

Abstract
A sterically demanding unsymmetrical pentafluorophenyl-triisopropylphenyl-λ3-iodane was developed as an effective reagent for the electrophilic pentafluorophenylation of various β-keto esters and a β-keto amide. 17 examples of α-pentafluorophenylated 1,3-dicarbonyl compounds 3 having a quaternary carbon center are provided. The resulting compounds were nicely transformed into chiral α-pentafluorophenyl ketones with an all-carbon stereogenic center in high yields and high enantioselectivities using asymmetric organocatalysis (up to 98 % ee) or asymmetric metal catalysis (up to 82 % ee).
Keywords: asymmetric organocatalysis, diaryl-λ3-iodanes, electrophilic pentafluorophenylation, palladium
Trifluoromethylation and fluorination are now key technologies in innovations in pharmaceutical, agrochemical and advanced material industries.[1] On the other hand, direct pentafluorophenylation is still undeveloped, despite the potential use of the pentafluorophenyl (C6F5) group in liquid crystal materials, organic semiconductors and bioactive compounds.[2] Looking at the success of chiral drug industries[3] as well as fluorinated pharmaceuticals,[1] we are interested in molecules containing the C6F5 group attached to a Csp³ center as drug candidates in future markets.
Due to the strong electron-accepting property of the C6F5 group with a planar π system, characteristic stacking effects by polar π interactions have been observed between the C6F5 group and non-fluorinated counterparts.[4] Therefore, bioactive compounds with a C6F5 moiety are expected to interact with ubiquitous aromatic moieties of biomolecules such as amino acids and nucleic acids.[5] Indeed, Gellman and co-workers disclosed that the pentafluorophenyl variants of phenylalanine in a small protein enhance the stability of its tertiary structure.[5a,b] Nucleoside analogues containing a C6F5 instead of natural nucleobases have been used as tools for studying base pairing and DNA replication factors.[5c,d] The C6F5 group is also a universal base in peptide nucleic acids (PNAs).[5e] Although introduction of the C6F5 group into a Csp² center has been actively investigated by using radical and transition metal-catalyzed cross-coupling reactions,[6] these are not applicable for the construction of a stereogenic Csp³ center. Instead, silylated or metal-C6F5 reagents are a suitable choice for this purpose, and their utility is limited to the nucleophilic pentafluorophenylation of carbonyls, imines and related compounds.[7] Pentafluorophenylboronate serves as an aryl donor for a rhodium-catalyzed cross-coupling reaction with α-aryldiazoacetate, producing an α-pentafluorophenyl carboxylic acid ester.[8] On the other hand, methods for the electrophilic pentafluorophenylation of a stereocenter are rare, except for a nucleophilic attack on hexafluorobenzene.[9] In this method, however, the scope of the substrate is limited due to the need for drastic conditions (high temperature and a strong base).[9]
As part of an on-going synthetic program focused on organofluorine reagents,[10] we herein disclose our studies describing the synthesis and use of aryl-pentafluorophenyl-λ3-iodanes 1 (aryl pentafluorophenyl iodonium salts) as effective electrophilic pentafluorophenylating reagents. A variety of β-keto esters 2 is nicely α-pentafluorophenylated by 1 providing α-pentafluorophenyl-1,3-dicarbonyl compounds 3 with a quaternary carbon center in good yields under mild conditions. In particular, a structurally new and sterically demanding unsymmetrical diaryl-λ3-iodane 1 d was found to be most effective. The resulting α-pentafluorophenyl β-keto esters 3 were transformed into chiral α-pentafluorophenyl ketones with an all-carbon stereogenic center by organocatalysis or metal catalysis, that is, by decarboxylation followed by enantioselective alkylation under cinchona alkaloids catalysis, or asymmetric decarboxylative allylation under the Tsuji–Trost condition using palladium(II) catalysis (Scheme 1).
Scheme 1.
