

Abstract
Objectives
To discuss the role of clinical trials in the changing landscape of cancer care resulting in individualized cancer treatment plans including a discussion of several innovative randomized studies designed to evaluate multiple targeted therapies in molecularly defined subsets of individuals.
Data Sources
Medical and nursing literature, research articles, and clinicaltrials.gov.
Conclusion
Recent advancements in cancer biomarkers and biomedical technology have begun to transform fundamentals of cancer therapeutics and clinical trials through innovative adaptive trial designs. The goal of these studies is to learn not only if a drug is safe and effective but also how it is best delivered and who will derive the most benefit.
Implications for Nursing Practice
Implementation of clinical trials in the cancer biomarker era requires knowledge, skills, and expertise related to the use of biomarkers and molecularly defined processes underlying a malignancy, as well as an understanding of associated ethical, legal, and social issues to provide competent, safe, and effective health care and patient communication.
Keywords: Precision medicine, Clinical trial, Adaptive design, Biomarker, Molecular targets, Research ethics
Introduction
Precision medicine is an approach to deliver optimal patient outcomes by integrating clinical and molecular patient data to understand the biological basis of the disease.1 This approach guides selection of the most appropriate targeted therapy based on distinctive patient characteristics and unique molecular features of a malignancy. The aim of this strategy is to optimize patient outcomes, while providing more favorable safety profiles than traditional population-based cancer chemotherapy.
Clinical trials are the means by which investigational agents, devices, or biologics, such as chemotherapy agents, blood products, or gene therapies are scientifically evaluated in human volunteers for safety and efficacy. Typically, candidate therapeutic agents progress through a carefully regulated and lengthy multi-phase clinical trial process before receiving US Food and Drug Administration (FDA) approval. Moving forward, significant modifications to current clinical trial designs will be necessary to progress toward a more personalized approach. Adaptive trial design is an example of an innovative accelerated effort for evaluating targeted therapies. This design allows researchers to analyze accumulating study data at prospective interim time points and to alter the course of an individual’s study plan or the trial itself.2 Common types of trial adaptations are listed in Table 1.2 This paper will describe adaptive design, and present examples of studies currently being conducted using this novel approach, as well as discuss ways in which genomic and biomarker research advances precision medicine.
TABLE 1.
The Most-Common Types of Adaptive Settings in Modern Clinical Trials
| Stopping early (or late, that is, extending accrual) with a conclusion of either superiority or futility |
| Adaptively assigning doses to more-efficiently assess the dose-outcome relationship |
| Dropping arms or doses |
| Seamless phases of drug development within a single trial |
| Changing the proportion of patients randomized to each arm |
| Adaptively homing in on an indication or responder population |
| Adding arms or doses |
| Changing accrual rate |
Reprinted with permission from Macmillan Publishers Ltd: Nature Reviews Clinical Oncology © 2012.2
Adaptive design trials have the ability to answer multiple questions in a single trial structure.2,3 The paradigm in oncology is shifting to use trials to learn not only if a drug is safe and effective but also how it is best delivered and who will derive the most benefit. Adaptive trials use a strategy in which results of an interim analysis can influence the treatment arms offered to patients subsequently enrolled. Below we discuss two adaptive clinical trials programs as examples.
I-SPY
I-SPY 1 (ClinicalTrials.gov numbers: NCT00043017) is a neoadjuvant trial of women with locally advanced breast cancer, which are assessed for estrogen receptor (ER), progesterone receptor, human epidermal growth factor 2 (HER2), and Mammaprint (Agendia, Irvine, CA), a 70-gene predictive signature of distant recurrence prior to treatment (or randomization).4,5 The trial evaluates molecular biomarkers of treatment and response and breast imaging to guide “adaptive” (ie, subsequent otpimal treatments). Initial studies were used to develop and validate optimal metrics of treatment response in I-SPY1.
