

Abstract
BACKGROUND AND PURPOSE
Pancreatic cancer is characterized by alterations in several key signalling proteins, including increased expression and activity of the Src tyrosine kinase and focal adhesion kinase (FAK), which have been linked to its chemoresistance. Sustained Src inhibition reactivates survival pathways regulated by the transcription factor STAT3, also leading to resistance. Therefore, simultaneously targeting Src/FAK and STAT3 signalling could provide an important strategy for treating pancreatic cancer. Recently, we described novel quinazolinediones that increased generation of reactive oxygen species (ROS) and were cytotoxic in pancreatic cancer cells. Here, we have investigated effects of our lead compound, QD232, on Src/FAK and STAT3 signalling.
EXPERIMENTAL APPROACH
The major signalling pathways affected by QD232 in pancreatic cancer cell lines were identified by Kinexus proteomic analysis. Changes in key signalling proteins were confirmed by Western blotting. Cell migration was assessed by Boyden chamber and wound healing assays. Direct inhibition of kinase activity in vitro was assayed with a panel of 92 oncogenic kinases. Safety and efficacy of QD232 were determined in a xenograft mouse model of pancreatic cancer.
KEY RESULTS
QD232 potently inhibited Src/FAK and STAT3 phosphorylation, decreasing pancreatic cancer cell viability and migration. Furthermore, QD232 arrested cell cycle progression and induced apoptosis in these cells at low micromolar concentrations. Effects of QD232 on Src/FAK and STAT3 phosphorylation were blocked by N-acetylcysteine or glutathione.
CONCLUSIONS AND IMPLICATIONS
QD232 is a novel compound with a unique, ROS-dependent mechanism, effective in drug-resistant cancer cell lines. This compound shows potential as therapy for pancreatic cancer.
Tables of Links
| TARGETS |
|---|
| Src tyrosine kinase |
| FAK, focal adhesion kinase |
| LIGANDS |
|---|
| Dasatinib |
| Gemcitabine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Pancreatic cancer is one of the most lethal forms of cancer worldwide. It has been estimated that in 2014, around 46 420 people will be diagnosed and 39 590 people will die of pancreatic cancer in the United States (http://cancer.gov, 2014a). Similarly, 41 300 people will die of pancreatic cancer in the European Union (Malvezzi et al., 2014). Late-stage detection and chemoresistance make it difficult to treat pancreatic cancer effectively, resulting in poor prognosis. These grim statistics clearly demonstrate the urgent need for development of agents with novel mechanisms of action to combat pancreatic cancer. FOLFIRINOX [a combination regime consisting of folinic acid, 5-fluorouracil (5-FU), irinotecan and oxaliplatin] is the most commonly used drug combination for the treatment of pancreatic cancer (http://cancer.gov, 2014b).
Pancreatic cancer is characterized by numerous key mutations in genes of several signal transduction pathways including signalling by the cytosolic, non-receptor tyrosine kinase, Src. Src kinase is overactivated in several forms of cancer where it plays an important role in regulation of cell adhesion, motility, invasion, proliferation, survival and angiogenesis (Lutz et al., 1998; Summy and Gallick, 2003; Hilbig, 2008; Kim et al., 2009). Recent studies have successfully validated Src and its phosphorylated form (pSrc) as novel biomarkers for pancreatic cancer (Yokoi et al., 2011). Increased expression, phosphorylation and activity of Src have been reported in gemcitabine-resistant pancreatic cancer cells (Duxbury et al., 2004a,b). Src inhibition effectively slows the growth of human pancreatic cancer xenografts in mice (Rajeshkumar et al., 2009), suppresses invasiveness (Ito et al., 2003), and reverses resistance to 5-FU and gemcitabine (Duxbury et al., 2004a,b; Ischenko et al., 2008). Combined use of Src inhibitors, such as the pyrazolopyrimidine PP2 or AZM475271, and gemcitabine results in decreased tumour growth and inhibition of metastatic spread in orthotopic xenografts of pancreatic cancer (Duxbury et al., 2004b; Yezhelyev et al., 2004). Additionally, Src inhibition by PP2 can augment sensitivity to 5-FU in 5-FU-resistant pancreatic cancer cells (Ischenko et al., 2008).
The effort to develop drugs for late-stage and highly resistant pancreatic cancer led to the evaluation of dasatinib, a potent Src inhibitor, in clinical trials for metastatic and locally advanced pancreatic cancer (http://clinicaltrials.gov, 2014a –g). Studies have shown that dasatinib inhibits cell proliferation, migration, invasion, and anchorage independent growth resulting in tumour growth reduction in vivo. However, dasatinib treatment also leads to resistance because of the lack of inhibition of signalling via the STAT3 and MAPK pathways (Nagaraj et al., 2010). STAT3 reactivation following Src inhibition occurs in several forms of cancer resulting in resistance to Src inhibitors (Byers et al., 2009; Sen et al., 2009). Therefore, there is a need to develop safe and potent drugs with a new mechanism of action that can overcome resistance induced by the current Src inhibitors.
Recently, we screened a large set of highly diverse compounds in an assay specific for cellular respiration and discovered a novel class of compounds that induced oxygen consumption and were cytotoxic by Akt-directed generation of reactive oxygen species (ROS), in a panel of pancreatic cell lines (Pathania et al., 2014). Here, we have shown that the lead compound, QD232 (referred to as compound 3b in Pathania et al., 2014), caused a rapid and sustained inactivation of Src, focal adhesion kinase (FAK) and STAT3. Our central hypothesis is that QD232 inhibited phosphorylation and activation of three substrates, Src, FAK and STAT3 and thus was able to overcome resistance to Src inhibitors. Furthermore, QD232 is a novel agent with unique targets and has biodistribution, safety and efficacy characteristics necessary for further development.
Methods
Cell culture
Pancreatic cancer cell lines (MIA PaCa-2, PANC-1 and BxPC-3) were purchased from the American Type Culture Collection (Manassas, VA, USA). The pancreatic cancer cell line (ASPC-1) was kindly provided by Dr Alan L. Epstein (Keck School of Medicine, University of Southern California, Los Angeles, CA, USA). Human foreskin fibroblast cell lines (HFF-1) were kindly provided by Dr Carla Grandori (Fred Hutchinson Cancer Research Center, Seattle, WA, USA) (Wang et al., 2011). Resistant cell lines, MIA PaCa-2-GR (gemcitabine resistant) and MIA PaCa-2-GTR (gemcitabine and erlotinb resistant) were kindly provided by Dr Sarkar (Department of Pathology, Wayne State University School of Medicine, Detroit, MI, USA) (Soubani et al., 2012). All cell lines used were maintained in culture under 35 (10 for HFF-1) passages and tested regularly for Mycoplasma contamination using Plasmo Test™ (InvivoGen, San Diego, CA, USA). Cell lines were maintained in the appropriate growth media [DMEM (Cellgro, Mediatech, Manassas, VA, USA) for PANC-1, MIA PaCa-2, and RPMI-1640 (Cellgro) for ASPC-1, BxPC-3 and HFF-1] containing 10% heat-inactivated FBS (Gemini-Bioproducts, West Sacramento, CA, USA) at 37°C in a humidified atmosphere of 5% CO2. MIA PaCa-2-GR and MIA PaCa-2-GTR were maintained in DMEM (with 10% FBS) supplemented with 200 nM gemcitabine, and 200 nM gemcitabine and 2 μM erlotinib (every other week), respectively. For subculture and experiments, cells were washed with 1 × Dulbecco's PBS (DPBS, Cellgro), detached using 0.025% trypsin-EDTA (Cellgro), collected in growth media and centrifuged. All experiments were performed in growth media using sub-confluent cells in the exponential growth phase.
Cytotoxicity assay
Cytotoxicity was assessed by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described (Pathania et al., 2014) (details in Supporting Information Appendix S1).
