

Abstract
Autologous dermal fibroblasts (dFbs) are promising candidates for enhancing muscle regeneration in Duchenne muscular dystrophy (DMD) due to their ease of isolation, immunological compatibility, and greater proliferative potential than DMD satellite cells. We previously showed that mouse fibroblasts, after MyoD-mediated myogenic reprogramming in vivo, engraft in skeletal muscle and supply dystrophin. Assessing the therapeutic utility of this system requires optimization of conversion and transplantation conditions and quantitation of engraftment so that these parameters can be correlated with possible functional improvements. Here, we derived dFbs from transgenic mice carrying mini-dystrophin, transduced them by lentivirus carrying tamoxifen-inducible MyoD, and characterized their myogenic and engraftment potential. After cell transplantation into the muscles of immunocompetent dystrophic mdx4cv mice, tamoxifen treatment drove myogenic conversion and fusion into myofibers that expressed high levels of mini-dystrophin. Injecting 50,000 cells/µl (1 × 106 total cells) resulted in a peak of ~600 mini-dystrophin positive myofibers in tibialis anterior muscle single cross-sections. However, extensor digitorum longus muscles with up to 30% regional engraftment showed no functional improvements; similar limitations were obtained with whole muscle mononuclear cells. Despite the current lack of physiological improvement, this study suggests a viable initial strategy for using a patient-accessible dermal cell population to enhance skeletal muscle regeneration in DMD.
Introduction
Skeletal muscle has an inherent capacity for regeneration, where upon injury, resident stem cells called satellite cells participate in the repair of damaged myofibers. While this process is robust in normal, healthy tissue, pathologies of skeletal muscle can lead to higher susceptibility to contraction-induced injury in muscle fibers and reduced ability to repair damage.1 In the case of Duchenne muscular dystrophy (DMD), this leads to ongoing cycles of degeneration and regeneration and ultimately much of the skeletal muscle is replaced with adipose and fibrotic connective tissue.2
DMD is an X-linked recessive disorder occurring in about 1 in 3,500 live male births. It is caused by mutations in the 2.1 Mb dystrophin gene, producing severe, progressive muscle wasting, requiring assisted mobility by the early teens, and is lethal in the twenties to thirties due to cardiac and respiratory muscle failure.2 While no effective treatments currently exist to halt DMD progression, experimental gene replacement and exon-skipping therapies are advancing rapidly.
Cell-based therapies show potential as an effective approach for gene delivery and restoration of skeletal muscle regeneration, especially if used in conjunction with viral gene delivery and/or exon skipping strategies.3 Many cell types have been explored for their potential to contribute to skeletal muscle in the context of muscle wasting disorders.4 While engraftment has been demonstrated, efficiency is reduced due to immune clearance, hypoxia, apoptosis, and minimal migration from the site of injection.5 Cell source also remains an issue; for example, muscle-derived satellite cells are difficult to isolate and expand, and cells from dystrophic tissue may have intrinsic properties, such as low population doubling capacities and precocious differentiation, that limit their therapeutic applications.6,7 In addition, the clinical efficacy and long-term benefit of cell-based therapies to muscle function is yet unclear; thus further basic studies in this area are beneficial.
For autologous-derived cell transplantations, fibroblasts have shown promise in a variety of tissues, including bone, neural, cardiac, and skeletal muscle.8 Importantly, dermal fibroblasts (dFbs) are readily accessible in patients, expandable in vitro after a minimally invasive dermal biopsy,9 and possess increased mitotic activity and resistance to differentiation into myofibroblasts compared with fibroblasts from other tissues.10
Fibroblasts from various origins can undergo myogenic conversion with the addition of the basic helix-loop-helix skeletal muscle-specific transcription factor MyoD.11–14 However, forced expression of MyoD can lead to cell growth arrest and differentiation,13,15 thereby reducing the engraftment capacity of donor cells into host skeletal muscle. Improved control over the timing of myogenic differentiation has been achieved by MyoD activation systems, such as tetracycline-induced MyoD expression, and by the MyoD-estrogen receptor fusion protein that is transported into nuclei in response to estradiol.16,17 These systems are not well suited for in vivo studies due to the need for delivery of the tetracycline transactivator protein and because of the ability of natural estrogens to activate the estrogen receptor.16 The development of estrogen receptor mutations allowing selective binding of the drug 4-hydroxytamoxifen (4OHT) has given rise to the MyoD-ER(T) fusion protein; the 4OHT-mediated inducible system provides greatly increased posttranslational control of MyoD activity in an in vivo environment.18,19
We previously demonstrated in vivo myogenic conversion of neonatal tail tip fibroblasts from mdx4cv mice using the inducible MyoD-ER(T) system.19 Here, we investigated conversion and transplantation conditions and described muscle-wide quantitation of engraftment of myogenically converted dFbs carrying a therapeutically relevant mini-dystrophin transgene.20,21
Results
dFbs efficiently convert into the myogenic lineage
To explore the myogenic conversion of dFbs isolated from mdx4cv mice expressing a therapeutic dystrophin, we transduced cells isolated from the whole skins of neonatal transgenic miniDys-eGFP/mdx4cv (miniDys-GFP)22 mice with a lentiviral vector expressing MyoD-ER(T) (Figure 1a). Beginning 48 hours posttransduction, MyoD-ER(T) was activated by three daily additions of 4OHT to the culture media (Figure 1b). After several days, myosin heavy chain (MHC) and miniDys-GFP expressing cells were observed (Figure 1c). Converted dFbs were compared to converted mouse 10T1/2 cells, a positive control for myogenic conversion after addition of MyoD, and to MM14 mouse myoblasts under conditions that promote differentiation (see Materials and Methods) (Figure 2a). A similar percentage of dFbs and 10T1/2 cells expressed MyoD, as detected by immunofluorescence, after transduction with MyoD-ER(T) (Figure 2b). Following 4OHT treatment, ~80% of dFbs became MHC+, and ~50% fused into multinucleated myotubes. This was roughly twice the efficiency observed for 10T1/2 cells (Figure 2b); but not as high as the >90% of MM14 cells that became MHC+ and the ~80% that fused into multinucleated myotubes (data not shown). Similarly, when individual clones were evaluated for stable myogenic conversion, only ~30% of 10T1/2 clones were myogenic while nearly 80% of the dFb clones expressed MHC (Figure 2c). Importantly, >90% of the MHC+ dFbs in high density cultures expressed the miniDys-GFP transgene (Figures 1c and 2d). In the absence of 4OHT, only ~0.3% of MyoD-ER(T)-transduced dFbs became MHC+, while cells not treated with 4OHT and not transduced displayed less than 0.1% MHC expression (see Supplementary Figure S1). Similarly, in dFbs not transduced with MyoD-ER(T), treatment with 4OHT also resulted in ~0.1% MHC expression. These data illustrate the extremely low spontaneous myogenic conversion rates of wildtype and MyoD-ER(T) dFbs in the absence of 4OHT.
Figure 1.
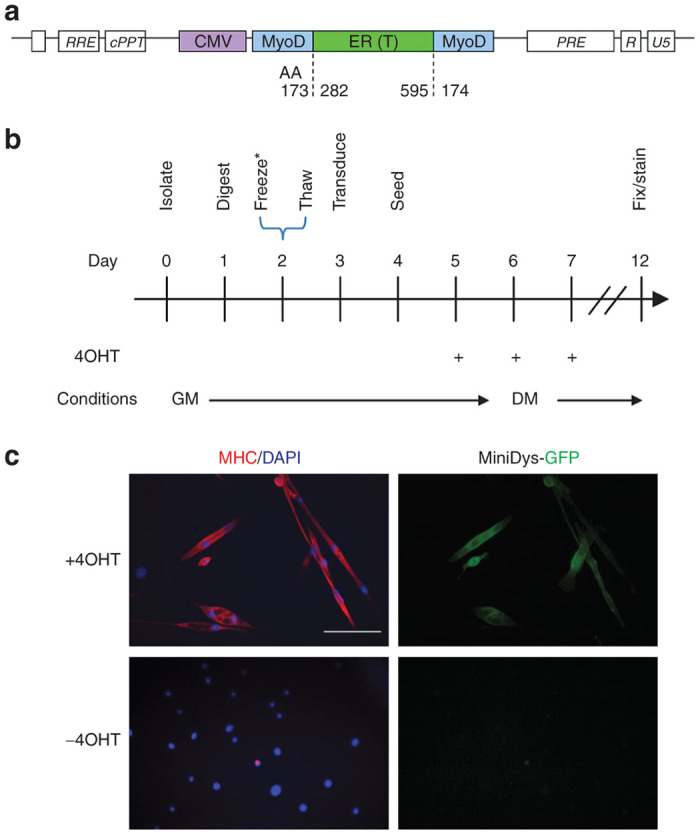
Conversion of the dermal cell population (dFb) into the myogenic lineage. (a) Illustration of the MyoD-ER(T) cassette within lentiviral packaging elements, showing the amino acid (AA) junctions for insertion of the ER(T) segment into MyoD. (b) In vitro dFb conversion timeline. *Timeline represents a typical experiment; see the Materials and Methods section for slight variations and protocol details. 4OHT, 4-hydroxytamoxifen; GM, growth medium; DM, differentiation medium. (c) Immunofluorescence detection of myosin heavy chain (MHC) and miniDys-GFP, two indicators of myogenic differentiation, with and without 4OHT treatment of dFbs transduced with MyoD-ER(T) and converted as in b. Scale bar = 100 µm.