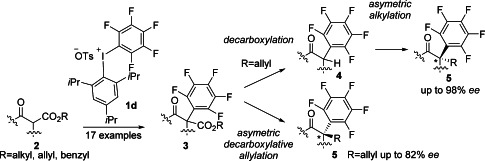
Electrophilic pentafluorophenylation by 1 and transformations to chiral α-pentafluorophenyl ketones having an all-carbon stereocenter.
It is a well-known fact that diaryl-λ3-iodanes react with enolates providing α-arylated carbonyl compounds.[11] While one of the two aryl groups of diaryl-λ3-iodanes is transferred into the substrates, a more electron-deficient aryl group plays a fundamental role in the arylation of enolates with unsymmetrical diaryl-λ3-iodanes.[12] Despite of a large number of papers on the arylation of enolates using this concept, pentafluorophenylation using diaryl-λ3-iodanes has surprisingly never been reported.[11, 12] This fact encouraged us to design a series of unsymmetrical C6F5-containing diaryl-λ3-iodanes as electrophilic pentafluorophenylation reagents for enolates. The reasons why unsymmetrical diaryl-λ3-iodanes are targeted are indicated next. Due to the electron-deficient nature of the C6F5 group, another set of aryl ligands were easily designed to stay attached to the parent iodanes. Furthermore, electron-poor symmetrical salts of bispentafluorophenyl-λ3-iodanes are difficult to prepare.[13] The five unsymmetrical diaryl-λ3-iodanes designed in this study were prepared in three steps from iodopentafluorobenzene via a Koser-type reagent, hydroxy(tosyloxy)iodopentafluorobenzene 6[14] according to a modified version of the published procedure, including oxidation and the Friedel–Crafts reaction (Scheme 2).[15]
Scheme 2.

Reagents and conditions: a) Oxone, TFA/CHCl3, RT, 2 h; b) TsOH⋅H2O, CH3CN, RT, 3 h (87 % over 2 steps); c) ArH, CF3CH2OH or TFA, RT, overnight.
We first examined the pentafluorophenylation of methyl indanone carboxylate 2 a using 1 (Table 1). The use of diaryl-λ3-iodane 1 a with a mesityl (2,4,6-Me3C6H2) group as a dummy ligand[16] was expected to be highly suitable in chemoselective pentafluorophenylation, but the outcome was disappointing with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)/CH2Cl2, yielding 6 % of 3 a and a detectable amount of an α-mesitylation side-product (Entry 1, Table 1). Attempts to improve the disappointing yield by changing the base, solvent, and additive were all unsuccessful (Entries 2–4). We next fine-tuned both the electronic and steric effects of the dummy ligand on 1. However, diaryl-λ3-iodanes 1 b–c with electron-donating methoxy group(s) on the benzene ring were also poor reagents for this transformation (Entries 5 and 6). Hence, sterically highly demanding triisopropyl-substituted diaryl-λ3-iodane 1 d was examined.[17] The yield improved slightly to 17 % in the presence of K2CO3 and tetrabutylammonium iodide in N,N-dimethylformamide (DMF; Entry 7). In an attempt to further improve the yield, the effect of the base and solvent on the reaction was examined. The combination of organic or inorganic bases with solvents had an impact on yield (Entries 8–17), which was finally improved to 72 % when the reaction of 2 a with 1 d was carried out in the presence of K3PO4 in CH2Cl2 (Entry 18). Re-optimization of reagents 1 under the best reaction conditions of K3PO4 in CH2Cl2 could not improve the yield (Entries 19–22, Table 1).
Table 1.
