

Abstract
Purpose.
The cell surface receptor CD40 is required for the development of retinopathies induced by diabetes and ischemia/reperfusion. The purpose of this study was to identify signaling pathways by which CD40 triggers proinflammatory responses in retinal cells, since this may lead to pharmacologic targeting of these pathways as novel therapy against retinopathies.
Methods.
Retinal endothelial and Müller cells were transduced with vectors that encode wild-type CD40 or CD40 with mutations in sites that recruit TNF receptor associated factors (TRAF): TRAF2,3 (ΔT2,3), TRAF6 (ΔT6), or TRAF2,3 plus TRAF6 (ΔT2,3,6). Cells also were incubated with CD40-TRAF2,3 or CD40-TRAF6 blocking peptides. We assessed intercellular adhesion molecule-1 (ICAM-1), CD40, monocyte chemoattractant protein-1 (MCP-1), VEGF, and prostaglandin E2 (PGE2) by fluorescence-activated cell sorting (FACS), ELISA, or mass spectrometry. Mice (B6 and CD40−/−) were made diabetic using streptozotocin. The MCP-1 mRNA was assessed by real-time PCR.
Results.
The CD40-mediated ICAM-1 upregulation in endothelial and Müller cells was markedly inhibited by expression of CD40 ΔT2,3 or CD40 ΔT6. The CD40 was required for MCP-1 mRNA upregulation in the retina of diabetic mice. The CD40 stimulation of endothelial and Müller cells enhanced MCP-1 production that was markedly diminished by CD40 ΔT2,3 or CD40 ΔT6. Similar results were obtained in cells incubated with CD40-TRAF2,3 or CD40-TRAF6 blocking peptides. The CD40 ligation upregulated PGE2 and VEGF production by Müller cells, that was inhibited by CD40 ΔT2,3 or CD40 ΔT6. All cellular responses tested were obliterated by expression of CD40 ΔT2,3,6.
Conclusions.
Blockade of a single CD40-TRAF pathway was sufficient to impair ICAM-1, MCP-1, PGE2, and VEGF upregulation in retinal endothelial and/or Müller cells. Blockade of CD40-TRAF signaling may control retinopathies.
Keywords: diabetes, CD40, intercellular adhesion molecules, chemokine
Cell surface receptor CD40 drives diabetic and ischemic retinopathies. We report that inhibition of the CD40-TRAF2,3 or CD40-TRAF6 pathway impairs proinflammatory responses thought to mediate the development of these retinopathies. Inhibition of CD40-TRAF signaling may represent an approach to control retinopathies.
Introduction
The cell surface receptor CD40 is a receptor constitutively expressed on antigen-presenting cells that also can be present on nonhematopoietic cells.1 Its counter-receptor, CD154 (CD40 ligand) is expressed on activated CD4+ T cells and platelets.1 The receptor CD154 also exists as a biologically active soluble protein present in plasma.2 The CD40-CD154 interaction promotes the development of various inflammatory and autoimmune disorders.1,3
Nonhematopoietic cells are either CD40− or express low levels of CD40 under basal conditions. However, CD40 is induced or upregulated in these cells during inflammation.4–6 We reported that retinal endothelial cells, Müller cells, ganglion neurons, retinal microglia, and RPE cells express CD40 at low levels (corrected mean fluorescence intensity between 100 and 160).7–10 Moreover, retinal CD40 mRNA expression increases in diabetic mice and in mice subjected to retinal ischemia/reperfusion7,9 (and Portillo et al., unpublished observations, 2008). In the case of diabetes, CD40 upregulation occurs in retinal endothelial cells, Müller cells, and microglia.9 Study of the regulation of CD40-mediated proinflammatory responses in retinal cells is important because of the pathogenic role of CD40 in retinopathies with an inflammatory component. Indeed, CD40−/− mice are protected from ischemia/reperfusion-induced retinopathy and early diabetic retinopathy.7,9
The role of the CD40-CD154 pathway in various disorders with an inflammatory component made it an attractive therapeutic target. Administration of blocking anti-CD154 mAb showed therapeutic efficacy in mice.11 However, clinical trials of anti-CD154 mAb administration for Crohn's disease, lupus nephritis, and idiopathic thrombocytopenic purpura were stopped due to thromboembolic events.12 Since CD40 is the major receptor for CD154, blockade of signaling downstream of CD40 may represent an alternative approach to inhibit the CD40-CD154 pathway. Therapeutic strategies to block this pathway must take into account that CD40-CD154 signaling also is central for protection against a broad variety of pathogens.1 Thus, the approaches to inhibit CD40 signaling ideally should be selective enough to impair proinflammatory responses while minimizing the risk of immunosuppression.