In I-SPY 1, chemotherapy was administered before surgery, and biomarkers were compared with tumor response on the basis of magnetic resonance imaging (MRI), pathologic residual disease at the time of surgical excision, and 3-year disease-free survival. The study found that pathologic complete response (pCR), defined as no invasive tumor present in either the breast or axillary lymph nodes, differed by molecular subset; hormone receptor-positive/HER2-negative carcinomas were associated with the lowest pCR (9%) and hormone receptor-negative/HER2-positive had the highest pCR (45%).4 I-SPY 1 also indicated that pCR was predictive of recurrence free survival within a molecular subset.4 The study showed that MRI volume was the best predictor of residual disease after chemotherapy.5 This study established the infrastructure to integrate biomarkers and imaging with shared methods and real-time access to study data which will be leveraged for I-SPY 2.
I-SPY 2 (investigation of serial studies to predict your therapeutic response with imaging and molecular analysis 2) (ClinicalTrials.gov numbers: NCT01042379) is an adaptive design trial using Bayesian statistics comparing novel drugs in combination with standard chemotherapy with the efficacy of standard therapy alone. The trials schema is shown in Figure 1. Acceptability criteria for novel drugs include: compatibility with taxane therapy and for HER2-directed therapy, comparability with taxane plus trastuzumab; rational for efficacy in breast cancer; targeting key pathways/molecules in breast cancer: HER2, insulin-like growth factor receptor (IGFR), phosphatidylinositol 3-kinase (PI3K), macrophages, Akt, Akt and mitogen-activated protein kinase (MAPK), PI3K and mitogen-activated protein/extracellular signal-related kinase kinase (MEK), death receptor, cMET, mammalian target of rapamycin (mTOR); and fitting the strategic model for single/multiple molecular targeting in combination with chemotherapy.6 The study will have two control arms consisting of standard neoadjuvant chemotherapy (weekly paclitaxel or paclitaxel plus trastuzumab for HER2+ patients) followed by doxorubicin and cyclophosphamide. Each experimental drug will be tested in a minimum of 20 patients and a maximum of 120 patients.6 Patients must present with at least a 3-cm lesion and have a core biopsy, MRI, and blood sample draw to access eligibility. Eligible patients must either be MammaPrint high-risk, MammaPrint low-risk and ER-negative, or Mammaprint low-risk and ER-positive and HER2-positive.6 The agents currently under investigation in the trial are Neratinib (Her2 inhibitor), ABT-888 (a PARP inhibitor), AMG 386 (angiopoietin 1 and 2 neutralizing peptibody), AMG 479 (monoclonal antibody against IGFR1) plus metformin, MK-2206 (Akt inhibitor) with or without trastuzumab, AMG 386 and Trastuzumab, T-DM1 (trastuzumab conjugated to cytotoxic agent mertansine) and pertuzumab (monoclonal antibody targeted against HER2), and pertuzumab plus trastuzumab.6 Following determination of eligibility patients are randomized to treatment assignment, have 3 weeks of either novel therapy or standard therapy, undergo a repeat MRI and core biopsy, and then continue treatment for an additional 9 weeks followed by another MRI and core biopsy before initiating standard chemotherapy with doxorubicin and cyclophosphamide.6 A blood sample and additional MRI are obtained before surgery. The patient is assessed at surgery for pCR and followed for disease-free and overall survival. Based on the biomarkers assessed at baseline, the relationship between pCR and signatures of interest are modeled with the randomization probability changing to account to accumulating data.6 Agents that do well within a specific molecular signature subgroup of interest will progress through the trial more quickly and be graduated with their corresponding biomarker to be tested in a small phase III trial. Agents that are not efficacious compared with standard treatment in any of the molecular signature will be dropped for futility. As agents are graduated or dropped new agents can be added to the trial. Recently, one of the agents, ABT-888, was graduated showing promising results in patients with triple-negative breast cancer. Patients with triple-negative breast cancer had a 52% pCR receiving ABT-888 plus standard chemotherapy compared with 26% pCR in those receiving standard chemotherapy alone.7
FIGURE 1.

I-SPY 2 Schema trial design for I-SPY 2 (investigation of serial studies to predict your therapeutic response with imaging and molecular analysis 2). HER2, human epidermal growth factor receptor 2; MRI, magnetic resonance imaging; AC, anthracycline (doxo-rubicin) and cyclophosphamide (Cytoxan).