Initial studies on pentafluorophenylation of β-keto ester 2 a.[a]
 | ||||||
|---|---|---|---|---|---|---|
| Entry | 1 | Base | Additive | Solvent | Time [h] | Yield [%][b] |
| 1 | 1 a | DBU | – | CH2Cl2 | 2 | 6 |
| 2 | 1 a | DABCO | – | CH2Cl2 | 18 | 6 |
| 3 | 1 a | tBuOK | – | CH2Cl2 | 23 | 7 |
| 4[c] | 1 a | K2CO3 | Bu4NI | DMF | 8 | 9 |
| 5[c] | 1 b | K2CO3 | Bu4NI | DMF | 4 | 6 |
| 6[c] | 1 c | K2CO3 | Bu4NI | DMF | 42 | trace |
| 7[c] | 1 d | K2CO3 | Bu4NI | DMF | 2 | 17 |
| 8 | 1 d | NaH | – | DMF | 2 | 48 |
| 9 | 1 d | NaH | – | toluene | 4 | 28 |
| 10 | 1 d | NaH | – | THF | 20 | 8 |
| 11 | 1 d | NaH | – | CH2Cl2 | 4 | 58 |
| 12 | 1 d | iPr2NEt | – | CH2Cl2 | 11 | 21 |
| 13 | 1 d | K2CO3 | – | CH2Cl2 | 6 | 58 |
| 14 | 1 d | Cs2CO3 | – | CH2Cl2 | 2 | 53 |
| 15 | 1 d | NaOH | – | CH2Cl2 | 6 | 44 |
| 16 | 1 d | tBuOK | – | CH2Cl2 | 4 | 50 |
| 17 | 1 d | KF | – | CH2Cl2 | 8 | 49 |
| 18 | 1 d | K3PO4 | – | CH2Cl2 | 2 | 72 |
| 19 | 1 a | K3PO4 | – | CH2Cl2 | 2 | 20 |
| 20 | 1 b | K3PO4 | – | CH2Cl2 | 2 | 10 |
| 21 | 1 c | K3PO4 | – | CH2Cl2 | 2 | 5 |
| 22 | 1 e | K3PO4 | – | CH2Cl2 | 2 | 11 |
Reagents and conditions: a) 1 a–e (1.1 equiv), base (1.2 equiv), additive (10 mol %), solvent, RT, time. All reactions were carried out on a 19.0 mg (0.100 mmol) scale.
Isolated yield.
K2CO3 (3.0 equiv) was used.
With optimized reaction conditions in hand, the substrate scope was explored (Table 2). Reagent 1 d was identified as the most practical reagent for the electrophilic pentafluorophenylation of various substrates 2 in the presence of K3PO4 in CH2Cl2 at room temperature. When the reaction was carried out with β-keto esters 2 a–m derived from 1-indanones, it proceeded efficiently affording desired products 3 a–m in good yields. Substrates with sterically demanding tert-butyl ester 2 c and transformable allyl ester 2 d were reacted to give desired products 3 c and 3 d. The electronic or steric nature of the benzene ring and ester moiety of the substrates might not have affected the yields of 3 that much. The yields derived from tetralone 2 n and 2 o or cyclopentanone 2 p were slightly lower due to the less enolizable nature of the substrates. Notably, β-keto amido 2 q was also reactive giving 3 q in 41 % yield (see Table 2).
Table 2.
Substrate scope on pentafluorophenylation of β-keto esters 2[a]
 |
|---|
 |
The reaction of 2 with 1 d (1.1 equiv) was carried out in the presence of K3PO4 (1.2 equiv) in CH2Cl2 at RT, unless noted otherwise.
[b] 1 d (1.6 equiv) and K3PO4 (1.7 equiv) were used. [c] Reaction time was 22 h.
The allyl α-pentafluorophenyl-β-keto esters 3 d and 3 o that were obtained were easily transformed into α-pentafluorophenyl ketones 4 d and 4 o by palladium(II)-catalyzed decarboxylation using tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3), 1,2-bis(diphenylphosphino)ethane (dppe) and Meldrums acid as a proton source[18] in tetrahydrofuran (THF) with 96 % and 93 % yield, respectively (Scheme 3).
