (Figure 5A, B). Moreover, we found that MNs were excluded from the cells and that all MNs were positive for fluorescence (Figure 5C). By counting the number of MNs, we found that the frequency of MN production and the frequency of MNs with γ-H2AX signals were significantly higher in the MAPK–ERK1/2 inhibitor groups compared to the control group (2.33- and 1.81-fold, respectively) (Table3). These results suggest that DM DNA is prone to breakage, resulting in DM aggregation, entrapment in MNs and loss from the cells.
Figure 5.
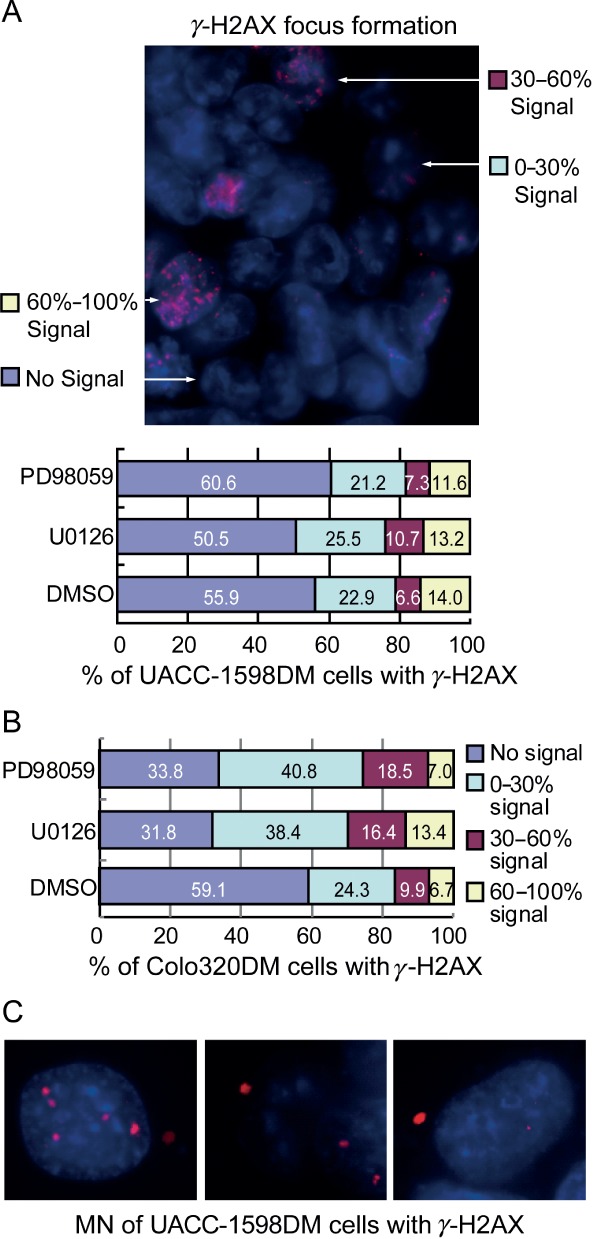
Tumour cells produce γ-H2AX foci under ERK1/2 inhibitor treatment. (A, B) γ-H2AX foci in UACC-1598DM or Colo320DM cells were divided into four groups, according to the area of fluorescent signal. (C) Micronuclei (MN) with γ-H2AX fluorescent signal in UACC-1598DM cells
Table 3.