This trial is a public–private partnership managed through the Foundation for the National Institutes of Health Biomarkers Consortium and involves collaboration from the National Cancer Institute, FDA, more than 20 academic centers, multiple pharmaceutical companies, laboratories, non-profit organizations, and advocates. The advocates have been involved in reviewing the consent documents and protocol design, helping to develop patient materials, and staffing a hotline/e-mail inbox in which potential patients and patients currently on trial can get access to a counselor with special training about the trial.8 Additionally, there is a trial website (http://www.ispy2.org), which explains the trial, trial procedures, who should participate, and gives contact information for the study sites.9 This integrated team is hoping to continue to move novel agents through the drug development pipeline to speed the approval process, including designing an I-SPY 3 trial with international collaborators that will look at both pCR and long-term survival.10
Battle
The phase II Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE) program (ClinicalTrials.gov numbers: NCT00409968, NCT00411671, NCT00411632, NCT00410059, and NCT00410189) is a second example of a clinical trial to determine regimes for precision or personalized medicine (Fig. 2). Bio-markers have emerged as an important factor in planning treatment for non–small cell lung cancer (NSCLC) because of knowledge that specific epidermal growth factor receptor (EGFR) mutations lead to improved outcomes with EGFR tyrosine kinase inhibitors (TKI).11–15 The BATTLE program consists of an umbrella trial plus four phase II protocols.16 These phase II protocols used agents directed against promising molecular targets at the time the study began in 2005. The targets included EGFR (erlotinib), KRAS/BRAF (sorafinib), retinoid-EGFR signaling (bexarotene and erlotinib), and vascular endothelial growth factor receptor (VEGFR) (vantetanib). The primary endpoint of the study was the 8-week disease control rate (DCR) defined as complete or partial response or stable disease via Response Evaluation Criteria in Solid Tumors (RECIST).16 A 30% DCR in similar patients was used as a control, with treatment efficacy defined as a greater than 80% probability of achieving greater than 30% DCR.16
FIGURE 2.
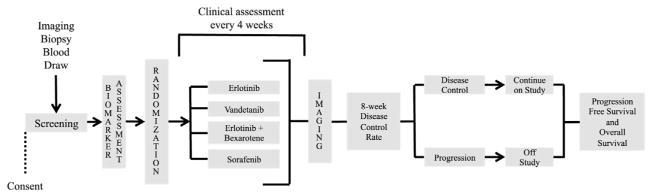
Battle schema trial design for Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE).
Patients enrolled in the umbrella trial underwent tumor biopsy and biomarker analysis for 11 biomarkers: mutations in EGFR, KRAS, and BRAF; copy numbers of EGFR and the Cyclin D1 gene (CCND1); and protein expression level of VEGF, VEGF-2, RXRs α, β, and γ, and Cyclin D1.16 The biomarker analysis was completed with day 14 biopsy; patients and investigators were blinded to biomarker results until the patient went off study. The results of the biomarker analysis were used to classify patients into one of five groups: 1) EGFR mutation and/or amplification; 2) KRAS or BRAF mutation; 3) VEGF and/or VEGF-2 overexpression; 4) RXRs α, β, and γ, and/or Cyclin D1 overexpression and/or CCND1 amplification; or 5) negative for biomarker panel.16 Those patients who were positive for more than one marker were assigned to a treatment group based on the marker with highest predictive value. During the first part of the study patients were enrolled randomly to each of the four phase II studies except for patients with prior erlotinib treatment who were excluded from the erlotinib-containing study arms.16 The results of this randomized portion of the study were used to assess the association between a given marker group and disease control. For example, patients with an EGFR mutation and/or amplification had a certain probability of disease control with each of the treatment regimens. For the second part of the trial this probability was incorporated into a Bayesian adaptive algorithm to randomly assign patients to an optimally predicted treatment arm. The probability of disease control was updated throughout the trial based on accumulating data.