The CD40 receptor signals via adaptor proteins, such as TNF receptor–associated factors (TRAF) and JAK3.13,14 The TRAF factors are key mediators of CD40 signaling.13 Receptor CD40 has domains that directly bind TRAF2 and TRAF315,16 (TRAF3 typically inhibits CD40 signaling), and a domain that binds TRAF6.15 Although there can be overlap in the cellular responses induced by the TRAF2,3 and TRAF6 binding sites, responses triggered by these sites can be distinct. The TRAF6 binding site drives IL-12 secretion by dendritic cells, dendritic cell maturation, TNF-α, IL-1β, IL-6, and nitric oxide synthase 2 (NOS2) upregulation in macrophages, autophagy-mediated antimicrobial activity in macrophages, IL-6 production by B cells, and plasma cell formation.17–24 On the other hand, the TRAF2,3 binding site promotes immunoglobulin isotype switch.25
The role of CD40-TRAF signaling in retinal cells is unknown. We generated retroviral vectors that encode wt CD40 or CD40 with mutations in the TRAF2,3 (ΔT2,3), TRAF6 (ΔT6), or TRAF2,3 plus TRAF6 binding sites (ΔT2,3,6) reported to prevent recruitment of the appropriate TRAFs.26–28 This approach is well suited to studying TRAF signaling downstream of CD40.18,25,28,29 Using human retinal endothelial cells, Müller cells, and a rat Müller cell line transduced with these retroviral vectors, we examined the role of TRAF binding sites in the upregulation of intercellular adhesion molecule-1 (ICAM-1), monocyte chemoattractant protein-1 (MCP-1), VEGF, and prostaglandin E2 (PGE2). These molecules were studied since they are upregulated after retinal ischemia-reperfusion and/or in the diabetic retina, and are considered to have a pathogenic role in these retinopathies.30–35
Materials and Methods
Cells
Primary human retinal endothelial and Müller cells were obtained as described.9 Endothelial cells were cultured in gelatin-coated tissue culture flasks containing Dulbecco's modified Eagle medium (DMEM) plus 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA) supplemented with endothelial cell growth supplement from bovine pituitary (15 μg/mL; Sigma-Aldrich, St. Louis, MO, USA) and insulin/transferrin/selenium (Sigma-Aldrich). Cell identity was confirmed by incorporation of acetylated low-density lipoprotein (>90%). Human Müller cells were cultured in DMEM/F12 containing 20% FBS. Cultures were >95% pure for Müller cells (vimentin+, CRALBP+, and GFAP− by immunofluorescence). Human retinal cells were used between passages 3 to 6. The rat Müller cell line rMC-1 was a gift from Dr V. R. Sarthy (Northwestern University, Chicago, IL, USA).36
In Vitro Stimulation
To induce CD40 stimulation, cells were treated with multimeric human CD154 (gift from Richard Kornbluth, MD, PhD, Multimeric Biotherapeutics, Inc., La Jolla, CA, USA) for 24 hours as described.7 Specificity was confirmed by detecting >95% neutralization of the responses after addition of antihuman CD154 mAb (Ancell Corporation, Bayport, MN, USA). As controls we used omission of CD154 or incubation with a nonfunctional CD154 mutant (T147N).37 Endothelial cells also were incubated with IFN-γ (500 IU/mL; PeproTech, Rocky Hill, NJ, USA) plus TNF-α (500 IU/mL; PeproTech). In some experiments Celecoxib (1 μM; Sigma-Aldrich) was added to cells 1 hour before stimulation with CD154.
Retroviral Vectors and Transductions
The cDNA for wild-type human CD40, CD40Δ22 (a mutant that ablates binding to TRAF2 and TRAF3; ΔTRAF2, 3), CD40EEAA (a mutant that prevents binding to TRAF6; ΔTRAF6), and CD40Δ55 (a mutant that ablates binding to TRAF2, TRAF3, and TRAF6; ΔTRAF2, 3, 6) were gifts from Gail Bishop, PhD (University of Iowa, Iowa City, IA, USA).28,38 The CD40 cDNA were cloned into the murine stem cell virus-based bicistronic retroviral vector MIEG3 that encodes enhanced GFP (EGFP)39 as described previously.40 Ecotropic retroviral supernatants were generated by transfecting the Phoenix-gp cell line with MIEG3-based retroviral vectors and plasmids encoding envelop (RD114; gift from Yasu Takeuchi, University College London, London, UK) and gag-pol using the calcium phosphate transfection kit (Invitrogen Life Technologies, Carlsbad, CA, USA) as described.40 Cells were incubated overnight with retrovirus in the presence of polybrene (8 μg/mL; Sigma-Aldrich). Cells were washed and used at least 48 hours after infection with retroviruses.
Cell-Permeable Peptides
Peptides consisted of the amino acid sequence of the TRAF2,3 or TRAF6 binding site of CD40 followed by the TAT47–57 cell penetrating peptide. The sequences for the CD40-TRAF2,3 and the CD40-TRAF6 blocking peptides were NH2-NTAAPVQETLHG YGRKKRRQRRR-OH and NH2-KQEPQEIDFPDD YGRKKRRQRRR-OH. The TAT47–57 sequence is underlined. Control peptides were TAT47–57 or TAT47–57 linked to scrambled peptide. No differences in the effects between control peptides were noted. Peptides were manufactured by Proteintech Group (San Diego, CA, USA) and were low in endotoxin and >98% pure by HPLC. Peptides were added to cells 3 hours before stimulation with CD154.
Flow Cytometry
Retinal cells were incubated with human IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) followed by staining with anti-human CD40 PE (BD Biosciences, San Jose, CA, USA), anti-human ICAM-1 PE (eBiosciences, San Diego, CA, USA), anti-rat ICAM-1 PerCP-eFluor 710 (eBiosciences), or appropriate isotype control mAbs. After fixation with 1% paraformaldehyde, flow cytometry data acquisition was performed using a LSR II and running FACSDiva software (Becton Dickinson, San Jose, CA, USA). FlowJo software (Tree Star, Inc., Ashland, OR, USA) was used for data analysis. Expression of CD40 and ICAM-1 is expressed as corrected mean fluorescence intensity (cMFI).
Cytokine ELISA
Cell-free supernatants were collected at 24 hours and used to measure concentrations of MCP-1 and VEGF (R & D Systems, Minneapolis, MN, USA).
Measurement of PGE2
Culture supernatants were subjected to solid phase extraction.41 The PGE2 concentrations were analyzed by reverse-phase HPLC and electrospray ionization mass spectrometry as previously described.41 The PGE2 also was measured by ELISA (Cayman Chemical, Ann Arbor, MI, USA).
Statistical Analysis
All results were expressed as the mean ± SEM. Data were analyzed by 2-tailed Student's t-test and ANOVA. Differences were considered statistically significant at P < 0.05.