Scheme 3.
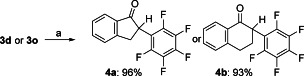
Palladium-catalyzed decarboxylation of β-keto esters 3 d and 3 o to α-pentafluorophenyl ketones 4 a and 4 b. Reagents and conditions: a) Pd2(dba)3 (2.5 mol %), dppe (6.25 mol %), Meldrums acid (2.5 equiv), THF, RT or 50 °C, 1–2 h.
With α-pentafluorophenyl ketones 4 in hand, we were ready to investigate the enantioselective functionalization of 4 to create a quaternary chiral carbon center bearing a C6F5 group by asymmetric organocatalysis. Asymmetric alkylation of α-phenyl ketones has been actively researched in recent years.[19] However, there are no examples of the use of α-pentafluorophenyl ketones as substrates. Fortunately, a brief survey of reaction conditions consisting of chiral catalysts, bases and solvents for allylation (see table S1 in the Supporting Information) led to the rapid selection of cinchoninium bromide as the phase-transfer catalyst of choice in aqueous toluene in the presence of KOH at −40 °C. The scope of the asymmetric alkylation of 4 is shown in Table 3. In all cases, high yields with excellent enantioselectivities up to 98 % ee were accomplished almost independent of both the electronic and steric natures of electrophiles R−Br, and substrates 4 (indanone 4 a and tetralone 4 b). The absolute configuration of 5 e was determined as R by X-ray crystallography (Figure 1), and the stereochemistry of 5 a–d, 5 f–i was tentatively assigned by analogy.
Table 3.
Creation of an all-carbon stereogenic center with C6F5: asymmetric alkylation of α-pentafluorophenyl ketones 4 with R−Br under organocatalysis[a]
 | ||||||
|---|---|---|---|---|---|---|
| Entry | 4, n | R | Time [h] | 5 | Yield[b] [%] | ee[c] [%] |
| 1[d] | 4 a, n=1 | allyl | 96 | 5 a | 95 | 87 (−) |
| 2 | 4 a, n=1 | benzyl | 24 | 5 b | 95 | 96 (+) |
| 3 | 4 a, n=1 | CH2-p-Tol | 48 | 5 c | 91 | 96 (−) |
| 4[e] | 4 a, n=1 | CH2-p-MeOPh | 72 | 5 d | 84 | 95 (−) |
| 5 | 4 a, n=1 | CH2-p-BrPh | 48 | 5 e | 87 | 98 (−) |
| 6 | 4 a, n=1 | CH2-1-Naphthyl | 96 | 5 f | 83 | 92 (+) |
| 7 | 4 a, n=1 | CH2-2-Naphthyl | 24 | 5 g | 89 | 94 (−) |
| 8 | 4 b, n=2 | benzyl | 48 | 5 h | 93 | 96 (−) |
Reagents and conditions: a) R−Br (2.0 equiv), 7 (10 mol %), aq KOH (50 wt %), toluene, −40 °C, time. All reactions were carried out using 7 a as a catalyst unless noted otherwise.
Isolated yield.
Determined by HPLC analysis.
Catalyst 7 b was used instead of 7 a.
R−Br (1.2 equiv) was employed.
Figure 1.

X-ray crystallographic structure of (R)-5 e (CCDC 1009021).
In another approach to chiral pentafluorophenyl compounds, direct asymmetric transformation of allyl-α-pentafluorophenyl-β-keto esters 3 d and 3 o to α-allyl-α-pentafluorophenyl-ketones 5 a and 5 i was achieved by palladium(II)-catalyzed asymmetric decarboxylative allylation[20] in the presence of the Trost ligand with high enantioselectivities (82 % and 80 % ee, respectively; Scheme 4). The absolute stereochemistry of 5 obtained by this procedure is opposite to that obtained by direct alkylation mentioned in Table 3.
Scheme 4.