Figure 2.
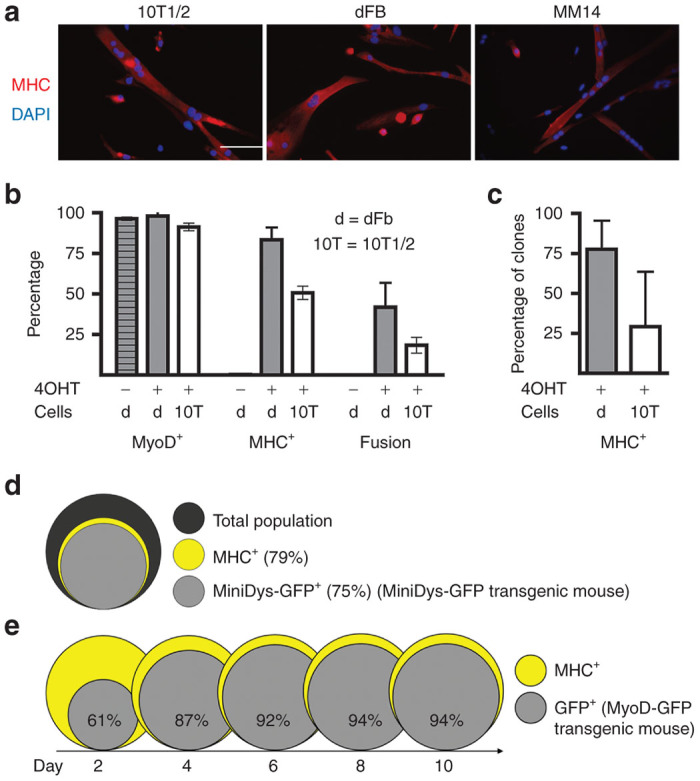
Myogenic markers in converted dFbs. Both dFbs and 10T1/2 mouse embryonic fibroblasts were transduced with a lentiviral vector carrying MyoD-ER(T) (MOI 10) and treated with 4OHT, then were compared to the differentiated MM14 myogenic cell line. (a) 10T1/2s and dFbs become MHC+ and fuse into multinucleated myotubes following myogenic conversion, similar to MM14 MHC+ myotubes. (b) Quantification of the percentage of each cell type that expressed MyoD and MHC and fuse following myogenic conversion for dFbs (d) and 10T1/2s. For 10T1/2s, 91% ± 6% expressed MyoD, 51% ± 10% expressed MHC, and 18% ± 12% fused; for dFbs, 98% ± 4% expressed MyoD, 83% ± 8% expressed MHC, and 49% ± 8% fused. Both MHC expression and fusion are statistically significantly different between 10T1/2s and dFbs (P < 0.05). (c) Quantification of the percentage of clones that expressed MHC following myogenic conversion. About 30% of the 10T1/2 clones and nearly 80% of the dFb clones contained MHC+ cells, a statistically significant difference (P < 0.05). (d) Venn diagram representing the percentage of MHC+ cells (yellow) and miniDys-GFP+ cells (light gray) in the total population (dark gray) of converted dFbs derived from the miniDys-GFP mouse, where >90% of MHC+ cells are also GFP+. The muscle-specific human alpha skeletal actin promoter drives expression of miniDys-GFP in the transgenic mouse. (e) Venn diagrams showing activity at the MyoD promoter over a time course following conversion of dFbs derived from the MyoD-GFP mouse. Since the MyoD antibody does not distinguish between MyoD-ER(T) and endogenous MyoD, we used these transgenic mice to test MyoD promoter activity, detected via expression of a cytosolic GFP reporter, following activation of MyoD-ER(T). Expression from the MyoD promoter is shown in gray, with percentages indicating cells expressing the reporter relative to MHC+ cells (yellow). Cells were fixed for staining 10 days after the initial 4OHT treatment (day 15 in Figure 1b). For all conditions and cell types, data represent averages from 5 to 10 fields per well across at least three wells. Data were pooled from two independent experiments for 10T1/2 cells, and three independent experiments for dFbs. Scale bar = 100 µm.
To track myogenic conversion over time and the activation of MyoD expression in converting cells, we isolated dFbs from MyoD-GFP mice. This population was then transduced with the MyoD-ER(T) lentivirus, converted to myogenic cells, and differentiation was monitored for 10 days after initiating 4OHT treatment. By day 4, nearly 90% of the MHC+ cells were also GFP+, coincident with maximum culture density and peak conversion (Figure 2e). These phenotypes persisted for at least 10 days even though 4OHT treatment ceased on day 2. Since MyoD is known to have a short half-life,23 these data indicate that sustained 4OHT treatment and MyoD-ER(T) activity are not required to maintain myogenic differentiation.
To determine the minimum multiplicity of infection (MOI) of the MyoD-ER(T) lentivirus required to obtain optimal transduction and conversion in dFbs, cultures were transduced with various MOIs of MyoD-ER(T) lentivirus and separately transfected with the CK8e-luciferase plasmid (a muscle-specific M-creatine kinase-based reporter construct, expressed only in differentiated striated muscle cells) as a means of detecting converted cells. Normalized luciferase activity increased linearly between MOIs of 1 and 10 (see Supplementary Figure S2). At an MOI of 10, >90% of the cells expressed MyoD (Figure 2b), and qPCR indicated a population average of at least one MyoD-ER(T) lentiviral integration event per cell (data not shown). Transductions with MOIs between 10 and 100 led to similar levels of luciferase activity with maximal expression levels attained between MOIs of 10 and 20, and decreasing levels at an MOI of 200. At MOIs of 100 and 200, it was also noted that cell proliferation slowed dramatically compared with lower MOIs (data not shown).
Conditions affecting myogenic conversion
We additionally tested whether conditions that promote differentiation of myogenic cells also promote conversion of dFbs. Self-depletion of mitogens from the medium during conversion resulted in a higher percentage of MHC+ cells than if cells were maintained in 2% fetal bovine serum (FBS) via medium change every 2 days. Concurrent treatment (through day 6) of 4OHT and basic fibroblast growth factor (bFGF), a known mitogen for both myoblasts and fibroblasts, doubled the percentage of MHC+ cells in both dFb and 10T1/2 cells (Figure 1b), but only when cultures were also periodically refed with medium containing 2% FBS (data not shown). This suggests that transplanted MyoD-ER(T) dFb populations could potentially expand in vivo if sufficient mitogens were present in the graft environment.
Since it is known that cell density can influence myogenic differentiation of various cell types,24–26 we explored cell density influences on MyoD-ER(T) dFb differentiation. We started with a seeding density that limited cell–cell contact, and then increased cell density over a 20-fold range, and found no significant effect on myogenic conversion in the entire culture dish or within individual microscopic fields that had between 1 and >40 nuclei per field (see Supplementary Figure S3). Similar results were obtained with 10T1/2 cells (data not shown). This does not exclude the possibility of soluble factors subtly influencing 4OHT-mediated reprogramming in vivo, but it suggests that myogenic conversion does not require direct cell contacts.
Donor miniDys-GFP cells are not rejected in a syngeneic setting
Since this system includes expression of an immunologically novel protein, we next asked whether donor cells carrying the miniDys-GFP transgene might be rejected by the mdx4cv hosts. To avoid any potential effect of tamoxifen treatment on the immune system function, we transplanted 5 × 105 mononuclear cells from whole muscle isolated from miniDys-GFP mice into tibialis anterior (TA) muscles of mdx4cv hosts. TAs were harvested at 8 or 16 weeks and the total number of GFP+ fibers were counted in multiple cross-sections throughout each transplanted muscle. For both time points GFP+ fibers were retained, and the average number did not differ significantly between 8 and 16 weeks (see Supplementary Figure S4). These data indicate that expression of the transgene was stable, and that miniDys-GFP+ fibers were not cleared by the immune system.
Transplantation of dFbs does not lead to fibrosis
Given the known role of fibroblasts in the formation of fibrotic lesions in dystrophic muscle,27,28 we examined whether transplantation of dFbs resulted in ectopic collagen deposition or structural abnormalities after in vivo myogenic conversion (Figure 3a). Figure 3b shows a typical cross-section with engrafted muscle fibers 4 weeks after transplantation and in vivo conversion of 5 × 105 miniDys-GFP donor dFbs into mdx4cv TA muscles. Examination by hematoxylin and eosin staining revealed no gross abnormalities in muscle morphology with only occasional foci of mononuclear cells near engraftment sites (Figure 3c). These mononuclear cells may represent immune cell infiltration, a common feature in mdx4cv mice,29 as similar concentrations of mononuclear cells were observed at locations outside the engraftment site in both injected and control sections. Importantly, no collagen accumulation beyond control levels was detected at engraftment sites by Sirius red staining (Figure 3c). Furthermore, the transverse sections indicate that the engrafted muscle fibers are oriented parallel to the adjacent endogenous fibers.