Number of micronuclei (MN) in ERK1/2 inhibitor-treated UACC-1598DM cells with γ-H2AX immunofluorescence
|
MN-DM+ |
|||||||
|---|---|---|---|---|---|---|---|
| UACC-1598DM cells | No. of cells | No. of cells with MNs | MN frequency (×10−2) | No. of MN-DM+ | MN-DM+/ MN (%) | Frequency (×10−2) | Fold change over control |
| DMSO | 140 | 6 | 4.29 | 6 | 100.00 | 4.29 | 1.00 |
| U0126 | 180 | 18 | 10.00 | 18 | 100.00 | 10.00 | 2.33 |
| PD98059 | 168 | 13 | 7.74 | 13 | 100.00 | 7.74 | 1.81 |
Dephosphorylation of ERK1/2 decreases malignant cell behaviour via DM elimination
We further tested the effect of the decrease in DMs on cellular behaviour. The growth rates of ERK1/2-dephosphorylated tumour cells were lower compared to control cells (Figure 6A, B), the numbers of colonies for the experimental groups were significantly less than the control group (Figure 6C, D) and the number of invading cells decreased with MAPK–ERK1/2 inhibitor treatment compared to control (Figure 6E). Taken together, the inhibition of ERK1/2 phosphorylation decreases malignant cell behaviour and is accompanied by the reduction of DMs, as well as DM-carried oncogene amplification and expression.
Figure 6.

ERK1/2 inhibitors decrease malignant cell properties. (A, B) MTS analysis of growth rate of UACC-1598 and Colo320DM cells with ERK1/2 inhibitor treatment compared to DMSO controls (mean OD value ± SD). (C) Colony-forming assay. (D) Statistical analysis of the colony-forming assay (mean number of colonies ± SD). (E) Statistical analysis of cell invasion assay of UACC-1598 with the addition of ERK1/2 inhibitors compared to DMSO (mean number of invading cells ± SD); *p < 0.05, **p < 0.01, ***p < 0.001, compared to the control group, by ANOVA and Dunnett's multiple comparison post-test
ERK1 and ERK2 are involved in DM production in tumour cells
To assess whether ERK1 or ERK2 participate in DM production, we established UACC-1598 stable knock-down cell lines, which express sh-ERK1-1 and sh-ERK2-1 and have lower levels of ERK1 or ERK2 compared to the sh-control, sh-ERK1-2 and sh-ERK2-2 cell lines (see supplementary material, Figure S2A, B). The number of DMs was dramatically reduced in sh-ERK1-1 and sh-ERK2-1 cells compared to the control, sh-ERK1-2 and sh-ERK2-2 cells (see supplementary material, Figure S2C). Furthermore, the amplification levels of DM-carried genes, MCL1, MYCN and EIF5A2, were all significantly reduced in sh-ERK1-1 and sh-ERK2-1 cells but not in sh-ERK1-2 and sh-ERK2-2 cells (see supplementary material, Figure S2D–F), which was confirmed by FISH analysis (see supplementary material, Figure S2G). In response to decreased ERK1 and ERK2 expression, the coverage area of fluorescence signals (indicating the amplification level of DM-carried genes) was reduced, and the percentage of the cells with strongly hybridized signals was also reduced, implying that reduction of ERK1 and ERK2 decreases the copy number of DM-carried genes.
Moreover, the frequency of MNs with fluorescent signals was higher and we found increased formation of HSRs with fluorescent signals in ERK1/2 knock-down cells than in control (see supplementary material, Figure S2H, I). FACS data revealed that more cells stayed in S/G2 phases when ERK1 and ERK2 were down-regulated (see supplementary material, Figure S2J). These results suggest that DMs are lost from cells after being entrapped in MNs or transformed into HSRs during S/G2 during ERK1 and ERK2 knock-down. In addition, along with the decrease of DMs, cell growth rates, cell colony formation and cell invasion were all decreased compared to the control group (see supplementary material, Figure S3A–C). Thus, inhibition of ERK1 and ERK2 by siRNA decreases malignant cell behaviour, accompanied by the reduction of DMs and DM-carried genes.