Creation of an all-carbon stereogenic center with C6F5: Palladium-catalyzed asymmetric decarboxylative allylation of 3 d and 3 o using the Trost ligand.
In conclusion, this study discloses aryl-pentafluorophenyl-λ3-iodanes 1 as effective reagents for the electrophilic pentafluorophenylation of β-keto esters and amide 2. Among several reagents designed, an unsymmetrical diaryl iodonium salt 1 d with a highly sterically demanding 2,4,6-triisopropyl phenyl group as a dummy ligand was most effective for the electrophilic pentafluorophenylation reaction. In the presence of K3PO4 in CH2Cl2 at room temperature, the pentafluorophenylation of nucleophiles proceeded smoothly to yield the desired products. The resulting products were transformed into chiral α-pentafluorophenyl ketones bearing an all-carbon stereogenic center by asymmetric palladium(II) catalysis and phase-transfer organocatalysis. New studies on the potential of these reagents are now in progress.[21]
Acknowledgments
This study was financially supported in part by the Platform for Drug Discovery, Informatics, and Structural Life Science, Scientific Research (B) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan (25288045) and the Advanced Catalytic Transformation (ACT-C) from the Japan Science and Technology (JST) Agency. We thank Mr. Tatsuya Ochiai, Professor Tomohiro Ozawa and Professor Hideki Masuda (Nagoya Institute of Technology) for their help in X-ray crystallographic analysis.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
References
- 1a.Ojima I. J. Org. Chem. 2013;78:6358–6383. doi: 10.1021/jo400301u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b.Purser S, Moore PR, Swallow S, Gouverneur V. Chem. Soc. Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 1c.Shibata N, Mizuta S, Kawai H. Tetrahedron: Asymmetry. 2008;19:2633–2644. [Google Scholar]
- 2a.Kishikawa K, Oda K, Aikyo S, Kohmoto S. Angew. Chem. Int. Ed. 2007;46:764–768. doi: 10.1002/anie.200603594. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2007;119:778–782. [Google Scholar]
- 2b.Yoon M-H, Facchetti A, Stern CE, Marks TJ. J. Am. Chem. Soc. 2006;128:5792–5801. doi: 10.1021/ja060016a. [DOI] [PubMed] [Google Scholar]
- 2c.Babudri F, Farinola GM, Naso F, Ragni R. Chem. Commun. 2007:1003–1022. doi: 10.1039/b611336b. [DOI] [PubMed] [Google Scholar]
- 2d.Tsuzuki T, Shirasawa N, Suzuki T, Tokito S. Adv. Mater. 2003;15:1455–1458. [Google Scholar]
- 2e.Sakamoto Y, Suzuki T, Miura A, Fujikawa H, Tokito S, Taga Y. J. Am. Chem. Soc. 2000;122:1832–1833. [Google Scholar]
- 2f.Kitamura T, Wada Y, Yanagida S. J. Fluorine Chem. 2000;105:305–311. [Google Scholar]
- 2g.de Leval X, Ilies M, Casini A, Dogne J-M, Scozzafava A, Masini E, Mincione F, Starnotti M, Supuran CT. J. Med. Chem. 2004;47:2796–2804. doi: 10.1021/jm031116j. [DOI] [PubMed] [Google Scholar]
- 3.Voutchkova AM, Osimitz TG, Anastas PT. Chem. Rev. 2010;110:5845–5882. doi: 10.1021/cr9003105. [DOI] [PubMed] [Google Scholar]