Results
Role of the CD40-TRAF2,3 and the CD40-TRAF6 Binding Sites in CD154-Induced Upregulation of ICAM-1 in Human Retinal Endothelial Cells (HRECs)
Primary HRECs were transduced with bicistronic retroviral vectors that encode EGFP and either wt CD40 or CD40 mutants lack binding to TRAF2,3 (ΔT2,3), TRAF6 (ΔT6), or TRAF2,3,6 (ΔT2,3,6).26–28 The percentages of transduced cells (EGFP+) and the cMFI for CD40 on EGFP+ cells were similar for all vectors (Figs. 1A, 1B; P > 0.5). The CD154 markedly upregulated ICAM-1 on transduced (EGFP+) HREC that expressed wt CD40 (Fig. 1C). This effect was specific since it was obliterated by a neutralizing anti-CD154 mAb (>95% inhibition; data not shown). Consistent with the low CD40 expression in HREC under basal conditions, HREC transduced with the empty retroviral vector (MIEG3) exhibited less pronounced upregulation of ICAM-1 (mean cMFI Ctr = 58; CD154 = 103) in response to CD154. Thus, cellular responses in transduced HAEC are driven mainly by retroviral-induced CD40. Expression of either CD40 ΔT2,3 or CD40 ΔT6 markedly inhibited ICAM-1 upregulation, while the expression of CD40 ΔT2,3,6 obliterated this response (Fig. 1D). The effects of the mutations were specific, since upregulation of ICAM-1 in response to IFN-γ/TNF-α was similar regardless of the retroviral vector used (Fig. 1E, P > 0.1). Thus, a mutation that prevents CD40-TRAF2,3 or CD40-TRAF6 interaction is sufficient to inhibit ICAM-1 upregulation in HREC.
Figure 1.

Role of CD40-TRAF binding sites on ICAM-1 upregulation in HREC. The HREC were transduced with MIEG3-based retroviral vector that encode EGFP and either wt CD40, CD40 ΔT2,3, CD40 ΔT6, CD40 ΔT2,3,6. (A) Percentages of HREC that became EGFP+ after incubation with retroviral vectors. (B) Expression of CD40 on gated EGFP+ cells shown as corrected mean fluorescence intensity (cMFI). (C) Dot plots of HREC transduced with wt CD40-encoding retroviral vector depicting expression of ICAM-1 and EGFP at 24 hours post-incubation with or without CD154. (D) The HREC transduced with the retroviral vectors were incubated with or without CD154 and expression of ICAM-1 (cMFI) on gated EGFP+ cells was assessed by flow cytometry at 24 hours. (E) The HREC transduced with the retroviral vectors were incubated with IFN-γ plus TNF-α and expression of ICAM-1 on gated EGFP+ cells was assessed by flow cytometry at 24 hours. Results are shown as mean ± SEM and are representative of 3 to 4 experiments. *P < 0.05, ***P < 0.001 represent comparison to cells that express wt CD40.
CD40 Drives MCP-1 Upregulation in the Retina of Diabetic Mice
Before examining whether CD40-TRAF signaling in retinal cells regulates MCP-1 production, we tested whether CD40 drives retinal MCP-1 upregulation in vivo. Male B6 and CD40−/− mice were rendered diabetic by administration of streptozotocin. Throughout the study B6 and CD40−/− mice exhibited similar blood glucose concentrations (B6 = 364 ± 13 mg/mL; CD40−/− = 361 ± 7 mg/mL) as well as hemoglobin A1c (HbA1c) levels (B6 = 8.7 ± 0.3%; CD40−/− = 8.4 ± 0.3%; P > 0.5). Diabetic B6 mice, but not diabetic CD40−/− mice, upregulated MCP-1 (Table 1).
Table 1.
Changes in mRNA Levels of MCP-1 in the Retinas of Diabetic Mice
|
B6 Control |
B6 Diabetic |
CD40−/− Control |
CD40−/− Diabetic |
|
| MCP-1 mRNA, relative levels | 1.0 ± 0.15 | 2.2 ± 0.32* | 1.06 ± 0.1 | 1.13 ± 0.1 |
At 2 months of diabetes, retinas from diabetic B6 and CD40−/− mice as well as from control (nondiabetic) animals were collected and used for mRNA extraction. The mRNA levels of MCP-1 were assessed by real-time quantitative PCR using 18S rRNA as internal control. Data are expressed as fold-increase in diabetic mice compared to retinas from nondiabetic controls. Data shown represent mean ± SEM (10 to 15 animals per group).
Diabetic B6 mice showed significantly higher MCP-1 upregulation (P < 0.05) compared to control B6 mice and diabetic CD40−/− mice.
Role of the CD40-TRAF2,3 and the CD40-TRAF6 Binding Sites in CD154-Induced Upregulation of MCP-1 in HRECs
The CD154 stimulated MCP-1 production by HREC that expressed wt CD40 (Fig. 2). The expression of CD40 ΔT2,3,6 obliterated the MCP-1 production induced by CD154 (Fig. 2). Expression of CD40 ΔT2,3 or CD40 ΔT6 also markedly inhibited MCP-1 production (Fig. 2). Thus, a mutation in either the TRAF2,3 or TRAF6 binding sites is sufficient to impair MCP-1 production by HREC.
Figure 2.
