

Abstract
BACKGROUND AND PURPOSE
Neovascularization occurring in atherosclerotic lesions may promote plaque expansion, intraplaque haemorrhage and rupture. Oxidized LDL (oxLDL) are atherogenic, but their angiogenic effect is controversial; both angiogenic and anti-angiogenic effects have been reported. The angiogenic mechanism of oxLDL is partly understood, but the role of the angiogenic sphingolipid, sphingosine 1-phosphate (S1P), in this process is not known. Thus, we investigated whether S1P is involved in the oxLDL-induced angiogenesis and whether an anti-S1P monoclonal antibody can prevent this effect.
EXPERIMENTAL APPROACH
Angiogenesis was assessed by capillary tube formation by human microvascular endothelial cells (HMEC-1) cultured on Matrigel and in vivo by the Matrigel plug assay in C57BL/6 mice.
KEY RESULTS
Human oxLDL exhibited a biphasic angiogenic effect on HMEC-1; low concentrations were angiogenic, higher concentrations were cytotoxic. The angiogenic response to oxLDL was blocked by the sphingosine kinase (SPHK) inhibitor, dimethylsphingosine, by SPHK1-siRNA and by an anti-S1P monoclonal antibody. Moreover, inhibition of oxLDL uptake and subsequent redox signalling by anti-CD36 and anti-LOX-1 receptor antibodies and by N-acetylcysteine, respectively, blocked SPHK1 activation and tube formation. In vivo, in the Matrigel plug assay, low concentrations of human oxLDL or murine oxVLDL also triggered angiogenesis, which was prevented by i.p. injection of the anti-S1P antibody.
CONCLUSION AND IMPLICATIONS
These data highlight the role of S1P in angiogenesis induced by oxLDL both in HMEC-1 cultured on Matrigel and in vivo in the Matrigel plug model in mice, and demonstrate that the anti-S1P antibody effectively blocks the angiogenic effect of oxLDL.
Tables of Links
| TARGETS | |
|---|---|
| GPCRsa | Transportersc |
| S1P receptors | ABC |
| S1P1 receptor | Enzymesd |
| Catalytic receptorsb | p38MAPK |
| VEGFR-2 | SPHK1 |
| SPHK2 |
| LIGANDS |
|---|
| Allopurinol |
| H2O2 |
| Nitric oxide (NO) |
| Sphingosine |
| Sphingosine 1-phosphate |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a,b,c,d).
Introduction
Angiogenesis is a physiological process required for embryonic vascular development, which is also involved in wound healing and the pathophysiological progress of diseases such as diabetic retinopathy, cancer and atherosclerosis (Carmeliet and Jain, 2011). In the normal arterial wall, the vasa vasorum constitute a microvascular network in the adventitia. While no capillaries are found in the intima and the media of normal arteries, neovascularization is seen in the intima of human atherosclerotic lesions (Kolodgie et al., 2003; Moreno et al., 2006). These neocapillaries are thought to favour the progression of the plaque and to promote plaque instability and intraplaque haemorrhage and finally to increase the risk of athero-thrombotic events (Khurana et al., 2005; Moreno et al., 2006; Michel et al., 2011). Hypoxia, ischaemia and oxidative stress, which are common events in atherosclerotic lesions, play a key role in angiogenesis by activating hypoxia-inducible transcription factors that induce the expression of angiogenic factors (Ushio-Fukai and Alexander, 2004). Many factors, including VEGF, PDGF, TGFβ, ephrin, angiopoietin and lipid mediators, including sphingosine 1-phosphate (S1P), lysophosphatidic acid and PGs, stimulate endothelial cell migration, proliferation and angiogenesis (Spiegel and Milstien, 2003; Shibuya, 2008; Carmeliet and Jain, 2011).
Sphingolipid mediators, such as ceramide and S1P, are bioactive second messengers with opposing biological properties. For instance, ceramide is pro-apoptotic, while S1P is involved in survival and cell proliferation (Hannun and Obeid, 2008).
Ceramide, generated by de novo biosynthesis or by sphingomyelin hydrolysis by sphingomyelinases, is degraded by ceramidases into sphingosine, which is phosphorylated into S1P by sphingosine kinases 1 and 2 (SPHK1, SPHK2; Hannun and Obeid, 2008). S1P is involved in embryonic development, and participates in physiological and pathological vascular biology by regulating endothelial integrity, migration and proliferation, angiogenesis, vascular tone and leukocyte recruitment (Hla, 2003; Spiegel and Milstien, 2003; Daum et al., 2009; Pitson, 2011). S1P, which acts as an extracellular auto/paracrine mediator, through GPCRs S1P1-S1P5 (Hla, 2003) and as an intracellular mediator (Spiegel and Milstien, 2003), is considered a promising therapeutic target in various diseases, such as cancer, cardiovascular diseases and pathological angiogenesis, inflammatory and immune diseases (Pyne and Pyne, 2011).
Oxidized low-density lipoproteins (oxLDL) are thought to play a role in atherogenesis (Witztum and Steinberg, 1991; Tsimikas and Miller, 2011). Moderate oxLDL concentrations trigger cell migration, proliferation and inflammatory signalling, whereas higher concentrations are toxic and apoptotic (Witztum and Steinberg, 1991; Salvayre et al., 2002). OxLDL and oxidized phospholipids have been shown to be angiogenic (Bochkov et al., 2006; Dandapat et al., 2007; Yu et al., 2011), but also anti-angiogenic by inhibiting endothelial cell growth and endothelial progenitor cell differentiation (Murugesan et al., 1993; Chen et al., 2000). The angiogenic effect of a low oxLDL concentration is mediated by the LOX-1 receptor, oxidative stress, p38MAPK (Dandapat et al., 2007) and by the PI3 kinase/Akt pathway (Yu et al., 2011). We previously reported that activation of SPHK1 by oxLDL and the resulting generation of S1P is involved in the mitogenic response of smooth muscle cells to oxLDL (Augé et al., 1999). This led us to investigate whether the SPHK1/S1P pathway plays a role in oxLDL-induced angiogenesis. We found that S1P is required for oxLDL-induced capillary tube formation by human microvascular endothelial cell-1 (HMEC-1) and in vivo oxLDL-induced angiogenesis in the murine Matrigel plug model and these events were effectively prevented, in vitro and in vivo, by the anti-S1P mAb.
Methods
Lipoprotein preparation and lipid oxidation content determination
LDL were prepared from human pooled sera and mildly oxidized by UV-C irradiation, as reported (Escargueil-Blanc et al., 2001). OxLDL contained 63–97 nmol lipid hydroperoxide·mg−1 apoB, 5.6–8.3 nmol TBARS·mg−1 apoB and 8–11 nmol 4-HNE·mg−1 apoB. Murine VLDL were prepared by ultracentrifugation from plasma of hypercholesteromic apoE-/- mice (Plump et al., 1992) and were oxidized as human LDL.
Cell culture
HMEC-1 (Dr. Candal, CDC, Atlanta; Ades et al., 1992) were grown in MCDB-131 containing fetal calf serum (FCS, 10%), glutamine (40 μmol·L−1) and antibiotics (100 U·mL−1 penicillin, 100 mg·L−1 streptomycin). Cells were starved for 24 h in serum-poor medium (0.1% FCS), before the experiments.
Capillary tube formation
Capillary tube formation was performed by the Matrigel assay, used by Dandapat et al. (2007), under the following conditions. HMEC-1 were seeded (30 000 cells per well) on 24-well plates coated with Matrigel and grown in MCDB-131 supplemented with 0.1% FCS (negative control) and when indicated with native or oxLDL and inhibitors (at the indicated concentration, as mg apoB·L−1), in a 5% CO2 humidified incubator, at 37°C. Culture in 2.5% FCS was used as positive control. After 18 h, the cells were labelled with calcein-AM (1 μmol·L−1) for 30 min, observed by fluorescence microscopy (exc.496/em.516) and microphotographies were captured (10 different pictures per well). Then, all cells were counted per picture, and the percentage of linked cells was determined. The statistical analysis was done on the results of 6 to 8 separate experiments, as indicated in the legends to the figures.
Cell migration
The cell migration assay was performed using a Boyden chamber (8 μm pore size, Costar transwells permeable support, Corning Incorporated Life Science, Tewksbury, MA, USA). HMEC-1 (2 × 105 cells per well) in 0.1% FCS-containing MCDB-131 medium, were seeded in the upper chamber of Transwell inserts (8 μm pore size polycarbonate membrane) and the agents were placed in the lower chamber. After 12 h incubation, cells were stained with 0.1% crystal violet, and HMEC-1 having moved through the pores to the lower side of the membrane were observed by microscopy. Ten pictures were captured for each condition and migrating cells were counted. Alternatively, crystal violet staining was dissolved in acetic acid and the absorbance was measured by spectrophotometry (OD 570 nm). The results (counting and absorbance) are expressed as % of the unstimulated control (100%).
SPHK1 activity and S1P determination
SPHK1 activity was measured according to Olivera and Spiegel (1993), under the conditions used by Augé et al. (1999).
S1P was determined in HMEC-1 using the S1P elisa kit, following the manufacturer's instructions (Echelon Bioscience, Salt Lake City, UT, USA).