Cell cycle analysis
Sub-confluent cells were seeded in 60 mm tissue culture dishes at a density of 2 × 105 cells per plate in growth media and allowed to adhere overnight. The following day, cells were treated with QD232 or dasatinib or DMSO as vehicle control. Upon completion of treatment, cells were detached with trypsin, and both media and cells were collected by centrifugation. Cells were washed and re-suspended in 1× DPBS, and then fixed in 70% ethanol overnight at −20°C. For determination of DNA content, fixed cells were washed with 1× DPBS, treated with 100 μg·mL−1 RNase A (Sigma-Aldrich, St. Louis, MO, USA), and stained with propidium iodide (50 μg·mL−1, Sigma-Aldrich). DNA content of the samples was analysed by flow cytometry using the BD LSR II flow cytometer (BD Biosciences, San Jose, CA, USA).
Western blotting analyses
Cells were seeded and allowed to adhere overnight. After the desired treatment, cells were washed with 1× DPBS and lysed using RIPA lysis buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) in the presence of protease inhibitor (SIGMAFAST™ protease inhibitor cocktail tablet, EDTA-free, Sigma-Aldrich) and phosphatase inhibitor (sodium orthovanadate, VWR International, Radnor, PA, USA). Cell lysates were sonicated and centrifuged at 13 500× g for 10 min at 4°C. Protein concentration of the whole cell lysates was measured using BCA protein assay and equal amounts of total protein were resolved on 10% polyacrylamide via SDS–PAGE. The separated proteins were electroblotted onto nitrocellulose membrane and blocked in 5% milk in tris-buffered saline with Tween® 20 for 1 h at room temperature. The membrane was probed with primary antibodies to Src, p-Src, STAT3, p-STAT3, FAK, p-FAK, p-p130CAS, p-paxillin, Bcl-2, survivin and Bax (Cell Signaling Technology, Danvers, MA, USA) at 4°C overnight. HRP-conjugated secondary antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) in combination with SuperSignal West Dura (ThermoFisher Scientific, Waltham, MA, USA) were used to visualize proteins of interest with a ChemiDoc Imaging System (Bio-Rad Laboratories).
Annexin V-FITC apoptosis assay
MIA PaCa-2 cells (2.5 × 105 cells per 60 mm dish) were seeded and allowed to adhere overnight. After the indicated treatments, both floating and attached cells were collected, stained with the Annexin V-FITC apoptosis detection kit (Dyes: Annexin V-FITC and propidium iodide; BioVision, Milpitas, CA, USA) according to the manufacturer's protocol. Cells were analysed in an LSR II flow cytometer (BD Biosciences).
siRNA transfection
Sub-confluent MIA PaCa-2 cells (5 × 105 cells per dish) were transfected in 35 mm dishes with 80 nM STAT3, Src or control siRNA for 24 h according to the manufacturer's protocol (Santa Cruz Biotechnology). Cells were treated with QD232 (5 μM, 4 h), lysed and then blotted for desired proteins as described earlier.
In vitro wound healing (scratch) assay
For BxPC-3 and MIA PaCa-2 cells: Sub-confluent cells (7 × 104 cells per well) were plated in a 96-well plate, allowed to adhere overnight, and then serum-starved for 24 h. Wounds were made with a 200 μL pipette tip. Cells were treated with QD232 (0, 0.05, 0.1, 0.5, 1, 2.5 and 5 μM) in medium containing 10% FBS. Negative control wells received serum-free medium. After 24 h, cells were fixed with 100% methanol for 10 min and stained with Giemsa nuclear stain (10% Giemsa, 10% methanol and 80% distilled water) for 30 min at room temperature and washed with distilled water. Stained cells were imaged using BD Pathway 435 High-Content Bioimager (BD Biosciences) using 4× objective.
For ASPC-1 and PANC-1 cells (Li et al., 2004): 96-well plates were pre-coated with collagen (45 μg·mL−1 dissolved in 0.2 N acetic acid) overnight at 4°C, followed by BSA blocking (2 mg·mL−1 in DPBS for 1 h) at room temperature. Sub-confluent cells were seeded in serum-free medium and allowed to adhere overnight. Wounds and treatments were performed using the protocol described above.
In vitro migration assay
Overnight serum-starved cells (MIA PaCa-2, 75 000 cells per well) were plated in serum-free medium on the top chamber of the 24-well plate cell culture inserts fitted with transparent PET membranes with 8 μm pores (BD Biosciences) and allowed to lightly adhere overnight. Next day, cells were treated with QD232 (0.1 and 1.0 μM) in serum-free medium in the upper chamber. Cells were stimulated to migrate by adding fresh medium with 10% FBS to the lower chamber for 24 h. Negative control wells received serum-free medium in the lower chamber. Cells that did not migrate and remained adherent to the upper surface of the membrane at 24 h were lightly scraped off using a Q-tip. Cells that migrated to the lower surface of the membrane were fixed with 100% methanol for 10 min, stained with Giemsa nuclear stain for 30 min at room temperature and washed with deionized water. Images from representative fields of the stained membranes were captured using Nikon DAIPHOT 300 inverted microscope (Nikon Instruments, Melville, NY, USA) with a 10× objective. Images were quantified using ImageJ software (http://rsb.info.nih.gov/).
Imaging
Sub-confluent cells were seeded in a 60 mm dish (MIA PaCa-2; 5 × 105 cells per well) or a 384-well plate (MIA PaCa-2; 5 × 103 cells per well; HFF-1; 8 × 103 cells per well) and allowed to adhere overnight. MIA PaCa-2 cells in 60 mm dish were treated with 5 μM QD232 for increasing time periods (10 or 30 min, 1, 4, 8, 12, or 24 h) and imaged live using Nikon inverted microscope with a 10× objective. Cells in 384-well plates were treated with 5 or 10 μM of QD232 for 4 h, fixed with 100% methanol for 10 min and stained with Giemsa nuclear stain (10% Giemsa, 10% methanol and 80% distilled water) for 30 min at room temperature and washed with distilled water. Stained cells were imaged using BD Pathway 435 High-Content Bioimager (BD Biosciences) with 10× objective.
Xenograft studies
All animal care and experimental procedures were in accordance with the USC institutional guidelines, which were in agreement with the Guidelines for the Care and Use of Laboratory Animals and were approved by the USC Animal Care and Use Committee, under protocol number 11458. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 10 animals were used in the experiments described here.
MIA PaCa-2 cells (1 × 106 cells in a 100 μL suspension of 50% Matrigel/50% DPBS, v/v) were injected subcutaneously into the dorsal flank of 8-week-old female athymic nude mice (Simonsen Laboratories, Gilroy, CA, USA). Tumour size was monitored three times a week through caliper measurement using the following equation: V = d2 × D/2, where d represents width and D represents length of the tumour. Mice were randomly grouped when average tumour size reached 100 mm3. Treatment was administered by i.p. injection in a 100 μL vehicle (5% DMSO, 10% propylene glycol and 85% saline, v/v) five times weekly. Mice in the treatment group (n = 4) received doses of QD232 starting from 10 mg·kg−1 body weight and later increased to 15 mg·kg−1 (from day 10) and 20 mg·kg−1 (from day 19). Mice in the control group (n = 6) received the vehicle. The study was concluded when average tumour size in the control group reached 1000 mm3. Mice exhibiting excessive tumour burden (>1.0 cm3) were euthanised using CO2 gas. Routine health checks were performed. Body weight of each mouse was measured using a weighing scale.
Data analysis
Results are shown as means ± SD or means ± SEM as stated in the figure legends or table subscripts. Unpaired t-test was performed for data analysis using the online version of graphpad software (http://www.graphpad.com/quickcalcs/ttest1.cfm).
Materials
Stock solutions of all compounds were prepared in DMSO (EMD Chemicals, Gibbstown, NJ, USA) and stored at −20°C. Further dilutions were made fresh in DPBS or cell culture media. Gemcitabine hydrochloride and dasatinib monohydrate were purchased from LKT Laboratories (St. Paul, MN, USA). Control samples in all the experiments were treated with vehicle (0.1% DMSO).