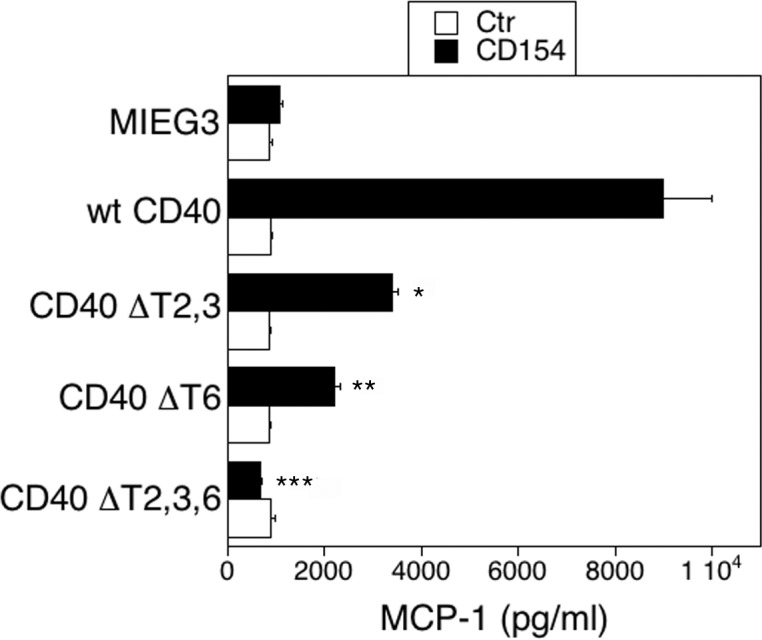
Role of CD40-TRAF binding sites on MCP-1 production in HREC. The HREC transduced with the retroviral vectors were incubated with or without CD154 for 24 hours and MCP-1 concentrations in supernatants determined by ELISA. Results are shown as mean ± SEM and are representative of 3 to 4 experiments. *P < 0.05, **P < 0.01, ***P < 0.001 represent comparison to cells that express wt CD40.
Effects of Pharmacologic Inhibition of CD40-TRAF Signaling in CD154-Induced Upregulation of ICAM-1 in HRECs
We reported that cell permeable peptides that include the amino acid sequence of the TRAF2,3 or TRAF6 binding site of CD40 block appropriate CD40-TRAF signaling.23 We incubated untransduced HREC with peptides that consisted of the amino acid sequence of the TRAF2,3 or the TRAF6 binding sites of CD40 linked to TAT47–57. The HRECs then were stimulated with CD154. The CD40-TRAF2,3 and CD40-TRAF6 blocking peptides impaired upregulation of ICAM-1 in response to CD154 (Fig. 3). Taken together, not only genetic but also a pharmacologic approach to block CD40-TRAF2,3 or CD40-TRAF6 signaling inhibit a CD40-mediated proinflammatory response in HREC.
Figure 3.
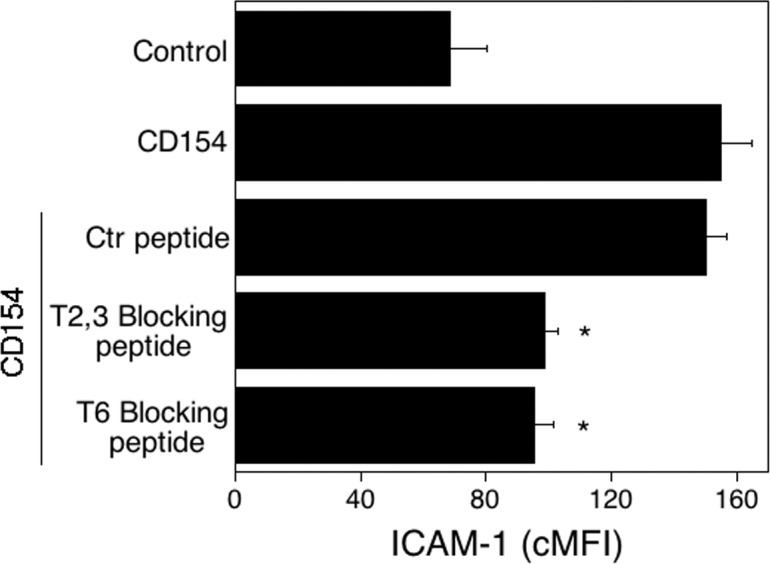
Effects of CD40-TRAF blocking peptides on ICAM-1 upregulation in HREC stimulated with CD154. Three hours before the onset of stimulation with CD154, HREC were incubated with control, CD40–TRAF2,3, or CD40-TRAF6 blocking peptides (1 μM). Cells then were incubated with or without CD154 for 24 hours. The ICAM-1 expression was examined by flow cytometry. Results are shown as mean ± SEM and are representative of 3 experiments. *P < 0.05, represent comparison to cells treated with control peptide.
Role of CD40-TRAF Binding Sites in ICAM-1 and MCP-1 by Human Retinal Müller Cells (HRMCs)
The HRMCs were transduced with the retroviral vectors that encode wt or CD40 mutants. The percentages of transduced cells (EGFP+) and the cMFI for CD40 on transduces cells were similar for all groups (Figs. 4A, 4B; P > 0.5). Incubation with CD154 upregulated ICAM-1 and MCP-1 production in HRMC that expressed wt CD40 (Figs. 4C, 4D). These responses were obliterated in HRMC that expressed CD40 ΔT2,3,6 (Fig. 5C). Expression of CD40 ΔT2,3 or CD40 ΔT6 were sufficient to markedly inhibit upregulation of ICAM-1 and MCP-1 (Figs. 4C, 4D).
Figure 4.