The study enrolled 341 patients between November 30, 2006 and October 28, 2009.16 The first 97 patients were randomly assigned, with an additional 158 patients undergoing adaptive randomization.16 The study population had a mean age of 62 and was heavily pretreated with 45% having prior erlotinib treatment and a median of two regimens of cytotoxic chemotherapy.16 Overall, 46% of patients achieved an 8-week DCR with individual study arm DCRs of 34% (erlotinib), 33% (vandetanib), 50% (erlotinib and bexarotene), and 58% (sorafenib).16 Treatment/biomarker pairs were considered efficacious if there was a greater than 80% probability of achieving a DCR in over 30% patients: erlotinib in group 3; erlotinib and bexarotene in groups 1, 4, and 5; and sorafenib in groups 2, 3, and 5.16 In addition to the prespecified biomarker groups, the investigators also examined the effects of individual biomarkers on treatment efficacy. EGFR mutation was predictive of erlotinib response, overexpression of VEGF-2 was predictive of vandetanib response, and over-expression of Cyclin D1 was predictive of erlotinib and bexarotene combination response.16
This trial demonstrated the feasibility of conducting a biopsy-mandated, real-time biomarker-based adaptively randomized clinical trial in heavily pretreated advanced lung cancer patients. Traditionally, biomarker profiles are determined at the original diagnosis but may not reflect the tumor status during progression. The trial confirmed the prespecified hypotheses of treatment efficacy in biomarker-positive patients treated with agents, which have a related mechanism of action. The study also found that the biomarkers groups were less predictive than the individual markers.
In a follow-up study, which is currently open, BATTLE-2 (Clinicaltrials.gov number: NCT01248247), the initial 200 patients will be tested for a limited number of biomarkers including KRAS and EGFR mutation and EML4-ALK gene fusion.16 Patients with EGFR mutation and EML4-ALK gene fusion will be excluded unless they failed treatment with an inhibitor specifically targeting the related pathway.17 Investigators will also conduct prospective testing of biomarkers or combinations of biomarkers (signatures) using mutation analysis, protein expression, and mRNA signatures. The best biomarkers will then be used to guide patient assignment using an adaptive design for an additional 200 patients. This trial will examine erlotinib; sorafenib; a combination of erlotinib plus an Akt inhibitor (MK-2206), thought to overcome EGFR resistance; and a combination of a MEK inhibitor (AZD6244) plus an Akt inhibitor (MK-2206), thought to target KRAS.18 This study will also be multi-institutional, enrolling patients both at M.D. Anderson Cancer Center and Yale University. The goal of the study is similar to the original BATTLE trial, which will define a specific biomarker population, which predicts response to targeted agent using 8-week DCR as the primary endpoint.
The investigators will also be expanding the adaptive design into patient receiving front-line therapy in BATTLE-FL (Clinicaltrials.gov number: NCT01263782) and BATTLE-Prevention (under development).19 BATTLE-FL will examine bio-markers to better define what drug (bevacizumab, cetuximab, or cixutumumab) given in combination with carboplatin or pemetrexed will improve progression-free survival. BATTLE-Prevention will use information from advanced cancer treatment to bring agents and biomarkers into the adjuvant setting to treat patients with stage I-IIIA NSCLC to prevent recurrence.19,20
Ethical and Regulatory Issues
The ethical and regulatory issues in clinical trials for precision medicine share many aspects with clinical trials in general. This includes disclosure policy, privacy and data sharing, confidentiality, and good clinical practice. As the use of adaptive design trials has grown, the FDA issued a draft guidance in 2010 entitled “Adaptive Design Clinical Trials for Drugs and Biologics” discussing aspects of adaptive design trials, when and how to interact with the FDA during planning, and issues to consider during analysis.21 The major regulatory issue when planning adaptive design trials is that all adaptations need to be specified before trial initiation.2 Because an adaptive-design trial can evolve as data accumulates it is important to limit data access during the trial.3 The probability of a patient being assigned to one arm versus another must remain confidential to maintain the integrity of the trial. For example, if patients with biomarker “X” are having a superior response to drug “Y”, patients with biomarker “X” would have a higher probability of being assigned study arm contain drug “Y”. If the confidentiality of this superior response were to be known by the blinded study investigators during the course of the trial patients could possibly be treated outside of the trial with drug “Y”.
New biomarkers tested in clinical trials along with drug efficacy to define a responder population are called in vitro (IVD) companion diagnostic device. An IVD companion diagnostic device requires separate approval from the FDA. The research related to the IVD companion diagnostic device is conducted under an investigational device exemption. The FDA issued a draft guidance in 2011 entitled “Guidance for Industry and Food and Drug Administration Staff – In Vitro Companion Diagnostic Devices”.22 This guidance discusses both co-development of a IVD companion diagnostic device in which the development of the therapeutic product depend on the biomarker as well as development of an IVD companion diagnostic device after drug approval. For example, vemurafenib (a kinase inhibitor indicated for the treatment of unresectable or meta-static melanoma) was approved with an IVD companion diagnostic device (cobas 4800 BRAF V600 mutation test [Roche, IN]). Vemurafenib is not indicated for patients with wild-type BRAF.