Figure 3.
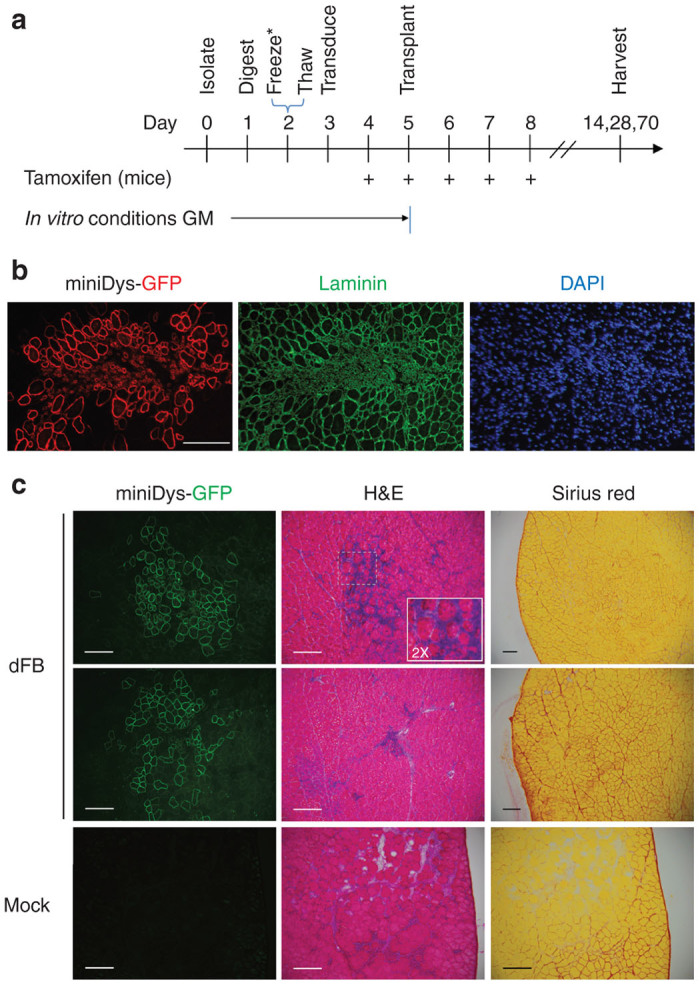
In vivo conversion of dFbs derived from miniDys-GFP transgenic donors. (a) In vivo dFb conversion timeline. *Timeline represents a typical experiment; see the Materials and Methods section for slight variations and protocol details. GM, growth medium (see Materials and Methods). (b) A typical engraftment site in a TA muscle following in vivo conversion of dFbs 4 weeks posttransplantation, showing immunostaining for GFP to detect localization of miniDys-GFP, laminin to detect all muscle fibers and the structural characteristics at the engraftment site, and DAPI for detection of nuclei. (c) Features of engraftment sites. The left column is direct imaging of miniDys-GFP at engraftment sites. The middle and right columns are hematoxylin and eosin (H&E) and Sirius red staining, respectively, and show no gross abnormalities or ectopic collagen deposition following in vivo dFb conversion (top two rows). The inset in the top row H&E is twice the magnification of the original image. H&E and Sirius red staining of dFb transplanted muscles was similar to that of mock transplantations with saline (bottom row). Scale bars = 200 µm.
Engraftment versus donor dFb cell dose
The efficiency of dFb cell engraftment in conjunction with in vivo conversion was determined by injecting graded doses of miniDys-GFP donor-derived dFbs in a fixed volume of 20 µl into mdx4cv TA muscles, with the in vivo conversion scheme shown in Figure 3a. Muscles were harvested 4 weeks posttransplantation, and the GFP+ fibers within the single transverse section containing the maximum number of GFP+ fibers were then counted (Figure 4a). The results indicate more total GFP+ fibers at higher dFb doses up to 1 × 106 cells per transplant, or 50,000 cells/µl, whereas at 2 × 106 cells (100,000 cells/µl), some muscles had reduced GFP+ fibers. Representing the data in terms of the number of injected cells per single GFP+ fiber shows that efficiency, using single needle track deliveries, plateaued between doses of 1 × 105 and 1 × 106 cells, and then became orders of magnitude less efficient at 2 × 106 cells (Figure 4b).
Figure 4.
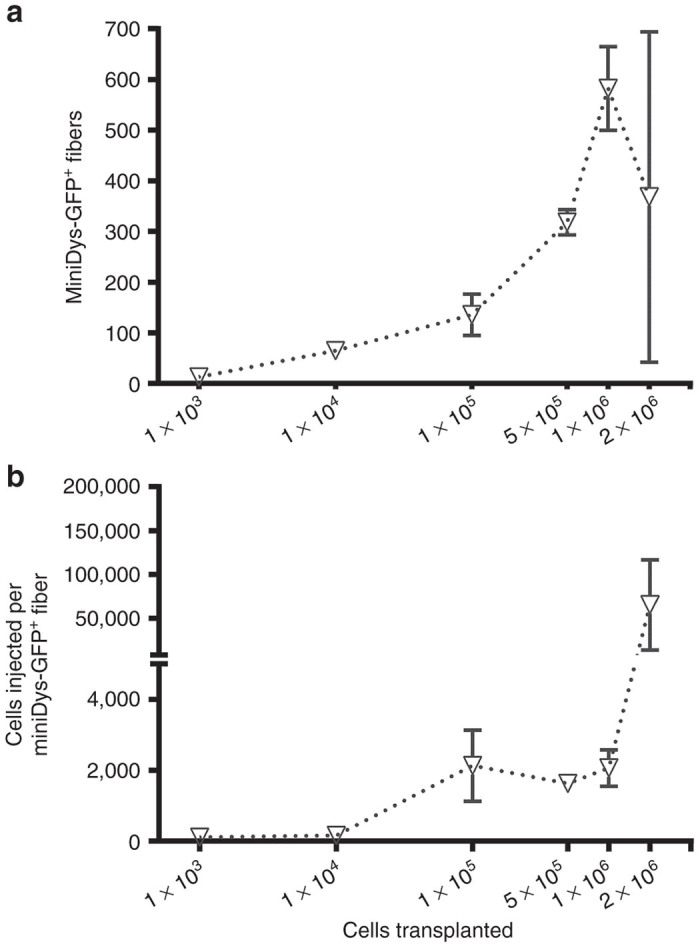
Engraftment of dFbs at various cell doses following in vivo conversion into the myogenic lineage. All cells were transplanted in 20 µl into TAs of mdx4cv hosts. Engraftment sites were detected by immunostaining for GFP, and the highest engrafted section was used to represent each muscle for each cell dose. (a) Cell dose versus number of positive fibers, revealing around 1 × 106 cells as a limit for reliable engraftment. (b) Representation of the data in terms of the number of cells required to achieve a single engrafted fiber. Results include pooled data obtained from two independent experiments, with n = 3–7 at each dose. Error bars are mean ± SEM.
Engrafted fiber distribution in host muscles
We next sought to quantify the extent of engraftment that could be obtained by dFb transplantation. For these studies 5 × 105 dFbs from miniDys-GFP donors were transplanted into recipient TA muscles as previously described (Figure 3a). At 2 and 4 weeks posttransplantation, injected muscles were harvested, and cryosections were evaluated at approximately 0.5 mm and 0.1 mm intervals, respectively, throughout the muscle length. Each section was directly imaged for GFP+ fibers, and these were counted and plotted corresponding to the respective proximal-to-distal location of each section (Figure 5). TAs harvested at both time points exhibited longitudinal distributions of miniDys-GFP+ fibers, but at 4 weeks, the average positive fiber numbers were ~21% higher compared with 2 weeks. Additionally, the percentage of engrafted area in each transplanted TA muscle was determined by comparing the engrafted area to the total muscle cross-sectional area. In all sections tested, less than 10% of the total muscle cross-sectional area was engrafted (not shown).
Figure 5.