Role of CD40-TRAF binding sites on ICAM-1 upregulation and MCP-1 production in Müller cells. (A–D) The HRMC were transduced with MIEG3-based retroviral vector that encode either wt CD40, CD40 ΔT2,3, CD40 ΔT6, or CD40 ΔT2,3,6. Percentages of HRMC that became EGFP+ after incubation with retroviral vectors (A) and expression of CD40 (cMFI) on gated EGFP+ cells (B). The HRMC transduced with the retroviral vectors were incubated with or without CD154 for 24 hours. Expression of ICAM-1 (cMFI) on gated EGFP+ cells was assessed by flow cytometry (C) and MCP-1 concentrations in supernatants determined by ELISA (D). (E, F) Rat Müller cells (rMC1) were transduced with MIEG3-based retroviral vector that encode EGFP and either wt human-mouse CD40, human-mouse CD40 ΔT2,3, human-mouse CD40 ΔT6, or human-mouse CD40 ΔT2,3,6. Expression of CD40 (cMFI) on gated EGFP+ cells was assessed by flow cytometry (E). (F) Cells were incubated with or without human CD154 for 24 hours. Expression of ICAM-1 (cMFI) on gated EGFP+ cells was assessed by flow cytometry at 24 hours. Results are shown as mean ± SEM and are representative of 3 to 4 experiments. *P < 0.05, **P < 0.01, ***P < 0.01 represent comparison to cells that express wt CD40.
Figure 5.
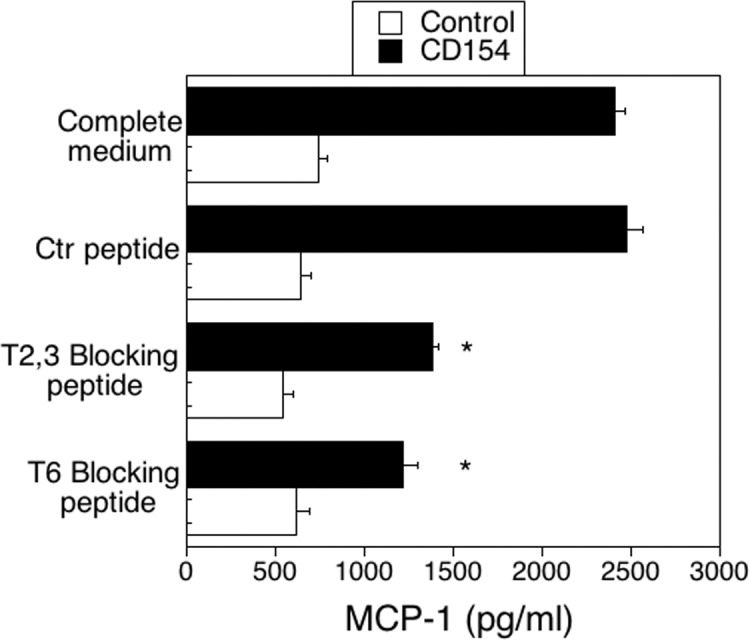
Effects of CD40-TRAF blocking peptides on MCP-1 production in HRMC stimulated with CD154. Three hours before the onset of stimulation with CD154, HRMC transduced with wt CD40-encoding retroviral vector were incubated with control, CD40–TRAF2,3 or CD40-TRAF6 blocking peptides (10 μM). The MCP-1 concentrations in supernatants collected at 24 hours were determined by ELISA. Results are shown as mean ± SEM and are representative of 3 experiments. *P < 0.05, represent comparison to cells treated with control peptide.
We determined whether CD40-TRAF signaling also controls a proinflammatory response in rodent Müller cells. We used a rat Müller cell line transduced with retroviral vectors that encode a chimera of the extracellular domain of human CD40 and the intracellular domain of mouse CD40 (hmCD40). Addition of human CD154 to rodent cells that express this chimera results in functional CD40 signaling.22 Moreover, the TRAF binding motives are similar among various species, such as mouse, rat, humans (TRAF2,3 proximal: PxQxT; TRAF2 distal SVxE; TRAF6: PxExxAr/Ac), and CD40 can interact and signal through TRAFs of different species (for example human to mouse).15,16,40 Rat Müller cells (rMC1) were transduced with retroviral vectors that encode hmCD40 chimeras with mutations in the TRAF2,3, TRAF6, or TRAF2,3 plus TRAF6 binding sites.22 Transduced cell lines were >95% EGFP+. The levels of CD40 expression were similar for all vectors (Fig. 4E). The ICAM-1 upregulation was TRAF binding site–dependent, since it was impaired in Müller cells that expressed hmCD40 ΔT2,3 or hmCD40 ΔT6 (Fig. 4F).
The HRMC transduced with wt-CD40-encoding vector were incubated with the CD40-TRAF2,3 blocking peptides, CD40-TRAF6 blocking peptide, or control peptide followed by stimulation with CD154. The CD40-TRAF2,3 and CD40-TRAF6 blocking peptides inhibited MCP-1 production by CD154-treated HRMC (Fig. 5). Taken together, inhibition of CD40-TRAF2,3 or CD40-TRAF6 via genetic or pharmacologic approaches impair CD40-induced proinflammatory responses in Müller cells.
Role of CD40-TRAF Binding Sites in PGE2 and VEGF Production by Retinal Müller Cells
It is not known if CD40 stimulates PGE2 and VEGF production by Müller cells. The HRMC that express wt CD40 upregulated production of PGE2, an effect that was obliterated by the COX-2 inhibitor celecoxib (Fig. 6A). The PGE2 production was inhibited in HRMC that expressed CD40 ΔT2,3 or CD40 ΔT,6 and was obliterated in those that expressed CD40 ΔT2,3,6 (Fig. 6B). Similarly, CD40 ligation caused VEGF upregulation that was dependent of the CD40-TRAF2,3 and CD40-TRAF6 binding sites (Fig. 6C). Table 2 summarizes the inhibitory effects of the mutations in the TRAF binding sites. Taken together, blockade of CD40-TRAF2,3 or CD40-TRAF6 signaling was sufficient to markedly inhibit proinflammatory responses in retinal endothelial and Müller cells.
Figure 6.