Adaptive design trials are complex, with operational issues related to logistic and procedural implementation. Some institutional review boards may be unfamiliar with the design and questions still exist about how best to ensure adequate informed consent.23 A major consideration is the additional start-up costs required to invest in integrating the process and information technology infrastructure.24 Although once this has been setup it can be leveraged for additional studies. Adaptive design clinical trials such as those described earlier have lower enrollment by virtue of their response-adaptive randomization, and thus have less opportunity to cause harm by virtue of randomizing a higher proportion of patients to study arms in which patients like them have previously responded.25
Nursing Implications
Recent discoveries in basic science have transformed our understanding of cancer, as well as our approach to patient care and expectations for patient outcomes. As the practice of precision oncology medicine becomes a reality, nurses are increasingly called upon to implement biomarker-based care into all aspects of nursing practice, including clinical trials. Providing safe, competent, and effective care for individual’s participating in modern clinical trials requires knowledge, skills, and expertise related to imaging, appropriate treatment options (both standard of cancer and potential clinical trials), and necessary and/or supplemental disease biomarkers. Nursing practice in this context also requires knowledge of appropriate referrals for counseling and interdisciplinary services and patient follow-up, as well as responsibilities related to the conduct of clinical trials.
To prepare nurses to adequately to perform in the new era of clinical trials and the human genome, nursing organizations such as the American Nurses Association, the Oncology Nursing Society (ONS), and the International Society of Nurses in Genetics (ISONG) have developed competencies, curricula guidelines, and in some cases outcome indicators to incorporate the genetic, genomic, and clinical trials perspective into nursing education, practice, and research. Table 2 combines the aspects of ONS’ Oncology Clinical Trials Nurse Competencies26 and a set of genetic and genomic nurse competencies from the American Nurses Association.27 Both sets of competencies were developed in direct response to the needs of professional nurses for comprehensive curriculum on their specific content area and for standardization of role expectation. These competencies involve aspects of protocol compliance such as good clinical practice and understanding the relationship of genetics/genomics to a clinical trial; informed consent process helping the patients understand all aspects of the trial; subject recruitment; trial-related communication; management of research subjects; ethical issues; and professional development. As imaging plays a larger role in determining treatment effectiveness as in I-SPY 2 in which MRI volume correlates with risk of residual disease and the importance of image guided-biopsy in both the I-SPY trials and BATTLE trials, it will become more important for oncology nurses to understand imaging as it relates to the clinical trial they are conducting or the type of imaging necessary for their patient care responsibilities. This includes: preparing the patient effectively, supporting the patient through the procedure, safely caring for the patient post imaging, assisting with imaging preparation when necessary, maintaining a safe environment, and performing specific interventions during imaging when required. These competencies were recently highlighted in a Royal College of Nursing publication.28 The nursing societies have not yet developed competencies directly related to bio-markers, which may be more directly relevant to adaptive design trials and could be developed as the field progresses. The oncology nurse is an integral part of the interdisciplinary team required for efficient clinical trials in the era of precision medicine.
TABLE 2.