Discussion
According to the Mitelman database, the overall frequency of DMs is 1.4% for primary cancers, with DMs having the highest frequency in adrenal carcinoma (28.6%, by topography) and neuroblastoma (31.7%, by morphology) 4. DMs are malignant cytogenetic markers and the presence of DMs is closely correlated with tumour progression and drug resistance 4. Many chromosome regions carrying oncogenes or drug-resistance genes have been reported to be amplified via DMs in malignant tumour cells and drug-resistant cells 33–37. The elaboration of the molecular structure and the underlying molecular mechanism of DMs have emerged through work on the complex connection of amplicons and non-coding sequences 34,38,39. Matrix attachment regions (MARs) carried on DMs exert functional regulation on their target genes and are potentially involved in DM-mediated oncogene activation 40. Indeed, the importance of DMs is becoming clearer with detailed molecular research.
Snapka and Varshavsky 41 were the first to report that a low concentration of hydroxyurea (HU) could reduce the number of DMs and reverse the malignant behaviour of cells. Von Hoff et al 43 and other groups 42,44 have also confirmed that DMs could be slowly lost during selection. In view of the natural tendency of DMs to be lost during cell division, the elimination of DMs from tumour cells might be a novel strategy for DM-positive cancer therapy. Thus, the elucidation of the factors relevant to DMs is a major goal in cancer therapy. Our study was based on the approach of identifying the common abnormal events in malignant tumours. Using different DM-containing tumour cell lines as the platform, we found that the MAPK family ERK1/2 protein kinases were constitutively phosphorylated in ovarian cancer UACC-1598, its subclone UACC-1598DM, and Colo320DM cells. Using specific inhibitors, U0126 and PD98059, we report here that the dephosphorylation of constitutive p-ERK1/2, but not JNK1/2/3 and p38, reduced the number of DMs, degree of amplification and expression of DM-carried genes. We confirmed that the constitutive phosphorylation of ERK1/2 contributes to the production or maintenance of DMs in tumour cells. Moreover, we elaborated the mechanism of DMs loss through blockage of ERK1/2 phosphorylation, showing that DM DNA is prone to breakage, and reveal that DMs cluster, become entrapped in MNs and are lost from tumour cells or transformed into HSRs during S/G2 phase. With the loss of DMs and the decreased copy number of DM-carried genes, the cell's malignant behaviour can be reversed. Further, we identified that both ERK1 and ERK2 are involved in DM production and DM-carried gene amplification in tumour cells. We are currently collecting clinical samples to verify the association of ERK1/2 activation and DMs. As the frequency of DMs in tumours is limited, these studies remain for the future.
In conclusion, our data provide evidence that DM production and maintenance is strongly associated with constitutively phosphorylated EKR1/2 in tumour cells. The discovery adds new insight into our knowledge of the well-studied ERK1 and ERK2 kinases. In view of the data presented here and previous reports, we recommend that ERK1/2 inhibitors be considered for the specific treatment of DM-containing tumours with constitutive ERK1/2 phosphorylation.
Acknowledgments
This study was supported by the Programme for Changjiang Scholars and Innovative Research Team in University (Grant No. IRT1230, to YJ); the International Science and Technology Cooperation Programme of China (Grant No. 2013DFA31610, to SF); the National Natural Science Foundation of China (Grant Nos 31271347 and 31000626, to WS; Grant No. 81201761, to LY); the New Century Support Programme for the Excellent Scholar, Ministry of Education of China (Grant No. NCET-10-0149, to WS; Grant No. NCET-11-0954, to YY; Grant No. NCET-13-0758, to CZ); and Foundation for University Key Youth Scholar by Heilongjiang Province of China (Grant No. 1154G29, to YH).
Author contributions
WS, CQ, YH, JY and SF conceived and designed the study; WS, CQ, YH, WJ, LY, XL, YZ, ZZ, HZ, QL, PX, YF, LL, YZ and YC performed the experiments; WS, CQ, YH, LY, XL, XJ, XM, CZ, YJ, JB, JY, YY, JY and SF analysed the data; and WS, CQ, YH, LY, JY and SF prepared the manuscript. All authors revised and approved the final draft.