Role of CD40-TRAF binding sites on PGE2 and VEGF production by HRMC. (A) The HRMC were transduced with MIEG3-based retroviral vector that encode EGFP with or without human wt CD40. Cells were incubated with or without CD154 for 24 hours in the presence of absence of celecoxib (1 μM). The PGE2 was measured by mass spectrometry or ELISA. ***P < 0.001 represent comparison to cells with wt CD40 that were incubated with CD154 alone. (B, C) Production of PGE2 (B) and VEGF (C) by HRMC transduced with MIEG3-based retroviral vector that encode EGFP and either human wt CD40, CD40 ΔT2,3, CD40 ΔT6, CD40 ΔT2,3,6. Cells were incubated with or without CD154 for 24 hours. The VEGF concentrations were determined by ELISA. Results are shown as mean ± SEM and are representative of 3 experiments. *P < 0.05, **P < 0.01 represent comparison to cells that express wt CD40.
Table 2.
Effects of Expression of CD40 with Mutations in CD40-TRAF Binding Sites on Upregulation of Various Proinflammatory Responses on HREC, HRMC and Rat Müller Cells
|
ΔTRAF2,3 |
ΔTRAF6 |
ΔTRAF2,3,6 |
|
| HERCs | |||
| ICAM-1 | 56.5 ± 6.3 | 51.0 ± 6.5 | 99.6 ± 0.4 |
| MCP-1 | 64.7 ± 7.0 | 69.0 ± 8.5 | 99.9 ± 0.1 |
| HRMCs | |||
| ICAM-1 | 59.6 ± 5.3 | 54.9 ± 7.6 | 99.1 ± 0.4 |
| MCP-1 | 75.4 ± 6.1 | 60.6 ± 8.0 | 97.3 ± 2.7 |
| PGE2 | 83.0 ± 5.0 | 91.9 ± 7.8 | 97.1 ± 2.9 |
| VEGF | 81.5 ± 7.1* | 54.6 ± 2.3 | 97.4 ± 1.6 |
| Rat Müller cells | |||
| ICAM-1 | 77.5 ± 4.8 | 68.3 ± 2.0 | 98.1 ± 1.9 |
Data are expressed as mean inhibition compared to cells that expressed wt CD40. Results are averages ± SEM from 3 to 4 experiments. Inhibitory effects of the ΔT2,3 and ΔT6 mutations were similar with the exception of the effects on VEGF production by Müller cells.
P < 0.05.
Discussion
The CD40 receptor stimulates inflammatory responses in the retina and is a major driver of early diabetic retinopathy and ischemia/reperfusion-induced retinopathy.7,9 The signaling cascades through which CD40 induces proinflammatory responses in retinal cells remained unknown. Using cells that express CD40 with mutations in TRAF binding sites we report that ICAM-1 upregulation and MCP-1 production by retinal endothelial cells and Müller cells as well as PGE2 and VEGF production by Müller cells are dependent on TRAFs. The TRAF2,3 and the TRAF6 binding sites of CD40 are required for the optimal induction of these responses. Importantly, blockade of one of these signaling pathways is sufficient to markedly inhibit ICAM-1 upregulation and production of MCP-1, PGE2, and VEGF. We also report that CD40-TRAF blocking peptides impaired these CD40-induced proinflammatory responses. The fact that in vivo delivery of cell permeable peptides to various retinal cells occurs after intravitreal injection42 raises the possibility that a pharmacologic approach to prevent CD40-TRAF interaction may prove useful for the management of retinopathies driven by CD40.
Studies with bone marrow transplants indicate that leukocytes have an important role in the development of diabetic retinopathy.43 However, nonhematopoietic cells also are important in the development of inflammatory disorders. Using bone marrow transplants in a mouse model of ischemia/reperfusion-induced retinopathy, we reported that absence of CD40 in the retina inhibited ICAM-1 upregulation, leukocyte recruitment to the retina and neurovascular degeneration.7 The CD40 is operative in both hematopoietic and nonhematopoietic cellular compartments. Indeed, studies in an animal model of arterial injury revealed that expression of CD40 at the level of leukocytes and vascular wall cells is critical for neointima formation and vascular inflammation.44,45
The proinflammatory responses driven by the CD40-TRAF binding sites are likely of pathogenic relevance to retinopathies. The ICAM-1 is upregulated in the retina and in retinal endothelial cells after ischemia/reperfusion, as well as in the diabetic retina, phenomena that are dependent on CD40.7–9 Blockade or deficiency of ICAM-1 diminishes retinal injury following ischemia30 and capillary degeneration in diabetes.32 We report that CD40 mediates the upregulation of MCP-1 in the retina of diabetic mice, a chemokine that appears to have a pathogenic role in proliferative diabetic retinopathy in humans.31 The CD40-TRAF signaling enhances PGE2 and VEGF production by Müller cells, molecules that are elevated in diabetic and ischemic retinopathies and are considered to drive microvascular complications and neovascularization that accompany these diseases.33–35
One of the concerns with indiscriminate inhibition of CD40 signaling is that it would cause susceptibility to various infectious diseases. The CD40-TRAF6 signaling in MHC class II+ cells promotes vascular inflammation in vivo.44,46 Indeed, blockade of this pathway has been proposed as an approach to control CD40-mediated inflammatory disorders. However, the CD40-TRAF6 pathway also drives immune responses required for protection against infections. The TRAF6 binding site of CD40 is essential for CD40-mediated production of IL-12 in dendritic cells and dendritic cell maturation.19,24 This binding site also is responsible for autophagy-dependent and NOS2-dependent induction of macrophage antimicrobial activity22,23,40 as well as production of IL-1β, IL-6, and TNF-α.20 In addition, the CD40-TRAF6 binding site also regulates humoral immunity, since it promotes affinity maturation and the generation of long lived plasma cells.18 In contrast, while the proximal CD40-TRAF2,3 binding site drives immunoglobulin isotype switch,25 the TRAF2,3 binding sites do not have an appreciable role in the cellular immune responses described above.19,20,22,23,40 Our findings suggested that blockade of the CD40-TRAF2,3 pathway may represent an effective way to dampen CD40-mediated inflammation while likely having less inhibitory effect on CD40-mediated host protection than blockade of CD40-TRAF6 signaling. The development of CD40-TRAF2,3 inhibitors may lead to a novel therapeutic approach to control retinopathies, such as those induced by diabetes and ischemia.