Results
QD232 affects several critical cell-signalling pathways governing cell migration and invasion
In order to analyse the effects of QD232 on cellular signal transduction pathways, we performed a Kinex™ (Kinexus, Vancouver, Canada) antibody microarray proteomic analysis. Ingenuity Pathway Analysis (IPA) was used to further analyse the results obtained from the antibody microarray, and the canonical pathways were ranked in the order of statistical significance (Supporting Information Fig. S1). It is important to note that treatment with QD232 did not indiscriminately affect all survival and migration pathways. This was of significance because QD232 did not exert organ toxicity in the whole animal studies (see below). Treatment of MIA PaCa-2 pancreatic cancer cells with QD232 (1 μM for 24 h) affected critical cell-signalling pathways involving the Src/FAK complex (Supporting Information Fig. S1).
QD232 decreases activating phosphorylation of Src and FAK in pancreatic cancer cells
In order to confirm the results obtained from the Kinex™ antibody array analysis, we analysed the effect of QD232 on Src and FAK phosphorylation. As expected, treatment of MIA PaCa-2 and PANC-1 cells with QD232 decreased the activating phosphorylations of FAK at Y576/Y577, Y397, Y861 and Y925 in a concentration- and time-dependent manner (Figure 1A,F,G and Figure 2A,D,E; Supporting Information Tables S1 and S2). Additionally, treatment with QD232 resulted in decreased phosphorylation of Src at Y416 in concentration- and time-dependent manner (Y416 is the activating phosphorylation of Src) (Figure 1B,F, Figure 2B,D,E, and Supporting Information Fig. S2, Tables S1 and S2). However, QD232 did not affect the inactivating phosphorylation of Src (Y527, Figure 1C). Interestingly, the inhibitory effects of QD232 on Src and FAK were greater and slightly faster in onset in MIA PaCa-2 cells than in PANC-1 cells. This was in accordance with our findings reported earlier. It is important to note that the basal levels of p-Src are lower in PANC-1 cells than in MIA PaCa-2 cells. As we had shown that QD232 was slightly more potent in MIA PaCa-2 cells (Pathania et al., 2014); we selected this cell line for further mechanistic evaluation of QD232.
Figure 1.

QD232 decreases phosphorylation of signalling proteins involved in cell migration in pancreatic cancer cells. Treatment of MIA PaCa-2 cells with QD232 (5 μM) results in inhibition of activating phosphorylations of FAK (A) Src (B), STAT3 (D), p130CAS and paxillin (E) in a time-dependent manner. Moreover, it does not affect the inactivating phosphorylation of Src (C). QD232 treatment causes a concentration-dependent decrease in Src, FAK and STAT3 phosphorylation (F and G). Schematic representation of the effects of QD232 on selected cell-signalling pathways (H). (Y419 and Y530 are activating and inactivating phosphorylation sites for human c-Src, respectively. Text and figure refer to Y419 as Y416 and Y530 as Y527, as labelled on the antibodies received from Cell Signaling Technology, Danvers, MA, USA. Representative blots of three independent experiments are shown. Summary data are provided in Supporting Information Table S1.
Figure 2.
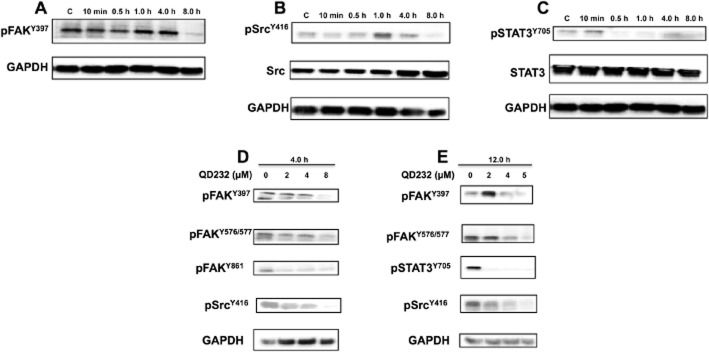
Treatment with QD232 decreases phosphorylation of FAK, Src and STAT3 in PANC-1 cells. Treatment of PANC-1 cells with QD232 (5 μM) inhibits activating phosphorylations of FAK (A) Src (B) and STAT3 (C) in a time-dependent manner. (D and E) QD232 treatment results in a concentration-dependent decrease in Src, FAK and STAT3 phosphorylation. Representative blots of three independent experiments are shown. Summary data are provided in Supporting Information Table S2.
Activation of Src promotes the interaction and phosphorylation of target proteins, such as paxillin and CAS family proteins that also bind FAK. Activation and phosphorylation of p130CAS and paxillin influences focal adhesion turnover, supporting cell migration. Treatment of MIA PaCa-2 cells with QD232 resulted in decreased phosphorylation of both p130 CAS and paxillin suggesting inhibition of cell migration (Figure 1E).
QD232 treatment results in changes in cell morphology and inhibits cell migration
QD232 blocks several pathways that support metastasis in pancreatic cancer cells (Figures 1 and 2 and Supporting Information Fig. S2). As expected, QD232 treatment resulted in morphological changes in pancreatic cancer cells within 4 h (Supporting Information Fig. S3A), while similar treatments did not induce significant morphological changes in HFF-1 cells (Supporting Information Fig. S3B).
Because cell migration precedes metastasis, we tested the ability of QD232 to inhibit cell migration in the Boyden chamber. Decreased migration of MIA PaCa-2 cells was observed in the presence of 1 μM QD232 (Figure 3A,B; Supporting Information Table S3). Inhibition of migration was confirmed in the wound healing assay, and this difference was not due to cytotoxicity as determined in cells treated in parallel (Figure 3C; Supporting Information Table S3). However, at a higher concentration of 5 μM, the increase in wound size can be attributed to the toxicity of the compound. QD232 decreased the ability of a panel of serum-starved pancreatic cancer cells (MIA PaCa-2, PANC-1, BxPC-3 and ASPC-1) to close the wound when stimulated with 10% FBS (Figure 3D–G and Supporting Information Fig. S4 and Table S3).
Figure 3.

QD232 inhibits cell migration. (A) A 24 h treatment with QD232 (1 μM) resulted in decreased migration of serum-starved MIA PaCa-2 cells through the membrane of a Boyden chamber in the presence of 10% FBS stimulation. Cells were imaged after fixing with methanol and staining with Giemsa using a Nikon microscope with 10× objective. (B) Quantification of data (means ± SD) shown in (A); effect induced by both concentrations of QD232 were significantly different from control (10% FBS only); P < 0.05. (C) Cell viability (means ± SD) as determined by MTT assay of MIA PaCa-2 cells treated with QD232 concentrations that were used in the cell migration assays under identical conditions. (D, E) Treatment with QD232 for 24 h inhibited closure of wounds stimulated by 10% FBS in serum-starved MIA PaCa-2 (D) and PANC-1 (E) cells in a dose-dependent manner. Media without FBS was used as a control for the inhibition of wound closure. Cells were imaged after fixing with methanol and staining with Giemsa using BD Pathway 435 High-Content Bioimager with a 4× objective. Representative images of three independent experiments are shown. (F, G). Quantification of images (means ± SD) shown in D and E respectively. Images were quantified using ImageJ software (http://rsb.info.nih.gov/). All concentrations of QD232, except the lowest (0.05μM in F, and 0.05 and 0.1 μM in G), had significant effects, compared with 10% FBS only; P < 0.05.
QD232 inhibits cell cycle progression
The Src/FAK complex governs cell cycle progression by regulating several downstream targets (Zhou et al., 2011). QD232 inhibits activity of the Src/FAK complex and therefore should block cell cycle progression. Treatment of MIA PaCa-2 cells with QD232 (1 μM, 24 h) resulted in arrest of cells in S-phase (and slight G2/M). These effects were more prominent at 2.5 μM of QD232. Cell cycle arrest was sustained for 48 h (Figure 4A; Supporting Information Table S3). Similar results were observed in PANC-1 cells (Figure 4B; Supporting Information Table S3).
Figure 4.