Supplementary material on the internet
The following supplementary material may be found in the online version of this article:
Figure S1. The numbers of double minute chromosomes (DMs) and copies of DM-carried genes decrease after inhibition of ERK1/2 phosphorylation.
Figure S2. Double minute chromosomes (DM) production and copy number of DM-carried genes decrease with knock-down of ERK1 and ERK2.
Figure S3. The malignancy of tumour cells is decreased with ERK1 and ERK2 knock-down.
Table S1. siRNA sequence
Table S2. Primers for qPCR
Table S3. Number of micronuclei (MN) in ERK1/2 inhibitor-treated UACC-1598DM cells
References
- Biedler JL, Spengler BA. A novel chromosome abnormality in human neuroblastoma and antifolate-resistant Chinese hamster cell lives in culture. J Natl Cancer Inst. 1976;57:683–695. doi: 10.1093/jnci/57.3.683. [DOI] [PubMed] [Google Scholar]
- Stark GR, Debatisse M, Giulotto E, et al. Recent progress in understanding mechanisms of mammalian DNA amplification. Cell. 1989;57:901–908. doi: 10.1016/0092-8674(89)90328-0. [DOI] [PubMed] [Google Scholar]
- Dahlberg PS, Jacobson BA, Dahal G, et al. ERBB2 amplifications in esophageal adenocarcinoma. Ann Thorac Surg. 2004;78:1790–1800. doi: 10.1016/j.athoracsur.2004.05.037. [DOI] [PubMed] [Google Scholar]
- Fan Y, Mao R, Lv H, et al. Frequency of double minute chromosomes and combined cytogenetic abnormalities and their characteristics. J Appl Genet. 2011;52:53–59. doi: 10.1007/s13353-010-0007-z. [DOI] [PubMed] [Google Scholar]
- Bradham C, McClay DR. p38 MAPK in development and cancer. Cell Cycle. 2006;5:824–828. doi: 10.4161/cc.5.8.2685. [DOI] [PubMed] [Google Scholar]
- Galabova-Kovacs G, Kolbus A, Matzen D, et al. ERK and beyond: insights from B-Raf and Raf-1 conditional knockouts. Cell Cycle. 2006;5:1514–1518. doi: 10.4161/cc.5.14.2981. [DOI] [PubMed] [Google Scholar]
- Kohno M, Pouyssegur J. Targeting the ERK signaling pathway in cancer therapy. Ann Med. 2006;38:200–211. doi: 10.1080/07853890600551037. [DOI] [PubMed] [Google Scholar]
- Torii S, Yamamoto T, Tsuchiya Y, et al. ERK MAP kinase in G cell cycle progression and cancer. Cancer Sci. 2006;97:697–702. doi: 10.1111/j.1349-7006.2006.00244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Gibson TB, Robinson F, et al. MAP kinases. Chem Rev. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Hagan S, Rath O, et al. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- Golding SE, Rosenberg E, Neill S, et al. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res. 2007;67:1046–1053. doi: 10.1158/0008-5472.CAN-06-2371. [DOI] [PubMed] [Google Scholar]
- Tanami H, Imoto I, Hirasawa A, et al. Involvement of overexpressed wild-type BRAF in the growth of malignant melanoma cell lines. Oncogene. 2004;23:8796–8804. doi: 10.1038/sj.onc.1208152. [DOI] [PubMed] [Google Scholar]
- Lopez-Gines C, Gil-Benso R, Benito R, et al. The activation of ERK1/2 MAP kinases in glioblastoma pathobiology and its relationship with EGFR amplification. Neuropathology. 2008;28:507–515. doi: 10.1111/j.1440-1789.2008.00911.x. [DOI] [PubMed] [Google Scholar]
- Saavedra HI, Fukasawa K, Conn CW, et al. MAPK mediates RAS-induced chromosome instability. J Biol Chem. 1999;274:38083–38090. doi: 10.1074/jbc.274.53.38083. [DOI] [PubMed] [Google Scholar]
- Cui Y, Borysova MK, Johnson JO, et al. Oncogenic B-Raf (V600E) induces spindle abnormalities, supernumerary centrosomes, and aneuploidy in human melanocytic cells. Cancer Res. 2010;70:675–684. doi: 10.1158/0008-5472.CAN-09-1491. [DOI] [PubMed] [Google Scholar]