Oncology Nursing Competencies for Clinical Trials in the Era of Precision Medicine
| Functional Area | Competencies |
|---|---|
| Protocol compliance | Facilitates compliance with research protocol and good clinical practice* |
| Demonstrates an understanding of the relationship of genetics/genomics to a clinical trial† | |
| Trial-related communication | Utilizes multiple communication methods to facilitate implementation of clinical trials* |
| Facilitates referrals for specialized genetic/genomic services for clients as needed and as allowed per protocol† | |
| Informed consent process | Demonstrates leadership in ensuring patient comprehension and safety during initial and ongoing clinical trial informed consent discussions* |
| Advocates for the rights of clients for autonomous, informed genetic/genomic related decision-making and voluntary action† | |
| Management of research subjects | Uses a variety of resources and strategies to manage the care of study participants, ensuring compliance with protocol procedures, assessments, and reporting requirements, as well as management of symptoms* |
| Identifies credible, accurate, appropriate, and current genetic/genomic information, resources, services, and/or technologies, per protocol† | |
| Demonstrate importance of tailoring genetic/genomic information and services to clients based on their culture, religion, knowledge level, literacy, and preferred language† | |
| Documentation | Ensures collection of source data and completion of documentation that validates the integrity of the study* |
| Collects personal, health, and developmental histories that consider genetic, environmental, and genomic influences and risks, per protocol† | |
| Subject recruitment | Utilizes a variety of recruitment strategies to enhance recruitment while being mindful of the needs of the study populations* |
| Assesses potential study subjects’ knowledge, perceptions, and responses to genetic/genomic information† | |
| Identifies study subjects who may benefit from specific genetic/genomic information and/or services based on assessment data, per protocol† | |
| Ethical issues | Demonstrates leadership in ensuring adherence to ethical practices during the trial in order to protect the rights, well-being, and privacy of participants and the collection of quality data* |
| Identifies ethical/ancestral, religious, legal, fiscal, and societal issues related to genetic/genomic information and technologies† | |
| Defines issues that undermine the rights of all clients for autonomous, informed genetic and genomic-related decision-making and voluntary action† | |
| Professional development | Assumes responsibility for identifying ongoing professional development needs and seeks resources and opportunities to meet need, ie, membership in nursing, oncology or research organizations* |
| Examines competency of practice regularly, identifying areas of strength, as well as areas in which professional development related to genetics/genomics would be beneficial† | |
| Incorporate genetic and genomic technologies/information into professional practice† | |
| Recognizes when one’s own attitude and values related to genetic/genomic science may affect care provided to clients† |
Contributor Information
Brandy M. Heckman-Stoddard, Division of Cancer Prevention, National Cancer Institute, Bethesda, MD.
Judith J. Smith, Division of Cancer Prevention, National Cancer Institute, Bethesda, MD.
References
- 1.Desmond-Hellmann S. Toward precision medicine: a new social contract? Sci Transl Med. 2012;4:129ed123. doi: 10.1126/scitranslmed.3003473. [DOI] [PubMed] [Google Scholar]
- 2.Berry DA. Adaptive clinical trials in oncology. Nat Rev Clin Oncol. 2012;9:199–207. doi: 10.1038/nrclinonc.2011.165. [DOI] [PubMed] [Google Scholar]
- 3.Berry DA. Adaptive clinical trials: the promise and the caution. J Clin Oncol. 2011;29:606–609. doi: 10.1200/JCO.2010.32.2685. [DOI] [PubMed] [Google Scholar]
- 4.Esserman LJ, Berry DA, DeMichele A, Carey L, Davis SE, Buxton M, et al. Pathologic complete response predicts recurrence-free survival more effectively by cancer subset: results from the I-SPY 1 TRIAL–CALGB 150007/150012, ACRIN 6657. J Clin Oncol. 2012;30:3242–3249. doi: 10.1200/JCO.2011.39.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hylton NM, Blume JD, Bernreuter WK, Pisano ED, Rosen MA, Morris EA, et al. Locally advanced breast cancer: MR imaging for prediction of response to neoadjuvant chemotherapy–results from ACRIN 6657/I-SPY TRIAL. Radiology. 2012;263:663–672. doi: 10.1148/radiol.12110748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barker AD, Sigman CC, Kelloff GJ, Hylton NM, Berry DA, Esserman LJ. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin Pharmacol Ther. 2009;86:97–100. doi: 10.1038/clpt.2009.68. [DOI] [PubMed] [Google Scholar]
- 7.I-SPY 2: Novel Study Design; San Antonio Breast Cancer Symposium; San Antonio, TX. 2013. [Google Scholar]
- 8.I-SPY2. [Accessed December 18, 2013];I-SPY2advocategoalsandassessmentplan. Available at: http://www.gemini-grp.com/ISPY/AdvocateGoalsAssessmentPlan.pdf.
- 9.I-SPY2. [Accessed December 18, 2013];I-SPY 2 is a clinical trial for women with newly diagnosed locally advanced breast cancer. Available at: http://www.ispy2.org.