- 4.Meyer EA, Castellano RK, Diederich F. Angew. Chem. Int. Ed. 2003;42:1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2003;115:1244–1287. [Google Scholar]
- 5a.Woll MG, Hadley EB, Mecozzi S, Gellman SH. J. Am. Chem. Soc. 2006;128:15932–15933. doi: 10.1021/ja0634573. [DOI] [PubMed] [Google Scholar]
- 5b.Cornilescu G, Hadley EB, Woll MG, Markley JL, Cellman SH, Cornilescu CC. Protein Sci. 2007;16:14–19. doi: 10.1110/ps.062557707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c.Lai JS, Qu J, Kool ET. Angew. Chem. Int. Ed. 2003;42:5973–5977. doi: 10.1002/anie.200352531. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2003;115:6155–6159. [Google Scholar]
- 5d.Mathis G, Hunziker J. Angew. Chem. Int. Ed. 2002;41:3203–3205. doi: 10.1002/1521-3773(20020902)41:17<3203::AID-ANIE3203>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2002;114:3335–3338. [Google Scholar]
- 5e.Das BK, Shibata N, Takeuchi Y. J. Chem. Soc. Perkin Trans. 1. 2002:197–206. [Google Scholar]
- 6a.Chen Q-Y, Li Z-T. J. Org. Chem. 1993;58:2599–2604. For examples of radical coupling reaction of C6F5 group. [Google Scholar]
- 6b.Frohn HJ, Klose A, Bardin VV. J. Fluorine Chem. 1993;64:201–215. [Google Scholar]
- 6c.Shang R, Fu Y, Wang Y, Xu Q, Yu H-Z, Liu L. Angew. Chem. Int. Ed. 2009;48:9350–9354. doi: 10.1002/anie.200904916. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2009;121:9514–9518. For examples of palladium-catalyzed coupling reaction of C6F5 group. [Google Scholar]
- 6d.Korenaga T, Kosaki T, Fukumura R, Ema T, Sakai T. Org. Lett. 2005;7:4915–4917. doi: 10.1021/ol051866i. [DOI] [PubMed] [Google Scholar]
- 6e.Lafrance M, Rowley CN, Woo TK, Fagnou K. J. Am. Chem. Soc. 2006;128:8754–8756. doi: 10.1021/ja062509l. [DOI] [PubMed] [Google Scholar]
- 6f.Do H-Q, Daugulis O. J. Am. Chem. Soc. 2008;130:1128–1129. doi: 10.1021/ja077862l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6g.He C-Y, Fan S, Zhang X. J. Am. Chem. Soc. 2010;132:12850–12852. doi: 10.1021/ja106046p. [DOI] [PubMed] [Google Scholar]
- 6h.Fan S, Chen F, Zhang X. Angew. Chem. Int. Ed. 2011;50:5918–5923. doi: 10.1002/anie.201008174. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2011;123:6040–6045. [Google Scholar]
- 6i.Nakatani A, Hirano K, Satoh T, Miura M. Org. Lett. 2012;14:2586–2589. doi: 10.1021/ol300886k. [DOI] [PubMed] [Google Scholar]
- 7a.Nishimine T, Fukushi K, Shibata N, Taira H, Tokunaga E, Yamano A, Shiro M, Shibata N. Angew. Chem. Int. Ed. 2014;53:517–520. doi: 10.1002/anie.201308071. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2014;126:527–530. [Google Scholar]
- 7b.Bernardi L, Indrigo E, Pollicino S, Ricci A. Chem. Commun. 2012;48:1428–1430. doi: 10.1039/c0cc05777k. [DOI] [PubMed] [Google Scholar]
- 7c.Zemtsov AA, Levin VV, Dilman AD, Struchkova MI, Belyakov PA, Tartakovsky VA, Hu J. Eur. J. Org. Chem. 2010:6779–6785. [Google Scholar]
- 7d.Fujita M, Obayashi M, Hiyama T. Tetrahedron. 1988;44:4135–4145. [Google Scholar]