- Kasiappan R, Shih HJ, Chu KL, et al. Loss of p53 and MCT-1 overexpression synergistically promote chromosome instability and tumorigenicity. Mol Cancer Res. 2009;7:536–548. doi: 10.1158/1541-7786.MCR-08-0422. [DOI] [PubMed] [Google Scholar]
- Kabil A, Silva E, Kortenkamp A. Estrogens and genomic instability in human breast cancer cells – involvement of Src/Raf/Erk signaling in micronucleus formation by estrogenic chemicals. Carcinogenesis. 2008;29:1862–1868. doi: 10.1093/carcin/bgn138. [DOI] [PubMed] [Google Scholar]
- Al-Mulla F, Hagan S, Al-Ali W, et al. Raf kinase inhibitor protein: mechanism of loss of expression and association with genomic instability. J Clin Pathol. 2008;61:524–529. doi: 10.1136/jcp.2007.046987. [DOI] [PubMed] [Google Scholar]
- Roh M, Gary B, Song C, et al. Overexpression of the oncogenic kinase Pim-1 leads to genomic instability. Cancer Res. 2003;63:8079–8084. [PubMed] [Google Scholar]
- Guan XY, Sham JS, Tang TC, et al. Isolation of a novel candidate oncogene within a frequently amplified region at 3q26 in ovarian cancer. Cancer Res. 2001;61:3806–3809. [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Tan Q, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Draghici S, Khatri P, Tarca AL, et al. A systems biology approach for pathway level analysis. Genome Res. 2007;17:1537–1545. doi: 10.1101/gr.6202607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Yu Y, Dotti G, et al. PPM1A and PPM1B act as IKKβ phosphatases to terminate TNFα-induced IKKβ–NF-κB activation. Cell Signal. 2009;21:95–102. doi: 10.1016/j.cellsig.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XY, Trent JM, Meltzer PS. Generation of band-specific painting probes from a single microdissected chromosome. Hum Mol Genet. 1993;2:1117–1121. doi: 10.1093/hmg/2.8.1117. [DOI] [PubMed] [Google Scholar]
- Guan XY, Fung JM, Ma NF, et al. Oncogenic role of eIF-5A2 in the development of ovarian cancer. Cancer Res. 2004;64:4197–4200. doi: 10.1158/0008-5472.CAN-03-3747. [DOI] [PubMed] [Google Scholar]
- Camps J, Nguyen QT, Padilla-Nash HM, et al. Integrative genomics reveals mechanisms of copy number alterations responsible for transcriptional deregulation in colorectal cancer. Genes Chromosomes Cancer. 2009;48:1002–1017. doi: 10.1002/gcc.20699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Zhao Y, Quan C, et al. Gemcitabine eliminates double minute chromosomes from human ovarian cancer cells. PLoS One. 2013;8:e71988. doi: 10.1371/journal.pone.0071988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech M, Kirsch-Volders M, Natarajan AT, et al. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 2011;26:125–132. doi: 10.1093/mutage/geq052. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Misaka N, Utani K. Nonselective DNA damage induced by a replication inhibitor results in the selective elimination of extrachromosomal double minutes from human cancer cells. Genes Chromosomes Cancer. 2007;46:865–874. doi: 10.1002/gcc.20473. [DOI] [PubMed] [Google Scholar]
- Lobrich M, Shibata A, Beucher A, et al. γH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010;9:662–669. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- Streubel B, Valent P, Jager U, et al. Amplification of the MLL gene on double minutes, a homogeneously staining region, and ring chromosomes in five patients with acute myeloid leukemia or myelodysplastic syndrome. Genes Chromosomes Cancer. 2000;27:380–386. [PubMed] [Google Scholar]