QD232 induces cell cycle arrest and cell death in pancreatic cancer cells. Exposure of MIA PaCa-2 (A) or PANC-1 (B) cells to QD232 resulted in S-phase (and slight G2/M) arrest in a concentration- and time-dependent manner. Dasatinib was used as a positive control for inducing cell cycle arrest in pancreatic cancer cells. Treatment of MIA PaCa-2 cells with QD232 resulted in decreased expression of cell survival proteins Bcl-2 (C and E) and survivin (D), and increased expression of pro-apoptotic Bax (C) in a concentration- and time-dependent manner. (F) Apoptotic cell death induced by QD232 in MIA PaCa-2 cells was further confirmed by AnnexinV assay. Control and QD232-treated MIA PaCa-2 cells were stained with Annexin V-FITC and PI and analysed by flow cytometry. Annexin V-negative and PI-negative cells were viable. Annexin V-positive and PI-negative cells were in early stages of apoptosis, and cells positive for both Annexin V and PI represented cell death by late stages of apoptosis and necrosis. 5 μM of QD232 was used for time course experiments in panel E. Representative data of three independent experiments are shown here. Summary data for these experiments are provided in Supporting Information Table S3.
QD232 induces apoptotic cell death
The Src/FAK complex regulates several cell-signalling pathways governing cell survival, proliferation and death. Apoptosis signalling is one of the canonical pathways most affected by QD232 treatment (Supporting Information Fig. S1). QD232 induced rapid and sustained inhibition of cancer cell proliferation resulting in cell death (Pathania et al., 2014). Here, QD232 (5 μM) caused cell death by decreasing the expression of major cell survival proteins including Bcl-2 and survivin after treatment of MIA PaCa-2 cells for at least 12 h (Figure 4C,D,E; Supporting Information Table S3). Additionally, QD232 increased expression of the pro-apoptotic protein Bax (Figure 4E; Supporting Information Table S3). Apoptotic activity of QD232 was further confirmed by Annexin V/PI staining, which showed that QD232 concentration-dependently increased the apoptotic population of MIA PaCa-2 cells (Figure 4F; Supporting Information Table S3).
QD232 inhibits tumour growth in vivo
QD232 was tested in an athymic nude mice xenograft model in order to determine its in vivo efficacy. The maximum tolerated dose of QD232 was determined by a dose-escalating experiment in athymic nude mice and no significant toxicity was observed at the highest dose (40 mg·kg−1). Subcutaneous human pancreatic cancer xenografts (MIA PaCa-2) were established on the dorsal flank of the immune-deficient mice, and treated with QD232 or vehicle until average tumour size in the control group reached 1000 mm3 (31 days). There was no significant response to QD232 treatment at a dose of 10 mg·kg−1. Therefore, we increased the dose to 20 mg·kg−1. Treatment with QD232 at 20 mg·kg−1 significantly suppressed growth of tumours as compared with the vehicle control group (Figure 5A). Along the course of the 31 day treatment, average tumour volume increased by 960% in the control group, while the average tumour volume only increased by 337% in the QD232-treated group, suggesting significant antitumour activity of QD232 in vivo. Mice were also monitored daily and weighed twice a week to detect potential drug-related toxicity (Figure 5B). No systemic symptoms of toxicity such as weakness, weight loss or lethargy were detected. Major organs were collected after euthanising the mice (day 31) in order to assess the potential drug-related toxicities at the cellular level. Samples were fixed in 10% neutral buffered formalin, embedded in paraffin, and stained with H&E for histological analysis. Careful examination of these tissue sections confirmed no signs of histological abnormalities in tissue samples taken from QD232-treated mice, compared with those from control mice (Figure 5C).
Figure 5.
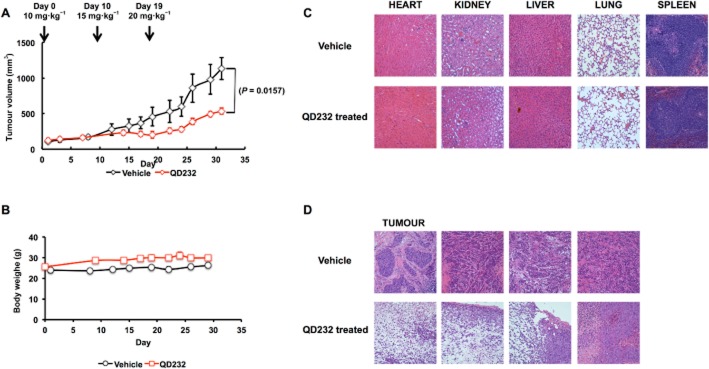
QD232 suppresses tumour growth in a mouse xenograft model of pancreatic cancer and does not exhibit significant signs of systemic toxicity. (A) The plot shows the tumour volumes (means ± SD) among the treatment groups over the course of the experiment. Vehicle control and QD232 were administered five times a week (5 days on, 2 days off regimen). Tumour volumes were measured three times a week. (B) Treatment with QD232 does not result in loss in body weight. (C) Representative micrographs of haematoxylin and eosin (H&E)-stained organ sections. No significant microscopic changes were detected in major organs after QD232 treatment. Images were taken with Olympus IX73 inverted microscope at 20× magnification. (D) Representative micrographs of H&E-stained tumour sections. Tumour samples showed obvious areas of necrosis.
QD232 is efficacious in resistant forms of cancer
Increased Src and FAK phosphorylation are associated with the inherent and acquired resistance to gemcitabine in pancreatic cancer cells (Duxbury et al., 2004b; Zhou et al., 2011). QD232 treatment inhibits Src/FAK-mediated cell signalling and is effective in inhibiting cell proliferation in MIA PaCa-2 cells either resistant to gemcitabine alone (MIA PaCa-2-GR; IC50 = 2.7 μM) or to both gemcitabine and erlotinib (MIA PaCA-2-GTR; IC50 = 2.3 μM). These values are similar to its IC50 (2.3 ± 0.2 μM) in parental MIA PaCa-2 cells (Pathania et al., 2014).
The lack of inhibition of STAT3 phosphorylation is one of the reasons for resistance to dasatinib therapy (Nagaraj et al., 2010). In our experiments, MIA PaCa-2 cells treated with QD232 showed a clear decrease in p-STAT3 after 30 min of drug exposure and of p-Src in 4 h. However, treatment of the same cell line with dasatinib significantly decreased levels of p-Src and not of p-STAT3, after 12 h of exposure (Figure 6A; Supporting Information Table S4).
Figure 6.
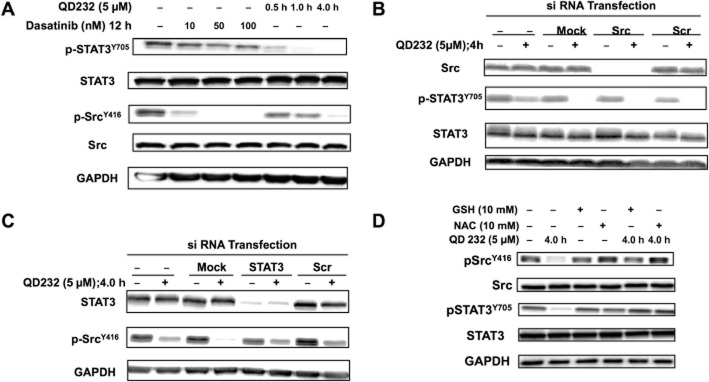
QD232 exhibits ROS-mediated effects on cell-signalling proteins. (A) Unlike dasatinib, QD232 can cause robust decrease in phosphorylation of Src as well as STAT3. Cells were treated with increasing concentrations of dasatinib for 12 h or with QD232 (5 μM) for 0.5–4.0 h. In cells treated with dasatinib, there is a clear inhibition of Src phosphorylation, but no effect on p-STAT3. However, QD232 treatment results in complete inhibition of STAT3 phosphorylation. (B) QD232 decreases STAT3 phosphorylation in Src knocked down MIA PaCa-2 cells suggesting that its effect on STAT3 is independent of its effect on Src. (C) QD232 decreases Src phosphorylation in STAT3 knocked down MIA PaCa-2 cells. (D) Treatment with QD232 in the presence of antioxidants (NAC or GSH) blocked the ability of QD232 to inhibit Src, FAK or STAT3 phosphorylation. Cells were pretreated for 2 h with antioxidants (10 mM), washed with 1× DPBS and then exposed to QD232 (5 μM, 4 h). Representative blots of three independent experiments are shown here and summary data for these experiments are shown in Supporting Information Table S4.