Acknowledgments
The authors thank Richard Kornbluth, Vijay Sarthy, Gail Bishop, and Yasu Takeuchi for providing reagents.
Supported by National Institutes of Health (NIH; Bethesda, MD, USA) Grant EY019250 (CSS), Grants 5-2008-233 and 1-2009-204 from the Juvenile Diabetes Foundation International (CSS), Helen Weil Ross Memorial Grant from the Dietrich Diabetes Research Institute (CSS) and NIH Grant P30 EY11373, as well as a postdoctoral fellowship from the Ohio Lions Eye Research Foundation (J-ACP).
Disclosure: J.-A.C. Portillo, None; I. Schwartz, None; S. Zarini, None; R. Bapputty, None; T.S. Kern, None; R.A. Gubitosi-Klug, None; R.C. Murphy, None; M.C. Subauste, P; C.S. Subauste, P
References
- 1. van Kooten C, Banchereau J. CD40-CD40 ligand. J Leuk Biol. 2000; 67: 2–17. [DOI] [PubMed] [Google Scholar]
- 2. Xu H, Zhang X, Mannon RB, Kirk AD. Platelet-derived or soluble CD154 induces vascularized allograft rejection independent of cell-bound CD154. J Clin Invest. 2006; 116: 769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol. 2009; 21: 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yellin MJ, D'Agati V, Parkinson G, et al. Immunohistologic analysis of renal CD40 and CD40L expression in lupus nephritis and other glomerulonephritis. Arthritis Rheum. 1997; 40: 124–134. [DOI] [PubMed] [Google Scholar]
- 5. Battaglia E, Biancone L, Resegotti A, Emanuelli G. Ruggero Fronda G, Camussi G. Expression of CD40 and its ligand, CD40L, in intestinal lesions of Crohn's disease. Am J Gastroenterol. 1999; 94: 3279–3284. [DOI] [PubMed] [Google Scholar]
- 6. Hakkinen T, Karkola K, Yla-Herttualla S. Macrophages, smooth muscle cells, endothelial cells, and T-cells express CD40 and CD40L in fatty streaks and more advanced human atherosclerotic lesions. Virch Archiv. 2000; 437: 396–405. [DOI] [PubMed] [Google Scholar]
- 7. Portillo J-AC, Van Grol J, Zheng L, et al. CD40 mediates retinal inflammation and neuro-vascular degeneration. J Immunol. 2008; 181: 8719–8726. [DOI] [PubMed] [Google Scholar]
- 8. Portillo J-AC, Okenka G, Kern TS, Subauste CS. Identification of primary retinal cells and ex vivo identification of pro-inflammatory molecules in retinal cells using flow cytometry. Mol Vis. 2009; 15: 1383–1389. [PMC free article] [PubMed] [Google Scholar]
- 9. Portillo J-AC, Greene JA, Okenka G, et al. CD40 promotes the development of early diabetic retinopathy. Diabetologia. 2014; 57: 2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Grol J, Muniz-Feliciano L, Portillo J-AC, Bonilha VL, Subauste CS. CD40 induces anti-Toxoplasma gondii activity in non-hematopoietic cells dependent on autophagy proteins. Infect Immun. 2013; 81: 2002–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signaling. Nature. 1998; 394: 200–203. [DOI] [PubMed] [Google Scholar]
- 12. Boumpas DT, Furie R, Manzi S, et al. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003; 48: 719–727. [DOI] [PubMed] [Google Scholar]
- 13. Bishop GA, Hostager BS, Brown KD. Mechanisms of TNF receptor-associated factor (TRAF) regulation in B lymphocytes. J Leuk Biol. 2002; 72: 19–23. [PubMed] [Google Scholar]
- 14. Hanissian SH, Geha R. Jak3 is associated with CD40 and is critical for CD40 induction of gene expression in B cells. Immunity. 1997; 6: 379–388. [DOI] [PubMed] [Google Scholar]
- 15. Pullen SS, Miller HG, Everdeen DS, Dang TT, Crute JJ, Kehry MR. CD40-tumor necrosis factor receptor-associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry. 1998; 37: 11836–11845. [DOI] [PubMed] [Google Scholar]
- 16. Lu LF, Cook WJ, Lin LL, Noelle RJ. CD40 signaling through a newly identified tumor necrosis factor receptor-associated factor 2 (TRAF2) binding site. J Biol Chem. 2003; 278: 45414–45418. [DOI] [PubMed] [Google Scholar]
- 17. Baccam M, Bishop GA. Membrane-bound CD154, but not anti-CD40 mAb, induces NF-κB independent B cell IL-6 production. Eur J Immunol. 1999; 29: 3855–3866. [DOI] [PubMed] [Google Scholar]
- 18. Ahonen CL, Manning EM, Erickson LD, et al. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat Immunol. 2002; 3: 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mackey MF, Wang Z, Eichelberg K, Germain RN. Distinct contributions of different CD40 TRAF binding sites to CD154-induced dendritic cell maturation and IL-12 secretion. Eur J Immunol. 2003; 33: 779–789. [DOI] [PubMed] [Google Scholar]
- 20. Mukundan L, Bishop GA, Head KZ, Zhang L, Wahl L, Suttles J. TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages. J Immunol. 2005; 174: 1081–1090. [DOI] [PubMed] [Google Scholar]
- 21. Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006; 116: 2366–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Subauste CS, Andrade RM, Wessendarp M. CD40-TRAF6 and autophagy-dependent anti-microbial activity in macrophages. Autophagy. 2007; 3: 245–248. [DOI] [PubMed] [Google Scholar]