- Vogt N, Lefevre SH, Apiou F, et al. Molecular structure of double-minute chromosomes bearing amplified copies of the epidermal growth factor receptor gene in gliomas. Proc Natl Acad Sci USA. 2004;101:11368–11373. doi: 10.1073/pnas.0402979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel F, Bris MJ, Herry A, et al. Double minutes containing amplified bcr–abl fusion gene in a case of chronic myeloid leukemia treated by imatinib. Eur J Haematol. 2003;70:235–239. doi: 10.1034/j.1600-0609.2003.00046.x. [DOI] [PubMed] [Google Scholar]
- Kawamata N, Zhang L, Ogawa S, et al. Double minute chromosomes containing MYB gene and NUP214–ABL1 fusion gene in T cell leukemia detected by single nucleotide polymorphism DNA microarray and fluorescence in situ hybridization. Leuk Res. 2009;33:569–571. doi: 10.1016/j.leukres.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CY, Feng YX, Yu Y, et al. The molecular mechanism of resistance to methotrexate in mouse methotrexate-resistant cells by cancer drug resistance and metabolism SuperArray. Basic Clin Pharmacol Toxicol. 2006;99:141–145. doi: 10.1111/j.1742-7843.2006.pto_470.x. [DOI] [PubMed] [Google Scholar]
- Storlazzi CT, Fioretos T, Surace C, et al. MYC-containing double minutes in hematologic malignancies: evidence in favor of the episome model and exclusion of MYC as the target gene. Hum Mol Genet. 2006;15:933–942. doi: 10.1093/hmg/ddl010. [DOI] [PubMed] [Google Scholar]
- Zhu J, Yu Y, Meng X, et al. De novo-generated small palindromes are characteristic of amplicon boundary junction of double minutes. Int J Cancer. 2013;133:797–806. doi: 10.1002/ijc.28084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Liu Z, Cao W, et al. Novel functional MAR elements of double minute chromosomes in human ovarian cells capable of enhancing gene expression. PLoS One. 2012;7:e30419. doi: 10.1371/journal.pone.0030419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapka RM, Varshavsky A. Loss of unstably amplified dihydrofolate reductase genes from mouse cells is greatly accelerated by hydroxyurea. Proc Natl Acad Sci USA. 1983;80:7533–7537. doi: 10.1073/pnas.80.24.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazka P, Hrabeta J, Vicha A, et al. Expulsion of amplified MYCN from homogeneously staining chromosomal regions in neuroblastoma cell lines after cultivation with cisplatin, doxorubicin, hydroxyurea, and vincristine. Cancer Genet Cytogenet. 2010;196:96–104. doi: 10.1016/j.cancergencyto.2009.08.005. [DOI] [PubMed] [Google Scholar]
- Von Hoff DD, McGill JR, Forseth BJ, et al. Elimination of extrachromosomally amplified MYC genes from human tumor cells reduces their tumorigenicity. Proc Natl Acad Sci USA. 1992;89:8165–8169. doi: 10.1073/pnas.89.17.8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Nakamura H, Kadota T, et al. Loss of amplified c-myc genes in the spontaneously differentiated HL-60 cells. Cancer Res. 1994;54:3561–3567. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The numbers of double minute chromosomes (DMs) and copies of DM-carried genes decrease after inhibition of ERK1/2 phosphorylation.
Figure S2. Double minute chromosomes (DM) production and copy number of DM-carried genes decrease with knock-down of ERK1 and ERK2.
Figure S3. The malignancy of tumour cells is decreased with ERK1 and ERK2 knock-down.
Table S1. siRNA sequence
Table S2. Primers for qPCR
Table S3. Number of micronuclei (MN) in ERK1/2 inhibitor-treated UACC-1598DM cells