QD232 exerts ROS-mediated effects
STAT3 is one of the downstream targets of Src and is directly activated by Src (Kim et al., 2009). Other signalling pathways can also affect p-STAT3 levels (Debnath et al., 2012). QD232 induced a robust decrease in Src and STAT3 phosphorylation, in our experiments. In order to determine if the decrease in STAT3 phosphorylation by QD232 was due to its effects on Src, we knocked down Src and re-assessed the effects of QD232 on STAT3 phosphorylation. In MIA PaCa-2 cells, STAT3 is activated even in the absence of Src, indicating that in this model, other pathways may be responsible for p-STAT3 levels (Figure 6B; Supporting Information Table S4). Interestingly, QD232 was able to decrease STAT3 phosphorylation in Src knocked down MIA PaCa-2 pancreatic cancer cells (Figure 6B). This suggests that the effect of QD232 on STAT3 was independent of its effect on Src. Moreover, QD232 was able to decrease Src phosphorylation in STAT3 knocked down cells (Figure 6C; Supporting Information Table S4), suggesting lack of a feedback loop from STAT3 to Src. In a cell-free, in vitro, kinase activity assay of 92 oncogenic kinases (including Src and FAK), QD232 at 10 μM caused only <25% inhibition or stimulation of the tested targets. It did not inhibit Src or FAK in this kinase activity assay (Supporting Information Fig. S5). These data suggest that the effect of QD232 on Src and STAT3 activation is mechanistically different from dasatinib.
We have previously shown that QD232 rapidly increased the cellular content of ROS (Pathania et al., 2014). ROS are known to act as essential second messengers in cells and can influence many signal transduction pathways with different kinetics, including Src/FAK signalling (Cunnick et al., 1998; Tang et al., 2005; Kemble and Sun, 2009). Therefore, we analysed the effects of QD232 on Src, FAK and STAT3 in the presence of the antioxidants, N-acetylcysteine (NAC) or glutathione (GSH). QD232 was not able to decrease phosphorylation of Src, FAK or STAT3 in MIA PaCa-2 cells in the presence of antioxidants (Figure 6D). This suggests that QD232 exerts ROS-mediated effects on Src/FAK/STAT3 signalling in pancreatic cancer cells.
Discussion and conclusions
Pancreatic cancer is one of the deadliest and most aggressive diseases with poor prognosis and limited therapeutic options for most patients (http://cancer.gov, 2014a). Therefore, there is a pressing need for more effective treatment strategies for this disease. Expression and activation of Src and FAK kinases have been linked to clinicopathological characteristics of pancreatic ductal adenocarcinoma (Lutz et al., 1998; Chatzizacharias et al., 2010). Src and FAK act as a complex and regulate many cellular events including cell proliferation, survival, adhesion, migration and invasion linked to cancer progression and metastasis (Summy and Gallick, 2003; Kim et al., 2009; Bolos et al., 2010). Therefore, targeting pathways leading to inhibition of the Src/FAK complex and its downstream effects is an attractive strategy for developing anticancer therapies.
Treatment of pancreatic cancer cells with QD232 resulted in a robust decrease in phosphorylation of the Src/FAK complex at several sites (Figures 1 and 2 and Supporting Information Fig. S2). Autophosphorylation of FAK at Y397 results in its activation and creates a docking site for Src. Once activated by FAK, Src can further phosphorylate FAK at various sites resulting in a feedback activation loop (Zhou et al., 2011). Together Src and FAK function as a protein complex for regulation of several cellular signal transduction pathways. QD232 decreased phosphorylation of FAK at several sites including the autophosphorylated Y397 that acts as a docking site for Src (Figures 1A,F,G and 2A,D,E). Association of Src with FAK results in full activation of Src. In turn, activated Src phosphorylates FAK at Y576/577 (catalytic domain) and Y925 (docking site for growth-factor-receptor-bound protein 2, mediates RAS-MAPK signalling, and Src-induced epithelial mesenchymal transition) (McLean et al., 2005). QD232 decreased Src phosphorylation at Y416 (activating phosphorylation) and Src-directed FAK phosphorylations at Y576/577 and Y925 (Figures 1A,B,F,G and 2B,D,E). Moreover, QD232 decreased phosphorylation of FAK at Y861 (Figures 1A,F and 2D), a site that influences tumour vasculature by governing the interaction of FAK with integrins and regulates FAK's interaction with p130CAS, promoting cell invasion (McLean et al., 2005). Decreased FAK-Y861 phosphorylation, following treatment with by QD232, led to decreased phosphorylation of its substrate p130CAS (Figure 1E). QD232 also decreased phosphorylation of paxillin (Figure 1E), a substrate of the Src/FAK complex that acts as an adaptor for several proteins involved in cell adhesion (Bolos et al., 2010).
The decreased Src/FAK phosphorylation was accompanied by morphological changes in cancer cells (Supporting Information Fig. S3A). These morphological changes were not observed in normal fibroblasts, suggesting that QD232 is more effective in cancer cells (Supporting Information Fig. S3B). Several studies have shown that cells lacking FAK expression migrate poorly compared with normal fibroblasts (Ilic et al., 1995; Owen et al., 1999; Sieg et al., 2000). QD232 decreased Src/FAK activation (Figures 1 and 2 and Supporting Information Fig. S2) and hence, inhibited cell migration in pancreatic cancer cells (Figure 3A,D,E and Supporting Information Fig. S4).
Src and FAK also play an important role in the regulation of the cell cycle progression (Reiske et al., 2000; Shen and Guan, 2001). Like other inhibitors of Src/FAK, QD232 blocks cell cycle progression in cancer cells (Figure 4A,B). Furthermore, inhibition of cellular FAK and Src has been linked to cell detachment that results in apoptosis (Hungerford et al., 1996; Xu et al., 1996; Beierle et al., 2010). Treatment with QD232 results in detachment of cancer cells (Supporting Information Fig. S3) leading to cell death (Figure 4F; Pathania et al., 2014). This mechanism is supported by QD232-induced decreased expression of pro-survival proteins including Bcl-2 and survivin (Figure 4C,D,E), and increased expression of Bax (Figure 4E). Moreover, we previously showed that QD232 activates Fas/FasL and caspases, which further validates that QD232 treatment results in apoptotic cell death (Pathania et al., 2014). More significantly, QD232 delays tumour growth in a mouse xenograft model of pancreatic cancer without inducing significant deleterious side effects (Figure 5).
A major limitation of pancreatic cancer therapy is the development of resistance to currently available therapies including gemcitabine. The expression and activation of Src increases from normal pancreas to pancreatitis to pancreatic adenocarcinoma, in accordance with its role in cancer progression and metastasis (Lutz et al., 1998; Summy and Gallick, 2003; Hilbig, 2008). Decreasing Src/FAK signalling has been shown to restore sensitivity to gemcitabine and 5-FU in pancreatic cancer cells (Duxbury et al., 2004a,b; Ischenko et al., 2008). QD232 inhibited cell proliferation in gemcitabine-resistant MIA PaCa-2 cells. Furthermore, Src inhibitors like dasatinib are being developed for treatment of pancreatic cancer, but they have limited efficacy as monotherapy in clinical settings (http://clinicaltrials.gov, 2014a–g). Recent studies have shown that dasatinib-resistant pancreatic cancer cells have activated STAT3 (Nagaraj et al., 2010). Additionally, STAT3 reactivation has been linked with sustained Src inhibition in other types of cancer (Sen et al., 2009). Our studies demonstrated that QD232 inhibited phosphorylation of both STAT3 and Src, and that activation of STAT3 was also inhibited in the absence of Src (Figures 1, 2 and 6B and Supporting Information Fig. S2). Therefore, QD232 treatment might be beneficial in resistant forms of pancreatic cancer exhibiting STAT3 activation.