Protein concentration was measured using the Bradford reagent (BioRad, Life Science, Marnes La Coquette, France).
siRNA transfection
HMEC-1 were transfected with human SPHK1 (L-004172) ON-TARGETplus SMARTpool siRNA (Dharmacon, Waltham, MA, USA), and esiRNAEGFP (EHUEGFP, Sigma-Aldrich France, L'Isle d'Abeau Chesnes, France) as control (100 nmol L−1 final concentration for each), using OptiMEM (Life Technologies SAS, Saint Aubin, France) and HiPerFect reagent according to the manufacturer's recommendations (Quiagen SAS, Courtaboeuf, France).
Quantitative RT-PCR analysis
Total RNA extracted from harvested cells was used for real-time quantitative PCR analysis to evaluate the expression of VEGF mRNAs in basal conditions and upon treatment of HMEC-1 with oxLDL or thapsigargin, as described previously (Oskolkova et al., 2008).
Evaluation of the cytotoxicity – MTT assay, Syto13/PI live-dead assay
The overall cytotoxicity was evaluated by the MTT assay and by the Syto13/PI live-dead assay performed under the previously reported conditions (Sanson et al., 2008).
Intracellular reactive oxygen species (ROS) evaluation
Intracellular ROS were determined using the fluorescent probe carboxy-DCFDA acetoxymethyl-ester (AM). At the end of the incubation with the agents, the cells were washed, lysed and the fluorescence was measured (ex.495/em.525 nm; Negre-Salvayre et al., 2002).
In vivo Matrigel plug angiogenesis assays
The Matrigel plug angiogenesis assay in mice was used to investigate the pro-angiogenic effect of oxLDLs in vivo and the effect of inhibitors (Passaniti et al., 1992). C57BL/6 mice (a total number of 45 animals were used in this study) were injected s.c. into the two flanks with 400 μL Matrigel containing native LDL or oxLDLs, under anaesthesia induced by isoflurane inhalation (CAM 1.15% vol.). When indicated, mice were injected i.p. with the anti-S1P mAb or isotype-matched control mAbs (50 mg·kg−1 body weight, every 3 days). After 2 weeks, the animals were injected in the vein tail with 100 μL FITC-Dextran (MW = 2,000,000, 25 g·L−1 in PBS), 10 min before the mice were killed (by an i.p. injection of 150 mg·kg−1 ketamin and 10 mg·kg−1 xylazine and cervical dislocation). The Matrigel plugs were removed, photographed and observed by use of fluorescence microscopy (exc.496/em.516 respectively). Angiogenesis was evaluated by image analysis (Adobe Photoshop System Software, Dublin, Ireland).
Alternatively, angiogenesis was evaluated by the haemoglobin content in the plug. Briefly, the Matrigel plug was digested with Dispase (BD Biosciences, Le-Pont-de-Claix, France) and the haemoglobin content was determined using the Drabkin reagent kit according to the manufacturer's protocol (Sigma-Aldrich France). The absorbance was measured in a microplate reader (Tecan France, Lyon, France) at 540 nm, and normalized to the plug weight.
Experimental animal protocols were approved by the University and INSERM Institutional Committee for animal experiments (Protocol n° 13-1048-1017). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Immunohistochemistry and immunofluorescence
Fixed frozen cryo-sections (7 μm thick) of Matrigel plugs were incubated with a rat anti-mouse CD31 monoclonal antibody and a fluorescent anti-rat IgG antibody DyLight-549 Conjugated, and counterstained with DAPI. Images were captured by Explora-Nova-Morpho-Expert software (Explora Nova, La Rochelle, France). Immunostaining was quantified with Explora-Nova-Macro-Writer software.
Statistical analyses
The results are presented as means ± SEM of n experiments. Estimates of statistical significance were performed by t-test or ANOVA (SigmaStat 3.5, Systat Software). When test for normality and equal variance (Kolmogorov–Smirnov) passed, differences between mean values were evaluated by Student's unpaired t-test (two groups) or by one-way anova (several experimental groups) followed by multiple comparisons by the Holm–Sidak test. Values of P < 0.05 were considered significant.
Chemicals
Matrigel was from BD Biosciences. Corning Transwell Polycarbonate Membrane 24-wells, calcein-AM and fluorescein isothiocyanate-dextran (average MW 2,000,000), polyethylene glycol (PEG)-conjugated catalase, Vas-2870 and diphenylene iodonium (DPEI), L-nitro-arginine methyl ester (NO biosynthesis inhibitor), allopurinol (xanthine oxidase inhibitor), the cytochrome P-450 inhibitor ketoconazole, myxothiazol, dimethylsphingosine (DMS), S1P, SU1498, propidium iodide, MTT, Crystal violet, Drabkin's Reagent were from Sigma-Aldrich. The rat anti-mouse CD31 mAb and Dispase (5,000 Caseinolytic units) were from BD Pharmingen (San Jose, CA, USA). The fluorescent anti-rat IgG Ab DyLight549-Conjugated was from Tebu-Bio SAS (Le Perray en Yvelines, France). [33P]-ATP was from Perkin-Elmer (Villebon sur Yvette, France). Syto-13, DCFDA-acetoxymethylester and DAF were from Invitrogen, mitoPy1 was from Tocris BioScience (R&D France, Lille, France). The anti-CD36-blocking antibody and the isotype control were from Abcam (Burlingame, CA, USA) and the anti-LOX-1 antibody and the isotype control were from R&D Systems. Anti-S1P mAb (Sphingomab, LT1002) and the isotype-matched non-specific IgG1 mAb (LT1017) were from Lpath (San Diego, CA, USA).
Results
Low oxLDL concentration elicits capillary tube formation and migration of HMEC-1: inhibition by the anti-S1P mAb
HMEC-1 cell lines were used in the angiogenesis experiments because of their microvascular origin, immortalization and stability over time, in contrast to primary endothelial cells (e.g. HUVEC), which come from multiple donors and exhibit limited lifespans and, sometimes, exhibit phenotypic changes within the time of the culture. Figure 1A demonstrates that, as reported for HCAEC (Dandapat et al., 2007), HMEC-1 cells exhibited a biphasic effect in response to oxLDL. At low concentrations (10–50 mg apoB·L−1), oxLDL exhibited a pro-angiogenic effect, that came back to baseline at a concentration around 100 mg apoB·L−1, and was even toxic at higher oxLDL concentrations (Supporting information Figure S1; Salvayre et al., 2002). Differences in the optimal pro-angiogenic concentration reported in the literature result from differences in LDL oxidation levels since we used mildly oxidized LDL, whereas Dandapat et al. (2007) utilized highly oxidized LDL. Moreover, the dual concentration-dependent effect of oxLDL may explain the apparently contradictory effect (angiogenic vs. anti-angiogenic) of oxLDL (Murugesan et al., 1993; Chen et al., 2000; Yu et al., 2011).
Figure 1.
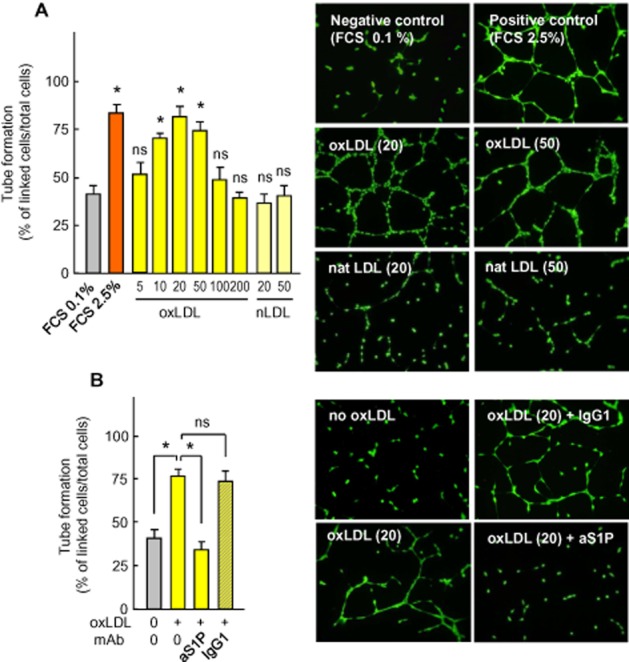
Capillary tube formation by HMEC-1 is stimulated by low concentration of oxLDL and inhibited by anti-S1P monoclonal antibody. (A) Quantification of capillary tubes formed by HMEC-1 grown on Matrigel with 0.1% FCS (negative control) or 2.5% FCS (positive control) or oxLDL or native LDL at the indicated concentrations (mg apoB·L−1). After an 18 h incubation, the cells were stained with calcein (1 μmol·L−1, 30 min) and photographed (Nikon Coolpix 995 camera) under a fluorescence microscope. Tube formation was expressed as linked cells per 100 cells. Results are means ± SEM of 6 to 8 experiments. Statistical analysis by anova and Holm–Sidak (by comparison to the negative control, FCS 0.1%). *P < 0.05; ns, not significant. (B) Effect of the anti-S1P monoclonal antibody (anti-S1P mAb) Sphingomab™ (aS1P, 10 mg·L−1) or control isotype-matched IgG1 mAb (IgG1, 10 mg·L−1) on capillary tube formation induced by oxLDL (20 mg apoB·L−1). Results are means ± SEM of six experiments; *P < 0.05; ns, not significant. In (A and B), right panels, representative pictures of the experiments.