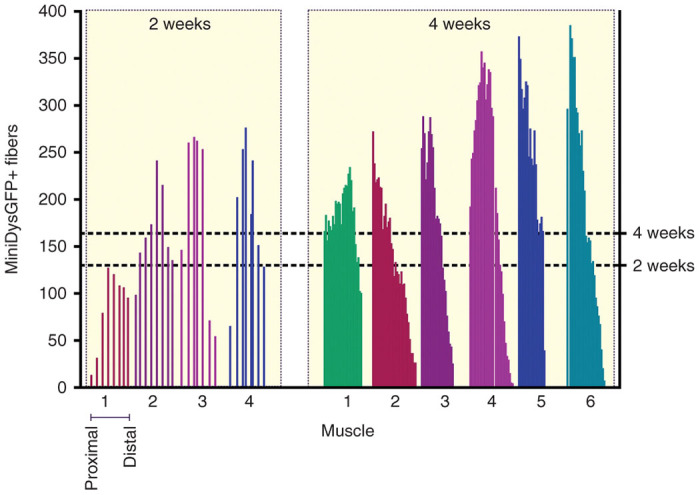
Profiles of engraftment for in vivo converted dFbs across TA muscles of mdx4cv hosts at 2 and 4 weeks. Each numbered bar plot on the x axis represents a single muscle, where each bar within plots indicates the number of GFP+ fibers per cross section. Cryosections were collected throughout each muscle, and counted approximately every 0.5 mm for the 2-week time point and every 0.1 mm for the 4-week time point and plotted from proximal (left) to distal (right). Note that the placement of each bar within plots reflects the actual section counted, and thus distances between bars are occasionally slightly variable (2-week time point), and in rare cases missing (4-week time point), due to unscorable sections. The right axis and dotted lines show mean positive fibers for each time point, where the 4-week time point had a slightly higher average number (164 positive fibers) than the 2-week time point (130 positive fibers), determined for each by averaging across all sections of all muscles.
To investigate long-term stability of engraftment and to increase the overall engrafted area so as to create a system conducive to whole muscle functional evaluation, 5 × 105 dFbs from miniDys-GFP donors were injected into the much smaller extensor digitorum longus (EDL) muscle. Ten weeks after transplantation, transverse sections of injected EDL muscles exhibited miniDys-GFP+ fibers in 10 to ~30% of the muscle cross sectional area, and the average percent engrafted area across all sections of all muscles was ~14% (Figure 6a). Importantly, the persistence of miniDys-GFP expression 10 weeks after dFb injection implies relatively long-term survival of donor dFb-derived muscle fibers. Comparable engraftment was observed when 5 × 105 whole muscle mononuclear (WMM) cells (which include satellite cells) were transplanted into EDL muscles (Figure 6b). The individual sections with the highest percentage area engrafted are shown for each transplanted EDL and cell type (Figure 6c).
Figure 6.

Engraftment comparison of dFbs and whole muscle mononuclear (WMM) cells derived from the miniDys-GFP transgenic donor after transplantation into EDL muscles of mdx4cv hosts. Engraftment levels were determined by direct imaging of GFP and quantification of the percentage area containing miniDys-GFP+ fibers in each of seven sections for each muscle 10 weeks after transplantation (see Materials and Methods). (a) Plots representing engraftment of dFbs in three transplanted EDL muscles, where each numbered bar plot on the x axis within the graphs represents an individual muscle, and each bar indicates the area containing GFP+ fibers in a single cross section. Sections were quantified approximately every 0.5 mm throughout each muscle and represented proximal (left) to distal (right). Dotted lines show the average percentage area engrafted across all three muscles, determined by averaging the percentage area across all sections of all muscles. (b) Plots representing engraftment of WMM cells in three transplanted EDL muscles, as in part a. (c) The sections with the maximum engraftment for each muscle are shown for dFbs and WMM cells, with percentage area indicated. Scale bars = 200 µm.
Contractile properties of mdx4cv EDL muscles were not improved following dFb transplantation
We next tested whether the transplantation of dFbs or WMM cells into dystrophic muscle improves force development or protects whole muscle from injury. Ten weeks after transplantation, EDL muscles were dissected and subjected to in vitro force measurements followed by an eccentric contraction injury protocol (see Materials and Methods). We found no improvement in maximum isometric force or specific force. The masses were equivalent in all injected muscles, with none showing reversal of the characteristic mdx4cv hypertrophy. No significant differences were observed in protection from contraction-induced injury in cell-transplanted muscles compared to saline-injected controls (Figure 7). Average whole muscle engraftment of dFbs across all muscles in the physiology data was ~10%, with a range of ~3–20%. No correlation was found for percent whole muscle area engrafted and protection from contraction-induced injury or other measurements of contractile function (data not shown), though the EDL with 20% engraftment retained slightly more force in the contraction-induced injury assay compared with the lower engrafted muscles. These data suggest that transplants into adult dystrophic muscles may require significantly higher levels of widespread engraftment to obtain clear improvement in mechanical properties.
Figure 7.
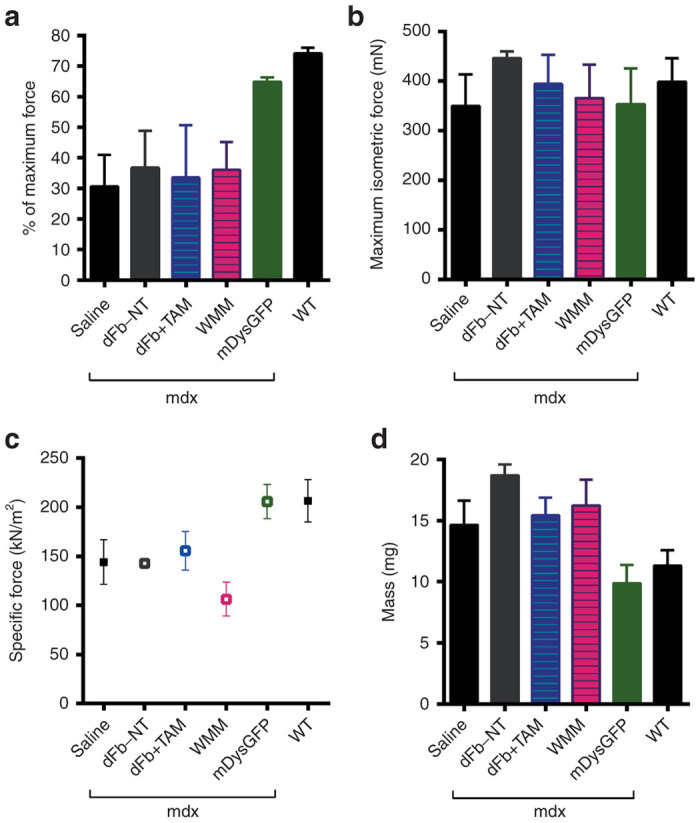
Whole muscle physiology after transplantation and in vivo conversion of dFb (dFb+TAM), compared to whole muscle mononuclear cells (WMM). Ten weeks after cell transplantation, in vitro force measurements were carried out on whole EDL muscles, followed by an eccentric contraction protocol. (a) Percentage of original force development after the fifth eccentric contraction in transplanted muscles with negative (saline, dFb-NT) and positive (mDysGFP, WT) controls. No significant differences were observed between transplanted muscles and negative controls. (b) Maximum isometric force was unchanged across all mice. (c) Specific force generating capacity was unchanged between transplanted muscles and negative controls, except for a slightly reduced specific force for WMM transplanted muscles. (d) In all injected mdx muscles, mass was equivalent and was significantly greater than WT or mDysGFP muscles. Means and standard deviations in each assay were calculated from n = 3–11 EDL muscles for each treatment type, except for n = 2 EDLs for measurements of contraction induced injury in the mDysGFP group. Abbreviations: mdx, mdx4cv background where indicated; dFb-NT, injected cells equivalent to dFb+TAM but without tamoxifen treatment; mDysGFP, noninjected control transgenic mouse EDLs; WT, noninjected C57BL/6.
Discussion
We investigated conversion and transplantation conditions and describe muscle-wide quantitation of engraftment of myogenically converted dFbs carrying a mini-dystrophin transgene. A major goal was to begin developing a clinically viable cell-mediated strategy for the treatment of dystrophic skeletal muscle. We, therefore, avoided pretreatment of recipient muscles with toxins and irradiation, two common protocols in mice that initiate extensive regeneration and can eliminate host satellite cells,30,31 as neither of these procedures would be feasible in DMD patients. Additionally, donor cells were transplanted into immunocompetent, syngeneic mice to mimic the autologous immune system setting that would be encountered in DMD treatment.
One clinically relevant component of autologous cell-based transplantation for DMD therapy not directly tested in the present study was the ex vivo transduction of donor dFbs with a dystrophin expression cassette. We instead used myogenically convertible mdx4cv dFbs that already carried a miniDys-GFP transgene. Technical parameters of this essential portion of the treatment protocol are being investigated in on-going studies. Moreover, dual lentiviral transduction was previously demonstrated in tail-tip fibroblasts with micro-dystrophin-GFP and MyoD-ER(T).19 It will additionally be important in all genetic correction strategies to apply recent advances in gene editing and vector technologies that may, for example, allow human autologous cells to carry full-length dystrophin.4,32
We have confirmed efficient in vitro conversion, similar to previous studies using inducible19 and constitutively active MyoD.13 We additionally established that lentiviral delivery of the cassette expressing MyoD-ER(T) at MOI 10–20 is effective for myogenic conversion, and additional vectors, while possibly maximizing MHC+ cells, are unnecessary. Minimizing the essential vector number is clinically important, as it decreases the probability of insertional mutagenesis, and reduces the potential for premature differentiation due to MyoD overexpression.