- 7e.Dilman AD, Gorokhov VV, Belyakov PA, Struchkova MI, Tartakovsky VA. Russian. Chemical. Bulletin. 2007;56:1522–1525. [Google Scholar]
- 7f.Li Z, Gevorgyan V. Angew. Chem. Int. Ed. 2012;51:1225–1227. doi: 10.1002/anie.201106969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angew. Chem. 2012;124:1251–1253. [Google Scholar]
- 8.Tsoi Y-T, Zhou Z, Yu W-Y. Org. Lett. 2011;13:5370–5373. doi: 10.1021/ol2022577. [DOI] [PubMed] [Google Scholar]
- 9a.Plevey RG, Sampson P. J. Chem. Soc. Perkin Trans. 1. 1987:2129–2136. [Google Scholar]
- 9b.Amii H, Uneyama K. Chem. Rev. 2009;109:2119–2183. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]
- 10.Liang T, Neumann CN, Ritter T. Angew. Chem. Int. Ed. 2013;52:8214–8264. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2013;125:8372–8423. [Google Scholar]
- 11a.Merritt EA, Olofsson B. Angew. Chem. Int. Ed. 2009;48:9052–9070. doi: 10.1002/anie.200904689. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2009;121:9214–9234. For recent review on diaryl-λ3-iodanes. [Google Scholar]
- 11b.Zhdankin VV. Chem. Rev. 2008;108:5299–5358. doi: 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c.Dohi T, Kita Y. Chem. Commun. 2009:2073. doi: 10.1039/b821747e. For examples of α-arylation of enolates with diaryl-λ3-iodanes. [DOI] [PubMed] [Google Scholar]
- 11d.Guo J, Dong S, Zhang Y, Kuang Y, Liu X, Lin Li, Feng X. Angew. Chem. Int. Ed. 2013;52:10245–10249. doi: 10.1002/anie.201303602. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2013;125:10435–10439. [Google Scholar]
- 11e.Ito M, Itani I, Toyoda Y, Morimoto K, Dohi T, Kita Y. Angew. Chem. Int. Ed. 2012;51:12555–12558. doi: 10.1002/anie.201206917. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2012;124:12723–12726. [Google Scholar]
- 11f.Harvey JS, Simonovich SP, Jamison CR, MacMillan DWC. J. Am. Chem. Soc. 2011;133:13782–13785. doi: 10.1021/ja206050b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11g.Bigot A, Williamson AE, Gaunt MJ. J. Am. Chem. Soc. 2011;133:13778–13781. doi: 10.1021/ja206047h. [DOI] [PubMed] [Google Scholar]
- 11h.Allen AE, MacMillan DWC. J. Am. Chem. Soc. 2011;133:4260–4263. doi: 10.1021/ja2008906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11i.Norrby P-O, Petersen TB, Bielawski M, Olofsson B. Chem. Eur. J. 2010;16:8251–8254. doi: 10.1002/chem.201001110. [DOI] [PubMed] [Google Scholar]
- 11j.Aggarwal VK, Olofsson B. Angew. Chem. Int. Ed. 2005;44:5516–5519. doi: 10.1002/anie.200501745. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2005;117:5652–5655. [Google Scholar]
- 11k.Ochiai M, Kitagawa Y, Takayama N, Takaoka Y, Shiro M. J. Am. Chem. Soc. 1999;121:9233–9234. [Google Scholar]
- 11l.Oh CH, Kim JS, Jung HH. J. Org. Chem. 1999;64:1338–1340. [Google Scholar]
- 12.Ochiai M, Kitagawa Y, Toyonari M. ARKIVOC (Gainesville, FL, U.S.) 2003;2003:43–48. [Google Scholar]
- 13a.Bielawski M, Olofsson B. Org. Synth. 2009;86:308–314. [Google Scholar]