Several signalling pathways, such as ROS, p38MAPK, ERK1/2 and VEGF, mediate the angiogenic effect of oxLDL (Dandapat et al., 2007), but the role of S1P, a bioactive and potentially angiogenic sphingolipid mediator (Spiegel and Milstien, 2003), has never been investigated in oxLDL-induced angiogenesis. Accordingly, we tested whether or not the angiogenic effect of oxLDL on HMEC-1 tube formation was dependent on S1P, by employing a highly specific anti-S1P monoclonal antibody, Sphingomab (Figure 1B). HMEC-1 tube formation elicited by oxLDL was strongly inhibited by the anti-S1P mAb (Sphingomab), whereas the non-specific isotype-matched IgG1 mAb had no effect (Figure 1B). Under these conditions, the anti-S1P mAb was not toxic and did not potentiate the toxicity of angiogenic oxLDL concentrations (Supporting information Figure S2).
As endothelial cell migration is required for angiogenesis, we investigated whether oxLDL also induced HMEC-1 migration through a S1P-dependent pathway. As expected, in the Boyden's chamber assay, oxLDL (20 mg apoB·L−1) stimulated HMEC-1 migration, which was inhibited by the anti-S1P mAb (Figure 2).
Figure 2.
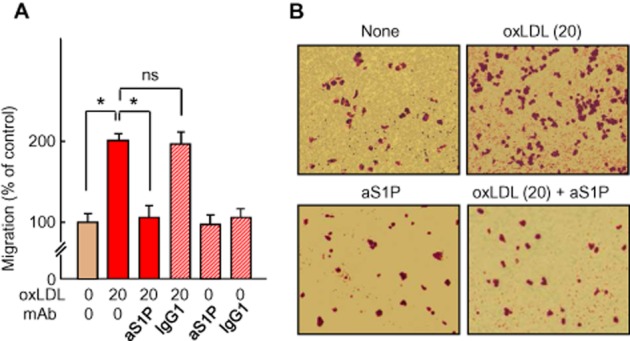
Migration of HMEC-1 induced by oxLDL and inhibition by anti-S1PmAb. (A) Cell migration in Boyden chamber. HMEC-1 were seeded in the upper chamber of Transwell inserts. The agents, oxLDL (20 mg apoB·L−1), anti-S1P mAb (aS1P, 10 mg·L−1) or non-specific IgG1 mAb (IgG1, 10 mg·L−1) were added to the lower chamber. After a 12 h incubation, migratory cells were stained with 0.5% crystal violet, and counted (data are expressed as % of the unstimulated control). Results are means ± SEM of eight experiments; *P < 0.05; ns, not significant. (B) Representative microphotographies of Transwell inserts stained by crystal violet.
It may be noted that the addition of exogenous S1P elicited capillary tube formation by HMEC-1 (Figure 3A), which was completely inhibited by the anti-S1P mAb (Figure 3B and C).
Figure 3.
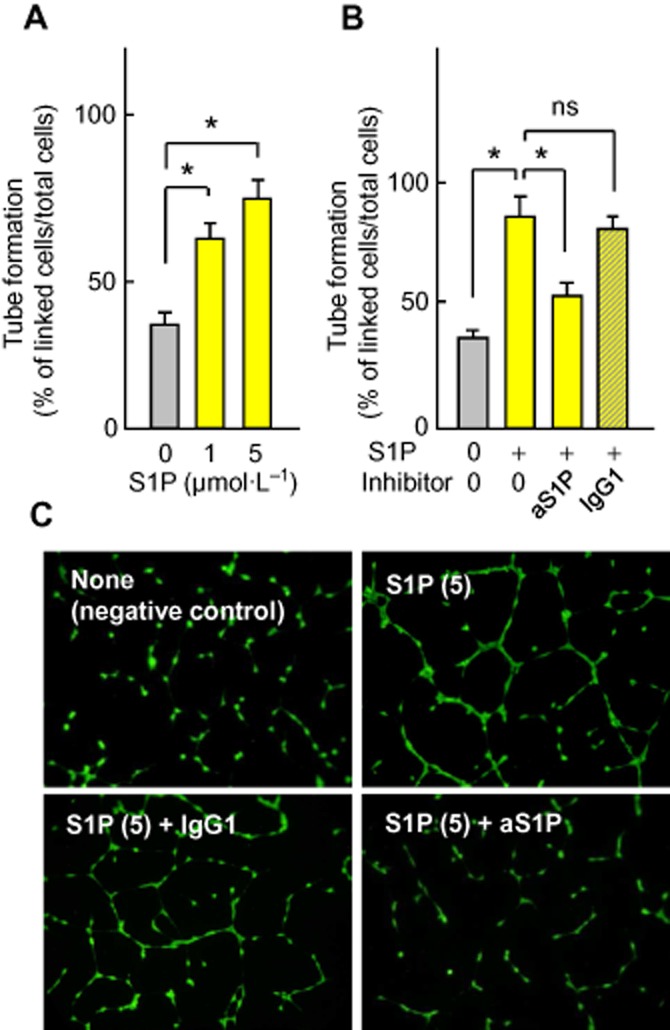
Exogenous S1P stimulates capillary tube formation. Inhibition by anti-S1P mAb. (A, B) HMEC-1 were seeded on Matrigel, incubated with exogenous S1P and anti-S1P mAb or control IgG1 mAb, and capillary tube formation was evaluated. In (A), effect of increasing S1P concentration (0–5 μmol·L−1) on capillary tube formation. Results are means ± SEM of six experiments. In (B), effect of anti-S1P (aS1P, 10 mg·L−1) or inactive IgG1 mAb (IgG1, 10 mg·L−1) on capillary tube formation induced by S1P (5 μmol·L−1). Results are means ± SEM of 6 to 8 experiments; *P < 0.05; ns, not significant. (C) Representative pictures of the experiments.
Altogether, these data suggest that extracellular S1P is required for the angiogenic effect of oxLDL, which can be effectively inhibited by the anti-S1P mAb.
SPHK1/S1P is required for capillary tube formation elicited by oxLDL
In our system, LDL-associated S1P cannot explain the angiogenic effect of oxLDL since native LDL are not angiogenic, under the conditions used, and since oxLDL contain a lower level of S1P than native LDL (Kimura et al., 2001). Thus, we investigated whether oxLDL triggered cellular S1P generation through SPHK1 activation (Spiegel and Milstien, 2003). In HMEC-1 activated by oxLDL, SPHK1 was activated in a time- and dose-dependent manner and S1P was concomitantly produced (Figure 4A–C). When SPHK was inhibited by the SPHK inhibitor, DMS, or was down-regulated by specific SPHK1-siRNA, the HMEC-1 tube formation by oxLDL was completely blocked (Figure 4D–G), suggesting that SPHK1 is required for oxLDL-induced angiogenesis.
Figure 4.
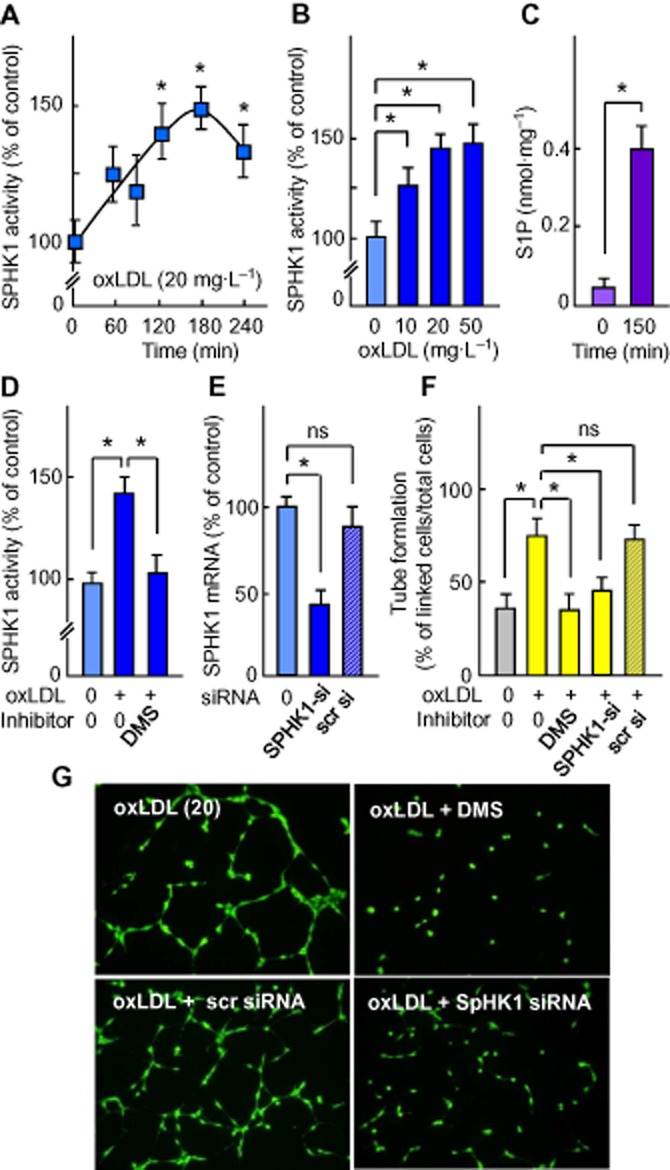
Activation of the SPHK1/S1P pathway by oxLDL and inhibition of capillary tube formation by DMS and SPHK1-siRNA. (A, B) OxLDL-induced SPHK1 activation (expressed as % of the unstimulated control). (A) Time-course of SPHK1 activation by oxLDL (20 mg apoB·L−1). (B) Dose-response for effect of oxLDL on SPHK1 activation measured after 150 min incubation. (C) S1P content in HMEC-1 stimulated by oxLDL (20 mg apoB·L−1) for 150 min. (D–E) Effect of DMS and siRNA specific to SPHK1 or scrambled on SPHK1 activation by oxLDL. Cells were pre-incubated with the SPHK inhibitor DMS (1 μmol·L−1, 30 min) or SPHK1 siRNA for 24 h, then oxLDL (20 mg apoB·L−1) were added for 150 min. (D) SPHK1 activity; (E) qPCR evaluation of SPHK1 mRNA. (F–G) Effect of DMS and SPHK1-siRNA (pre-incubated with cells as in D and E) on HMEC-1 tube formation induced by oxLDL (20 mg apoB·L−1). (G) Representative pictures of the experiments. In (A–F), results are means ± SEM of 6 to 8 experiments; *P < 0.05; ns, not significant.
As HMEC-1 express CD36 (Ades et al., 1992) and LOX-1 receptors (Dandapat et al., 2007), which may participate in the uptake of oxLDL by endothelial cells (Adachi and Tsujimoto, 2006), and promote ROS formation and angiogenesis (Dandapat et al., 2007; Silverstein and Febbraio, 2009), we investigated whether these receptors play a role in oxLDL-induced SPHK1-mediated angiogenesis. The angiogenic concentration of oxLDL induced a rise of DCFDA fluorescence reflecting an increase in intracellular ROS (Figure 5A). Anti-CD36 and anti-LOX-1 blocking antibodies (but not non-specific control antibodies – ‘IgG irr’) inhibited the uptake of oxLDL by 37% and 30%, respectively, and reduced ROS generation (Figure 5B). It is noteworthy that anti-CD36 and anti-LOX-1 antibodies inhibited SPHK1 activation and tube formation elicited by oxLDL (Figure 5C–E). This suggests a role for CD36 and LOX-1 in the uptake of oxLDL and in subsequent ROS generation, in agreement with previous reports (Dandapat et al., 2007; Li et al., 2010).
Figure 5.
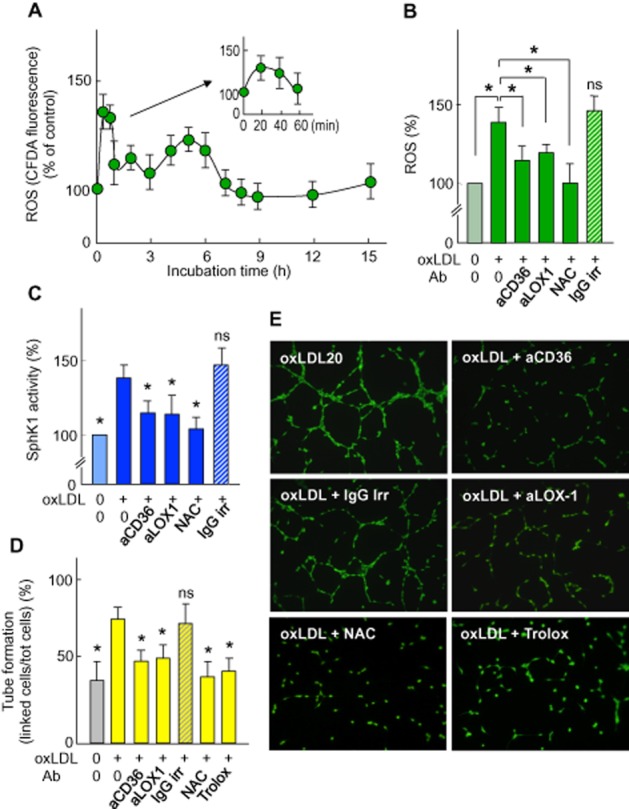
Effect of anti-CD36 and anti-LOX-1 on ROS generation, SPHK1 activation and capillary tube formation induced by oxLDL. (A) Time course of intracellular ROS triggered by oxLDL (mg apoB·L−1) in HMEC-1 preloaded with the fluorescent carboxy-dichlorodihydrofluorescein diacetate acetoxymethyl ester (DCFDA-AM) probe. (B) Intracellular ROS in HMEC-1 pre-incubated with anti-CD36 antibody (2.5 mg·L−1) or anti-LOX-1 antibody (5 mg·L−1) for 2 h or with NAC (1 mmol·L−1 for 1 h), then incubated with oxLDL (20 mg apoB·L−1) for 20 min. (C) SPHK1 activity in extracts from cells pre-incubated with anti-CD36 or anti-LOX-1 Ab or with NAC, as in (B), and incubated with oxLDL (20 mg apoB·L−1) for 150 min. (D) Effect of anti-CD36 (2.5 mg·L−1) or anti-LOX-1 Ab (5 mg·L−1), with NAC (1 mmol·L−1) or trolox (10 μmol·L−1), on tube formation induced by oxLDL (20 mg apoB·L−1). Mean ± SEM of 6 to 8 experiments, *P < 0.05; ns, not significant. (E) Representative pictures of the experiments used to obtain the results shown in (D).
To identify the nature and the source of cellular ROS generated by HMEC-1 treated with an angiogenic concentration of oxLDL, we utilized two H2O2-sensitive fluorescent probes (DCFDA and MitoPy1), and DAF, a NO-sensitive probe, as a role for NO has been reported in S1P-induced angiogenesis (Rikitake et al., 2002) and pharmacological inhibitors. As reported in the Supporting Information Table S2, ROS were detected by DCFDA and MitoPY1, but not by DAF, thus indicating that H2O2 is the predominant ROS generated. This was confirmed by the inhibitory effect of PEG-catalase, which degrades specifically H2O2 (Supporting Information Table S2). ROS generation by oxLDL was significantly inhibited by Vas-2870 and by DPEI, two NAD(P)H oxidase inhibitors, indicating a role for this enzyme, in agreement with Dandapat et al. (2007; Supporting Information Table S2). In addition, the inhibitory effect of myxothiazol suggests that mitochondria participate in the oxLDL-induced ROS generation (Supporting Information Table S2). This is consistent with the recently recognized crosstalk between mitochondria and NADPH oxidase that leads to a mutual reinforcement of ROS production (Daiber, 2010; Dikalov, 2011; Jiang et al., 2011).
As H2O2 may trigger SPHK1 activation (Cinq-Frais et al., 2013), which is inhibited by N-acetylcysteine (NAC; You et al., 2007; Ader et al., 2008), we tested the effect of NAC, which neutralized ROS and inhibited both SPHK1 activation and HMEC-1 tube formation induced by oxLDL (Figure 5B–E). Altogether, these data support the hypothesis that the uptake of oxLDL mediated through CD36 and LOX-1 receptors is coupled to the generation of ROS that activate the SPHK1/S1P pathway and HMEC-1 tube formation.
OxLDL are angiogenic in the mouse Matrigel plug assay: inhibition by anti-S1P mAb
The angiogenic effect of oxLDL in vivo, and the role of S1P were evaluated in the mouse Matrigel plug assay (Passaniti et al., 1992). Matrigel plugs, containing oxLDL or native LDL, or vehicle were s.c. injected into mouse flanks. After 2 weeks, capillary neoformation in the plugs was analysed. Red blood-coloured neo-capillaries were quantified by two morphometric methods (Supporting Information Figure S3 and S4), by determining the haemoglobin content in the plug. Capillaries were also visualized on plug sections by fluorescence microscopy and by immunostaining of the endothelial cells with anti-CD31 antibody (Figures 6 and 7). Negative control plugs appeared pale and gray transparent and contained only few blood-coloured capillaries, whereas oxLDL-containing Matrigel plugs were more intensely red-coloured (Figure 6 and Supporting Information Figure S3–S5) and contained more haemoglobin (Figure 7).
Figure 6.
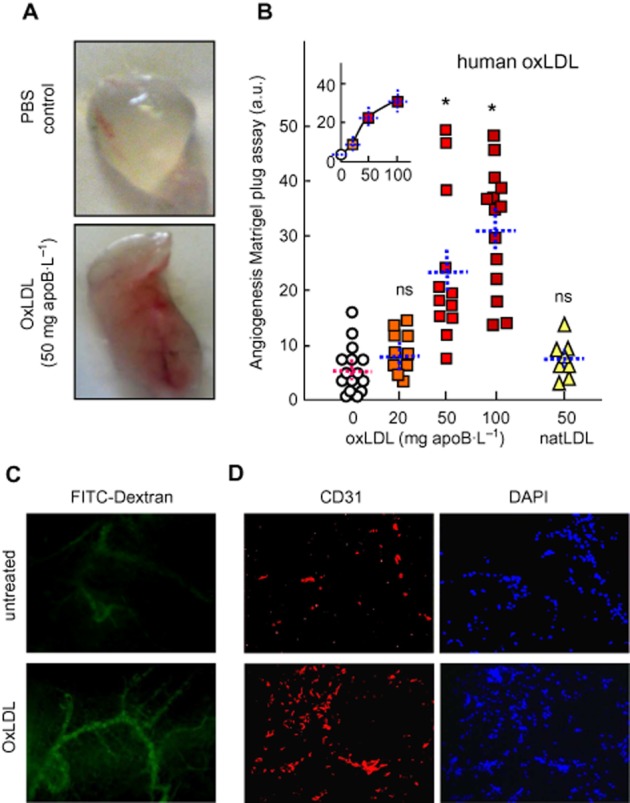
Human oxLDL stimulate angiogenesis in the murine Matrigel plug model. (A, B) C57BL/6 mice (8–14 per group), were injected s.c. with 0.4 mL Matrigel containing human native (natLDL) or oxLDL. Plugs were removed after 2 weeks and photographed. (A) Representative macrophotographs of plugs. Angiogenesis was evaluated by image analysis (Supporting information Figure S3 and S4). (B) Quantitative evaluation of angiogenesis in plugs containing PBS (circles), oxLDL (squares) or natLDL (triangles). Each point represents the value of angiogenesis in one plug. Means ± SEM are indicated by the dotted lines. Statistical analysis by one-way anova and Holm–Sidak (compared with PBS (0) group). *P < 0.05; ns, not significant. (C) Representative microphotographs of capillaries visualized by fluorescent FITC-dextran (mice injected i.v. with FITC-dextran 10 min before killing), and (D), plug sections labelled with anti-CD31 antibody and counterstained with DAPI.
Figure 7.
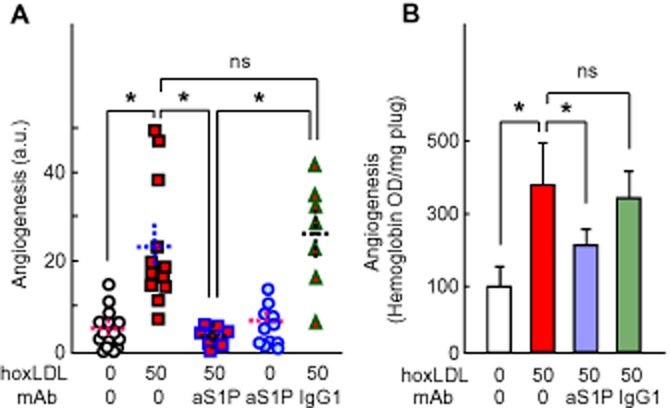
Effect of anti-S1P mAb on oxLDL-induced angiogenesis in murine Matrigel plug. Mice were injected with Matrigel containing 50 mg apoB·L−1 of human oxidized LDL (hoL50), then were injected, i.p., every 3 days with the anti-S1P mAb (50 mg·kg−1 body weight) or the non-specific isotype-matched IgG1 (50 mg·kg−1 body weight) for 2 weeks. Angiogenesis was quantified by image analysis, as in Figure 6B (A) or by haemoglobin determination (B). The haemoglobin content in Matrigel plugs was determined using the Drabkin reagent kit and was normalized to the plug weight. In (A), each point represents the angiogenesis score in one plug. In each column, mean ± SEM is indicated by the dotted lines. (B) The results are expressed as mean of haemoglobin OD·mg−1 plug ± SEM. Statistical analysis was done by one-way anova followed by Holm–Sidak test (SigmaStat sofware) *P < 0.05; ns, not statistically significant.
Human oxLDL induced a dose-dependent angiogenic response (0–100 mg apoB·L−1), whereas native LDL (50 mg apoB·L−1) were only slightly angiogenic (Figure 6). Finally, murine oxidized VLDL were angiogenic like human oxLDL (Supporting Information Figure S5), thus suggesting that the in vivo angiogenic effect of human oxLDL results from the oxidation process and not from an immune response of C57BL/6 mice against human oxLDL antigens.
The anti-S1P mAb blocked angiogenesis elicited by human oxLDL or murine oxVLDL (both used at 50 mg·L−1) in the Matrigel plug assay, while the non-specific isotype-matched IgGκ1 had no effect (Figure 7 and Supporting Information Figure S5). Similar results were observed when quantifying the haemoglobin content, which reflects the blood vessel formation in the Matrigel plugs (Figure 7B). These data suggest that oxLDL-induced angiogenesis in the Matrigel plug model requires extracellular S1P, and is effectively blocked by anti-S1P mAb.
Discussion
The data reported indicate for the first time that oxLDL-induced angiogenesis requires SPHK1/S1P signalling both in vitro in a HMEC-1 tube formation model, and in vivo in the murine Matrigel plug model, and is effectively prevented by SPHK1 inhibitors and by an anti-S1P mAb Sphingomab that neutralizes extracellular S1P.
Since sphingolipid mediators are generated upon stimulation of vascular cells by oxLDL (Augé et al., 2000) and since S1P is a known to be angiogenic (Lee et al., 1999), we hypothesized that S1P may contribute to oxLDL-induced angiogenesis. The crucial role of extracellular S1P is supported by the inhibition of oxLDL-induced HMEC-1 migration and tube formation by the anti-S1P mAb, which reduces specifically the bioavailability of extracellular S1P (Sabbadini, 2011). Interestingly, at low oxLDL concentrations, the anti-S1P mAb did not increase the oxLDL toxicity. Moreover, when administered to C57BL/6 mice implanted with oxLDL-containing Matrigel plugs, this antibody also prevented the angiogenic effect of oxLDL in vivo. This is consistent with the efficacy of Sphingomab, and its humanized form sonepcizumab™, to block VEGF-induced angiogenesis in the murine Matrigel plug, retinal neovascularization and tumour angiogenesis in animal models in which S1P exerts cell-protective, tumorigenic and angiogenic effects (Visentin et al., 2006; Sabbadini, 2011).
Extracellular S1P can be secreted by endothelial cells or/and provided by blood (platelets and HDL) (Spiegel and Milstien, 2003). In our experiments, HMEC-1 were starved in serum-poor medium containing only low levels of S1P that could account for the basal tube formation, but not for the angiogenic effect of oxLDL. Similarly, S1P contained in LDL cannot explain the oxLDL-induced angiogenesis since native LDL are not angiogenic and since oxidation reduces the level of LDL-associated S1P (Kimura et al., 2001). Thus, it is more likely that S1P is biosynthesized and secreted by HMEC-1 since (i) oxLDL trigger a time- and dose-dependent activation of SPHK1 associated with the concomitant generation of S1P; and (ii) the inhibition of SPHK1 by DMS or by SPHK1-specific siRNA blocked the oxLDL-induced HMEC-1 tube formation. According to the classical ‘inside-out’ signalling, S1P generated by cells can be exported by S1P transporters, such as SPNS2 and ABC transporter family (Takabe et al., 2008; Nishi et al., 2014). Finally, in our HMEC-1 model, the SPHK1/S1P pathway activated by oxLDL is apparently the main source of extracellular S1P, which mediates oxLDL-induced angiogenesis as it was blocked by anti-S1P mAb.
Another issue is the relationship between ROS and SPHK1 activation. In HMEC-1, the uptake of oxLDL is associated with the generation of ROS through LOX-1 and CD36 receptors. Our findings are consistent with previous studies showing that oxLDL trigger ROS generation through a LOX-1 receptor-dependent activation of NADPH oxidase (Dandapat et al., 2007) and that the interaction of oxidized lipids or oxLDL with CD36 elicits a cellular oxidative stress (Sukhanov et al., 2006; Li et al., 2010). The detection of ROS by DCFDA and MitoPY1, and their inhibition by PEG-catalase strongly suggest that H2O2 is the predominant ROS. The inhibitory effect of Vas-2870, a NADPH oxidase inhibitor, and myxothiazol, an inhibitor of the mitochondrial respiratory chain, probably results from the recently reported crosstalk between mitochondria and NADPH oxidases, which can lead to a mutual reinforcement of ROS production (Daiber, 2010; Dikalov, 2011; Jiang et al., 2011). Intracellular ROS were generated in HMEC-1 during 5 to 6 h upon addition of low angiogenic oxLDL concentrations, as previously observed with higher oxLDL concentrations (Robbesyn et al., 2005).
In HMEC-1, the antioxidant NAC prevented both the oxLDL-induced increase in ROS and SPHK1 activation, thus suggesting that ROS (and more precisely H2O2) mediate SPHK1 activation, in agreement with the ROS-induced activation of SPHK1 by hyperglycaemia (You et al., 2007), hypoxia (Ader et al., 2008) and exogenously added H2O2 (Cinq-Frais et al., 2013). Moreover, NAC and trolox (a hydrophilic analogue of tocopherol) also inhibited angiogenesis triggered by oxLDL, thus supporting the hypothesis that the binding of oxLDL with CD36 or the LOX-1 receptor elicits ROS (H2O2) generation, which mediates SPHK1 activation, S1P generation and the angiogenic response.
In our HMEC-1 model, the angiogenic effect is apparently independent of VEGF since oxLDL did not induce either VEGF mRNA production (Supporting Information Figure S6A) or increase the level of VEGF (determined by the Human VEGF elisa Kit of Signosis; data not shown). This discrepancy with the data of Dandapat et al. (2007) may result from differences in the endothelial cell type (HMEC-1 vs. HCAEC) and LDL oxidation (mild UV-induced LDL oxidation vs. copper-induced LDL oxidation).
As a crosstalk between the VEGF and S1P signalling pathways has been reported (Tanimoto et al., 2002; Spiegel and Milstien, 2003), we investigated whether the VEGF receptor, VEGFR-2 was implicated in oxLDL-induced angiogenesis. Interestingly, the VEGFR-2 inhibitor SU-1498 blocked, in part, the angiogenic effect of oxLDL (Supporting Information Figure S6B) and of exogenously added S1P, thus suggesting that VEGFR-2 is involved in the angiogenic effect of S1P, possibly via a transactivation of VEGFR-2 by S1P (Tanimoto et al., 2002) or via the formation of a complex comprising the S1P receptor and VEGFR-2, and, perhaps, other growth factor receptors (Bergelin et al., 2010; Pyne and Pyne, 2010). This suggests that oxLDL may activate various angiogenic interconnected signalling pathways, for example, the SPHK1/S1P and VEGFR-2 pathways, even in the absence of secretion of VEGF. Of course, the possibility that VEGF may play a role in vivo, in the mouse Matrigel plug model, is not excluded. This does not conflict with the inhibitory effect of anti-S1P mAb, since the angiogenic signalling of VEGFR-2 is partly mediated by S1P (Spiegel and Milstien, 2003) and VEGF-supported angiogenesis is largely blocked by systemic administration of the anti-S1P mAb (Visentin et al., 2006). Of note, we did not detect any increase in TGFβ nor PDGF mRNAs, under the experimental conditions of oxLDL-induced HMEC-1 angiogenesis, indicating that these growth factors do not contribute to the angiogenesis induced by oxLDL in this model system.
In atherosclerotic plaques, the level of LDL oxidation is probably not uniform and the local oxLDL concentration varies during the evolution of the plaque. Indeed, LDL oxidation is a progressive process depending on lipid composition, residence time, rate of metabolic clearance of lipoproteins and intensity and duration of oxidative stress (Stocker and Keaney, 2004; Levitan et al., 2010). Our results and those of Dandapat et al. (2007) suggest that low concentrations of oxLDL may trigger angiogenesis or reinforce the effect of other angiogenic factors. Interestingly, under our experimental conditions, SPHK1 inhibitors and anti-S1P mAb effectively blocked the oxLDL-induced angiogenic effect, without increasing their toxicity, possibly because the oxLDL concentration was largely below the toxic threshold dose.
As oxLDL and neocapillaries could participate in atherosclerotic plaque progression and plaque instability (Kolodgie et al., 2003; Moulton et al., 2003; Michel et al., 2011) and blood levels of S1P are correlated with the extent of plaque formation in CAD patients (Deutschman et al., 2003), it may be speculated that drugs targeting the SPHK/S1P system (administered during angioplasty or by eluting stents) could be an additional approach in the treatment of CAD and restenosis after stenting. This is consistent with the efficacy of the SPHK inhibitor DMS to delay intimal hyperplasia (McDonald et al., 2010) and may help to counter the angiogenic effect of oxLDL in advanced atherosclerotic plaques.
Acknowledgments
The authors wish to thank C. Bernis, M. H. Grazide and P. Clerc for their excellent technical assistance, and Lpath for giving the anti-S1P mAb. This work was supported by INSERM, University Toulouse-3 and Agence Nationale pour la Recherche (ANR-12-BSV1-0016-01 ‘Carina’). M. Trayssac is granted by ‘Ministère de l'Enseignement Supérieur et de la Recherche’ (PhD fellowship) and by ‘Société Française de Transplantation’.
Glossary
- oxLDL
oxidized low-density lipoproteins
- S1P
sphingosine 1-phosphate
- HMEC-1
human microvascular endothelial cells
- SPHK
sphingosine kinase
- SPHK1
sphingosine kinase 1
- ROS
reactive oxygen species
Author contribution
C. C. participated in the design of, and performed, the experiments. M. T. performed, in part, the in vitro experiments. B. G.-S. helped with the in vivo angiogenesis studies. E. M. carried out the Q-PCR and siRNA studies. R. Sabbadini gave the anti-S1P mAb and contributed to the analysis and discussion of the results. R. Salvayre designed the experiments, analysed the data and wrote some of the manuscript. A. N.-S. designed the experiments, analysed the data and wrote part of the manuscript.
Conflicts of interest
Professor R. Sabbadini is the Founder and Vice President of Lpath, Inc. He is an inventor of the anti-S1P antibodies and has stock in Lpath. No other conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12897
Figure S1 Toxicity of increasing concentration of oxLDL in HMEC-1 grown on Matrigel. HMEC-1, grown on Matrigel, were incubated for 24 h with increasing concentration of oxLDL or native LDL (natLDL). The cytotoxicity was evaluated (A) by the MTT assay, expressed as % of the untreated (0) control and (B, C) by the live/dead assay. Cells were stained by two fluorescent intercalating compounds, the permeant green-coloured syto13 and the non-permeant red-coloured propidium iodide (PI; under conditions where the red fluorescence prevails over the green fluorescence when the two fluorescent probes enter the nucleus, i.e. cell membrane permeabilized). Living and dying cells were counted on the basis of staining and morphological features. In A, B, mean ± SEM of 6 to 10 separate experiments. Comparison with the untreated control by one-way anova and Holm–Sidak *P < 0.05; ns, not significant. (D) Higher magnification showing the structure of the nucleus. Left panel: living cells exhibit a loose green-coloured chromatin (PI does not enter the normal cell). Middle panel: primary apoptosis is characterized by condensed pyknotic or fragmented nucleus stained green/yellow by Syto13 (at this stage, apoptotic cells are not permeable to PI). During post-apoptotic necrosis, the nucleus exhibits similar apoptotic morphology but is stained red by PI (due to plasma membrane permeabilization in a late step of apoptosis). Right panel: primary necrosis is characterized by an early permeabilization of the plasma membrane allowing PI to enter the cell and stain red the loose chromatin of the nucleus.
Figure S2 Anti-S1P mAb is not toxic to HMEC-1 treated with low concentration of oxLDL. HMEC-1 were grown in 12 multiwell plates in MCDB-131 medium containing 10% FCS, then starved in serum poor (0.1% FCS) medium for 24 h before addition of oxLDL at the indicated concentrations (20 or 50 mg apoB·L−1) and anti-S1P mAb (aS1P, 10 mg·L−1). In A, evaluation by the MTT assay of the whole toxicity of oxLDL (20 and 50 mg·L−1) and aS1P. In B, C, live/dead assays using syto-13 (green) and PI (red) DNA probes, as in A, on HMEC-1 incubated with oxLDL and aS1P. In B, counting of living and dying cells. C, pictures representative of HMEC-1 viability in the presence of oxLDL (at the indicated concentrations) and aS1P. Mean ± SEM of eight separate experiments, *P < 0.05; ns, not significant.
Figure S3 Human oxLDL stimulate angiogenesis in vivo in the Matrigel plug model. (A, B) Angiogenesis in Matrigel plugs. C57/BL6 mice were injected subcutaneously with 0.4 mL Matrigel containing on one flank PBS and on the other flank human oxLDLs. Plugs were removed after 2 weeks and photographied. Representative macrophotographies of plugs containing PBS (control) or oxLDLs (50 mg apoB·L−1). Angiogenesis was quantified by image analysis of the red blood colour using Adobe photoshop software (upper right panel in A and B) or by manual drawing followed by image analysis (lower right panels in A and B).
Figure S4 Comparison of the two methods of quantification of angiogenesis on macrophotographies. A and B, Quantification of angiogenesis in Matrigel plugs obtained by image analysis of blood-coloured vessels using Adobe Photoshop software (A) and by manual drawing followed by image analysis (B). In C, comparison of the data by the two methods.
Figure S5 Effect of anti-S1P mAb on mouse oxVLDL (moV)-induced angiogenesis in the murine Matrigel plug assay. Mice were injected with Matrigel containing 0 or 50 mg apoB·L−1 murine oxVLDL (moV50). Mice were intraperitoneously injected every 3 days with (or without) the anti-S1P mAb (50 mg·kg−1 body weight) for 2 weeks before removing the plugs and quantification of angiogenesis. Each point represents the angiogenesis score in one plug. Mean ± SEM are indicated by the doted line. Comparison of groups was performed by one-way anova followed Holm–Sidak test (SigmaStat sofware) *P < 0.05; ns, not statistically significant.
Figure S6 Effect of oxLDL on VEGF expression (A) and effect of the VEGFR2 inhibitor SU1295 on angiogenesis elicited by oxLDL. (A) Effect of oxLDL on VEGF expression in HMEC-1. Cells were incubated in the presence of oxLDL (20 mg apoB·L−1) or thapsigargin (5 μmol·L−1) at the indicated time. VEGF mRNA level was evaluated by qPCR (normalized to TBP mRNA). (B and C) Effect of the VEGFR2 inhibitor SU1498 on angiogenesis induced by oxLDL (20 mg apoB·L−1) or by S1P (5 μmol·L−1). In A and B, mean ± SEM of six separate experiments, *P < 0.05; ns, not significant. In C, representative microphotographs of cells stained as in Figure 1.
Figure S7 Time course of inhibition of tube formation by trolox added after oxLDL. HMEC-1 were incubated with or without hoxLDL (20 mg apoB·L−1, added at time 0) and trolox (10 μmol·L−1), was added at the indicated time (either at time 0, or 1, 3, 5 or 9 h after oxLDL. Then, tube formation was evaluated at time 18 h (as in Figures 1, 3 and 4). Note that, under the used conditions, trolox added up to 7 h, inhibited tube formation, thus suggesting that ROS generated up to 7 h are required for tube formation. Mean ± SEM of six separate experiments. One-way anova followed by Holm–Sidak post hoc test. *P < 0.05,
Table S1 Evaluation of ROS induced by oxLDL (20 mg apoB·L−1) using various ROS-specific probes (expressed as percent of the unstimulated control).
Table S2 Effects of inhibitors on ROS generation elicited by oxLDLs.
References
- Adachi H, Tsujimoto M. Endothelial scavenger receptors. Prog Lipid Res. 2006;45:379–404. doi: 10.1016/j.plipres.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Ader I, Brizuela L, Bouquerel P, Malavaud B, Cuvillier O. Sphingosine kinase 1: a new modulator of HIF-1-alpha during hypoxia in human cancer cells. Cancer Res. 2008;68:8635–8642. doi: 10.1158/0008-5472.CAN-08-0917. [DOI] [PubMed] [Google Scholar]
- Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, et al. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic receptors. Br J Pharmacol. 2013b;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013c;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013d;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augé N, Nègre-Salvayre A, Salvayre R, Levade T. Sphingomyelin metabolites in vascular cell signaling and atherogenesis. Prog Lipid Res. 2000;39:207–229. doi: 10.1016/s0163-7827(00)00007-2. [DOI] [PubMed] [Google Scholar]
- Augé N, Nikolova-Karakashian M, Carpentier S, Parthasarathy S, Nègre-Salvayre A, Salvayre R, et al. Role of sphingosine 1-phosphate in the mitogenesis induced by oxidized LDL in smooth muscle cells via activation of sphingomyelinase, ceramidase, and sphingosine kinase. J Biol Chem. 1999;274:21533–21538. doi: 10.1074/jbc.274.31.21533. [DOI] [PubMed] [Google Scholar]
- Bergelin N, Löf C, Balthasar S, Kalhori V, Törnquist K. S1P1 and VEGFR-2 form a signaling complex with extracellularly regulated kinase 1/2 and protein kinase C-alpha regulating ML-1 thyroid carcinoma cell migration. Endocrinology. 2010;151:2994–3005. doi: 10.1210/en.2009-1387. [DOI] [PubMed] [Google Scholar]
- Bochkov VN, Philippova M, Oskolkova O, Kadl A, Furnkranz A, Karabeg E, et al. Oxidized phospholipids stimulate angiogenesis via autocrine mechanisms, implicating a novel role for lipid oxidation in the evolution of atherosclerotic lesions. Circ Res. 2006;99:900–908. doi: 10.1161/01.RES.0000245485.04489.ee. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Jiang W, Via DP, Luo S, Li TR, Lee YT, et al. Oxidized LDL inhibit endothelial cell proliferation by suppressing basic fibroblast growth factor expression. Circulation. 2000;101:171–177. doi: 10.1161/01.cir.101.2.171. [DOI] [PubMed] [Google Scholar]
- Cinq-Frais C, Coatrieux C, Grazide MH, Hannun YA, Nègre-Salvayre A, Salvayre R, et al. A signaling cascade mediated by ceramide, src and PDGFRβ coordinates the activation of the redox-sensitive neutral sphingomyelinase-2 and sphingosine kinase-1. Biochim Biophys Acta. 2013;1831:1344–1356. doi: 10.1016/j.bbalip.2013.04.014. [DOI] [PubMed] [Google Scholar]
- Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. 2010;1797:897–906. doi: 10.1016/j.bbabio.2010.01.032. [DOI] [PubMed] [Google Scholar]
- Dandapat A, Hu C, Sun L, Mehta JL. Small concentrations of oxLDL induce capillary tube formation from endothelial cells via LOX-1-dependent redox-sensitive pathway. Arterioscler Thromb Vasc Biol. 2007;27:2435–2442. doi: 10.1161/ATVBAHA.107.152272. [DOI] [PubMed] [Google Scholar]
- Daum G, Grabski A, Reidy MA. Sphingosine 1-phosphate: a regulator of arterial lesions. Arterioscler Thromb Vasc Biol. 2009;29:1439–1443. doi: 10.1161/ATVBAHA.108.175240. [DOI] [PubMed] [Google Scholar]
- Deutschman DH, Carstens JS, Klepper RL, Smith WS, Page MT, Young TR, et al. Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am Heart J. 2003;146:62–68. doi: 10.1016/S0002-8703(03)00118-2. [DOI] [PubMed] [Google Scholar]
- Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escargueil-Blanc I, Salvayre R, Vacaresse N, Jürgens G, Darblade B, Arnal JF, et al. Mildly oxidized LDL induces activation of platelet-derived growth factor beta-receptor pathway. Circulation. 2001;104:1814–1821. doi: 10.1161/hc4001.097179. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Hla T. Signaling and biological actions of sphingosine 1-phosphate. Pharmacol Res. 2003;47:401–407. doi: 10.1016/s1043-6618(03)00046-x. [DOI] [PubMed] [Google Scholar]
- Jiang F, Zhang Y, Dusting GJ. N ADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- Khurana R, Simons M, Martin JF, Zachary IC. Role of angiogenesis in cardiovascular disease: a critical appraisal. Circulation. 2005;112:1813–1824. doi: 10.1161/CIRCULATIONAHA.105.535294. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Sato K, Kuwabara A, Tomura H, Ishiwara M, Kobayashi I, et al. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J Biol Chem. 2001;276:31780–31785. doi: 10.1074/jbc.M104353200. [DOI] [PubMed] [Google Scholar]
- Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–2325. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–312. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- Levitan I, Volkov S, Subbaiah PV. Oxidized LDL: diversity, patterns of recognition, and pathophysiology. Antioxid Redox Signal. 2010;13:39–75. doi: 10.1089/ars.2009.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Febbraio M, Reddy SP, Yu DY, Yamamoto M, Silverstein RL. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J Clin Invest. 2010;120:3996–4006. doi: 10.1172/JCI42823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald RA, Pyne S, Pyne NJ, Grant A, Wainwright CL, Wadsworth RM. The sphingosine kinase inhibitor N,N-dimethylsphingosine inhibits neointimal hyperplasia. Br J Pharmacol. 2010;159:543–553. doi: 10.1111/j.1476-5381.2009.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel JB, Virmani R, Arbustini E, Pasterkamp G. Intraplaque haemorrhages as the trigger of plaque vulnerability. Eur Heart J. 2011;32:1977–1985. doi: 10.1093/eurheartj/ehr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno PR, Purushothaman KR, Sirol M, Levy AP, Fuster V. Neovascularization in human atherosclerosis. Circulation. 2006;113:2245–2252. doi: 10.1161/CIRCULATIONAHA.105.578955. [DOI] [PubMed] [Google Scholar]
- Moulton KS, Vakili K, Zurakowski D, Soliman M, Butterfield C, Sylvin E, et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci U S A. 2003;100:4736–4741. doi: 10.1073/pnas.0730843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugesan G, Chisolm GM, Fox PL. Oxidized LDL inhibits the migration of aortic endothelial cells in vitro. J Cell Biol. 1993;120:1011–1019. doi: 10.1083/jcb.120.4.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre-Salvayre A, Augé N, Duval C, Robbesyn F, Thiers JC, Nazzal D, et al. Detection of intracellular reactive oxygen species in cultured cells using fluorescent probes. Methods Enzymol. 2002;352:62–71. doi: 10.1016/s0076-6879(02)52007-3. [DOI] [PubMed] [Google Scholar]
- Nishi T, Kobayashi N, Hisano Y, Kawahara A, Yamaguchi A. Molecular and physiological functions of sphingosine 1-phosphate transporters. Biochim Biophys Acta. 2014;1841:759–765. doi: 10.1016/j.bbalip.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Olivera A, Spiegel S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 1993;365:557–560. doi: 10.1038/365557a0. [DOI] [PubMed] [Google Scholar]
- Oskolkova OV, Afonyushkin T, Leitner A, von Schlieffen E, Gargalovic PS, Lusis AJ, et al. ATF4-dependent transcription is a key mechanism in VEGF up-regulation by oxidized phospholipids: critical role of oxidized sn-2 residues in activation of unfolded protein response. Blood. 2008;112:330–339. doi: 10.1182/blood-2007-09-112870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passaniti A, Taylor RM, Pili R, Guo Y, Long PV, Haney JA, et al. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin and fibroblast growth factor. Lab Invest. 1992;67:519–528. [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitson SM. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci. 2011;36:97–107. doi: 10.1016/j.tibs.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Plump AS, Smith JD, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft JG, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- Pyne S, Pyne NJ. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- Pyne S, Pyne NJ. Translational aspects of sphingosine-1-phosphate biology. Trends Mol Med. 2011;17:463–472. doi: 10.1016/j.molmed.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Hirata K, Kawashima S, Ozaki M, Takahashi T, Ogawa W, et al. Involvement of endothelial nitric oxide in sphingosine-1-phosphate–induced angiogenesis. Arterioscler Thromb Vasc Biol. 2002;22:108–114. doi: 10.1161/hq0102.101843. [DOI] [PubMed] [Google Scholar]
- Robbesyn F, Augé N, Vindis C, Cantero AV, Barbaras R, Negre-Salvayre A, et al. HDL prevent the oxidized LDL-induced EGFR activation and subsequent matrix metalloproteinase-2 upregulation. Arterioscler Thromb Vasc Biol. 2005;25:1206–1212. doi: 10.1161/01.ATV.0000164805.73558.80. [DOI] [PubMed] [Google Scholar]
- Sabbadini RA. Sphingosine-1-phosphate antibodies as potential agents in the treatment of cancer and age-related macular degeneration. Br J Pharmacol. 2011;162:1225–1238. doi: 10.1111/j.1476-5381.2010.01118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvayre R, Auge N, Benoist H, Negre-Salvayre A. Oxidized low-density lipoprotein-induced apoptosis. Biochim Biophys Acta. 2002;1585:213–221. doi: 10.1016/s1388-1981(02)00343-8. [DOI] [PubMed] [Google Scholar]
- Sanson M, Ingueneau C, Vindis C, Thiers JC, Glock Y, Rousseau H, et al. Oxygen-regulated protein-150 prevents calcium homeostasis deregulation and apoptosis induced by oxidized LDL in vascular cells. Cell Death Differ. 2008;15:1255–1265. doi: 10.1038/cdd.2008.36. [DOI] [PubMed] [Google Scholar]
- Shibuya M. VEGF-dependent and -independent regulation of angiogenesis. BMB Rep. 2008;41:278–286. doi: 10.5483/bmbrep.2008.41.4.278. [DOI] [PubMed] [Google Scholar]
- Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behaviour. Sci Signal. 2009;2:re3. doi: 10.1126/scisignal.272re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–1478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- Sukhanov S, Higashi Y, Shai SY, Itabe H, Ono K, Parthasarathy S, et al. Novel effect of oxidized low-density lipoprotein: cellular ATP depletion via downregulation of glyceraldehyde-3-phosphate dehydrogenase. Circ Res. 2006;99:191–200. doi: 10.1161/01.RES.0000232319.02303.8c. [DOI] [PubMed] [Google Scholar]
- Takabe K, Paugh SW, Milstien S, Spiegel S. ‘Inside-out’ signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto T, Jin ZG, Berk BC. Transactivation of vascular endothelial growth factor receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase. J Biol Chem. 2002;277:42997–43001. doi: 10.1074/jbc.M204764200. [DOI] [PubMed] [Google Scholar]
- Tsimikas S, Miller YI. Oxidative modification of lipoproteins: mechanisms, role in inflammation and potential clinical applications in cardiovascular disease. Curr Pharm Des. 2011;17:27–37. doi: 10.2174/138161211795049831. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol Cell Biochem. 2004;264:85–97. doi: 10.1023/b:mcbi.0000044378.09409.b5. [DOI] [PubMed] [Google Scholar]
- Visentin B, Vekich JA, Sibbald BJ, Cavalli AL, Moreno KM, Matteo RG, et al. Validation of an anti-S1P antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell. 2006;9:225–238. doi: 10.1016/j.ccr.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Witztum JL, Steinberg D. Role of oxidized LDL in atherogenesis. J Clin Invest. 1991;88:1785–1792. doi: 10.1172/JCI115499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You B, Ren A, Yan G, Sun J. Activation of sphingosine kinase-1 mediates inhibition of vascular smooth muscle cell apoptosis by hyperglycemia. Diabetes. 2007;56:1445–1453. doi: 10.2337/db06-1418. [DOI] [PubMed] [Google Scholar]
- Yu S, Wong SL, Lau CW, Huang Y, Yu CM. Oxidized LDL at low concentration promotes in vitro angiogenesis and activates NO synthase through PI3K/Akt/eNOS pathway in human coronary artery endothelial cells. Biochem Biophys Res Commun. 2011;407:44–48. doi: 10.1016/j.bbrc.2011.02.096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Toxicity of increasing concentration of oxLDL in HMEC-1 grown on Matrigel. HMEC-1, grown on Matrigel, were incubated for 24 h with increasing concentration of oxLDL or native LDL (natLDL). The cytotoxicity was evaluated (A) by the MTT assay, expressed as % of the untreated (0) control and (B, C) by the live/dead assay. Cells were stained by two fluorescent intercalating compounds, the permeant green-coloured syto13 and the non-permeant red-coloured propidium iodide (PI; under conditions where the red fluorescence prevails over the green fluorescence when the two fluorescent probes enter the nucleus, i.e. cell membrane permeabilized). Living and dying cells were counted on the basis of staining and morphological features. In A, B, mean ± SEM of 6 to 10 separate experiments. Comparison with the untreated control by one-way anova and Holm–Sidak *P < 0.05; ns, not significant. (D) Higher magnification showing the structure of the nucleus. Left panel: living cells exhibit a loose green-coloured chromatin (PI does not enter the normal cell). Middle panel: primary apoptosis is characterized by condensed pyknotic or fragmented nucleus stained green/yellow by Syto13 (at this stage, apoptotic cells are not permeable to PI). During post-apoptotic necrosis, the nucleus exhibits similar apoptotic morphology but is stained red by PI (due to plasma membrane permeabilization in a late step of apoptosis). Right panel: primary necrosis is characterized by an early permeabilization of the plasma membrane allowing PI to enter the cell and stain red the loose chromatin of the nucleus.
Figure S2 Anti-S1P mAb is not toxic to HMEC-1 treated with low concentration of oxLDL. HMEC-1 were grown in 12 multiwell plates in MCDB-131 medium containing 10% FCS, then starved in serum poor (0.1% FCS) medium for 24 h before addition of oxLDL at the indicated concentrations (20 or 50 mg apoB·L−1) and anti-S1P mAb (aS1P, 10 mg·L−1). In A, evaluation by the MTT assay of the whole toxicity of oxLDL (20 and 50 mg·L−1) and aS1P. In B, C, live/dead assays using syto-13 (green) and PI (red) DNA probes, as in A, on HMEC-1 incubated with oxLDL and aS1P. In B, counting of living and dying cells. C, pictures representative of HMEC-1 viability in the presence of oxLDL (at the indicated concentrations) and aS1P. Mean ± SEM of eight separate experiments, *P < 0.05; ns, not significant.
Figure S3 Human oxLDL stimulate angiogenesis in vivo in the Matrigel plug model. (A, B) Angiogenesis in Matrigel plugs. C57/BL6 mice were injected subcutaneously with 0.4 mL Matrigel containing on one flank PBS and on the other flank human oxLDLs. Plugs were removed after 2 weeks and photographied. Representative macrophotographies of plugs containing PBS (control) or oxLDLs (50 mg apoB·L−1). Angiogenesis was quantified by image analysis of the red blood colour using Adobe photoshop software (upper right panel in A and B) or by manual drawing followed by image analysis (lower right panels in A and B).
Figure S4 Comparison of the two methods of quantification of angiogenesis on macrophotographies. A and B, Quantification of angiogenesis in Matrigel plugs obtained by image analysis of blood-coloured vessels using Adobe Photoshop software (A) and by manual drawing followed by image analysis (B). In C, comparison of the data by the two methods.
Figure S5 Effect of anti-S1P mAb on mouse oxVLDL (moV)-induced angiogenesis in the murine Matrigel plug assay. Mice were injected with Matrigel containing 0 or 50 mg apoB·L−1 murine oxVLDL (moV50). Mice were intraperitoneously injected every 3 days with (or without) the anti-S1P mAb (50 mg·kg−1 body weight) for 2 weeks before removing the plugs and quantification of angiogenesis. Each point represents the angiogenesis score in one plug. Mean ± SEM are indicated by the doted line. Comparison of groups was performed by one-way anova followed Holm–Sidak test (SigmaStat sofware) *P < 0.05; ns, not statistically significant.
Figure S6 Effect of oxLDL on VEGF expression (A) and effect of the VEGFR2 inhibitor SU1295 on angiogenesis elicited by oxLDL. (A) Effect of oxLDL on VEGF expression in HMEC-1. Cells were incubated in the presence of oxLDL (20 mg apoB·L−1) or thapsigargin (5 μmol·L−1) at the indicated time. VEGF mRNA level was evaluated by qPCR (normalized to TBP mRNA). (B and C) Effect of the VEGFR2 inhibitor SU1498 on angiogenesis induced by oxLDL (20 mg apoB·L−1) or by S1P (5 μmol·L−1). In A and B, mean ± SEM of six separate experiments, *P < 0.05; ns, not significant. In C, representative microphotographs of cells stained as in Figure 1.
Figure S7 Time course of inhibition of tube formation by trolox added after oxLDL. HMEC-1 were incubated with or without hoxLDL (20 mg apoB·L−1, added at time 0) and trolox (10 μmol·L−1), was added at the indicated time (either at time 0, or 1, 3, 5 or 9 h after oxLDL. Then, tube formation was evaluated at time 18 h (as in Figures 1, 3 and 4). Note that, under the used conditions, trolox added up to 7 h, inhibited tube formation, thus suggesting that ROS generated up to 7 h are required for tube formation. Mean ± SEM of six separate experiments. One-way anova followed by Holm–Sidak post hoc test. *P < 0.05,
Table S1 Evaluation of ROS induced by oxLDL (20 mg apoB·L−1) using various ROS-specific probes (expressed as percent of the unstimulated control).
Table S2 Effects of inhibitors on ROS generation elicited by oxLDLs.