- 23. Portillo J-AC, Muniz-Feliciano L, Subauste MC, Heinzel FP, Subauste CS. CD40 and TNF-α synergize to induce nitric oxide synthase in macrophages. Immunology. 2012; 135: 140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Portillo J-AC, Greene JA, Schwartz I, Subauste MC, Subauste CS. Blockade of CD40-TRAF2,3 or CD40-TRAF6 intercations is sufficient to impair pro-inflammatory responses in human aortic endothelial cells and human aortic smooth muscle cells [published online ahead of print July 22, 2014] Immunology. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jabara HH, Laouini D, Tsitsikov E, et al. The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity. 2002; 17: 265–276. [DOI] [PubMed] [Google Scholar]
- 26. Hostager BS, Hsing Y, Harms DE, Bishop GA. Different CD40-mediated signaling events require distinct CD40 structural features. J Immunol. 1996; 157: 1047–1053. [PubMed] [Google Scholar]
- 27. Hostager BS, Bishop GA. Cutting edge: contrasting roles of TNF receptor-associated factore 2 (TRAF2) and TRAF3 in CD40-activated B lymphocyte differentiation. J Immunol. 1999; 162: 6307–6311. [PubMed] [Google Scholar]
- 28. Jalukar SV, Hostager BS, Bishop GA. Characterization of the roles of TNF receptor-associated factor 6 in CD40-mediated B lymphocyte effector functions. J Immunol. 2000; 164: 623–630. [DOI] [PubMed] [Google Scholar]
- 29. Haxhinasto SA, Hostager BS, Bishop GA. Cutting edge: molecular mechanisms of synergy between CD40 and the B cell antigen receptor: role for TNF receptor-associated factor 2 in receptor interaction. J Immunol. 2002; 169: 1145–1149. [DOI] [PubMed] [Google Scholar]
- 30. Tsujikawa A, Ogura Y, Hiroshiba N, et al. Retinal ischemia-reperfusion injury attenuated by blocking of adhesion molecules of vascular endothelium. Invest Ophthalmol Vis Sci. 1999; 40: 1183–1190. [PubMed] [Google Scholar]
- 31. Mitamura Y, Takeuchi S, Matsuda A, Tagawa Y, Mizue Y, Nishihira J. Monocyte chemotactic protein-1 in the vitreous of patients with proliferative diabetic retinopathy. Ophthalmologica. 2001; 215: 415–418. [DOI] [PubMed] [Google Scholar]
- 32. Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004; 18: 1450–1452. [DOI] [PubMed] [Google Scholar]
- 33. Kern TS, Miller CM, Du Y, et al. Topical administration of nepafenac inhibits diabetes-induced retinal microvascular disease and underlying abnormalities of retinal metabolism and physiology. Diabetes. 2007; 56: 373–379. [DOI] [PubMed] [Google Scholar]
- 34. Sennlaub F, Valamanesh F, Vazquez-Tello A, et al. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation. 2003; 108: 198–204. [DOI] [PubMed] [Google Scholar]
- 35. Vinores SA, Youssri AI, Luna JD, et al. Upregulation of vascular endothelial growth factor in ischemic and non-ischemic human and experimental retinal disease. Histol Histopathol. 1997; 12: 99–109. [PubMed] [Google Scholar]
- 36. Sarthy VP, Brodjian SJ, Dutt K, Kennedy BN, French RP, Crabb JW. Establishment and characterization of a retinal Müller cell line. Invest Ophthalmol Vis Sci. 1998; 39: 212–216. [PubMed] [Google Scholar]
- 37. Bajorath J, Seyama K, Nonoyama S, Ochs HD, Aruffo A. Classification of mutations in the human CD40 ligand, gp39, that are associated with X-linked hyper IgM syndrome. Protein Sci. 1996; 5: 531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsing Y, Hostager BS, Bishop GA. Characterization of CD40 signaling determinants regulating nuclear factor-kappa B activation in B lymphocytes. J Immunol. 1997; 159: 4898–4906. [PubMed] [Google Scholar]
- 39. Williams DA, Tao W, Yang F, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocytic immunodeficiency. Blood. 2000; 96: 1646–1654. [PubMed] [Google Scholar]
- 40. Andrade RM, Wessendarp M, Portillo J-AC, et al. TRAF6 signaling downstream of CD40 primes macrophages to acquire anti-microbial activity in response to TNF-α. J Immunol. 2005; 175: 6014–6021. [DOI] [PubMed] [Google Scholar]
- 41. Talahalli R, Zarini S, Sheibani N, Murphy RC, Gubitosi-Klug RA. Increased synthesis of leukotrienes in the mouse model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2010; 51: 1699–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Diem R, Taheri N, Dietz GPH, et al. HIV-Tat-mediated Bcl-XL delivery protects retinal ganglion cells during experimental autoimmune optic neuritis. Neurobiol Dis. 2005; 20: 218–226. [DOI] [PubMed] [Google Scholar]
- 43. Li G, Veenstra AA, Talahalli RR, et al. Marrow-derived cells regulate the development of early diabetic retinopathy and tactile allodynia in mice. Diabetes. 2012; 61: 3294–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Donners MM, Beckers L, Lievens D, et al. The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood. 2008; 111: 4596–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Song Z, Jin R, Yu S, Nanda A, Granger DL, Li G. Crucial role of CD40 signaling in vascular wall cells in neointima formation and vascular remodeling after vascular interventions. Atheroscler Thromb Vasc Biol. 2012; 32: 50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lutgens E, Lievens D, Beckers L, et al. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. 2010; 207: 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]