- 13b.Helber J, Frohn H-J, Klose A, Scholten T. ARKIVOC (Gainesville, FL, U.S.) 2003;2003:71–82. [Google Scholar]
- 13c.Tyrra W, Butler H, Naumann D. J. Fluorine Chem. 1993;60:79–83. [Google Scholar]
- 13d.Schäfer S, Wirth T. Angew. Chem. Int. Ed. 2010;49:2786–2789. doi: 10.1002/anie.200907134. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2010;122:2846–2850. [Google Scholar]
- 13e.Hirschberg ME, Barthen P, Frohn H-J, Bläser D, Tobey B, Jansen G. J. Fluorine Chem. 2014;163:28–33. [Google Scholar]
- 14a.Zagulyaeva AA, Yusubov MS, Zhdankin VV. J. Org. Chem. 2010;75:2119–2122. doi: 10.1021/jo902733f. [DOI] [PubMed] [Google Scholar]
- 14b.Moriarty RM, Penmasta R, Prakash I. Tetrahedron Lett. 1987;28:877–880. [Google Scholar]
- 14c.Hossain MD, Kitamura T. Bull. Chem. Soc. Jpn. 2006;79:142–144. [Google Scholar]
- 15.Dohi T, Yamaoka N, Kita Y. Tetrahedron. 2010;66:5775–5785. [Google Scholar]
- 16a.Malmgren J, Santoro S, Jalalian N, Himo F, Olofsson B. Chem. Eur. J. 2013;19:10334–10342. doi: 10.1002/chem.201300860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b.Lancer KM, Wiegand GH. J. Org. Chem. 1976;41:3360–3364. [Google Scholar]
- 17.Phipps RJ, Grimster NP, Gaunt MJ. J. Am. Chem. Soc. 2008;130:8172–8174. doi: 10.1021/ja801767s. [DOI] [PubMed] [Google Scholar]
- 18a.Carroll MP, Müller-Bunz H, Guiry PJ. Chem. Commun. 2012;48:11142–11144. doi: 10.1039/c2cc36452b. [DOI] [PubMed] [Google Scholar]
- 18b.Marinescu SC, Nishimata T, Mohr JT, Stoltz BM. Org. Lett. 2008;10:1039–1042. doi: 10.1021/ol702821j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19a.Kano T, Hayashi Y, Maruoka K. J. Am. Chem. Soc. 2013;135:7134–7137. doi: 10.1021/ja403340r. [DOI] [PubMed] [Google Scholar]
- 19b.Dong X-Q, Teng H-L, Tong M-C, Huang H, Taoa H-Y, Wang C-J. Chem. Commun. 2010;46:6840–6842. doi: 10.1039/c0cc01987a. [DOI] [PubMed] [Google Scholar]
- 19c.Trost BM, Schroeder GM, Kristensen J. Angew. Chem. Int. Ed. 2002;41:3492–3495. doi: 10.1002/1521-3773(20020916)41:18<3492::AID-ANIE3492>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. 2002;114:3642–3645. [Google Scholar]
- 19d.Dolling U-H, Davis P, Grabowski EJJ. J. Am. Chem. Soc. 1984;106:446–447. [Google Scholar]
- 20a.Reeves CM, Eidamshaus C, Kim J, Stoltz BM. Angew. Chem. Int. Ed. 2013;52:6718–6721. doi: 10.1002/anie.201301815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angew. Chem. 2013;125:6850–6853. [Google Scholar]
- 20b.Trost BM, Xu J. J. Am. Chem. Soc. 2005;127:2846–2847. doi: 10.1021/ja043472c. [DOI] [PubMed] [Google Scholar]
- 20c.Nemoto T, Matsumoto T, Masuda T, Hitomi T, Hatano K, Hamada Y. J. Am. Chem. Soc. 2004;126:3690–3691. doi: 10.1021/ja031792a. [DOI] [PubMed] [Google Scholar]
- 20d.Chieffi A, Kamikawa K, Åhman J, Fox JM, Buchwald SL. Org. Lett. 2001;3:1897–1900. doi: 10.1021/ol0159470. [DOI] [PubMed] [Google Scholar]
- 21. The reaction of other substrates such as acyclic β-esters and pyrroles under similar conditions gave poor yields.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary