

Abstract
BACKGROUND AND PURPOSE
Two peptide agonists of the glucagon-like peptide-1 (GLP-1) receptor, exenatide and GLP-1 itself, exert anti-hypersensitive effects in neuropathic, cancer and diabetic pain. In this study, we have assessed the anti-allodynic and anti-hyperalgesic effects of the non-peptide agonist WB4-24 in inflammatory nociception and the possible involvement of microglial β-endorphin and pro-inflammatory cytokines.
EXPERIMENTAL APPROACH
We used rat models of inflammatory nociception induced by formalin, carrageenan or complete Freund's adjuvant (CFA), to test mechanical allodynia and thermal hyperalgesia. Expression of β-endorphin and pro-inflammatory cytokines was measured using real-time quantitative PCR and fluorescent immunoassays.
KEY RESULTS
WB4-24 displaced the specific binding of exendin (9–39) in microglia. Single intrathecal injection of WB4-24 (0.3, 1, 3, 10, 30 and 100 μg) exerted dose-dependent, specific, anti-hypersensitive effects in acute and chronic inflammatory nociception induced by formalin, carrageenan and CFA, with a maximal inhibition of 60–80%. Spinal WB4-24 was not effective in altering nociceptive pain. Subcutaneous injection of WB4-24 was also antinociceptive in CFA-treated rats. WB4-24 evoked β-endorphin release but did not inhibit expression of pro-inflammatory cytokines in either the spinal cord of CFA-treated rats or cultured microglia stimulated by LPS. WB4-24 anti-allodynia was prevented by a microglial inhibitor, β-endorphin antiserum and a μ-opioid receptor antagonist.
CONCLUSIONS AND IMPLICATIONS
Our results suggest that WB4-24 inhibits inflammatory nociception by releasing analgesic β-endorphin rather than inhibiting the expression of proalgesic pro-inflammatory cytokines in spinal microglia, and that the spinal GLP-1 receptor is a potential target molecule for the treatment of pain hypersensitivity including inflammatory nociception.
Tables of Links
| TARGETS |
|---|
| GPCRs |
| GLP-1 receptor |
| δ opioid receptor |
| κ opioid receptor |
| μ opioid receptor |
| LIGANDS | |
|---|---|
| CTAP | Naltrindole |
| Exendin (9-39) | Nor-binaltorphimine |
| GLP-1 | TNF-α |
| IL-1β | |
| IL-6 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
The activation of the glucagon-like peptide-1 (GLP-1) receptor by the endogenous incretin GLP-1 and its exogenous mimetic exenatide leads to a wide range of biological actions in the pancreas, such as the stimulation of glucose-dependent insulin synthesis and secretion, the reduction of glucagon levels and marked changes in β-cell proliferation and apoptosis (Drucker, 2007; Holst, 2007; Kim and Egan, 2008). Exenatide and GLP-1 have therapeutic value in type-2 diabetes mellitus (DeFronzo et al., 2005; Triplitt and Chiquette, 2006). We recently found that the intrathecal administration of two peptide GLP-1 receptor agonists, exenatide and GLP-1(7–36), produced specific and potent antinociception, by 60–90% in conditions of formalin-induced tonic hyperalgesia, mechanical allodynia induced by peripheral nerve injury and bone cancer, and painful diabetic neuropathy. The anti-hypersensitive effects of exenatide and GLP-1 were found to be completely prevented by GLP-1 receptor antagonists and GLP-1 receptor gene knockdown. Our results suggested that the activation of spinal GLP-1 receptors leads to specific antinociception in persistent pain states, notably in refractory neuropathic pain, cancer pain and painful diabetic neuropathy (Gong et al., 2014b). However, the effects of GLP-1 receptor activation on inflammatory nociception have not been examined using acute and chronic inflammatory models, such as that induced by carrageenan and complete Freund's adjuvant (CFA).
GLP-1 receptors are specifically expressed in spinal dorsal horn microglial cells and their up-regulation accompanies microglial proliferation and hypertrophy following peripheral nerve injury (Gong et al., 2014b). Spinal microglia, which play a crucial role in the initiation and development of chronic pain, including inflammatory pain (Tsuda et al., 2003; Raghavendra et al., 2004; Hua et al., 2005; Taves et al., 2013), are converted from their resting shape to an activated shape after peripheral inflammation or injury (Svensson et al., 2003; Hua et al., 2005). Activated microglia produce and release numerous neurotrophins and pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6 (Chauvet et al., 2001; Taves et al., 2013). The cytokines released by activated microglia can induce the central sensitization of neurons in the spinal dorsal horn by altering excitatory or inhibitory synaptic transmission, leading to hyperalgesia (Kawasaki et al., 2008; Zhang et al., 2011). In contrast, microglial activation is also involved in the production and release of analgesic endorphins and activation of their corresponding receptors (McCarthy et al., 2001; Pocock and Kettenmann, 2007). For instance, β-endorphin was synthesized and released by cultured microglia in response to corticotropin-releasing hormone (Sacerdote et al., 1993).
The findings of GLP-1 receptor activation on cytokine release are controversial. GLP-1 induced morphological changes in microglia and inhibited LPS-induced IL-1β, IL-6 and inducible NOS production in cultured astrocytes (Iwai et al., 2006). Exenatide also suppressed 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced expression of TNF-α and IL-1β (Kim et al., 2009). In contrast, exenatide increased the expression of IL-6 and IL-1β in the hypothalamus and the hindbrain (Shirazi et al., 2013). On the other hand, we showed that exenatide stimulated β-endorphin release from cultured microglia and the spinal cords of the neuropathic rats, and that the stimulatory effect was prevented by minocycline, a specific inhibitor of microglial activation (Gong et al., 2014b). Therefore, it would provide new knowledge to study the involvement of pro-inflammatory cytokines and analgesic endorphins, derived from microglia, in the antinociceptive effects of GLP-1 receptor agonists in models of inflammatory nociception.
The effects of GLP-1 receptor agonists with different molecular structures may help to confirm the hypothesis that the activation of GLP-1 receptors leads to antinociception in pain hypersensitivity, as this is a useful means of excluding structure-related actions that may not be relevant to the GLP-1 receptor activity. WB4-24, a dimer consisting of a cyclobutane core and two pairs of symmetrical side chains, is probably the most active non-peptide GLP-1 receptor orthosteric agonist with activity in the micromolar range (Liu et al., 2012). In this paper, we investigated the antinociceptive effects of WB4-24 in rat models of acute and chronic inflammatory nociception induced by formalin, carrageenan and CFA, and explored the underlying mechanisms of action and particularly the involvement of microglial β-endorphin release and cytokine expression.
Methods
Animals
All animal care and experimental procedures complied with the guidelines of the Animal Care and Welfare Committee of the Shanghai Jiao Tong University School of Pharmacy. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 248 animals were used in the experiments described here.
Male Wistar 1-day-old neonatal and adult rats (180–250 g) were obtained from the Shanghai Experimental Animal Institute for Biological Sciences (Shanghai, China). The animals were housed in a temperature- and humidity-controlled environment on a 12 h light/dark cycle (lights on at 07:00 h) with food and water ad libitum. They were acclimated to the laboratory environment for 3–5 days before entering the study. Experimental study groups were assigned randomly and the nociceptive responses were recorded without knowledge of the treatments.
Cell culture
The cortices of 1-day-old neonatal Wistar rats were collected and the cell suspension was plated in 75 cm2 tissue culture flasks (1 × 107 cells per flask) pre-coated with poly-L-lysine and incubated at 37°C in a humidified atmosphere of 95% O2 and 5% CO2. The dissociated cells were suspended in DMEM medium Gibco, BRL, Paisley, UK) supplemented with 10% FCS, penicillin (100 U·mL−1) and streptomycin (100 μg·mL−1). Fresh medium was provided every other day and 8 days later, microglial cells were prepared as floating cell suspensions by shaking the flasks at 260 rpm for 2 h. The aliquots were transferred to plates and unattached cells were removed by washing with serum-free DMEM. The purity of the harvested microglia was greater than 95%, as determined by measuring CD11b (OX42) immunoreactivity.
HEK293 and PC12 cells, which were purchased from the Shanghai Institute for Cell Biology Bank (Shanghai, China), were cultivated at 37°C in a 5% CO2/95% O2 humidified environment. The HEK293 cells were cultivated in DMEM supplemented with 10% FBS, 2.0 mM L-glutamine and hygromycin B (100 μg·mL−1). The PC12 cells were cultivated in RPMI 1640 medium supplemented with 10% FBS.
Ligand-binding assay
The binding assays were performed as described by Devaraj et al., (2005) and He et al., (2013). For the saturation-binding assay, exendin(9–39)-FITC was applied at concentrations from 1 to 300 nM. Total and non-specific binding were measured in the absence and presence of unlabelled GLP-1 in excess (100 μM). The cells were equilibrated for 1 h on ice then analysed for fluorescent intensity using a FACScan® flowcytometer (Becton Dickinson, Franklin Lakes, NJ, USA). The dissociation constant (Kd) was calculated for the specific binding using the following one-site binding hyperbola non-linear regression analysis equation: Bound = Bmax × [L]/([L] + Kd), where Bmax is the mean fluorescence intensity when the labelled ligand is fully bound to the binding sites, Kd is the concentration of the labelled ligand exendin(9–39)-FITC required to reach half-maximal binding and [L] is the concentration of the labelled ligand.
Hydrogen peroxide-induced oxidative damage
Cultured cells were grown on 96-well plates at a density of 1.6 × 104 cells per well. Twenty-four hours after incubation and the cells were attached; hydrogen peroxide (500 or 800 μM, final concentration) was added to PC12 cells for 15 min and to HEK293 cells for 5 min. The cells were washed, treated with WB4-24 or exenatide at different concentrations and cultured again for 12 h. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT, Amresco, Solon, OH, USA) assay was conducted to determine cell viability. The cells in the 96-well plates were rinsed with PBS and 0.5 mg·mL−1 MTT was added to each well. The microplate was incubated at 37°C for 4 h. The medium containing MTT was removed and 200 μL DMSO was added to each well. The plate was shaken on a microplate shaker to dissolve the blue MTT-formazan. The absorbance was read at 570 nm against a reference wavelength of 630 nm on a microplate reader (Multiskan MK3, Thermo Labsystems, Vantaa, Finland). The cell viability after exposure to hydrogen peroxide was expressed as a percentage of normal cell viability in the absence of hydrogen peroxide.
RNA extraction, reverse transcription and real-time quantitative PCR
TRIzol reagent (Invitrogen, Grand Island, NY, USA) was used to isolate the total RNA from rat spinal lumbar enlargements (L3–L5) and cultured microglia (Zhang et al., 2013). A sample of 1 μg of total RNA was reverse transcribed using a ReverTra Ace qPCR RT-Kit (Toyobo Co., Ltd., Osaka, Japan). The RNA extraction and real-time PCR procedures were conducted as described by Zhang et al., (2013). The 2−ΔΔCt method was used to calculate the fold change after normalization to GAPDH (Zhao et al., 2010). The primers were as follows: 5′-CCC TCC TGC TTC AGA CCT CCA-3′ [proopiomelanocortin (POMC) exon 2–3 forward], 5′-TCT CTT CCT CCG CAC GCC TCT-3′ (POMC exons 2–3 reverse) (Sitte et al., 2007; Busch-Dienstfertig et al., 2012); 5′-CCA AGG TCA TCC ATG ACA AC-3′ (GAPDH forward), 5′-TCC ACA GTC TTC TGA GTG GC-3′ (GAPDH reverse); 5′-CCC CGA CTA TGT GCT CCT CAC-3′ (TNF-α forward), 5′-AGG GCT CTT GAT GGC GGA-3′ (TNF-α reverse); 5′-GGA AGG CAG TGT CAC TCA TTG TG-3′ (IL-1β forward), 5′-GGT CCT CAT CCT GGA AGC TCC-3′ (IL-1β reverse); 5′-GGG ACT GAT GTT GTT GAC AGC C-3′ (IL-6 forward), 5′-CAT ATG TAA TTA AGC CTC CGA CTT GTG-3′ (IL-6 reverse) (Raghavendra et al., 2004).
β-endorphin measurement
The β-endorphin levels in the spinal cord (L3–L5) homogenates and the culture medium from primary microglial cells were determined using an enzyme-linked fluorescent immunoassay kit (Phoenix Pharmaceuticals, Burlingame, CA, USA). The assay was validated by the manufacturer for determining peptides within a linear range of 29.6–608 pg·mL−1. The cross-activity of the assay included α-endorphin (100%) and γ-endorphin (60%), but not met-enkephalin (0%) or leu-enkephalin (0%). The relative fluorescence units were read with a Fluorescence Microplate Reader (Thermo Labsystems) and the β-endorphin concentrations were determined at the same time by comparison with a calibration curve run.
Intrathecal catheterization and injection in rats
An 18 cm polyethylene catheter (PE-10: 0.28 mm i.d. and 0.61 mm o.d., Clay Adams, Parsippany, NJ, USA) with a volume of 13 μL was inserted into the lumbar level of the rat spinal cord as described elsewhere (Gong et al., 2011; Huang et al., 2012). Inhaled isoflurane anaesthesia (4% for induction and 1% for maintenance) controlled by an anaesthesia meter (Ugo Basile Gas Anesthesia System, Comerio, Italy) was administered. The placement of the catheter in the spinal cord was confirmed by administering 4% lidocaine (10 μL followed by 10 μL of normal saline for flushing) with a 50 μL micro-injector (Shanghai Anting Micro-Injector Factory, Shanghai, China) after recovery from anaesthesia. The lidocaine test was performed 5–7 days before the drug-testing sessions began. Only rats that exhibited no motor impairment before the intrathecal administration of lidocaine, but experienced bilateral paralysis of their hindlimbs after the lidocaine injection were selected for the study. The exclusion rate in our laboratory was zero. To administer the control and test articles intrathecally, 10 μL of each of the drugs was injected with a 50 μL micro-injector, followed by a normal saline flush with a volume of 10 μL.
Rat formalin test
The rats were acclimated individually to the observation cage for 30 min before testing. The formalin test was performed as previously described, but with minor modifications (Gong et al., 2011; 2014b,): 50 μL 5% formalin in 0.9% saline was injected s.c. into the dorsal side of the left hindpaw and the rat was then immediately placed in a 23 × 35 × 19 cm transparent polycarbonate box. Nociceptive behaviour was manually quantified by counting the number of times that the formalin-injected paw flinched in 1 min epochs. The measurements were taken at 10 min intervals beginning immediately after the formalin injection and ending 90 min later.
Rat carrageenan model of acute inflammatory nociception
Through s.c. intraplantar surface injection to the right hindpaws, 100 μL of 2% carrageenan in normal saline was administered to the rats. Thermal hyperalgesia and mechanical allodynia in both the carrageenan-injured and contralateral hindpaws were assessed at several time points before and after the control- and test-article administration (Gong et al., 2011).
Rat model of CFA-induced acute and chronic inflammatory nociception
CFA inflammatory nociception was induced using the method described by Butler et al. 1992), with adaptations. Briefly, 30 μL of a combination of IFA and heat-inactivated Mycobacterium tuberculosis (10 mg·mL−1) was injected into the tibiotarsal joint of the left hindpaw of each rat under inhaled isoflurane anaesthesia. The tibiotarsal joint injection of CFA produced immediate and long-lasting inflammation and pain hypersensitivity. The early phase (1–2 days after CFA injection) represents acute inflammatory pain, and the later phase (2–3 weeks after CFA injection) represents chronic rheumatoid arthritis disease.
Behavioural assessment of heat hyperalgesia and mechanical allodynia
The nociceptive reflex responses to thermal stimuli were assessed by placing the injured and contralateral hindpaw above a radiant heat source (at a low intensity of 45) and measuring the paw withdrawal latency in response to a noxious heat stimulus, using a 390 G Plantar Test (IITC Life Science Instruments, Woodland Hills, CA, USA). To prevent tissue damage, the latency cut-off was set at 30 s. The paw withdrawal latency was evaluated for no less than 30 min at each of several time points before and after control- and test-article administration. The result of each test was calculated as a mean of three repeated measurements.
To assess mechanical allodynia, the hindlimb withdrawal threshold evoked by stimulation of the ipsilateral injured and contralateral hindpaw with a 2391 electrical von Frey hair (IITC Life Science Instruments) was determined while the rat stood on a metal grid. The monofilament, which produced forces ranging from 0.1 to 65.0 G, was applied to the footpad with increasing force until the rat suddenly withdrew its hind limb. The lowest force producing a withdrawal response was considered the threshold. Triplicate measurements were made at intervals of approximately 30 s and the mean of these threshold values for each hindpaw at each time point was averaged.
Data analysis
The maximal possible effect (MPE) was calculated as follows: (post-drug threshold in ipsilateral hind limb-baseline threshold in ipsilateral hind limb)/(baseline threshold in contralateral hind limb-baseline threshold in ipsilateral hind limb) × 100 (Chaplan et al., 1994). The dose–response analysis, Scatchard analysis and Schild analysis were performed as described in Gong et al. (2012) and Zhang et al. (2013). The data were expressed as means ± SEM or with 95% confidence limits, and there were no missing data. The statistical significance was evaluated by one-way or two-way repeated-measures anova in Prism (version 5.01, GraphPad Software, Inc., San Diego, CA, USA). Post hoc Student–Newman–Keuls test was conducted when the effect of the drug (dose) [for the one-way anova, the factor was drug (dose); for the two-way anova, the factors were drug (dose), time and their interaction] was observed to be statistically significant. The probability values were two-tailed and the P value meeting the statistical significance criterion was 0.05.
Materials
WB4-24 was obtained from the National Center for Drug Screening (Shanghai, China) and dissolved in 1% DMSO and 19% PEG400 in saline. Heat-inactivated Mycobacterium tuberculosis was purchased from the Chengdu Institute of Biological Products (Chengdu, China). Minocycline and naltrindole were purchased from Sinopharm Group Chemical Reagent Co. (Shanghai, China) and Tocris Bioscience (Bristol, UK). CTAP, norbinaltorphimine (nor-BNI) and the rabbit polyclonal antibody neutralizing β-endorphin or dynorphin A were bought from Abcam (Cambridge, UK). Carrageenan, incomplete Freund's adjuvant (IFA) and LPS (Escherichia coli strain O26:B6) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Exendin(9–39)-FITC and GLP-1-FITC were commercially synthesized, conjugated and purified (≥90%) by China Peptides Co. (Shanghai, China).
Results
WB4-24 exerts GLP-1 receptor agonist effects in microglia
We previously showed that GLP-1 receptors were specifically expressed on microglia, but not on astrocytes or neurons (Gong et al., 2014b). Here, we used a binding assay to confirm the specific binding of GLP-1 receptors in primary microglia, in comparison with HEK293 cells that stably express human GLP-1 receptors and PC12 cells that express rat GLP-1 receptors. Exendin(9–39), a competitive antagonist that binds the GLP-1 receptor N-terminal ectodomain, but lacks the amino acids needed for interaction with the extracellular loop regions to induce intracellular signalling (Thorens et al., 1993; Lopez de Maturana et al., 2003), was used to probe GLP-1 receptors. Exendin(9–39) was then added in FITC form (from 1 to 300 nM) to the primary microglia. An equilibrium-saturation analysis revealed a single saturable site with a high affinity specifically for exendin(9–39)-FITC, with a Bmax value of 66.0 (median fluorescent intensity) and a Kd value of 54.0 nM (Figure 1A). The binding-saturation kinetics was also demonstrated in the HEK293 cells and PC12 cells, with Bmax values of 90.2 and 96.6 (median fluorescent intensity) and Kd values of 17.9 and 53.7 nM respectively (Figure 1B and C).
Figure 1.

Saturation kinetics of the binding of exendin(9–39)-FITC in rat microglial cells (A), HEK293 cells that express human GLP-1 receptor (B), and rat PC12 cells (C). The specific binding of exendin(9–39)-FITC was obtained by subtracting non-specific binding from total binding; the former was determined in the presence of 100 μM GLP-1. The insert is a Scatchard plot with the calculated Kd and Bmax values. The data are presented as means ± SEM (n = 3 in each treatment) from two independent studies.
Receptor-binding and cyclic AMP response element reporter gene assays have previously been used to identify WB4-24 as a novel cyclobutane class of non-peptide GLP-1 receptor agonists (Liu et al., 2012). To further confirm that the competitive-displacement mode of WB4-24 occurred at the same GLP-1 receptor binding site as GLP-1, a range of concentrations of WB4-24 were applied in a displacement-binding assay to both HEK293 and PC12 cells. In the HEK293 cells, co-treatment with WB4-24 (30, 100 and 300 μM) concentration-dependently shifted the binding curve of GLP-1-FITC to the right, without affecting maximum binding. A Schild analysis indicated that the slope was −0.91 and the pA2 was 4.92 (Figure 2A). WB4-24 manifested the same competitive-binding mode in the PC12 cells, with very similar values for the slope (−0.91) and pA2 (4.87) (Figure 2B). Co-treatment with GLP-1 (0.3, 1.0 and 3.0 μM) in both the HEK293 and the PC12 cells also concentration-dependently shifted the binding curve of GLP-1-FITC to the right without affecting maximum binding. Schild plots of GLP-1 identified slope values of −1.23 and −1.26 and pA2 values of 6.74 and 6.59 for the HEK293 and PC12 cells (Figure 2C and D) respectively.
Figure 2.

Competitive displacement by WB4-24 (A and B) and GLP-1 (C and D) of the GLP-1-FITC binding in HEK293 and PC12 cells. GLP-1-FITC at different concentrations was co-treated with WB4-24 (30, 100 and 300 μM) or GLP-1 (0.3, 1 and 3 μM). The insert is a Scatchard plot with the calculated slope and pA2 values. The concentration response was analysed using the non-linear least squares method. The data are presented as means ± SEM (n = 3 in each treatment) from two independent studies.
The potency of WB4-24 compared with that of GLP-1 in binding to GLP-1 receptors was tested further in primary microglial cells, HEK293 and PC12 cells using a displacement-binding assay. WB4-24 (30–3000 nM) or GLP-1 (0.3–30 nM) was added to compete with exendin(9–39)-FITC binding (30 nM, close to the Kd values) in microglial, HEK293 and PC12 cells. As shown in Figure 3, both GLP-1 and WB4-24 displaced the exendin(9–39)-FITC binding in a concentration-dependent manner. The maximum displacement was 100% and the IC50 values for GLP-1 and WB4-24 were 3.2 and 560.0 nM in the primary microglial cells (Figure 3A), 2.5 and 300 nM in the HEK293 cells (Figure 3B), and 1.7 and 260.0 nM in the PC12 cells (Figure 3C) respectively. Our data showed that WB4-24, although approximately 100-fold less potent than GLP-1, was a reversible and orthosteric agonist of microglial GLP-1 receptor with full intrinsic activity, and acted at the same recognition site as GLP-1. Neither WB4-24 nor GLP-1 showed cell specificity when binding GLP-1 receptors and neither was affected by the species differences between humans and rats.
Figure 3.

Concentration–displacement response curves of WB4-24 and GLP-1 in the binding of exendin(9–39)-FITC in microglial cells (A), HEK293 cells (B) and PC12 cells (C). Exendin(9–39)-FITC (30 nM) was added with increasing concentrations of WB4-24 or GLP-1. The concentration response was analysed using the non-linear least squares method. The data are presented as means ± SEM (n = 3 in each treatment) from two independent studies.
The relative potency of WB4-24 and exenatide in activating GLP-1 receptors was tested further in a functional assay – hydrogen peroxide oxidative damage assessment. The activation of GLP-1 receptors is known to protect against hydrogen peroxide-induced oxidative damage (Liu et al., 2007; Oeseburg et al., 2010). Neither exenatide (up to 3 × 10−8 M) nor WB4-24 (up to 10−5 M) treatments were associated with cellular dysfunction in HEK293 cells that stably expressed human GLP-1 receptors. Both WB4-24 (EC50 = 0.6 μM) and exenatide (EC50 = 2.6 nM) provided concentration-dependent and complete protection against hydrogen peroxide-induced oxidative damage (Figure 4A). The effects of WB4-24 and exenatide were examined further in PC12 cells. WB4-24 (EC50 = 1.2 μM) and exenatide (EC50 = 5.0 nM) conferred the same concentration-dependent protection against hydrogen peroxide-induced oxidative damage (Figure 4B).
Figure 4.
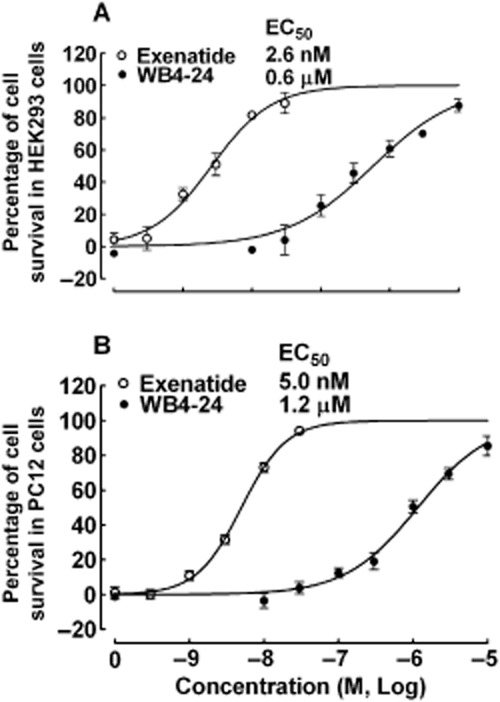
Protective effects of WB4-24 and exenatide against hydrogen peroxide-induced oxidative damage in HEK293 cells that stably express human GLP-1 receptor (A) and PC12 cells that express rat GLP-1 receptor (B). The cells were incubated in 96-well plates for 24 h, then treated with 500 or 800 μM hydrogen peroxide for 15 min (PC12 cells) or 5 min (HEK293 cells). The cells were treated with WB4-24 and exenatide for 12 h before the MTT assay. The data are expressed as means ± SEM (n = 3 in each treatment) from two to three independent studies.
Administered intrathecally, WB4-24 blocks inflammatory nociception
The spinal antinociceptive effects of WB4-24 were first examined with the formalin test. Six groups of rats (n = 6 per group) received a single intrathecal injection of the vehicle (10 μL) or WB4-24 (0.3, 1, 3, 10, or 30 μg) 30 min before a formalin injection. As shown in Figure 5A, the s.c. injection of formalin produced in the control rats a biphasic flinching response consisting of an acute phase (within 10 min) and a tonic phase (10–90 min). In the acute phase, the intrathecal injection of WB4-24 up to 30 μg did not significantly prevent the formalin-induced flinching response. However, WB4-24 blocked the tonic phase of the formalin-induced flinching response in a dose-dependent manner. The areas under the flinching–response curves from 10 to 90 min (AUC) were summed and the inhibitory percentage was calculated from the mean AUC value of vehicle-treated rats. An analysis of the dose response identified the ED50 as 2.0 μg (95% confidence limits: 1.3–2.8 μg) and the Emax as 84.7% (Figure 5B).
Figure 5.
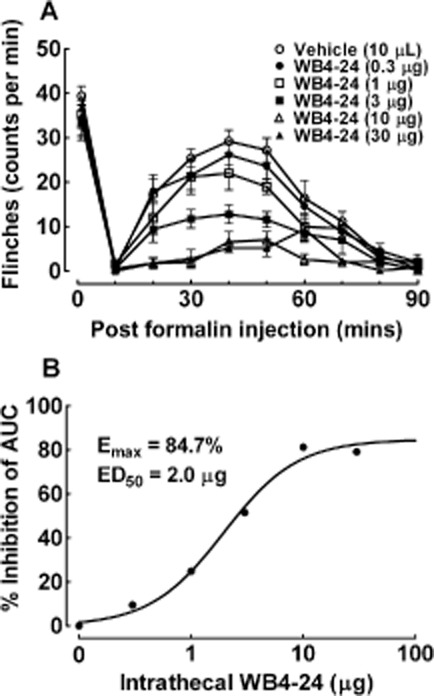
Antinociceptive effects of intrathecal injection of WB4-24 in the rat formalin test (A), Six groups of rats received intrathecal injections of either the vehicle (1% DMSO and 19% PEG400 in saline, 10 μL) or WB4-24 (0.3, 1, 3, 10, or 30 μg) 30 min before paw injections of 50 μL 5% formalin. Nociceptive behaviour was quantified by counting the number of flinches in the formalin-injected paws in 1 min epochs (B). The data are presented as means ± SEM (n = 6 per group). Dose–response analysis of WB4-24 on formalin-induced tonic flinching response.
Next, we examined the spinal antinociceptive effects of WB4-24 in the carrageenan model. Five groups of rats (n = 6 per group) first received intraplantar injections of carrageenan and 2.5 h later received intrathecal injections of the vehicle (10 μL) or WB4-24 (3, 10, 30 or 100 μg). Withdrawal responses to thermal and mechanical stimuli were measured in both their contralateral and ipsilateral hindpaws before carrageenan injection and at 0, 0.5, 1, 2 and 4 h after test article injection, which began 2.5 h after carrageenan injection. The intraplantar injection of carrageenan produced marked inflammation, as observed as swelling and redness, thermal hyperalgesia and mechanical allodynia (Figure 6A and B). The intrathecal injection of WB4-24 (up to 100 μg) did not significantly alter paw withdrawal latency in response to heat stimulation in the contralateral paws, but significantly blocked thermal hyperalgesia in the ipsilateral paws in a time- and dose-dependent manner (Figure 6A). The percentage of MPE of the WB4-24 antinociception from each rat was calculated at 1 h after injection and the inhibitory percentage was calculated from the mean MPE value of vehicle-treated rats. A dose–response analysis identified the Emax as 57.5% MPE and the ED50 as 15.3 μg (95% confidence limits: 12.8–18.3 μg) (Figure 6C). Furthermore, although WB4-24 did not significantly affect the thresholds of paw withdrawal in response to mechanical stimuli in the contralateral paws, it dose-dependently blocked carrageenan-induced mechanical allodynia in the ipsilateral paws (Figure 6B), with an Emax of 55.0% MPE and an ED50 of 21.7 μg (95% confidence limits: 14.5–32.5 μg) (Figure 6C).
Figure 6.

Antinociceptive effects of intrathecal injection of WB4-24 on thermal hyperalgesia and mechanical allodynia induced by carrageenan (A–C) and CFA (D–F for 1 day after injection, and G–I for 3 weeks after injection). In the carrageenan model, paw withdrawal responses were measured in both the contralateral and ipsilateral paws before and after an intraplantar injection of 100 μL 2% carrageenan. In the CFA model, paw withdrawal responses were measured in both the contralateral and ipsilateral paws 1 day and 3 weeks after a tibiotarsal joint CFA (30 μL) injection. The data are presented as means ± SEM (n = 6 per group). Dose–response analyses of WB4-24 on carrageenan- (C), CFA-induced acute (F, one day after injection) and chronic (I, 3 weeks after injection) induced thermal hyperalgesia and mechanical allodynia.
We measured the spinal antinociceptive effects of intrathecal WB4-24 in conditions of CFA-induced acute and chronic inflammatory nociception. Six groups of rats (n = 6 per group) received an intrathecal injection of the vehicle (10 μL) or WB4-24 (1, 3, 10, 30 or 100 μg) 1 day after CFA injection. The withdrawal responses to thermal and mechanical stimuli were measured in both the contralateral and ipsilateral hindpaws before and 0.5, 1, 2 and 4 h after test-article injection. The intrathecal injection of WB4-24 (up to 100 μg) did not significantly affect the paw withdrawal latency in response to heat stimulation or mechanical thresholds in the contralateral paws. However, WB4-24 significantly blocked thermal hyperalgesia and mechanical allodynia in the ipsilateral paws in a time- and dose-dependent manner (Figure 6D and E). The percentage of MPE of the WB4-24 antinociception from each rat was calculated at 1 h after injection and the inhibitory percentage was calculated from the mean MPE value of vehicle-treated rats. A dose–response analysis at 1 h after injection yielded Emax values of 85.7% and 85.0% MPE, and the ED50s values of 14.9 μg (95% confidence limits: 11.7–18.9 μg) and 23.1 μg (95% confidence limits: 11.0–48.3 μg) (Figure 6F).
To test antinociceptive effect of WB4-24 in CFA-induced chronic inflammatory nociception, the same six groups of rats received another intrathecal injection of the vehicle (10 μL) or WB4-24 (1, 3, 10, 30 or 100 μg) 3 weeks later. Again, the paw withdrawal responses to thermal and mechanical stimuli were measured before test-article injection and at 0.5, 1, 2 and 4 h after injection. The intrathecal injection of WB4-24 up to 100 μg did not significantly influence the paw withdrawal latency in response to heat stimulation or mechanical thresholds in the contralateral paws. However, WB4-24 significantly blocked thermal hyperalgesia or mechanical allodynia in the ipsilateral paws in a time- and dose-dependent manner (Figure 6G and H). The percentage of MPE of the WB4-24 antinociception from each rat was calculated at 1 h after injection and the inhibitory percentage was calculated from the mean MPE value of vehicle-treated rats. A dose–response analysis at 1 h after injection identified the Emaxs as 73.0% and 77.5% MPE, and the ED50s as 11.0 μg (95% confidence limits: 5.7–21.4 μg) and 13.6 μg (95% confidence limits: 6.0–31.0 μg) (Figure 6I).
Injected s.c., WB4-24 relieves CFA-induced acute inflammatory nociception
We determined the systemic antinociceptive effect of WB4-24 in CFA-induced acute inflammatory nociception following s.c. administration. One day after CFA injection, two groups of CFA-treated rats (n = 4 per group) received s.c. injections of the vehicle (5 mL·kg−1) and WB4-24 (100 mg·kg−1) respectively. The paw withdrawal responses to thermal and mechanical stimuli were measured before and 0.5, 1, 2 and 4 h after test-article injection. The s.c. injection of WB4-24, compared with that of the control article, did not significantly alter the paw withdrawal response in the contralateral paws during the 4 h observation period. However, this regimen significantly blocked thermal hyperalgesia and mechanical allodynia in the ipsilateral paws (P < 0.05 by anova followed by post hoc Student–Newman–Keuls test) (Figure 7A and B). At 1 h after injection, the MPE values were 52.6 and 48.6% for thermal hyperalgesia and mechanical allodynia respectively.
Figure 7.
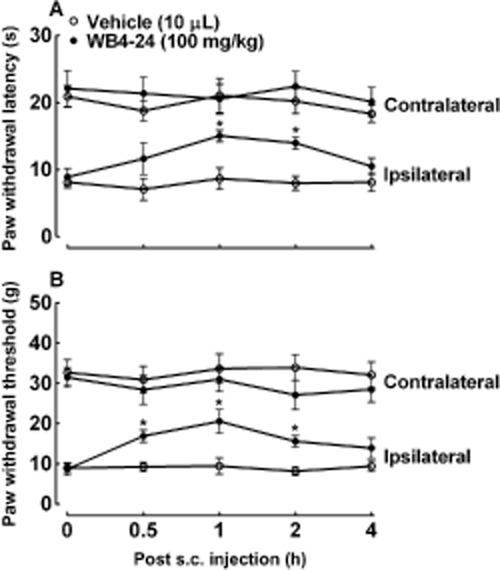
Antinociceptive effects of s.c. injection of WB4-24 during CFA-induced acute inflammatory nociception (A and B). The rats received an s.c. injection of the vehicle (5 mL·kg−1) or WB4-24 (100 mg·kg−1) 1 day after CFA injection. Paw withdrawal responses to thermal and mechanical stimuli were measured before test-article injection and at 0.5, 1, 2 and 4 h after injection. The data are presented as means ± SEM (n = 4 per group). *P < 0.05, significantly different from control vehicles; anova with post hoc Student–Newman–Keuls test).
WB4-24 stimulates microglial cells to release β-endorphin rather than inhibiting pro-inflammatory cytokine expression
We then determined the effects of WB4-24 on β-endorphin release or the expression of pro-inflammatory cytokines in microglial cells in the presence and absence of LPS. Primary microglial cells (1 × 105 cells per well) were treated with minocycline (60 μM) 1 h prior to LPS (3 ng·mL−1) and WB4-24 (1 μM) treatment, and the culture medium and microglial cells were collected 6 h later. As shown in Figure 8A, LPS (3 ng·mL−1) did not significantly change the baseline expression of the mRNA for the β-endorphin precursor POMC, relative to GAPDH in microglia, measured using real-time quantitative PCR. WB4-24 significantly increased the POMC expression by 2.8- and 1.5-fold in the presence and absence of LPS, respectively (P < 0.05 by anova followed by post hoc Student–Newman–Keuls test). Likewise, minocycline did not significantly alter the baseline level of POMC expression, but completely prevented any WB4-24-induced increase in POMC expression (P < 0.05). An elisa assay was used to confirm this observation by measuring β-endorphin protein level in the culture medium. WB4-24 significantly increased β-endorphin release from microglia with and without LPS stimulation, producing 2.9- and 1.7-fold increases, respectively, but did not affect the baseline β-endorphin secretion. Minocycline completely suppressed the stimulatory effect of WB4-24 in the presence and the absence of LPS stimulation, without altering the baseline β-endorphin release (P < 0.05) (Figure 8B).
Figure 8.
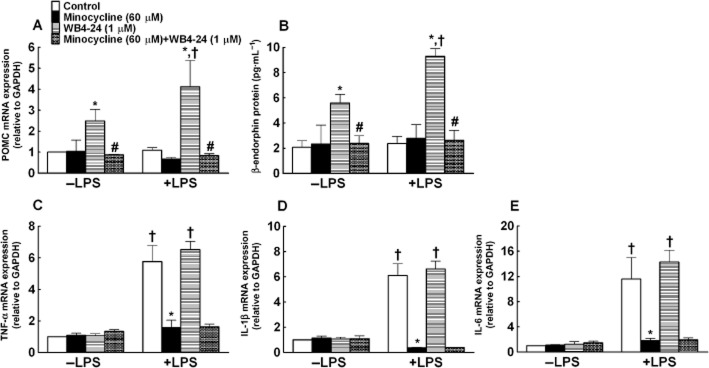
Effects of WB4-24 on β-endorphin release (A and B) and pro-inflammatory cytokine expression (C–E) in primary microglial cells in the presence and absence of LPS. The microglial cells were derived from the cortices of neonatal rats. Minocycline (60 μM) was treated 1 h before LPS (3 ng·mL−1) and WB4-24 (1 μM) treatment. The culture medium and microglial cells were collected 6 h later. The expression of POMC and pro-inflammatory cytokines was determined by real-time quantitative PCR and the β-endorphin level in the culture medium was measured using a specific fluorescent immunoassay kit. The data are presented as means ± SEM (n = 3 in each treatment) from three independent studies. *P < 0.05, significantly different from control; #P < 0.05, significantly different from WB4-24; one-way anova with post hoc Student–Newman–Keuls test). †P < 0.05, significantly different from control (absence of LPS); one-way anova with post hoc Student–Newman–Keuls test).
In contrast, LPS, as expected, produced a marked (5–11-fold) increase in the expression of TNF-α, IL-1β and IL-6 mRNA, relative to GAPDH, measured using the real-time quantitative PCR in the primary microglial cells, which was completely blocked by minocycline (P < 0.05). WB4-24 did not significantly inhibit the expression of TNF-α, IL-1β or IL-6 mRNA in the presence or absence of LPS stimulation (Figure 8C–E).
The effects of WB4-24 on β-endorphin release and the expression of cytokines were validated in vivo. One day after the vehicle or CFA injection, each of the two groups of rats (n = 6 per group) received a single intrathecal injection of the vehicle (10 μL) or WB4-24 (30 μg). They were killed 1 h after the intrathecal injection and ipsilateral spinal cord homogenates were obtained to measure the expression of POMC mRNA. As shown in Figure 9A, there were no significant differences in the POMC mRNA expression between the naive and CFA-treated rats. The intrathecal injection of WB4-24 increased the POMC expression by 1.2-fold in the naive rats, and its stimulatory effect was significantly (2.1-fold) greater in the CFA-treated rats (P < 0.05). It is noteworthy that the stimulatory effect of WB4-24 was also confirmed by measuring the β-endorphin protein levels. Intrathecal injection of WB4-24 significantly increased the amount of β-endorphin in the ipsilateral spinal cord, with a 0.4-fold increase and a 1.3-fold increase in the naive and CFA-treated rats, respectively (P < 0.05) (Figure 9B).
Figure 9.
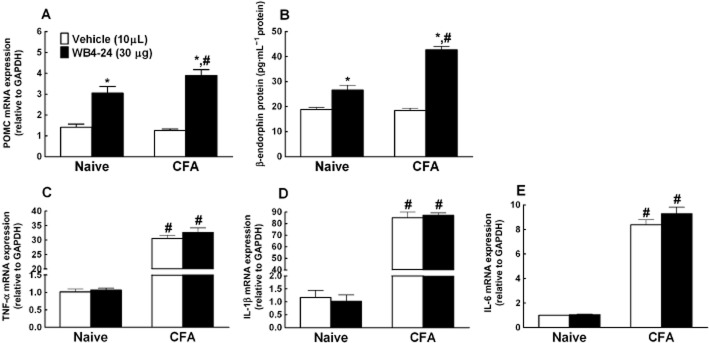
Effects of the intrathecal injection of WB4-24 on β-endorphin release (A and B) and pro-inflammatory cytokine expression (C–E) in naive rats and CFA-treated rats (1 day after injection). Measurements of spinal lumbar enlargement were obtained 1 h after the intrathecal injection of the vehicle (1% DMSO and 19% PEG400 in saline, 10 μL) or WB4-24 (30 μg). The expression of POMC and pro-inflammatory cytokines was determined by real-time quantitative PCR and the amount of β-endorphin in the culture medium was measured using a specific fluorescent immunoassay kit. The data are presented as means ± SEM (n = 6 per group). *P < 0.05, significantly different from vehicle; #P < 0.05, significantly different from corresponding values for naive rats; one-way anova with post hoc Student–Newman–Keuls test).
The same regimen was used to measure the expression of the cytokines. Compared with the results for the naive rats, CFA treatment was found to dramatically increase the expression of TNF-α, IL-1β and IL-6, with approximately 30-, 80- and 9-fold increases respectively (P < 0.05). The intrathecal injection of WB4-24 did not significantly reduce cytokine expression in the naive rats or the CFA-treated rats (Figure 9C–E).
Minocycline, β-endorphin antiserum and a μ-opioid receptor antagonist suppress WB4-24-induced antinociception
Two groups of CFA-treated (1 day after injection) rats (n = 6 per group) were first given an intrathecal injection of 10 μL saline or 100 μg minocycline, followed by a second intrathecal injection of 30 μg WB4-24 4 h later. Paw withdrawal thresholds were measured at 0.5, 1, 2 or 4 h after the second administration. As shown in Figure 10A and B, the intrathecal injection of WB4-24 produced time-dependent anti-hypersensitive effects in conditions of both heat hyperalgesia and mechanical allodynia. Minocycline completely prevented WB4-24 from blocking thermal hyperalgesia and mechanical allodynia in the ipsilateral paws (P < 0.05).
Figure 10.

Blockade effects of the intrathecally injected microglial inhibitor minocycline (A and B), a specific β-endorphin antiserum (C and D) and the selective μ-opioid receptor antagonist CTAP (E and F) on spinal WB4-24 antinociception in CFA-induced thermal hyperalgesia and mechanical allodynia in rats. Paw withdrawal latency and thresholds were measured 1 day after tibiotarsal joint CFA (30 μL) injection. Minocycline (100 μg) was intrathecally injected 4 h before spinal WB4-24 treatment. β-endorphin or dynorphin A antiserum (1:10, 10 μL) and the selective opioid receptor antagonists CTAP (30 μg), nor-BNI (30 μg) or naltrindole (30 μg) were intrathecally injected 30 min before intrathecal WB4-24 treatment. Paw withdrawal responses were measured in both the contralateral and ipsilateral paws before and 0.5, 1, 2 and 4 h after intrathecal WB4-24 administration. The data are presented as means ± SEM (n = 6 per group). *P < 0.05, significantly different from the saline plus WB4-24 group; two-way repeated-measures anova with post hoc Student–Newman–Keuls test).
Next, three groups of CFA-treated (1 day after injection) rats (n = 6 per group) received intrathecal injection of 10 μL blank rabbit serum, 1:10 β-endorphin antiserum or 1:10 dynorphin A antiserum. Based on the manufacturer's information, the β-endorphin antiserum was specific to β-endorphin and did not cross-react with methionine-enkephalin, leucine-enkephalin, dynorphin A or B, γ-endorphin, α-endorphin, ACTH or α-melanocyte-stimulating hormone. The dynorphin A antiserum was specific to dynorphin A without cross-reacting with β-endorphin, α-neoendorphin or leu-enkephalin. Each rat received a second intrathecal injection of 30 μg WB4-24 30 min later. The paw withdrawal responses were measured at 0.5, 1, 2 or 4 h after the second administration. The time and dose regimen of β-endorphin antiserum was based on the previous publication (Gong et al., 2014b). The intrathecal injection of WB4-24 produced time-dependent anti-hypersensitive effects in conditions of both heat hyperalgesia and mechanical allodynia in the ipsilateral paws. Pretreatment with the β-endorphin antiserum did not alter the basal withdrawal response in either paw, but completely prevented the anti-hyperalgesic and anti-allodynic effects exerted by WB4-24 in ipsilateral paws (P < 0.05). In contrast, the antiserum-neutralizing dynorphin A was not effective in reducing CFA-induced pain hypersensitivity in conditions of either thermal hyperalgesia or mechanical allodynia (Figure 10C and D).
The spinal antinociception in neuropathic pain elicited by exenatide was blocked by nonselective opioid receptor antagonist naloxone (Gong et al., 2014b). To determine which subtype of opioid receptors was responsible for WB4-24 antinociception, we tested the selective μ-opioid receptor antagonist CTAP (Steinmiller and Young, 2008), the κ-opioid receptor antagonist nor-BNI (Beardsley et al., 2010) and the δ-opioid receptor antagonist naltrindole (Drower et al., 1991). Four groups of CFA-treated (1 day after injection) rats (n = 6 per group) received the following pairs of intrathecal injections: saline (10 μL) + WB4-24 (30 μg); CTAP (10 μg) + WB4-24 (30 μg); nor-BNI (100 μg) + WB4-24 (30 μg); and naltrindole (5 μg) + WB4-24 (30 μg). The second treatment occurred 0.5 h after the first treatment and the paw withdrawal response was measured 0.5, 1, 2 or 4 h thereafter. As shown in Figure 10E and F, the intrathecal injection of WB4-24 induced time-dependent anti-hyperalgesia and anti-allodynia in the ipsilateral paws. Although the intrathecal injection of CTAP did not affect the withdrawal response in either the contralateral or the ipsilateral paws, it almost completely prevented the anti-hyperalgesic and anti-allodynic effects of WB4-24 in the ipsilateral paws (P < 0.05). In contrast, neither nor-BNI nor naltrindole significantly reduced the anti-hypersensitive effects of WB4-24 in either thermal hyperalgesia or mechanical allodynia.
Discussion
We recently demonstrated that two GLP-1 receptor peptide agonists, GLP-1(7–36) and exenatide, produced antinociception in neuropathic, bone cancer and diabetic pain models (Gong et al., 2014b). Low MW herbal iridoids, represented by geniposide and shanzhiside methylester, also alleviated neuropathic pain and bone cancer pain (Gong et al., 2014a; Zhu et al., 2014). The current study indicated that the non-peptide agonist WB4-24 was effective in blocking carrageenan- and CFA-induced acute and chronic mechanical allodynia and heat hyperalgesia with an efficacy of 60–80%, but not nociceptive pain in normal conditions. Our combined results show that GLP-1 receptor agonists specifically block pain hypersensitivity in a variety of animal models, regardless of the type of model and the type of stimulus used. This was further supported by the finding that WB4-24 suppresses CFA-induced acute inflammatory nociception (1 day after injection) and chronic inflammatory nociception (3 weeks after injection) to the same degree. The early phase represents acute inflammatory nociception, which is provoked by the release of a variety of peripheral proalgesic mediators, leading to peripheral sensitization and the release of spinal pro-inflammatory cytokines, which in turn cause central sensitization (Watanabe et al., 2005). In contrast, the immune system has a trophic role in the CFA-induced chronic phase (3 weeks), which represents models of rheumatoid arthritis (Honor et al., 1999; Helyes et al., 2004). In addition, there are differences in the biomolecules responsible for pain transmission and transduction in the peripheral nerves and the spinal cord between the two phases. For example, CFA was found to induce a marked increase in glutamate decarboxylase, the enzyme responsible for GABA synthesis, in the spinal dorsal horn at 3 weeks after injection, but not at 2 days after injection (Castro-Lopes et al., 1994).
The GLP-1 receptor is a 463-amino acid transmembrane-spanning protein in the family of B/secretin GPCRs. It mediates the effects of the endogenous GLP-1 peptides, and oxyntomodulin. The GLP-1 receptor has a long extracellular N-terminus with an α-helical region, five β-strands forming two antiparallel β-sheets and six conserved cysteine residues that form disulfide interactions (Bazarsuren et al., 2002; Underwood et al., 2010). Peptide ligands probably interact with GLP-1 receptors at two sites, the extracellular N-terminal ectodomain and the critical determinants in the receptor transmembrane regions, leading to full activation and signal transduction (Castro et al., 2005; Hoare, 2005; Laburthe et al., 2007). It is thus possible that non-peptide compounds may not be able of reacting with both N-terminal ectodomains and receptor transmembrane regions, due to insufficient mass or diameter.
WB4-24 displaced the specific binding of exendin(9–39) in microglia and provided 100% protection against hydrogen peroxide oxidative damage, with EC50 values in the micromolar range, whereas the peptide agonist exenatide (MW approximately 2500 Da) provided 100% protection with EC50 values in the nanomolar range. Shanzhiside methylester, 8-O-acetyl-shanzhiside methylester, geniposide, geniposidic acid, loganin and catalpol are herbal iridoids, each with a MW of approximately 300 Da. They are orthosteric, reversible and fully intrinsic agonists of GLP-1 receptors, with EC50 values in the submicromolar range, and produce antinociception by activating spinal GLP-1 receptors (Gong et al., 2014a,b,; Zhu et al., 2014). All suggest that the full activation of GLP-1 receptors may not require the simultaneous binding of the extracellular N-terminal ectodomain and the determinants in the transmembrane region, although the potency of the tested compounds as agonists of the GLP-1 receptor appears to be correlated with MW, i.e. exenatide > WB4-24 > iridoids. Moreover, the potency of GLP-1 receptor agonists is also associated with the compounds' antinociceptive activity, further suggesting that the spinal GLP-1 receptor is a potential therapeutic target for pain hypersensitivity. Furthermore, although WB4-24 has a molecular weight of approximately 1000 Da with a poor oral availability (He et al., 2012) and should have difficulty to penetrate the spinal cord, its positive antinociceptive effect after s.c. injection shows that it is possible to develop analgesics based on non-peptide or low MW GLP-1 receptor agonists.
Through the p38-MAPK pathway, minocycline, a broad-spectrum antimicrobial tetracycline compound, specifically inhibits the activation of microglia, but not the activation of astrocytes or neurons (Jung et al., 2003; Hua et al., 2005). Indeed, we observed that LPS and CFA in particular dramatically increased the expression of TNF-α, IL-1β and IL-6 in cultured microglial cells and spinal cord cells, producing five- and 11-fold increases in the cultured microglia and nine- and 80-fold increases in the spinal cord respectively. The increased expression of pro-inflammatory cytokines in the microglia was completely blocked by minocycline. Exenatide was recently reported to increase the expression of IL-6 and IL-1β in the hypothalamus and the hindbrain (Shirazi et al., 2013). In contrast, GLP-1 induced morphological changes in microglia and inhibited LPS-induced IL-1β, IL-6 and inducible NOS production in cultured astrocytes (Iwai et al., 2006). In addition, exenatide reduced MPTP-stimulated expression of TNF-α and IL-1β (Kim et al., 2009). It has been hypothesized that WB4-24 produces antinociception in part by inhibiting the expression of cytokines. However, our data do not support this hypothesis, as we did not find WB4-24 to inhibit the expression of the pro-inflammatory cytokines, stimulated by LPS or CFA, in either the spinal cord or the cultured microglial cells.
Instead, the antinociceptive effect of WB4-24 on inflammatory nociception was entirely due to the release of β-endorphin from the spinal microglia, whereas neither LPS nor CFA-induced β-endorphin expression/secretion in vitro or in vivo. β-endorphin is an endogenous opioid peptide neurotransmitter that specifically activates the opioid receptors located on neurons (Bach, 1997; Petraschka et al., 2007), although opioid receptors are also constitutively expressed in microglial cells (Chao et al., 1997). WB4-24 increased the spinal β-endorphin levels by 1.3-fold in CFA-treated rats and produced a 2.9-fold increase in β-endorphin release from LPS-stimulated cultured microglia. Furthermore, anti-allodynia and anti-hyperalgesia following spinal WB4-24 were completely prevented by the intrathecal injection of β-endorphin antiserum, but not dynorphin A antiserum, in the rat CFA model. In addition, the antinociceptive effects of WB4-24 were completely blocked by the selective μ-opioid receptor antagonist CTAP, but not by the κ-opioid receptor antagonist nor-BNI or the δ-opioid receptor antagonist naltrindole. The results are in agreement with our previous findings that antinociception in peripheral nerve injury-induced mechanical allodynia caused by exenatide was blocked by the nonselective opioid receptor antagonist naloxone (Gong et al., 2014b). This observation also extends our knowledge by demonstrating that β-endorphin, induced by GLP-1 receptor agonists, specifically activated μ-opioid receptors probably on neurons through microglial synapses interactions in the dorsal horn.
The production and release of β-endorphin were completely blocked by minocycline in the spinal cords of CFA-treated rats and in cultured microglia stimulated by LPS. The results suggest that the microglial p38-MAPK pathway is also involved in β-endorphin release. Recent studies have shown that microglia have a ‘protective’ state, known as alternative activation, in addition to the ‘destructive’ state as discussed earlier. Alternative activation is induced by IL-4, IL-13, IL-10 and TGF-β, which activate anti-inflammatory cascades or tissue repair mechanisms in microglia. However, there is little literature in this area and anti-inflammatory cascades in microglia remain poorly understood (Taves et al., 2013). Our data thus provide direct evidence that activated microglia release analgesic endorphins by a shared mechanism that activates the p38-MAPK pathway and stimulates the release of proalgesic cytokines and neurotrophic factors, thereby producing analgesia.
It is interesting to note that WB4-24 stimulated the release of basal β-endorphin in the spinal cords of naive rats and cultured microglia in the absence of LPS, although the amount of β-endorphin released was significantly smaller than that in CFA- or LPS-stimulated conditions. The results seem to suggest that GLP-1 receptor agonists could release β-endorphin in the presumed resting state. However, cultured microglia without LPS stimulation may not be in a ‘pure’ resting condition, as mechanical stress during the preparation of tissue and the culturing of cells may activate microglia, as shown by morphological changes in some cultured microglia even without LPS stimulation. It is known that almost any disturbance of homeostasis in the CNS microenvironment can trigger activation of microglia, characterized by morphological changes and up-regulation of a spectrum of intracellular molecules and surface antigens (Perry and Holmes, 2014). It may be reasonable to postulate that a small amount of β-endorphin could be released from activated microglia under normal conditions. Nevertheless, microglia must be activated for GLP-1 receptor agonists to release sufficient amounts of β-endorphin to produce analgesia, as intrathecal WB4-24 did not block formalin-induced acute flinch behaviour or withdrawal responses to mechanical or thermal stimuli in the contralateral paws of carrageenan- and CFA-treated rats, in contrast to its positive effect on β-endorphin release in naive rats. The ineffectiveness of WB4-24 in conditions of acute nociceptive pain may also be due to a lower sensitivity to exogenous and endogenous opioids in normal conditions, when microglial activation and central sensitization are not involved. As a result of this reduced sensitivity, the β-endorphin released (less than 10 pg·mg−1 protein) is insufficient to produce effective antinociception in conditions of acute nociceptive pain (Gong et al., 2014b).
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (No. 81374000 and No. 81202517).
Glossary
- CFA
complete Freund's adjuvant
- Emax
maximum effect
- GLP-1R
glucagon-like peptide-1 receptor
- MPE
maximal possible effect
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- POMC
proopiomelanocortin
Author contributions
H. F., N. G. and Y. X. W. conceived and designed the experiments. H. F., N. G., T. F. L. and A. N. M. performed the experiments. H. F. and Y. X. W. analysed the data. X. Y. W. and M. W. W. contributed reagents. H. F., Y. X. W., N. G. and M. W. W. wrote the paper.
Conflict of interest
The authors report no conflict of interest.
References
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to pharmacology 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach FW. Beta-endorphin in cerebrospinal fluid: relation to nociception. Dan Med Bull. 1997;44:274–286. [PubMed] [Google Scholar]
- Bazarsuren A, Grauschopf U, Wozny M, Reusch D, Hoffmann E, Schaefer W, et al. In vitro folding, functional characterization, and disulfide pattern of the extracellular domain of human GLP-1 receptor. Biophys Chem. 2002;96:305–318. doi: 10.1016/s0301-4622(02)00023-6. [DOI] [PubMed] [Google Scholar]
- Beardsley PM, Pollard GT, Howard JL, Carroll FI. Effectiveness of analogs of the kappa opioid receptor antagonist (3R)-7-Hydroxy-N-( (1S)-1-{ [ (3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide (JDTic) to reduce U50,488-induced diuresis and stress-induced cocaine reinstatement in rats. Psychopharmacology (Berl) 2010;210:189–198. doi: 10.1007/s00213-010-1846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch-Dienstfertig M, Labuz D, Wolfram T, Vogel NN, Stein C. JAK-STAT1/3-induced expression of signal sequence-encoding proopiomelanocortin mRNA in lymphocytes reduces inflammatory pain in rats. Mol Pain. 2012;8:83. doi: 10.1186/1744-8069-8-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler SH, Godefroy F, Besson JM, Weil-Fugazza J. A limited arthritic model for chronic pain studies in the rat. Pain. 1992;48:73–81. doi: 10.1016/0304-3959(92)90133-V. [DOI] [PubMed] [Google Scholar]
- Castro M, Nikolaev VO, Palm D, Lohse MJ, Vilardaga JP. Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc Natl Acad Sci U S A. 2005;102:16084–16089. doi: 10.1073/pnas.0503942102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Lopes JM, Tolle TR, Pan B, Zieglgansberger W. Expression of GAD mRNA in spinal cord neurons of normal and monoarthritic rats. Brain Res Mol Brain Res. 1994;26:169–176. doi: 10.1016/0169-328x(94)90088-4. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Shark KB, Sheng WS, Gekker G, Peterson PK. Activation of mu opioid receptors inhibits microglial cell chemotaxis. J Pharmacol Exp Ther. 1997;281:998–1004. [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chauvet N, Palin K, Verrier D, Poole S, Dantzer R, Lestage J. Rat microglial cells secrete predominantly the precursor of interleukin-1beta in response to lipopolysaccharide. Eur J Neurosci. 2001;14:609–617. doi: 10.1046/j.0953-816x.2001.01686.x. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28:1092–1100. doi: 10.2337/diacare.28.5.1092. [DOI] [PubMed] [Google Scholar]
- Devaraj S, Du Clos TW, Jialal I. Binding and internalization of C-reactive protein by Fcgamma receptors on human aortic endothelial cells mediates biological effects. Arterioscler Thromb Vasc Biol. 2005;25:1359–1363. doi: 10.1161/01.ATV.0000168573.10844.ae. [DOI] [PubMed] [Google Scholar]
- Drower EJ, Stapelfeld A, Rafferty MF, de Costa BR, Rice KC, Hammond DL. Selective antagonism by naltrindole of the antinociceptive effects of the delta opioid agonist cyclic[D-penicillamine2-D-penicillamine5]enkephalin in the rat. J Pharmacol Exp Ther. 1991;259:725–731. [PubMed] [Google Scholar]
- Drucker DJ. The role of gut hormones in glucose homeostasis. J Clin Invest. 2007;117:24–32. doi: 10.1172/JCI30076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong N, Gao ZY, Wang YC, Li XY, Huang JL, Hashimoto K, et al. A series of D-amino acid oxidase inhibitors specifically prevents and reverses formalin-induced tonic pain in rats. J Pharmacol Exp Ther. 2011;336:282–293. doi: 10.1124/jpet.110.172353. [DOI] [PubMed] [Google Scholar]
- Gong N, Wang YC, Wang HL, Ma AN, Hashimoto K, Wang YX. Interactions of the potent D-amino acid oxidase inhibitor CBIO with morphine in pain and tolerance to analgesia. Neuropharmacology. 2012;63:460–468. doi: 10.1016/j.neuropharm.2012.04.030. [DOI] [PubMed] [Google Scholar]
- Gong N, Fan H, Ma AN, Xiao Q, Wang YX. Geniposide and its iridoid analogs exhibit antinociception by acting at the spinal GLP-1 receptors. Neuropharmacology. 2014a;84C:31–45. doi: 10.1016/j.neuropharm.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Gong N, Xiao Q, Zhu B, Zhang CY, Wang YC, Fan H, et al. Activation of spinal glucagon-like peptide-1 receptors specifically suppresses pain hypersensitivity. J Neurosci. 2014b;34:5322–5334. doi: 10.1523/JNEUROSCI.4703-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HQ, Liao D, Wang ZG, Wang ZL, Zhou HC, Wang MW, et al. Functional characterization of three mouse formyl peptide receptors. Mol Pharmacol. 2013;83:389–398. doi: 10.1124/mol.112.081315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Guan N, Gao WW, Liu Q, Wu XY, Ma DW, et al. A continued saga of Boc5, the first non-peptidic glucagon-like peptide-1 receptor agonist with in vivo activities. Acta Pharmacol Sin. 2012;33:148–154. doi: 10.1038/aps.2011.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helyes Z, Szabo A, Nemeth J, Jakab B, Pinter E, Banvolgyi A, et al. Antiinflammatory and analgesic effects of somatostatin released from capsaicin-sensitive sensory nerve terminals in a Freund's adjuvant-induced chronic arthritis model in the rat. Arthritis Rheum. 2004;50:1677–1685. doi: 10.1002/art.20184. [DOI] [PubMed] [Google Scholar]
- Hoare SR. Mechanisms of peptide and nonpeptide ligand binding to class B G-protein-coupled receptors. Drug Discov Today. 2005;10:417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87:1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- Honor P, Menning PM, Rogers SD, Nichols ML, Basbaum AI, Besson JM, et al. Spinal substance P receptor expression and internalization in acute, short-term, and long-term inflammatory pain states. J Neurosci. 1999;19:7670–7678. doi: 10.1523/JNEUROSCI.19-17-07670.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur J Neurosci. 2005;22:2431–2440. doi: 10.1111/j.1460-9568.2005.04451.x. [DOI] [PubMed] [Google Scholar]
- Huang JL, Chen XL, Guo C, Wang YX. Contributions of spinal D-amino acid oxidase to bone cancer pain. Amino Acids. 2012;43:1905–1918. doi: 10.1007/s00726-012-1390-z. [DOI] [PubMed] [Google Scholar]
- Iwai T, Ito S, Tanimitsu K, Udagawa S, Oka J. Glucagon-like peptide-1 inhibits LPS-induced IL-1beta production in cultured rat astrocytes. Neurosci Res. 2006;55:352–360. doi: 10.1016/j.neures.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Jung KM, Park KS, Oh JH, Jung SY, Yang KH, Song YS, et al. Activation of p38 mitogen-activated protein kinase and activator protein-1 during the promotion of neurite extension of PC-12 cells by 15-deoxy-delta12,14-prostaglandin J2. Mol Pharmacol. 2003;63:607–616. doi: 10.1124/mol.63.3.607. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Moon M, Park S. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson's disease. J Endocrinol. 2009;202:431–439. doi: 10.1677/JOE-09-0132. [DOI] [PubMed] [Google Scholar]
- Kim W, Egan JM. The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev. 2008;60:470–512. doi: 10.1124/pr.108.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laburthe M, Couvineau A, Tan V. Class II G protein-coupled receptors for VIP and PACAP: structure, models of activation and pharmacology. Peptides. 2007;28:1631–1639. doi: 10.1016/j.peptides.2007.04.026. [DOI] [PubMed] [Google Scholar]
- Liu J, Yin F, Zheng X, Jing J, Hu Y. Geniposide, a novel agonist for GLP-1 receptor, prevents PC12 cells from oxidative damage via MAP kinase pathway. Neurochem Int. 2007;51:361–369. doi: 10.1016/j.neuint.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Liu Q, Li N, Yuan Y, Lu H, Wu X, Zhou C, et al. Cyclobutane derivatives as novel nonpeptidic small molecule agonists of glucagon-like peptide-1 receptor. J Med Chem. 2012;55:250–267. doi: 10.1021/jm201150j. [DOI] [PubMed] [Google Scholar]
- Lopez de Maturana R, Willshaw A, Kuntzsch A, Rudolph R, Donnelly D. The isolated N-terminal domain of the glucagon-like peptide-1 (GLP-1) receptor binds exendin peptides with much higher affinity than GLP-1. J Biol Chem. 2003;278:10195–10200. doi: 10.1074/jbc.M212147200. [DOI] [PubMed] [Google Scholar]
- McCarthy L, Wetzel M, Sliker JK, Eisenstein TK, Rogers TJ. Opioids, opioid receptors, and the immune response. Drug Alcohol Depend. 2001;62:111–123. doi: 10.1016/s0376-8716(00)00181-2. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeseburg H, de Boer RA, Buikema H, van der Harst P, van Gilst WH, Sillje HH. Glucagon-like peptide 1 prevents reactive oxygen species-induced endothelial cell senescence through the activation of protein kinase A. Arterioscler Thromb Vasc Biol. 2010;30:1407–1414. doi: 10.1161/ATVBAHA.110.206425. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY: An expert-driven knowledge base of drug targets and their ligands. Nucleic Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Holmes C. Microglial priming in neurodegenerative disease. Nat Rev Neurol. 2014;10:217–224. doi: 10.1038/nrneurol.2014.38. [DOI] [PubMed] [Google Scholar]
- Petraschka M, Li S, Gilbert TL, Westenbroek RE, Bruchas MR, Schreiber S, et al. The absence of endogenous beta-endorphin selectively blocks phosphorylation and desensitization of mu opioid receptors following partial sciatic nerve ligation. Neuroscience. 2007;146:1795–1807. doi: 10.1016/j.neuroscience.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock JM, Kettenmann H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007;30:527–535. doi: 10.1016/j.tins.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Sacerdote P, Denis-Donini S, Paglia P, Granucci F, Panerai AE, Ricciardi-Castagnoli P. Cloned microglial cells but not macrophages synthesize beta-endorphin in response to CRH activation. Glia. 1993;9:305–310. doi: 10.1002/glia.440090408. [DOI] [PubMed] [Google Scholar]
- Shirazi R, Palsdottir V, Collander J, Anesten F, Vogel H, Langlet F, et al. Glucagon-like peptide 1 receptor induced suppression of food intake, and body weight is mediated by central IL-1 and IL-6. Proc Natl Acad Sci U S A. 2013;110:16199–16204. doi: 10.1073/pnas.1306799110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitte N, Busch M, Mousa SA, Labuz D, Rittner H, Gore C, et al. Lymphocytes upregulate signal sequence-encoding proopiomelanocortin mRNA and beta-endorphin during painful inflammation in vivo. J Neuroimmunol. 2007;183:133–145. doi: 10.1016/j.jneuroim.2006.11.033. [DOI] [PubMed] [Google Scholar]
- Steinmiller CL, Young AM. Pharmacological selectivity of CTAP in a warm water tail-withdrawal antinociception assay in rats. Psychopharmacology (Berl) 2008;195:497–507. doi: 10.1007/s00213-007-0898-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, et al. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003;86:1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- Taves S, Berta T, Chen G, Ji RR. Microglia and spinal cord synaptic plasticity in persistent pain. Neural Plast. 2013;2013:1–10. doi: 10.1155/2013/753656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens B, Porret A, Buhler L, Deng SP, Morel P, Widmann C. Cloning and functional expression of the human islet GLP-1 receptor. Demonstration that exendin-4 is an agonist and exendin-(9–39) an antagonist of the receptor. Diabetes. 1993;42:1678–1682. doi: 10.2337/diab.42.11.1678. [DOI] [PubMed] [Google Scholar]
- Triplitt C, Chiquette E. Exenatide: from the Gila monster to the pharmacy. J Am Pharm Assoc (2003) 2006;46:44–52. doi: 10.1331/154434506775268698. quiz 53-45. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Underwood CR, Garibay P, Knudsen LB, Hastrup S, Peters GH, Rudolph R, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J Biol Chem. 2010;285:723–730. doi: 10.1074/jbc.M109.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Guo W, Zou S, Sugiyo S, Dubner R, Ren K. Antibody array analysis of peripheral and blood cytokine levels in rats after masseter inflammation. Neurosci Lett. 2005;382:128–133. doi: 10.1016/j.neulet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Zhang G, Jing X, Wang X, Shi W, Sun P, Su C, et al. Contribution of the proinflammatory cytokine IL-18 in the formation of human nasal polyps. Anat Rec (Hoboken) 2011;294:953–960. doi: 10.1002/ar.21385. [DOI] [PubMed] [Google Scholar]
- Zhang JY, Gong N, Huang JL, Guo LC, Wang YX. Gelsemine, a principal alkaloid from Gelsemium sempervirens Ait., exhibits potent and specific antinociception in chronic pain by acting at spinal alpha3 glycine receptors. Pain. 2013;154:2452–2462. doi: 10.1016/j.pain.2013.07.027. [DOI] [PubMed] [Google Scholar]
- Zhao WJ, Gao ZY, Wei H, Nie HZ, Zhao Q, Zhou XJ, et al. Spinal D-amino acid oxidase contributes to neuropathic pain in rats. J Pharmacol Exp Ther. 2010;332:248–254. doi: 10.1124/jpet.109.158816. [DOI] [PubMed] [Google Scholar]
- Zhu B, Gong N, Fan H, Peng CS, Ding XJ, Jiang Y, et al. Lamiophlomis rotata, an orally-available Tibetan herbal painkiller, specifically reduces pain hypersensitivity states through the activation of spinal GLP-1 Receptors. Anesthesiology. 2014;121:835–851. doi: 10.1097/ALN.0000000000000320. [DOI] [PubMed] [Google Scholar]