The alternative to direct reprogramming of fibroblasts, using patient-derived induced pluripotent stem cells that are then induced into myogenesis, for example, via PAX7 expression33 involves similar risks of insertional mutagenesis, as well as the possible elevated risk of immune reactions due to induced pluripotent stem cell changes during the isolation and expansion process.34 In addition, direct reprogramming of fibroblasts into a variety of other lineages, including neurons and cardiomyocytes, has been successful without a pluripotent intermediate.8 Further investigation with direct in vivo comparisons may help determine whether myogenic cells derived from various reprogramming strategies have equivalent properties and engraftment potential, and whether donor engrafted muscle fibers contain cells that are capable of regenerative responses.
In our in vitro conversion experiments, cells expressed myogenic genes as expected and became responsive to factors known to influence myoblast differentiation, such as mitogen deprivation. Additionally, the increase in percentage of MHC+ cells with addition of bFGF in vitro suggests that bFGF treatment differentially affects the converting population, possibly due to enhanced survival, selective mitogenic stimulation, or other signaling effects.35,36 The in vitro dFb conversion process does not appear to depend on cell contact or fusion (see Supplementary Figure S3), thus in vivo conversion should also have no specific requirement for contact with endogenous myogenic or other converting cells.
While the contribution of transplanted dFbs to dystrophin-positive fibers indicates their in vivo tamoxifen-mediated conversion into the myogenic lineage, an unknown proportion of cells may remain as fibroblasts. If these cells are present, it is unlikely that they interfere with muscle function; histological examination of dFb engraftment sites both with and without tamoxifen treatment indicated no gross morphological abnormalities or lesions in the recipient dystrophic muscle (Figure 3 and data not shown), and no functional decline is observed in non-tamoxifen treated, dFb transplanted controls (Figure 7). The absence of ectopic connective tissue in the current study is in contrast to observations in a pioneering MyoD-converted fibroblast study.14 This may be due to transplantation conditions such as prior notexin injection and irradiation of host limbs, in addition to low transduction efficiency and use of constitutively active MyoD fibroblasts in the earlier study.
Our studies suggest that the cell concentration for optimal engraftment of dFbs by single longitudinal injection into the TA muscles of an adult mouse is less than 100,000 cells/µl in a volume of 20 µl (Figure 4). Three major factors likely contribute to this limitation: i) oxygen and nutrient perfusion given the cell number and volume injected, ii) cell type-specific tolerance to transplantation stress, and iii) a possible cell type-specific interaction between transplantation stress and myogenic conversion or differentiation. Interestingly, for myoblasts transplanted into nonhuman primate muscles, 1 × 106 cells in 10 µl (100,000 cells/µl) or a larger volume of the same cell concentration (30–200 µl) injected at a single site resulted in ischemic central necrosis.5 These results support the idea that increasing volumes or cell numbers for single injections beyond a given limit may not improve engraftment. However, this limit must be determined for each cell type considered for cell-based therapy, since differential responses to stress and myogenic differentiation are likely, and must be understood in terms of both cell survival and long-term functional engraftment. Furthermore, multiple injections will likely be required for larger muscles. Two recent studies demonstrate the feasibility of a multiple injection protocol in human muscle. A clinical trial for oculopharyngeal muscular dystrophy showed improved muscle function with multiple injections of autologous myoblasts using similar cell concentrations.37 Furthermore, long-term follow up of one DMD patient after allotransplantation of myoblasts with immunosuppression in a high-density injection protocol showed toleration of the procedure with no detriment to muscle function.38 However, while improved strength was reported for the left first metacarpal flexor muscle, no improvement was detected in the biceps brachii.
When the distribution of engrafted fibers was evaluated in TA muscles 2 and 4 weeks after transplantation, the number of engrafted fibers was slightly higher at 4 weeks (Figure 5). This may be due to increased stability of mature engrafted fibers or further contribution from differentiating converted cells during the intervening two weeks. Analogous studies of EDL muscles 10 weeks after single longitudinal intramuscular injections indicated engraftment levels of up to 30% of the total area in transverse sections through the best transplants (Figure 6). The percent engraftment data and relatively restricted area occupied by miniDys-GFP+ fibers following the in vivo conversion and differentiation of transplanted dFbs (Figure 6), indicates that dFbs, like myoblasts, do not migrate significant distances throughout the muscle following their injection. The greater percent total muscle area of engraftment in EDL compared to TA muscles, coupled with the longitudinal distribution of engrafted cells throughout the entire muscle, suggests that EDLs may provide a better platform for whole muscle physiology studies.
Comparison of whole muscle EDL function following transplantation of WMM or in vivo-converted dFbs revealed that the cells did not protect against contraction induced injury (Figure 7); also, no improvement was observed in maximum isometric force, specific force, or mass among injected dystrophic muscles compared to wild type and transgenic mice carrying functional dystrophin. In contrast to our findings, it should be noted that improved muscle function has been reported following the transplantation of induced PAX7-myogenically converted human induced pluripotent stem cells into immunodeficient mdx4cv mouse TA muscles.33 This experimental protocol included cardiotoxin injections the day prior to cell transplantation, thereby causing an extensive regenerative response that would likely facilitate the formation of mosaic muscle fibers containing human donor cell dystrophin-positive as well as host mdx4cv myonuclei.
Much is still unknown about how engrafted fibers contribute to whole muscle function, and how de novo fibers (both hybrid and of donor origin) interact within the muscle during the regeneration period that follows intramuscular transplantation. Connections between engrafted fibers and the peripheral nervous system, myotendinous junctions, and other fibers laterally could affect individual fiber maturity and function as well as whole muscle lateral and longitudinal force transduction. Importantly, improvements in pathology have been observed despite nonuniform or mosaic dystrophin expression, emphasizing the importance of fiber connectivity, even between dystrophin positive and negative fibers.39,40 Limited transgene diffusion along mosaic fibers might also play a role.41
Compared to noncell gene transfer methods, cell-based therapy alone may require longer posttreatment intervals to achieve optimal transgene expression and structural maturity of muscle fibers. In this context, we found that whole muscle function in the donor transgenic miniDys-GFP mdx4cv mice is similar to that of wild type mice. Therefore, the absence of a rapid functional improvement in the recipient mdx4cv mice is not due to expression, localization, or functional problems associated with the donor miniDys-GFP (Figure 7). In addition, gross structural characteristics of converted dFb and WMM engrafted muscles were highly similar and showed no ectopic collagen deposition in TA or EDL muscles (Figure 3). However, we consistently observed regions of very small miniDys-GFP+ fibers after transplantation of either cell population (Figure 3b) that could indicate fiber immaturity or branching that affects function.42 In functional assays of dissected EDL muscles, this could obscure improvements that resulted from successful transgene delivery. Additional studies with longer time points may provide insight on the fate of such fibers.
The most compelling explanation for detecting no functional physiological benefit from our transplantation studies is that the mini-dystrophin-positive fibers contributed by donor cells, regardless of their origin, do not represent a sufficient overall percentage of the TA or EDL fibers to improve force or protect from injury. In the highest engrafted EDLs, myogenically converted dFb and WMM each achieved an average of 14% engraftment across whole muscles. Previous studies using viral vector-mediated gene transfer or exon skipping have shown that dystrophin levels must reach 20% of wild type levels, and similarly about 20% of fibers must be expressing dystrophin to achieve therapeutic efficacy.40,43 Predicting whether it is realistic to expect functional recovery thus requires an accurate quantitation of whole muscle engraftment. Fiber number and area analysis with consistent sampling throughout a transplanted muscle as shown in (Figures 5 and 6) is necessary, because even when regions containing two to three times higher than the mean levels of dystrophin-positive fibers are achieved, our experiments show that this is insufficient for whole muscle therapeutic benefit. In addition, it may be important to achieve the highest levels of transgene expression at sites that experience the highest stress during force development.44
Forced myogenic conversion using MyoD activates the expression of the endogenous MyoD gene as well as many other myogenic genes,35,45 such that converted cells maintain their myogenic properties following the withdrawal of tamoxifen. Our in vitro conversion data and the stability of engrafted muscle fibers up to 10 weeks after transplantation also support this conclusion. Therefore, it is unlikely that a similar strategy for inducing the myogenic conversion of engrafted autologous dFbs in human DMD patients would require more than transient treatment with tamoxifen. Furthermore, while most tamoxifen studies showing tolerable side effects have been carried out in female breast cancer patients, the analysis of tamoxifen-treated adolescent male gynecomastia patients have indicated no serious side effects.46 However, no tamoxifen studies of younger male humans have been reported. Side-effect issues would thus need to be fully understood in relation to the doses and treatment durations that would be required for tamoxifen use in DMD autologous cell myogenic conversion therapies.