Sustained Src inhibition results in STAT3 activation by induction of a homeostatic feedback loop leading to resistance to Src inhibitors (Sen et al., 2009). Conversely, complete inhibition of p-STAT3 results in Src activation as a result of induction of a feedback loop (Byers et al., 2009). More importantly, simultaneous blockade of Src and STAT3 activation has been shown to be a promising therapeutic approach for several forms of cancer including pancreatic cancer (Nagaraj et al., 2011). Our novel inhibitor, QD232, decreased STAT3 phosphorylation in Src knockdown cells and decreased Src phosphorylation in STAT3 knockdown cells (Figure 6B,C). Targeting three oncoproteins (Src/FAK and STAT3) and potentially overcoming chemoresistance by a single agent may lead to improved treatment for pancreatic cancer.
Our data demonstrated that the inhibition of the Src/FAK, and STAT3 by QD232 was mediated through ROS production and was not due to direct inhibition of kinase activity (Figure 6D and Supporting Information Fig. S5). ROS regulate several cellular events by governing a range of signal transduction pathways. For example, the natural product manumycin inhibits STAT3 by elevating intracellular ROS in glioma cells (Dixit et al., 2009). Similarly, phenethyl isothiocyanate inhibits STAT3 activation in prostate cancer cells by generating ROS (Gong et al., 2009). STAT3 deletion itself can sensitize cancer cells to oxidative stress (Barry et al., 2009). Additionally, inactivation of Src family tyrosine kinases by ROS has been reported (Cunnick et al., 1998; Tang et al., 2005; Kemble and Sun, 2009). Previously, we reported that QD232 treatment causes immediate and significant increase in cellular ROS production (Pathania et al., 2014). In this study, we demonstrated that treatment with QD232 in the presence of antioxidants did not result in decreased STAT3, Src or FAK phosphorylation (Figure 6D) suggesting that generation of ROS is essential for QD232 activity (Figure 7).
Figure 7.
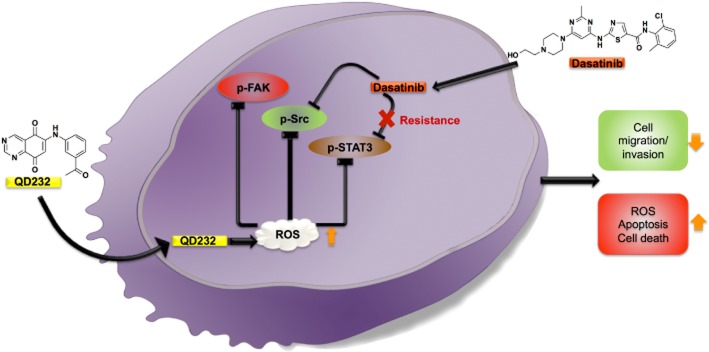
Proposed mechanism of action of QD232. Sustained Src inhibition by dasatinib results in STAT-3 reactivation and induces resistance to Src inhibitors. QD232 through ROS induction causes inhibition of Src, FAK and STAT3 activation leading to decreased cell proliferation, adhesion and migration followed by apoptotic cell death.
In conclusion, we have discovered a novel low MW compound that inhibited the growth of tumours in a mouse xenograft model of pancreatic cancer and exerted ROS-mediated decrease in Src/FAK and STAT3 phosphorylation, resulting in efficient inhibition of cell proliferation, adhesion and migration, ultimately leading to pancreatic cancer cell death by apoptosis (Figure 7). Although drugs that individually and selectively inhibit Src, FAK and STAT3 are under development, when used as single agents, they tend to induce resistance. For example, sustained inhibition of Src by dasatinib results in resistance mediated by STAT3 activation. Discovery of a single compound that can block the activation of these three critical signalling pathways simultaneously, will significantly contribute to more effective treatment and overcome resistance. Therefore, QD232 can serve not only as a unique pharmacological probe, but can also be developed into an effective therapeutic agent for pancreatic cancer, as monotherapy or in combination with other chemotherapeutic agents.
Acknowledgments
This study was supported in part by funds from the Sharon L. Cockrell Cancer Research Fund to N. N. and by financial support from the Regione Autonoma della Sardegna (grant CRP-25920), within the frame of ‘Legge regionale n. 7/2007, promozione della ricerca scientifica e dell'innovazione tecnologica in Sardegna, Annualità 2010’, to M. S. D. P. was a recipient of a USC Graduate School Dissertation Completion fellowship.
Glossary
- 5-FU
5-fluorouracil
- FAK
focal adhesion kinase
- IPA
Ingenuity Pathway Analysis
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAC
N-acetylcysteine
- QD232
6-[(3-acetylphenyl)amino]quinazoline-5,8-dione
- ROS
reactive oxygen species
Author contributions
D. P. and N. N. conceived and designed the experiments. D. P. and Y. K. performed the experiments. D. P., Y. K., M. S. and N. N. analysed the data. D. P. and N. N. wrote the paper.
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12855
Appendix S1 Methods.
Figure S1 QD232 affects several critical cell-signalling pathways governing cell migration and invasion. MIA PaCa-2 cells were treated with QD232 (1 μM) for 24 h, lysed following Kinexus's protocol, and screened for 378 pan-specific (protein abundance) and 273 phospho-site-specific antibodies. Control and treated cell lysates were labelled with fluorescent dye and analysed separately on the same chip spotted with the array of antibodies. Quantitative analysis of the fluorescence intensity signal for each target protein was performed for each sample, and fold-change ratios of treated-to-control were reported. These data were further analysed using Ingenuity Pathway Analysis (IPA) software and the plot displaying top canonical pathways affected by QD232 treatment was generated. −log p on the left Y-axis represents the statistical significance and the pathways are arranged in the order of their statistical significance. Ratio represents the ratio of number of signalling proteins affected by the treatment to the total number of signalling proteins in that pathway reported in IPA.
Figure S2 QD232 causes rapid decrease in p-Src in MIA PaCa-2 pancreatic cancer cells. QD232 (5 μM) results in decreasing phosphorylation of p-Src (A) in a time-dependent manner without affecting the expression of total Src (B). Cells were fixed with 0.4% formaldehyde and stained using primary antibodies for p-Src and Src followed by incubation with respective secondary antibodies and then counter-stained with DAPI nuclear dye. Cells were imaged using BD Pathway 435 High-Content Bioimager (BD Biosciences, San Jose, CA, USA) using 10× objective. Representative images of three independent experiments are shown.
Figure S3 QD232 causes rapid change in morphology of cancer cells, but not of normal cells. (A) 5 μM QD232 resulted in loss of adhesion and rounding up of MIA PaCa-2 pancreatic cancer cells within 4 h of treatment. Cells were imaged live using Nikon microscope with 10× objective. (B) However, no change in morphology was observed in normal HFF-1 fibroblasts even at 10 μM treatment with QD232. Cells were imaged after fixing with methanol and staining with Giemsa using BD Pathway 435 High-Content Bioimager (BD Biosciences, San Jose, CA, USA) using 10× objective. Representative images of three independent experiments are shown.
Figure S4 QD232 inhibits cell migration in cancer cells. QD232 inhibited closure of wounds stimulated by 10% FBS in serum-starved BxPC-3 (A) and ASPC-1 (B) pancreatic cancer cells in a concentration-dependent manner after 24 h treatment. Cells were imaged after fixing with methanol and staining with Giemsa using BD Pathway 435 High-Content Bioimager (BD Biosciences, San Jose, CA, USA) using 4× objective. (C, D) Quantification of data shown in A and B, respectively. Representative images of three independent experiments are shown. Images were quantified using ImageJ software (http://rsb.info.nih.gov/).
