

Abstract
BACKGROUND AND PURPOSE
Palmitoylethanolamide (PEA) acts via several targets, including cannabinoid CB1 and CB2 receptors, transient receptor potential vanilloid type-1 (TRPV1) ion channels, peroxisome proliferator-activated receptor alpha (PPAR α) and orphan G protein-coupled receptor 55 (GRR55), all involved in the control of intestinal inflammation. Here, we investigated the effect of PEA in a murine model of colitis.
EXPERIMENTAL APPROACH
Colitis was induced in mice by intracolonic administration of dinitrobenzenesulfonic acid (DNBS). Inflammation was assessed by evaluating inflammatory markers/parameters and by histology; intestinal permeability by a fluorescent method; colonic cell proliferation by immunohistochemistry; PEA and endocannabinoid levels by liquid chromatography mass spectrometry; receptor and enzyme mRNA expression by quantitative RT-PCR.
KEY RESULTS
DNBS administration caused inflammatory damage, increased colonic levels of PEA and endocannabinoids, down-regulation of mRNA for TRPV1 and GPR55 but no changes in mRNA for CB1, CB2 and PPARα. Exogenous PEA (i.p. and/or p.o., 1 mg·kg−1) attenuated inflammation and intestinal permeability, stimulated colonic cell proliferation, and increased colonic TRPV1 and CB1 receptor expression. The anti-inflammatory effect of PEA was attenuated or abolished by CB2 receptor, GPR55 or PPARα antagonists and further increased by the TRPV1 antagonist capsazepine.
CONCLUSIONS AND IMPLICATIONS
PEA improves murine experimental colitis, the effect being mediated by CB2 receptors, GPR55 and PPARα, and modulated by TRPV1 channels.
Tables of Links
| TARGETS |
|---|
| GPCRsa |
| CB1 receptor |
| CB2 receptor |
| GPR55 |
| Ion Channelsb |
| TRPV1 |
| Nuclear receptorsc |
| PPARα |
| Enzymesd |
| FAAH, fatty acid amide hydrolase |
| NAAA, N-acylethanolamine acid amidase |
| NAPE-PLD, N-arachidonyl-phosphatidylethanolamine PLD |
| LIGANDS |
|---|
| 2-AG, 2-arachidonoylglycerol |
| AM630 |
| Anandamide |
| Capsazepine |
| GW6471 |
| OEA, oleoylethanolamide |
| PEA, palmitoylethanolamide |
| Rimonabant |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,dAlexander et al., 2013a,b,c,d).
Introduction
Inflammatory bowel disease (IBD), which includes Crohn's disease and ulcerative colitis, is a chronic relapsing and destructive inflammatory disorder of the gastrointestinal tract. Significant progress in understanding IBD pathophysiology has led to the development of new therapies that target key molecules and immunological mechanisms (Löwenberg and D'Haens, 2013). However, the corticosteroids and the immunomodulatory drugs, which are the basis of treatment for the IBDs, do not always provide satisfactory outcomes (Burger and Travis, 2011; Ioannidis et al., 2011). Probably for this reason, the use of nutraceutical supplements by patients with IBD is widespread and growing. Recent studies agree that approximately 50% of patients with IBD try non-conventional remedies, which include the use of nutraceuticals (Opheim et al., 2012; Koning et al., 2013).
Palmitoylethanolamide (PEA) is a food component first discovered in the late 1950s when it was shown that the anti-allergic and anti-inflammatory activity exerted by egg yolk, peanut oil or soybean lecithin was due to a specific lipid fraction corresponding to PEA (Esposito and Cuzzocrea, 2013). In Italy and Spain, PEA is actually marketed under the brand name Normast (Epitech Srl, Milan, Italy) as a food component for special medical purposes to alleviate bowel complaints, although randomized clinical trials are lacking. Additionally, in the United States, PEA preparations are promoted for the treatment of IBD (proposed brand name Recoclix, CM&D Phaarma Ltd., Nestlé) (Keppel Hesselink et al., 2013).