The inducible MyoD system clearly requires testing in human adult dermal cells. In a similar system with constitutive MyoD expression in human fetal and adult dermal cells, the conversion was less efficient for human cells compared to mouse, and for fetal compared to adult, but the reductions were modest and human adult dermal cells still showed strong conversion ability.13 In addition, avoidance of premature differentiation using the tamoxifen-inducible MyoD tested in our studies may improve efficiency in the human cells. Another study that used a similar inducible MyoD system in mesenchymal stem cells demonstrated that human cells in vitro are receptive to tamoxifen-mediated induction.32
In summary, we have shown robust in vivo myogenic conversion, engraftment ability, and dystrophin delivery of syngeneic dFbs following transplantation. We also demonstrate the viability of the inducible MyoD system, and provide useful quantitation methods for evaluating engraftment. dFbs are therapeutically attractive, because they are safely accessible from patients and relatively easy to expand and transduce in vitro. Our studies support the utility of a strategy to use patient-accessible dermal cells to enhance regeneration of dystrophic skeletal muscle.
Materials and Methods
Construct and lentivirus preparation
The inducible MyoD construct19 included mouse MyoD and mouse ER(T) sequences driven by the cytomegalovirus promoter (GenBank #AF369966.1) within a lentiviral vector backbone as shown in Figure 1a. All recombinant DNA work was performed following guidelines for biosafety and containment measures provided by the University of Washington Institutional Biosafety Committee and the National Institutes of Health.
Vesicular stomatitis virus G pseudotyped self-inactivating lentiviruses were generated as described.47 Briefly, MyoD-ER(T) in a lentiviral transfer plasmid was cotransfected, along with three viral plasmids, by calcium phosphate precipitation into approximately 80% confluent human embryonic kidney 293D cells on 150 mm plates. The growth medium (GM) was refreshed 16 hours after transfection. Cells were cultured for an additional 48 hours, and the supernatant was collected. Viral supernatant was centrifuged at 400 × g for 10 minutes at 4 °C to remove cell debris, filtered once with a 0.45 µm surfactant-free cellulose acetate filter unit, once with a 0.20 µm surfactant-free cellulose acetate filter unit, then concentrated by centrifugation at 50,000 × g in a Beckman L8-70M ultracentrifuge for 2 hours at 4 °C. Supernatant was aspirated and virus gently resuspended in small volumes of phosphate-buffered saline (PBS). Titering was carried out by the transduction of NIH-3T3 cells with serial dilutions of vector preparations. After culturing for one week, proviral integration was evaluated by TaqMan real-time polymerase chain reaction (Applied Biosystems, Foster City, CA) of a virally packaged portion of the lentiviral transfer plasmid, and normalized to genomic DNA copies of the low density lipoprotein receptor to determine infectious units per ml of concentrated virus.
Primary cell isolation
Dermal cell populations were isolated from whole skins of 1–3-day-old neonatal mice. Two to four pups were anesthetized on ice before cervical dislocation, transferred to a sterile environment, immersed in Betadine solution (Purdue Products L.P., Stamford, CT) twice for 2 minutes each, rinsed in sterile deionized water, immersed in 70% ethanol twice for 2 minutes, then immersed in sterile phosphate-buffered saline (pH 7.2) (Gibco, Grand Island, NY), with penicillin/streptomycin (Gibco) (PBS-P/S) until dissection. The dermal skin layer was isolated as described.48 Briefly, limbs and tail were removed and the epidermis and dermis were carefully detached from the remaining tissue. Blood vessels or fat adhering to these layers was removed. Skins were then carefully floated dermis side down in 0.25% Trypsin-ethylenediaminetetraacetic acid at 4 °C overnight (~16 hours). The next day, the skins were laid epidermis side down, and the dermis was lifted away from epidermis with fine forceps, rinsed with PBS-P/S, minced lightly, and digested in 0.2% collagenase IV (Worthington Biochemical Corporation, Lakewood, NJ), 1.2 U dispase II (Worthington), and PBS-P/S in 5 ml total for 45–75 minutes at 37 °C, with trituration by 10 ml serological pipette every 15 minutes. Cells were centrifuged at 300 × g for 5 minutes, resuspended in GM, which consisted of Dulbecco's modified Eagle's medium (Gibco) with 10% fetal bovine serum (Thermo Scientific/HyClone, Logan, UT), 1% P/S, and 2 mmol/l l-glutamine (Gibco), and plated out at subconfluent density in GM with 10 ng/ml human bFGF (R&D Systems, Minneapolis, MN). Cells were trypsinized and cryopreserved in GM with 10% dimethyl sulfoxide 24–48 hours later. This heterogeneous dermal cell population, which we refer to as dFbs, was used for all in vitro and in vivo conversion experiments.
Whole muscle mononuclear cells were isolated from 6-week-old miniDys-GFP transgenic mouse hindlimb and diaphragm muscles as described previously.22 Briefly, muscles were dissected out and the majority of visible connective tissue removed, followed by mincing and 1 hour digestion at 37 °C in PBS-P/S with 0.2% collagenase IV and 1.2 U dispase II, with trituration by 10 ml serological pipette every 15 minutes. Tissue debris was filtered using 70 and 40 µm cell strainers, and cells were centrifuged and resuspended in 154 mmol/l NH4Cl red blood cell lysis buffer for 5 minutes at room temperature, followed by the addition of PBS-P/S to a total volume of 30 ml. Cells were centrifuged again and resuspended in Ham’s F10 medium (Gibco) with 15% horse serum (Thermo Scientific/Hyclone), 1 mmol/l CaCl2, 1% P/S and 0.5 µg/ml bFGF, counted, adjusted to 25,000 cells/µl and kept on ice for 30 minutes to 3 hours until transplantation.
Culture conditions
Media formulations were as follows. GM for fibroblasts was Dulbecco's modified Eagle's medium, 10% FBS, 1% P/S, and 2 mmol/l l-glutamine. GM was supplemented with 10 ng/ml bFGF once per day. Differentiation medium was equivalent to GM except with 2% FBS, supplemented with bFGF or with the vehicle, which consisted of Ham’s F10 medium, with 15% horse serum, 1 mmol/l CaCl2, and 1% P/S.
Dermal cells were thawed and expanded in GM with 10 ng/ml bFGF for 1 day prior to transduction with the lentiviral vector carrying MyoD-ER(T). Cells were transduced at a MOI of 10, or 10 infectious units per cell unless otherwise stated, in the presence of 8 µg/ml polybrene in 1 ml GM for 10 minutes, followed by plating of the cell/virus solution at 30–60% confluency in 10 ml GM per 150 cm plate. Transduction at other MOI was performed under identical conditions. Transduced cells were either plated for conversion experiments the next day or transplanted within 5 days, so that cells were used for myogenic conversion within 9 growth days of initial isolation (Figures 1b and 3a). For in vitro myogenic conversion of fibroblasts, unless otherwise specified, cells were plated at a density of 20,000 cells per well in six-well plates, and the next day treated with 4OHT at 2 µmol/l or the same volume of 95% ethanol in GM.
10T1/2 mouse embryonic fibroblasts and MM14 mouse myoblast49 cell lines were used for comparison to dFb conversion. 10T1/2 cells were treated with a conversion protocol identical to that used with dFbs, and MM14 myoblasts were grown in identical GM to WMM cells, including 5 ng/ml bFGF, then differentiated by allowing nutrients and growth factors in cultures to self-deplete as a positive control for dFb in vitro differentiation.
Luciferase assay
A firefly luciferase cDNA driven by modified enhancer and promoter regulatory elements from the mouse muscle creatine kinase gene (regulatory cassette CK8e) was subcloned into the polylinker of a lentiviral vector backbone47 and used for all luciferase experiments. CK8e is expressed only in differentiated skeletal and cardiac muscle cells (unpublished data). dFbs were transduced with the lentiviral vector carrying the MyoD-ER(T) cDNA as described in the conversion conditions section, transfected with the CK8e-luciferase cassette by calcium phosphate precipitation, and converted into the myogenic lineage as in Figure 1b. Cells were lysed using cell culture lysis buffer for luciferase assays or reporter lysis buffer for protein quantitation (Promega, Madison, WI). Lysates were evaluated using the Promega Luciferase Assay System and a Victor3 V plate reader, with automatic pump dispensing of Luciferase Assay Reagent (Promega). One transfected and converted well was evaluated in each of two independent experiments for each MOI shown in Supplementary Figure S2. Mean and standard error were calculated based on averaged readings across all luciferase assays, run in triplicate for lysates from each cell culture well. The Thermo Scientific Pierce Coomassie Plus Assay Reagent was used for total protein quantification on the Victor3 V.
Mice
Several different transgenic mouse lines were used in these studies. The mdx4cv mice have a point mutation resulting in a stop codon in exon 53 of the dystrophin gene, on the C57Bl/6 background,50 and were used as hosts in all transplantation experiments. MiniDys-GFP/mdx4cv mice contain a mini-dystrophin-enhanced green fluorescent protein (eGFP) fusion protein cDNA driven by human alpha-skeletal actin regulatory elements on the mdx4cv background.22 The MyoD-GFP mice contain a GFP cDNA driven by a 24 kb regulatory sequence upstream of the MyoD gene, on an enriched FVB background (a kind gift from Z. Yablonka-Reuveni, University of Washington). All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Washington.