Figure S5 QD232 does not cause any significant inhibition in activity of oncogenic kinases in vitro. QD232 (10 μM) does not significantly inhibit the activity of oncogenic kinases (including FAK and Src) in a kinase profiling in vitro substrate assay. Kinase Profiler™ assay was performed by EMD Millipore, Billerica, MA, USA. Concentration of ATP used in the assay was Km for each kinase. 10 μM ATP was used for kinases for which Km was not known.
Table S1 Quantification of data for Figure 1.
Table S2 Quantification of data for Figure 2.
Table S3 Quantification of data for Figure 4.
Table S4 Quantification of Data for Figure 6.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry SP, Townsend PA, McCormick J, Knight RA, Scarabelli TM, Latchman DS, et al. STAT3 deletion sensitizes cells to oxidative stress. Biochem Biophys Res Commun. 2009;385:324–329. doi: 10.1016/j.bbrc.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierle EA, Ma X, Trujillo A, Kurenova EV, Cance WG, Golubovskaya VM. Inhibition of focal adhesion kinase and src increases detachment and apoptosis in human neuroblastoma cell lines. Mol Carcinog. 2010;49:224–234. doi: 10.1002/mc.20592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolos V, Gasent JM, Lopez-Tarruella S, Grande E. The dual kinase complex FAK–Src as a promising therapeutic target in cancer. Onco Targets Ther. 2010;3:83–97. doi: 10.2147/ott.s6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers LA, Sen B, Saigal B, Diao L, Wang J, Nanjundan M, et al. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin Cancer Res. 2009;15:6852–6861. doi: 10.1158/1078-0432.CCR-09-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- cancer.gov. 2014a. Pancreatic cancer. Available at: http://www.cancer.gov/cancertopics/types/pancreatic/ (accessed 15/5/2014)
- cancer.gov. 2014b. Cancer topics. Available at: http://www.cancer.gov/cancertopics/druginfo/FOLFIRINOX (accessed 15/5/2014)
- 2014a. http://clinicaltrials.gov. Dasatinib added to gemcitabine for subjects with locally-advanced pancreatic cancer (LAPC). Available at: http://clinicaltrials.gov/ct2/show/NCT01395017 (accessed 5/1/2014)
- clinicaltrials.gov. 2014b. Gemcitabine hydrochloride, dasatinib, and erlotinib hydrochloride in treating patients with metastatic pancreatic cancer that cannot be removed by surgery. Available at: http://clinicaltrials.gov/ct2/show/NCT01660971 (accessed 5/1/2014)
- clinicaltrials.gov. 2014c. Dasatinib and gemcitabine hydrochloride or gemcitabine hydrochloride alone in treating patients with pancreatic cancer previously treated with surgery. Available at: http://clinicaltrials.gov/ct2/show/NCT01234935 (accessed 5/1/2014)
- clinicaltrials.gov. 2014d. Dasatinib in treating patients with stage IV pancreatic cancer. Available at: http://clinicaltrials.gov/ct2/show/NCT00544908 (accessed 5/1/2014)
- clinicaltrials.gov. 2014e. A phase I/expansion study of dasatinib (Gem/Dsat). Available at: http://clinicaltrials.gov/ct2/show/NCT00598091 (accessed 5/1/2014)
- clinicaltrials.gov. 2014f. Dasatinib in treating patients with metastatic pancreatic cancer. Available at: http://clinicaltrials.gov/ct2/show/NCT00474812 (accessed 5/1/2014)
- clinicaltrials.gov. 2014g. Phase II study of 5-FU, oxaliplatin plus dasatinib in metastatic pancreatic adenocarcinoma (FOLFOX-D). Available at: http://clinicaltrials.gov/ct2/show/NCT01652976 (accessed 5/1/2014)
- Chatzizacharias NA, Giaginis C, Zizi-Serbetzoglou D, Kouraklis GP, Karatzas G, Theocharis SE. Evaluation of the clinical significance of focal adhesion kinase and SRC expression in human pancreatic ductal adenocarcinoma. Pancreas. 2010;39:930–936. doi: 10.1097/MPA.0b013e3181d7abcc. [DOI] [PubMed] [Google Scholar]
- Cunnick JM, Dorsey JF, Standley T, Turkson J, Kraker AJ, Fry DW, et al. Role of tyrosine kinase activity of epidermal growth factor receptor in the lysophosphatidic acid-stimulated mitogen-activated protein kinase pathway. J Biol Chem. 1998;273:14468–14475. doi: 10.1074/jbc.273.23.14468. [DOI] [PubMed] [Google Scholar]
- Debnath B, Xu S, Neamati N. Small molecule inhibitors of signal transducer and activator of transcription 3 (Stat3) protein. J Med Chem. 2012;55:6645–6668. doi: 10.1021/jm300207s. [DOI] [PubMed] [Google Scholar]
- Ding W, You H, Dang H, LeBlanc F, Galicia V, Lu SC, et al. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology. 2010;52:945–953. doi: 10.1002/hep.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit D, Sharma V, Ghosh S, Koul N, Mishra PK, Sen E. Manumycin inhibits STAT3, telomerase activity, and growth of glioma cells by elevating intracellular reactive oxygen species generation. Free Radic Biol Med. 2009;47:364–374. doi: 10.1016/j.freeradbiomed.2009.04.031. [DOI] [PubMed] [Google Scholar]
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. siRNA directed against c-Src enhances pancreatic adenocarcinoma cell gemcitabine chemosensitivity. J Am Coll Surg. 2004a;198:953–959. doi: 10.1016/j.jamcollsurg.2004.01.037. [DOI] [PubMed] [Google Scholar]
- Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Inhibition of SRC tyrosine kinase impairs inherent and acquired gemcitabine resistance in human pancreatic adenocarcinoma cells. Clin Cancer Res. 2004b;10:2307–2318. doi: 10.1158/1078-0432.ccr-1183-3. [DOI] [PubMed] [Google Scholar]
- Gong A, He M, Krishna Vanaja D, Yin P, Karnes RJ, Young CY. Phenethyl isothiocyanate inhibits STAT3 activation in prostate cancer cells. Mol Nutr Food Res. 2009;53:878–886. doi: 10.1002/mnfr.200800253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbig A. Src kinase and pancreatic cancer. Recent Results Cancer Res. 2008;177:179–185. doi: 10.1007/978-3-540-71279-4_19. [DOI] [PubMed] [Google Scholar]
- Hungerford JE, Compton MT, Matter ML, Hoffstrom BG, Otey CA. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang-Verslues WW, Kuo WH, Chang PH, Pan CC, Wang HH, Tsai ST, et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS ONE. 2009;4:e8377. doi: 10.1371/journal.pone.0008377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Ischenko I, Camaj P, Seeliger H, Kleespies A, Guba M, De Toni EN, et al. Inhibition of Src tyrosine kinase reverts chemoresistance toward 5-fluorouracil in human pancreatic carcinoma cells: an involvement of epidermal growth factor receptor signaling. Oncogene. 2008;27:7212–7222. doi: 10.1038/onc.2008.326. [DOI] [PubMed] [Google Scholar]
- Ito H, Gardner-Thorpe J, Zinner MJ, Ashley SW, Whang EE. Inhibition of tyrosine kinase Src suppresses pancreatic cancer invasiveness. Surgery. 2003;134:221–226. doi: 10.1067/msy.2003.224. [DOI] [PubMed] [Google Scholar]
- Kemble DJ, Sun G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc Natl Acad Sci U S A. 2009;106:5070–5075. doi: 10.1073/pnas.0806117106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6:587–595. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Fan J, Chen M, Guan S, Sawcer D, Bokoch GM, et al. Mechanism of human dermal fibroblast migration driven by type I collagen and platelet-derived growth factor-BB. Mol Biol Cell. 2004;15:294–309. doi: 10.1091/mbc.E03-05-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz MP, Esser IB, Flossmann-Kast BB, Vogelmann R, Lührs H, Friess H, et al. Overexpression and activation of the tyrosine kinase Src in human pancreatic carcinoma. Biochem Biophys Res Commun. 1998;243:503–508. doi: 10.1006/bbrc.1997.8043. [DOI] [PubMed] [Google Scholar]