- 10.Winslow R. Wall Street Journal. New York, NY: Two experimental breast-cancer drugs pass major milestone in study. Available at: http://online.wsj.com/news/articles/SB10001424052702303293604579255831485652454. [Google Scholar]
- 11.Kim ES, Hirsh V, Mok T, Socinski MA, Gervais R, Wu YL, et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): a randomised phase III trial. Lancet. 2008;372:1809–1818. doi: 10.1016/S0140-6736(08)61758-4. [DOI] [PubMed] [Google Scholar]
- 12.Douillard JY, Shepherd FA, Hirsh V, Mok T, Socinski MA, Gervais R, et al. Molecular predictors of outcome with gefitinib and docetaxel in previously treated non-small-cell lung cancer: data from the randomized phase III INTEREST trial. J Clin On-col. 2010;28:744–752. doi: 10.1200/JCO.2009.24.3030. [DOI] [PubMed] [Google Scholar]
- 13.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 14.Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–967. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 15.Godin-Heymann N, Bryant I, Rivera MN, Ulkus L, Bell DW, Riese DJ, 2nd, et al. Oncogenic activity of epidermal growth factor receptor kinase mutant alleles is enhanced by the T790M drug resistance mutation. Cancer Res. 2007;67:7319–7326. doi: 10.1158/0008-5472.CAN-06-4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR, Jr, Tsao A, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discovery. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson NJ. Adaptive clinical trial design: has its time come? J Natl Cancer Inst. 2010;102:1217–1218. doi: 10.1093/jnci/djq319. [DOI] [PubMed] [Google Scholar]
- 18.Berry DA, Herbst RS, Rubin EH. Reports from the 2010 clinical and translational cancer research think tank meeting: design strategies for personalized therapy trials. Clin Cancer Res. 2012;18:638–644. doi: 10.1158/1078-0432.CCR-11-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gold KA, Kim ES, Lee JJ, Wistuba II, Farhangfar CJ, Hong WK. The BATTLE to personalize lung cancer prevention through reverse migration. Cancer Prev Res. 2011;4:962–972. doi: 10.1158/1940-6207.CAPR-11-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gold K, Kim E, Wistuba I, Hong W. Personalizing lung cancer prevention through a reverse migration strategy. In: Pezzuto JM, Suh N, editors. Natural products in cancer prevention and therapy. Heidelberg: Springer; 2013. pp. 221–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.US Food and Drug Administration. Guidance for industry adaptive design clinical trials for drugs and biologics - Draft Guidance. 2010 doi: 10.1080/10543406.2010.514453. Available at: http://www.fda.gov/downloads/Drugs/.../Guidances/UCM201790.pdf. [DOI] [PubMed]
- 22.US Food and Drug Administration. Guidance for industry and food and drug administration staff - in vitro companion diagnostic devices - Draft Guidance. 2011 Available at: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm262292.htm.
- 23.Gaydos B, Anderson KM, Berry D, Burnham N, Chuang-Stein C, Dudinak J, et al. Good practices for adaptive clinical trials in pharmaceutical product development. Drug Info J. 2009;43:539–556. [Google Scholar]
- 24.Quinlan J, Gaydos B, Maca J, Krams M. Barriers and opportunities for implementation of adaptive designs in pharmaceutical product development. Clin Trials. 2010;7:167–173. doi: 10.1177/1740774510361542. [DOI] [PubMed] [Google Scholar]
- 25.Lipsky AM, Lewis RJ. Response-adaptive decision-theoretic trial design: operating characteristics and ethics. Stat Med. 2013;32:3752–3765. doi: 10.1002/sim.5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oncology Nursing Society. [Accessed November 25, 2013];ONS oncology clinical trials nurse competencies. Available at: http://www.ons.org/media/ons/docs/publications/ctncompetencies.pdf.
- 27.Consensus Panel on Genetic/Genomic Nursing Competencies. Essentials of genetic and genomic nursing: competanies, curricula guidelines, and outcome indicators. 2. Silver Spring, MD: American Nurses Association; 2009. [Google Scholar]
- 28.Royal College of Nursing. [Accessed December 20, 2013];Core competencies for imaging nurses. 2012 Available from URL: http://www.rcn.org.uk/__data/assets/pdf_file/0004/474754/004_265.pdf.