Drug preparation and dosage
Tamoxifen (Sigma-Aldrich, St Louis, MO) was equilibrated to room temperature, then 65 °C preheated corn oil (Sigma-Aldrich) was added to the dry stock bottle at 100 mg/ml, and the bottle was then alternately heated to 56 °C and vortexed frequently for several hours or until most of the crystals were dissolved. The solution was then transferred and diluted to 40 mg/ml in preheated corn oil, and the heating/vortexing procedure was repeated until no crystals were visible. The tamoxifen preparation was then filter sterilized and aliquoted for storage at −20 °C. Working solutions were prepared by diluting aliquots to 20 mg/ml in sterile corn oil, and all mice were treated with a dose of 100 mg/kg per day by intraperitoneal injection for 5 days, beginning 1 day prior to cell transplantation. Vehicle-treated mice were intraperitoneally injected with an equal volume of sterile corn oil. For in vitro studies stock 4OHT (Sigma-Aldrich), the active form of the prodrug tamoxifen, was diluted to desired concentrations with 95% ethanol.
Transplantations
Cells were transplanted by open skin intramuscular injection into TA or EDL muscles in a single injection along the length of each muscle. dFbs were prepared for transplantation by dilution to desired concentrations in identical medium to the WMM cells, kept on ice for 30 minutes to 3 hours, and aspirated into a 25 µl Gastight Hamilton syringe equipped with a 32 gauge needle. By hand, needles were inserted into the distal ends of each muscle, about 2 mm from the tendon, and pushed longitudinally through the muscle to about 3 mm from the proximal tendon. Injections were performed concurrently with needle withdrawal at a rate of about 2 µl/second. One or both legs were injected with cells, and control legs were injected with saline. Mice were anesthetized with inhaled 1–5% isoflurane in oxygen and treated with standard postoperative care.
Cell and tissue processing and analysis
Antibodies used for immunofluorescence included mouse anti-MHC (MF20; Developmental Studies Hybridoma Bank, Iowa City, IA), rabbit anti-GFP (A-11122; Invitrogen/Molecular Probes, Grand Island, NY), rabbit anti-MyoD (M-318; Santa Cruz Biotechnology, Santa Cruz, CA), and rat anti-laminin (MAB 1914; Millipore/Chemicon, Billerica, MA). For in vitro studies, cells were fixed for 10 minutes in 2% paraformaldehyde with 1.5% sucrose at room temperature prior to staining. For transplantation studies, muscles were dissected out and frozen in optimal cutting temperature compound by floating on liquid nitrogen cooled isopentane. Two 20 µm thick transverse cryosections were mounted on glass slides every 100 µm throughout each transplanted muscle. Immunofluorescence was performed on unfixed sections, and hematoxylin and eosin and Picrosirius Red staining were performed on methanol and unfixed sections, respectively. GFP+ fibers were counted following direct imaging of GFP after confirmation of colocalization of the direct GFP signal with the signal detected by immunofluorescence using the antiGFP antibody. Percentage area calculations for EDL transplantations were performed using Image J software. The area of the engrafted site was obtained using the software’s region of interest feature to outline the outer edges of the region that contained GFP+ fibers, and inputting scale information into the software. The same procedure was used to determine total area, by outlining the entire EDL transverse section. The outlined engrafted area values were then divided by the outlined total area values to determine the percentage engrafted area. Note that muscles with an individual average across all sections of <10% area were excluded from this analysis; for both dFbs and WMM cells, 2/5 injected muscles were in this category.
Physiology
Mice were anesthetized with an initial dose of 300 mg/kg Avertin, followed by additional doses necessary to maintain deep anesthesia for the duration of the procedure. Contractile properties were measured as described.22 Briefly, proximal and distal EDL tendons were secured with 5-0 silk suture in situ, after which intact muscles were dissected and one tendon was tied to the lever arm of a servomotor and one to a force transducer for whole muscle physiology. Muscles were maintained in a 25 °C bath of buffered mammalian Ringer’s solution (121 mmol/l NaCl, 5 mmol/l KCl, 0.5 mmol/l MgCl2·6H20, 0.4 mmol/l NaH2PO4, 24 mmol/l NaHCO3, 1.8 mmol/l CaCl2·2H20, 5.5 mmol/l glucose), bubbled with 95% O2/5% CO2. A single tetanus at low frequency (100 Hz) was performed after securing the muscle onto the rig to ensure responsiveness to electrical stimulation and suture knot stability. Muscle length was adjusted to optimal length (Lo) for force development. Muscles were stimulated with a pulse duration of 2 ms, and a voltage that produced maximum twitch force. A stimulation frequency of 180 Hz for EDL muscles, with 300 ms duration, gave the maximum isometric tetanic force (Po). The susceptibility of muscles to contraction-induced injury was assessed by six lengthening contractions. The muscles were set at Lo, activated maximally, and then stretched through a strain of 30% of Lo at a velocity of 1 fiber length/s, and then returned at the same velocity to Lo, allowed a 10 s recovery period, then exposed to subsequent stretches of 30% each. Next, muscles were removed from the bath, trimmed to remove sutures and tendons, weighed, and frozen in liquid nitrogen cooled isopentane. Cross-sectional area (cm2) was calculated based on the measurements of optimal muscle length (mm), muscle mass (mg), a muscle density of 1.06 g/cm2 and a fiber length to optimal length ratio of 0.44. The specific Po (kN/m2) was determined by dividing Po (kN) by cross-sectional area (m2). The force deficit produced by the lengthening contraction protocol was assessed by expressing the Po (mN) measured after the each lengthening contraction as a percentage of the Po before injury.
Statistical analyses
Differences between samples were evaluated by unpaired t-test. All error values are plotted as one standard deviation unless stated otherwise.
Acknowledgments
We thank John K. Hall (Department of Neurology) for critical reading of the manuscript, and Zipora Yablonka-Reuveni (Department of Biological Structure) for provision of MyoD-GFP transgenic mice, and Rainer Ng for technical assistance with muscle physiology. This study was supported by NIH grant # P01-NS046788 (to J.S.C. and S.D.H.), by R37-AR40864 (to J.S.C.) and by RO1-AR18860 and the Muscular Dystrophy Association (to S.D.H.).
The authors declare no conflict of interest.
References
- Batchelor CL, Winder SJ. Sparks, signals and shock absorbers: how dystrophin loss causes muscular dystrophy. Trends Cell Biol. 2006;16:198–205. doi: 10.1016/j.tcb.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Emery AE, Muntoni F.2003Duchenne Muscular Dystrophy Oxford University Press; Oxford. [Google Scholar]
- Muir LA, Chamberlain JS. Emerging strategies for cell and gene therapy of the muscular dystrophies. Expert Rev Mol Med. 2009;11:e18. doi: 10.1017/S1462399409001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoni C. Emerging gene editing strategies for Duchenne muscular dystrophy targeting stem cells. Front Physiol. 2014;5:148. doi: 10.3389/fphys.2014.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuk D, Paradis M, Goulet M, Tremblay JP. Ischemic central necrosis in pockets of transplanted myoblasts in nonhuman primates: implications for cell-transplantation strategies. Transplantation. 2007;84:1307–1315. doi: 10.1097/01.tp.0000288322.94252.22. [DOI] [PubMed] [Google Scholar]
- Blau HM, Webster C, Pavlath GK. Defective myoblasts identified in Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 1983;80:4856–4860. doi: 10.1073/pnas.80.15.4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonka-Reuveni Z, Anderson JE. Satellite cells from dystrophic (mdx) mice display accelerated differentiation in primary cultures and in isolated myofibers. Dev Dyn. 2006;235:203–212. doi: 10.1002/dvdy.20602. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T, Wernig M. Molecular roadblocks for cellular reprogramming. Mol Cell. 2012;47:827–838. doi: 10.1016/j.molcel.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lortal B, Gross F, Peron JM, Pénary M, Berg D, Hennebelle I. Preclinical study of an ex vivo gene therapy protocol for hepatocarcinoma. Cancer Gene Ther. 2009;16:329–337. doi: 10.1038/cgt.2008.88. [DOI] [PubMed] [Google Scholar]
- van den Bogaerdt AJ, van Zuijlen PP, van Galen M, Lamme EN, Middelkoop E. The suitability of cells from different tissues for use in tissue-engineered skin substitutes. Arch Dermatol Res. 2002;294:135–142. doi: 10.1007/s00403-002-0305-3. [DOI] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- Gibson AJ, Karasinski J, Relvas J, Moss J, Sherratt TG, Strong PN. Dermal fibroblasts convert to a myogenic lineage in mdx mouse muscle. J Cell Sci. 1995;108:207–214. doi: 10.1242/jcs.108.1.207. [DOI] [PubMed] [Google Scholar]