- Malvezzi M, Bertuccio P, Levi F, La Vecchia C, Negri E. European cancer mortality predictions for the year 2014. Ann Oncol. 2014;25:1650–1656. doi: 10.1093/annonc/mdu138. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer – a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- Nagaraj NS, Smith JJ, Revetta F, Washington MK, Merchant NB. Targeted inhibition of SRC kinase signaling attenuates pancreatic tumorigenesis. Mol Cancer Ther. 2010;9:2322–2332. doi: 10.1158/1535-7163.MCT-09-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj NS, Washington MK, Merchant NB. Combined blockade of Src kinase and epidermal growth factor receptor with gemcitabine overcomes STAT3-mediated resistance of inhibition of pancreatic tumor growth. Clin Cancer Res. 2011;17:483–493. doi: 10.1158/1078-0432.CCR-10-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen JD, Ruest PJ, Fry DW, Hanks SK. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19:4806–4818. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania D, Sechi M, Palomba M, Sanna V, Berrettini F, Sias A, et al. Design and discovery of novel quinazolinedione-based redox modulators as therapies for pancreatic cancer. Biochim Biophys Acta. 2014;1840:332–343. doi: 10.1016/j.bbagen.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajeshkumar NV, Tan AC, De Oliveira E, Womack C, Wombwell H, Morgan S, et al. Antitumor effects and biomarkers of activity of AZD0530, a Src inhibitor, in pancreatic cancer. Clin Cancer Res. 2009;15:4138–4146. doi: 10.1158/1078-0432.CCR-08-3021. [DOI] [PubMed] [Google Scholar]
- Reiske HR, Zhao J, Han DC, Cooper LA, Guan JL. Analysis of FAK-associated signaling pathways in the regulation of cell cycle progression. FEBS Lett. 2000;486:275–280. doi: 10.1016/s0014-5793(00)02295-x. [DOI] [PubMed] [Google Scholar]
- Rountree CB, Ding W, He L, Stiles B. Expansion of CD133-expressing liver cancer stem cells in liver-specific phosphatase and tensin homolog deleted on chromosome 10-deleted mice. Stem Cells. 2009;27:290–299. doi: 10.1634/stemcells.2008-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen B, Saigal B, Parikh N, Gallick G, Johnson FM. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009;69:1958–1965. doi: 10.1158/0008-5472.CAN-08-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen TL, Guan JL. Differential regulation of cell migration and cell cycle progression by FAK complexes with Src, PI3K, Grb7 and Grb2 in focal contacts. FEBS Lett. 2001;499:176–181. doi: 10.1016/s0014-5793(01)02545-5. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–256. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- Soubani O, Ali AS, Logna F, Ali S, Philip PA, Sarkar FH. Re-expression of miR-200 by novel approaches regulates the expression of PTEN and MT1-MMP in pancreatic cancer. Carcinogenesis. 2012;33:1563–1571. doi: 10.1093/carcin/bgs189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- Tang H, Hao Q, Rutherford SA, Low B, Zhao ZJ. Inactivation of SRC family tyrosine kinases by reactive oxygen species in vivo. J Biol Chem. 2005;280:23918–23925. doi: 10.1074/jbc.M503498200. [DOI] [PubMed] [Google Scholar]
- Wang ML, Walsh R, Robinson KL, Burchard J, Bartz SR, Cleary M, et al. Gene expression signature of c-MYC-immortalized human fibroblasts reveals loss of growth inhibitory response to TGFbeta. Cell Cycle. 2011;10:2540–2548. doi: 10.4161/cc.10.15.16309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LH, Owens LV, Sturge GC, Yang X, Liu ET, Craven RJ, et al. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 1996;7:413–418. [PubMed] [Google Scholar]
- Yezhelyev MV, Koehl G, Guba M, Brabletz T, Jauch KW, Ryan A, et al. Inhibition of SRC tyrosine kinase as treatment for human pancreatic cancer growing orthotopically in nude mice. Clin Cancer Res. 2004;10:8028–8036. doi: 10.1158/1078-0432.CCR-04-0621. [DOI] [PubMed] [Google Scholar]
- Yokoi K, Hawke D, Oborn CJ, Jang JY, Nishioka Y, Fan D, et al. Identification and validation of SRC and phospho-SRC family proteins in circulating mononuclear cells as novel biomarkers for pancreatic cancer. Transl Oncol. 2011;4:83–91. doi: 10.1593/tlo.10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JH, Cheng HY, Yu ZQ, He DW, Pan Z, Yang DT. Resveratrol induces apoptosis in pancreatic cancer cells. Chin Med J (Engl) 2011;124:1695–1699. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Methods.
Figure S1 QD232 affects several critical cell-signalling pathways governing cell migration and invasion. MIA PaCa-2 cells were treated with QD232 (1 μM) for 24 h, lysed following Kinexus's protocol, and screened for 378 pan-specific (protein abundance) and 273 phospho-site-specific antibodies. Control and treated cell lysates were labelled with fluorescent dye and analysed separately on the same chip spotted with the array of antibodies. Quantitative analysis of the fluorescence intensity signal for each target protein was performed for each sample, and fold-change ratios of treated-to-control were reported. These data were further analysed using Ingenuity Pathway Analysis (IPA) software and the plot displaying top canonical pathways affected by QD232 treatment was generated. −log p on the left Y-axis represents the statistical significance and the pathways are arranged in the order of their statistical significance. Ratio represents the ratio of number of signalling proteins affected by the treatment to the total number of signalling proteins in that pathway reported in IPA.
Figure S2 QD232 causes rapid decrease in p-Src in MIA PaCa-2 pancreatic cancer cells. QD232 (5 μM) results in decreasing phosphorylation of p-Src (A) in a time-dependent manner without affecting the expression of total Src (B). Cells were fixed with 0.4% formaldehyde and stained using primary antibodies for p-Src and Src followed by incubation with respective secondary antibodies and then counter-stained with DAPI nuclear dye. Cells were imaged using BD Pathway 435 High-Content Bioimager (BD Biosciences, San Jose, CA, USA) using 10× objective. Representative images of three independent experiments are shown.
Figure S3 QD232 causes rapid change in morphology of cancer cells, but not of normal cells. (A) 5 μM QD232 resulted in loss of adhesion and rounding up of MIA PaCa-2 pancreatic cancer cells within 4 h of treatment. Cells were imaged live using Nikon microscope with 10× objective. (B) However, no change in morphology was observed in normal HFF-1 fibroblasts even at 10 μM treatment with QD232. Cells were imaged after fixing with methanol and staining with Giemsa using BD Pathway 435 High-Content Bioimager (BD Biosciences, San Jose, CA, USA) using 10× objective. Representative images of three independent experiments are shown.
Figure S4 QD232 inhibits cell migration in cancer cells. QD232 inhibited closure of wounds stimulated by 10% FBS in serum-starved BxPC-3 (A) and ASPC-1 (B) pancreatic cancer cells in a concentration-dependent manner after 24 h treatment. Cells were imaged after fixing with methanol and staining with Giemsa using BD Pathway 435 High-Content Bioimager (BD Biosciences, San Jose, CA, USA) using 4× objective. (C, D) Quantification of data shown in A and B, respectively. Representative images of three independent experiments are shown. Images were quantified using ImageJ software (http://rsb.info.nih.gov/).
Figure S5 QD232 does not cause any significant inhibition in activity of oncogenic kinases in vitro. QD232 (10 μM) does not significantly inhibit the activity of oncogenic kinases (including FAK and Src) in a kinase profiling in vitro substrate assay. Kinase Profiler™ assay was performed by EMD Millipore, Billerica, MA, USA. Concentration of ATP used in the assay was Km for each kinase. 10 μM ATP was used for kinases for which Km was not known.
Table S1 Quantification of data for Figure 1.
Table S2 Quantification of data for Figure 2.
Table S3 Quantification of data for Figure 4.
Table S4 Quantification of Data for Figure 6.