- Lattanzi L, Salvatori G, Coletta M, Sonnino C, Cusella De Angelis MG, Gioglio L. High efficiency myogenic conversion of human fibroblasts by adenoviral vector-mediated MyoD gene transfer. An alternative strategy for ex vivo gene therapy of primary myopathies. J Clin Invest. 1998;101:2119–2128. doi: 10.1172/JCI1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard C, Moisset PA, Dicaire A, Merly F, Tardif F, Asselin I. Transplantation of dermal fibroblasts expressing MyoD1 in mouse muscles. Biochem Biophys Res Commun. 1998;248:648–654. doi: 10.1006/bbrc.1998.8995. [DOI] [PubMed] [Google Scholar]
- Crescenzi M, Fleming TP, Lassar AB, Weintraub H, Aaronson SA. MyoD induces growth arrest independent of differentiation in normal and transformed cells. Proc Natl Acad Sci USA. 1990;87:8442–8446. doi: 10.1073/pnas.87.21.8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bo R, Torrente Y, Corti S, D’Angelo MG, Comi GP, Fagiolari G. In vitro and in vivo tetracycline-controlled myogenic conversion of NIH-3T3 cells: evidence of programmed cell death after muscle cell transplantation. Cell Transplant. 2001;10:209–221. [PubMed] [Google Scholar]
- Hollenberg SM, Cheng PF, Weintraub H. Use of a conditional MyoD transcription factor in studies of MyoD trans-activation and muscle determination. Proc Natl Acad Sci USA. 1993;90:8028–8032. doi: 10.1073/pnas.90.17.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian PS, White R, Hoare SA, Fawell SE, Parker MG. Identification of residues in the estrogen receptor that confer differential sensitivity to estrogen and hydroxytamoxifen. Mol Endocrinol. 1993;7:232–240. doi: 10.1210/mend.7.2.8469236. [DOI] [PubMed] [Google Scholar]
- Kimura E, Han JJ, Li S, Fall B, Ra J, Haraguchi M. Cell-lineage regulated myogenesis for dystrophin replacement: a novel therapeutic approach for treatment of muscular dystrophy. Hum Mol Genet. 2008;17:2507–2517. doi: 10.1093/hmg/ddn151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- Banks GB, Gregorevic P, Allen JM, Finn EE, Chamberlain JS. Functional capacity of dystrophins carrying deletions in the N-terminal actin-binding domain. Hum Mol Genet. 2007;16:2105–2113. doi: 10.1093/hmg/ddm158. [DOI] [PubMed] [Google Scholar]
- Li S, Kimura E, Ng R, Fall BM, Meuse L, Reyes M. A highly functional mini-dystrophin/GFP fusion gene for cell and gene therapy studies of Duchenne muscular dystrophy. Hum Mol Genet. 2006;15:1610–1622. doi: 10.1093/hmg/ddl082. [DOI] [PubMed] [Google Scholar]
- Thayer MJ, Tapscott SJ, Davis RL, Wright WE, Lassar AB, Weintraub H. Positive autoregulation of the myogenic determination gene MyoD1. Cell. 1989;58:241–248. doi: 10.1016/0092-8674(89)90838-6. [DOI] [PubMed] [Google Scholar]
- Clegg CH, Hauschka SD. Heterokaryon analysis of muscle differentiation: regulation of the postmitotic state. J Cell Biol. 1987;105:937–947. doi: 10.1083/jcb.105.2.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatori G, Lattanzi L, Coletta M, Aguanno S, Vivarelli E, Kelly R. Myogenic conversion of mammalian fibroblasts induced by differentiating muscle cells. J Cell Sci. 1995;108:2733–2739. doi: 10.1242/jcs.108.8.2733. [DOI] [PubMed] [Google Scholar]
- Pomerantz JH, Mukherjee S, Palermo AT, Blau HM. Reprogramming to a muscle fate by fusion recapitulates differentiation. J Cell Sci. 2009;122 Pt 7:1045–1053. doi: 10.1242/jcs.041376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal B, Serrano AL, Tjwa M, Suelves M, Ardite E, De Mori R. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 2008;22:1747–1752. doi: 10.1101/gad.465908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano AL, Muñoz-Cánoves P. Regulation and dysregulation of fibrosis in skeletal muscle. Exp Cell Res. 2010;316:3050–3058. doi: 10.1016/j.yexcr.2010.05.035. [DOI] [PubMed] [Google Scholar]
- Hartigan-O’Connor D, Kirk CJ, Crawford R, Mulé JJ, Chamberlain JS. Immune evasion by muscle-specific gene expression in dystrophic muscle. Mol Ther. 2001;4:525–533. doi: 10.1006/mthe.2001.0496. [DOI] [PubMed] [Google Scholar]
- Harris JB. Myotoxic phospholipases A2 and the regeneration of skeletal muscles. Toxicon. 2003;42:933–945. doi: 10.1016/j.toxicon.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Morgan JE, Hoffman EP, Partridge TA. Normal myogenic cells from newborn mice restore normal histology to degenerating muscles of the mdx mouse. J Cell Biol. 1990;111 6 Pt 1:2437–2449. doi: 10.1083/jcb.111.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves MA, Swildens J, Holkers M, Narain A, van Nierop GP, van de Watering MJ. Genetic complementation of human muscle cells via directed stem cell fusion. Mol Ther. 2008;16:741–748. doi: 10.1038/mt.2008.16. [DOI] [PubMed] [Google Scholar]
- Darabi R, Arpke RW, Irion S, Dimos JT, Grskovic M, Kyba M. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell. 2012;10:610–619. doi: 10.1016/j.stem.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481:295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seed J, Hauschka SD. Clonal analysis of vertebrate myogenesis. VIII. Fibroblasts growth factor (FGF)-dependent and FGF-independent muscle colony types during chick wing development. Dev Biol. 1988;128:40–49. doi: 10.1016/0012-1606(88)90264-3. [DOI] [PubMed] [Google Scholar]
- Tortorella LL, Milasincic DJ, Pilch PF. Critical proliferation-independent window for basic fibroblast growth factor repression of myogenesis via the p42/p44 MAPK signaling pathway. J Biol Chem. 2001;276:13709–13717. doi: 10.1074/jbc.M100091200. [DOI] [PubMed] [Google Scholar]
- Périé S, Trollet C, Mouly V, Vanneaux V, Mamchaoui K, Bouazza B. Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: a phase I/IIa clinical study. Mol Ther. 2014;22:219–225. doi: 10.1038/mt.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuk D, Goulet M, Roy B, Piette V, Côté CH, Chapdelaine P. First test of a “high-density injection” protocol for myogenic cell transplantation throughout large volumes of muscles in a Duchenne muscular dystrophy patient: eighteen months follow-up. Neuromuscul Disord. 2007;17:38–46. doi: 10.1016/j.nmd.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Phelps SF, Hauser MA, Cole NM, Rafael JA, Hinkle RT, Faulkner JA. Expression of full-length and truncated dystrophin mini-genes in transgenic mdx mice. Hum Mol Genet. 1995;4:1251–1258. doi: 10.1093/hmg/4.8.1251. [DOI] [PubMed] [Google Scholar]
- Chamberlain JS. Dystrophin levels required for genetic correction of Duchenne muscular dystrophy. Basic Appl Myol. 1997;7:251–255. [Google Scholar]
- Blaveri K, Heslop L, Yu DS, Rosenblatt JD, Gross JG, Partridge TA. Patterns of repair of dystrophic mouse muscle: studies on isolated fibers. Dev Dyn. 1999;216:244–256. doi: 10.1002/(SICI)1097-0177(199911)216:3<244::AID-DVDY3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Lovering RM, Michaelson L, Ward CW. Malformed mdx myofibers have normal cytoskeletal architecture yet altered EC coupling and stress-induced Ca2+ signaling. Am J Physiol Cell Physiol. 2009;297:C571–C580. doi: 10.1152/ajpcell.00087.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PS, Bye-a-Jee H, Wells DJ. Physiological characterization of muscle strength with variable levels of dystrophin restoration in mdx mice following local antisense therapy. Mol Ther. 2011;19:165–171. doi: 10.1038/mt.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering RM, McMillan AB, Gullapalli RP. Location of myofiber damage in skeletal muscle after lengthening contractions. Muscle Nerve. 2009;40:589–594. doi: 10.1002/mus.21389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- Lapid O, van Wingerden JJ, Perlemuter L. Tamoxifen therapy for the management of pubertal gynecomastia: a systematic review. J Pediatr Endocrinol Metab. 2013;26:803–807. doi: 10.1515/jpem-2013-0052. [DOI] [PubMed] [Google Scholar]
- Li S, Kimura E, Fall BM, Reyes M, Angello JC, Welikson R. Stable transduction of myogenic cells with lentiviral vectors expressing a minidystrophin. Gene Ther. 2005;12:1099–1108. doi: 10.1038/sj.gt.3302505. [DOI] [PubMed] [Google Scholar]
- Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc. 2008;3:799–810. doi: 10.1038/nprot.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkhart TA, Clegg CH, Hauschka SD. Control of mouse myoblast commitment to terminal differentiation by mitogens. J Supramol Struct. 1980;14:483–498. doi: 10.1002/jss.400140407. [DOI] [PubMed] [Google Scholar]
- Chapman VM, Miller DR, Armstrong D, Caskey CT. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc Natl Acad Sci USA. 1989;86:1292–1296. doi: 10.1073/pnas.86.4.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.