Apart from being present in the plant kingdom, PEA is biosynthesized and metabolized by different animal cell types (Keppel Hesselink et al., 2013). Chemically, PEA belongs to the family of acylethanolamides that include the endocannabinoid anandamide and the anorectic mediator oleoylethanolamide (OEA). Anandamide, OEA and PEA share anabolic and catabolic pathways with glycerophosphodiester PDE 1 (GDE1) and N-arachidonyl-phosphatidylethanolamine PLD (NAPE-PLD) being involved in their biosynthesis and fatty acid amide hydrolase (FAAH) in their degradation (Petrosino et al., 2010; Blankman and Cravatt, 2013). Additionally, N-acylethanolamine-hydrolysing acid amidase (NAAA) has been recently identified as a key specific enzyme involved in PEA degradation (Tsuboi et al., 2007). It is now established that PEA is biosynthesized to maintain cellular homeostasis when this is challenged by external stressors provoking inflammation, neuronal damage and pain (Skaper and Facci, 2012). PEA has been identified in the rodent (Capasso et al., 2001; Fu et al., 2007; Izzo et al., 2010; Diep et al., 2011) and human (Darmani et al., 2005; D'Argenio et al., 2007) digestive tract and, given exogenously (i.p.), inhibits intestinal transit in the inflamed gut (Capasso et al., 2001) and reduces intestinal injury caused by ischaemia-reperfusion (Di Paola et al., 2012). Proposed direct or indirect targets for PEA actions include a number of receptors, namely, cannabinoid (CB)1 and CB2 receptors (De Petrocellis et al., 2002; Smart et al., 2002), transient receptor potential vanilloid type-1 (TRPV1) ion channels (Ambrosino et al., 2013), PPARα (Lo Verme et al., 2005) and the orphan receptor GPR55 (Pertwee, 2007), which are involved (in the case of CB1, CB2, TRPV1 and PPARα) or are advocated to be involved (i.e. GPR55) in the mechanisms controlling intestinal inflammation (Cuzzocrea et al., 2004; Izzo and Camilleri, 2009; Holzer, 2011; Schicho et al., 2011).
Because PEA acts on key targets regulating intestinal inflammation and in the light of the widespread use of PEA-containing over-the-counter preparations for the relief of intestinal complaints, including IBD, we have investigated here the effect and the mode of action of this acylethanolamide in a murine model of colitis. Preliminary accounts of some of these results have been communicated to the XXXVI National Congress of the Italian Pharmacological Society (Petrosino et al., 2013).
Methods
Animals
All animal care and experimental procedures complied with the principles of laboratory animal care (NIH publication no.86-23, revised 1985) and the Italian D.L. no.116 of 27 January 1992 and associated guidelines in the European Communities Council Directive of 24 November 1986 (86/609/ECC). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 402 animals were used in the experiments described here.
Male ICR mice, weighing 25–30 g, were purchased by Harlan Italy (Corezzana, Milan, Italy) and housed in polycarbonate cages in isolators under a 12 h light/12 h dark cycle, temperature 23 ± 2°C and humidity 60%. Mice were fed ad libitum with standard food, except for the 24 h period immediately preceding the administration of 2,4,6-dinitrobenzenesulfonic acid (DNBS) and for the 2 h period preceding the administration of p.o. PEA.
Induction of experimental colitis and pharmacological treatment
Colitis was induced by the intracolonic administration of DNBS (Romano et al., 2013). Briefly, mice were anaesthetized with inhaled 5% isoflurane (Centro Agrovete Campania, Scafati, SA, Italy) and subsequently DNBS (150 mg·kg−1) was injected into the colon using a polyethylene catheter (1 mm in diameter) via the rectum (4.5 cm from the anus). Control mice received an intracolonical administration of water. Three days after DNBS or water administration, all animals were killed by asphyxiation with CO2, the mice abdomen was opened by a midline incision and the colon removed, isolated from surrounding tissues, opened along the anti-mesenteric border, rinsed, weighed and the length measured in order to determine the colon weight/colon lenght ratio (mg·cm−1) used as an indirect marker of inflammation. The body weight of mice was measured every day throughout the treatment period. All measurements were performed by workers who were unaware of the particular treatment. For biochemistry analysis, tissues were kept at −80°C until use, whereas for histological examination tissues were fixed in 10% formaldehyde. The dose of DNBS was selected on the basis of preliminary experiments showing colonic damage associated with high reproducibility and low mortality for a 150 mg·kg−1 dose. The time point of damage evaluation (i.e. 3 days after DNBS administration) was chosen because maximal DNBS-induced inflammation has been reported in mice after 3 days (Massa et al., 2004). PEA (0.1–10 mg·kg−1) was administered i.p. or p.o. three times (once a day) for three consecutive days starting 24 h after DNBS or vehicle inoculation (therapeutic protocol). Animals were killed 2 h after the third administration of PEA. In some experiments, the effect of PEA (1 mg·kg−1, p.o.) was evaluated in animals pretreated every day (i.p., 30 min before PEA) with the CB1 receptor antagonist rimonabant (3 mg·kg−1), the CB2 antagonist AM630 (10 mg·kg−1), the PPARα antagonist GW6471 (1 mg·kg−1), the GPR55 antagonist ML-191 (0.5 mg·kg−1) or the TRPV1 channels antagonist capsazepine (10 mg·kg−1). The doses of rimonabant, AM630, GW6471 and capsazepine were selected on the basis of previous published work concerning gastrointestinal pharmacology (De Schepper et al., 2008; Alhouayek et al., 2011; Capasso et al., 2014). The dose of ML-191 was selected on the basis of preliminary experiments showing that the antagonist, at 0.5 mg·kg−1 dose, did not affect, per se, DNBS-induced intestinal inflammation. A higher dose of ML-191 (1 mg·kg−1) affected, per se, DNBS-induced colitis (see Results).
Histology and immunohistochemistry
All histological and immunochemical evaluation was carried out without knowledge of the treatments. Such evaluations, performed 3 days after DNBS administration, were assessed on a segment of 1 cm of colon taken from 4 cm above the anal canal. After fixation for 24 h in saline 10% formaldehyde, samples were dehydrated in graded ethanol and embedded in paraffin. Thereafter, 5 mm sections were deparaffinized with xylene, stained with haematoxylin–eosin and observed in a DM 4000 B Leica microscope (Leica Microsystems, Milan, Italy). Microscopic scoring was performed as previously reported (Romano et al., 2013). Briefly, the colon was scored considering (i) submucosal infiltration (0, none; 1, mild; 2–3, moderate; 4–5 severe); (ii) crypt abscesses (0, none; 1–2 rare; 3–5, diffuse); and (iii) mucosal erosion (0, absent; 1, focus; 2–3, extended until the middle of the visible surface; 4–5, extended until the entire visible surface).
For immunohistochemical detection of Ki-67, paraffin-embedded slides were immersed in a Tris/EDTA buffer (pH 9.0), heated in a decloaking chamber at 125°C for 3 min and cooled at room temperature for 20 min. Sections were incubated for 10 min with 3% hydrogen peroxide, washed with Tris-buffered saline Tween 20 (Sigma-Aldrich, Milan, Italy) (pH 7.6) and incubated with rabbit monoclonal antibody to Ki-67 (1:100, Ventana Medical Systems, Tucson, AZ, USA) for 30 min at room temperature. The slides were washed three times with Tris-buffered saline Tween 20 and incubated with secondary antibody for 30 min. After, the slides were reacted with streptavidin for 20 min and 3,30-diaminobenzidine tetrahydrochloride for 5 min. Finally, the slides were counterstained with Mayer's haematoxylin. The intensity and localization of immunoreactivities against the primary antibody were examined on all sections with a microscope (Leica Microsystems). To ensure specificity of the immunohistochemical procedure, some tissues were processed without primary antibody to Ki-67.
Intestinal permeability assay
Intestinal permeability was evaluated using a FITC-labelled dextran method, as described previously (Borrelli et al., 2013). Briefly, 2 days after DNBS administration, mice were gavaged with 600 mg·kg−1 body weight of FITC-conjugated dextran (molecular mass 3–5 kDa). One day later, blood was collected by cardiac puncture, and the serum was immediately analysed for FITC-derived fluorescence (fluorescent microplate reader with 2104 EnVision Multilabel Plate Readers, [PerkinElmer Instruments, Walthan, MA, USA]; excitation wavelengths 485 ± 14 nm, emission wavelengths 520 ± 25 nm). Serial-diluted FITC dextran was used to generate a standard curve. Intestinal permeability was expressed as the concentrations of FITC (μM) found in the serum.
Myeloperoxidase (MPO) activity
MPO activity, a marker used to quantify the extent of neutrophil accumulation in whole-tissue colons, was determined as previously described (Borrelli et al., 2013). Full-thickness colons were homogenized in an appropriate lysis buffer (0.5% hexadecyltrimethylammonium bromide in 3-(N morpholino)propanesulfonic acid [MOPS] 10 mM) in the ratio of 50 mg tissue per mL MOPS. The samples were then centrifuged for 20 min at 15 000× g at 4°C. An aliquot of the supernatant was then incubated with NaPP (sodium phosphate buffer pH 5.5) and tetra-methylbenzidine 16 mM. After 5 min, H2O2 (9.8 M) in NaPP was added and the reaction stopped with acetic acid. The rate of change in absorbance was measured by a spectrophotometer at 650 nm. Different dilutions of human MPO enzyme of known concentration were used to obtain a standard curve. MPO activity was expressed as U·mg−1 of tissue.
Quantitative (real-time) RT-PCR analysis
The colons from animals treated with vehicles (control group), PEA, DNBS or DNBS plus PEA were removed (3 days after the administration of DNBS or water), collected in RNA later (Invitrogen, Carlsbad, CA, USA) and homogenized by a rotorstator homogenizer in 1.5 mL of Trizol® (Invitrogen). Total RNA was purified, quantified, characterized and retro-transcribed as previously described (Grimaldi et al., 2009). For all samples tested, the RNA integrity number (Bionalyzer 2100, Agilent) was greater than eight relative to a 0–10 scale. Quantitative real-time PCR was performed by an iCycler-iQ5® (Bio-Rad, Milan, Italy) in a 20 μL reaction mixture as described. Assays were performed in quadruplicate (maximum ΔCt of replicate samples <0.5), and a standard curve from consecutive fivefold dilutions (100–0.16 ng) of a cDNA pool representative of all samples was included for PCR efficiency determination. Optimized primers for SYBR Green analysis and optimum annealing temperatures were designed by the Allele-Id software version 7.0 (Biosoft International, Palo Alto, CA, USA) and were synthesized (HPLC purification grade) by MWG-Biotech (Ebersberg, Germany). For each target, all mRNA sequences at http://www.ncbi.nlm.nih.gov/gene/ were aligned and common primers were designed (see Supporting Information Table S1 for primer sequences). Relative expression calculation, correct for PCR efficiency and normalized with respect to reference genes β-actin and hypoxanthine-guanine phosphoribosyltransferase, was performed by the iQ5 software. Results are expressed as fold expression, compared with control (=1) (Aviello et al., 2012). Statistical significance was evaluated by the REST 2009 software (Pfaffl et al., 2002).
Identification and quantification of PEA, endocannabinoids and OEA
Full-thickness colons from animals receiving DNBS or vehicle (treated or not with PEA 1 mg·kg−1, p.o.) were removed (3 days after DNBS administration), immediately immersed into liquid nitrogen and stored at −80°C until extraction of endocannabinoids [anandamide and 2-arachidonoylglycerol (2-AG)] and related acylethanolamides (PEA and OEA). Tissues were extracted with chloroform/methanol (2:1, by volume) containing each 10 pmol of d8-anandamide, d4-PEA, d4-OEA and d5-2-AG, synthesized as described previously (for the former two compounds) (Di Marzo et al., 2008), or provided by Cayman Chemicals (for d5-2-AG, Ann Arbor, MI, USA). The lipid extracts were purified by silica column chromatography and the fractions containing anandamide, PEA and 2-AG were analysed by isotope dilution liquid chromatography–atmospheric pressure chemical ionization mass spectrometry (LC-MS). Results were expressed as picomoles per milligram of tissue.
Data analysis
Data are expressed as the mean ± SEM of n experiments. To determine statistical significance, Student's t-test was used for comparing a single treatment mean with a control mean, and a one-way anova followed by a Tukey–Kramer multiple comparisons test was used for the analysis of multiple treatment means. Values of P less than 0.05 were considered significant.
Materials
Ultramicronized PEA was kindly provided by Epitech Group (Saccolongo, Italy) and rimonabant by SANOFI Recherche, Montpellier, France. DNBS hydrate, MPO from human leucocytes and FITC-conjugated dextran (molecular mass 3–5 kDa) were purchased from Sigma Aldrich S.r.l. (Milan, Italy). Capsazepine, GW6471 and AM630 were supplied by Tocris (Space Import-Export SrL, Milan, Italy) and ML-191 by Cayman (Cabru SAS, Arcore, Italy). All reagents for cell culture and Western blot analysis were obtained from Sigma Aldrich S.r.l., Amersham Biosciences Inc. (Piscataway, NJ, USA), Bio-Rad Laboratories (Milan, Italy) and Microtech S.r.l. (Naples, Italy). All chemicals and reagents employed in this study were of analytical grade.
PEA was dissolved in ethanol/Tween 20/saline (1:1:8, 60 μL per mouse) for i.p. injection or suspended in carboxymethyl cellulose (1.5%, 150 μL per mouse) for p.o. administration. Rimonabant, ML-191 and capsazepine were dissolved in ethanol/Tween 20/saline (1:1:8, 60 μL per mouse), AM630 in DMSO/Tween 20/saline (1:1:8, 60 μL per mouse). DNBS was dissolved in 50% ethanol (0.15 mL per mouse). All vehicles had no significant effects on the responses under study.
Results
PEA reduced the impairment in body weight gain induced by colitis
Compared with control animals, DNBS administration caused significant weight loss. PEA (0.1–10 mg·kg−1; i.p., Figure 1A or p.o., Figure 1B), administered after the inflammatory insult, reduced in a dose-dependent manner the loss of body weight induced by colitis, the effect being significant starting from the 0.3 mg·kg−1 (i.p.) or 1 mg·kg−1 (p.o.) doses (Figure 1A and B).
Figure 1.

DNBS-induced colitis in mice. Changes in body weight (A,B) and colon weight/colon length ratio (C,D) from control and DNBS-treated mice in the presence or absence of i.p. (A,C) or p.o. (B,D) PEA. Mice were weighed before DNBS (or vehicle) administration and immediately before killing. Tissues were analysed 3 days after vehicle or DNBS administration. PEA (0.1–10 mg·kg−1) was administered once a day for three consecutive days starting 24 h after the inflammatory insult (therapeutic protocol). Bars are mean ± SEM of 12–15 mice for each experimental group. #P < 0.001 versus control (i.e. mice without intestinal inflammation). *P < 0.05, **P < 0.01 and ***P < 0.001 versus DNBS alone. The insert reports the percentage of inhibition of colon weight/colon length ratio after i.p. or p.o. PEA administration and demonstrates that i.p. PEA was significantly (P < 0.001) more active than p.o. PEA in the experimental model of colitis induced by DNBS.
PEA-reduced colon weight/colon lenght ratio
Intracolonic DNBS administration caused inflammatory damage, as indicated by the approximately 2.5-fold increase in colon weight/colon length ratio (a simple and reliable marker of intestinal inflammation and/or damage) (Figure 1C and D). PEA (0.1–10 mg·kg−1; i.p., Figure 1C or p.o., Figure 1D), administered after the inflammatory insult, significantly and in a dose-dependent manner, reduced the effect of DNBS on colon weight/colon length ratio. Although a significant inhibition of this ratio was achieved starting from the PEA 0.3 mg·kg−1 dose, irrespective of the route of administration, analysis of the curves representing the percentage of inhibition of inflammation showed a greater anti-inflammatory effect of PEA when given i.p. (see inset to Figure 1).
To confirm the anti-inflammatory activity of PEA, we performed MPO activity measurement, histological analysis, intestinal permeability measurement and immunohistochemistry using the submaximal dose of PEA of 1 mg·kg−1.
PEA reduced microscopic damage
Histological analysis of colonic mucosa of control mice showed an intact, unbroken epithelium (Figure 2A and B). In the DNBS group, colons showed tissue injury, as revealed by necrosis involving the full thickness of the mucosa, infiltrations of granulocytes into the mucosa/submucosa, oedema of submucosa and intramuscular and subserosal inflammation (Figure 2C and D). PEA (1 mg·kg−1, i.p. and p.o.) reduced the signs of colon injury. In the colon of animals given PEA (1 mg·kg−1) (Figure 2E and F), the oedema and the erosion areas were reduced in the mucosa and in the submucosa [microscopic score: control, 0 ± 0; DNBS, 9.2 ± 0.6#; DNBS + PEA (i.p.) 3.3 ± 1.0**; DNBS + PEA (p.o.) 5.2 ± 0.5**; n = 4 for each experimental group; #P < 0.01 vs. control; **P < 0.01 vs. DNBS].
Figure 2.
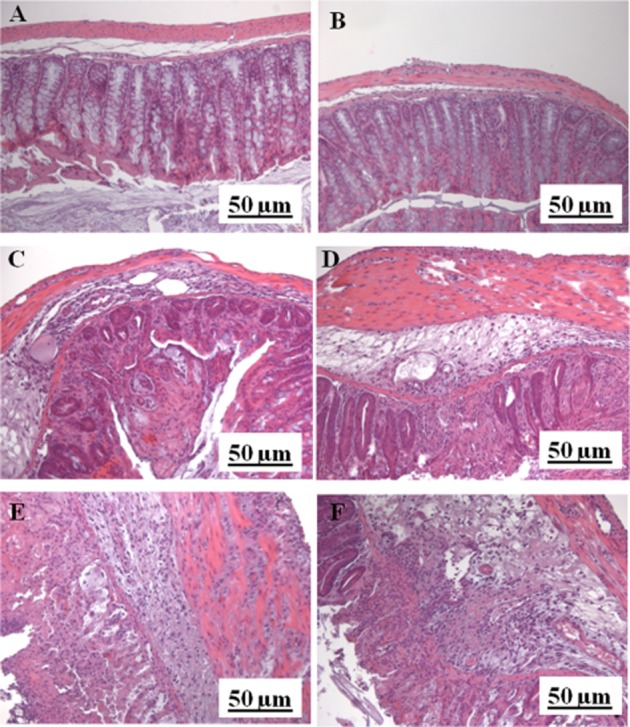
Histological evaluations of inflamed (DNBS treated) and non-inflamed colons: effect of PEA. No histological modification was observed in the mucosa and submucosa of control mice receiving the vehicle used to dissolve PEA i.p. (A) or p.o. (B); mucosal injury induced by DNBS administration in mice receiving the vehicle used to dissolve PEA i.p. (C) or p.o. (D); i.p. (E) and p.o. (F) PEA reduced the erosion and the inflammation area in the mucosa and in the submucosa. Histological analysis was performed 3 days after DNBS administration. PEA (1 mg·kg−1) was administered once a day for three consecutive days starting 24 h after the inflammatory insult (therapeutic protocol). Original magnification ×200. The Figure shows results representative of four experiments.
PEA reduced MPO activity
MPO activity is considered to be an index of neutrophil infiltration and it is largely used to quantify intestinal inflammation (Krawisz et al., 1984). DNBS-induced colitis was associated with a significant increase in MPO activity, which was reduced by i.p. (Figure 3A, 60% inhibition) or p.o. (Figure 3B, 64% inhibition) administration of PEA (1 mg·kg−1).
Figure 3.

Inhibitory effect of PEA on MPO (a marker of intestinal inflammation) activity (A,B) and on serum FITC–dextran concentration (a measure of intestinal permeability; C,D) in DNBS-induced colitis in mice. Permeability and MPO activity were measured on colonic tissues 3 days after vehicle or DNBS administration. PEA [1 mg·kg−1, i.p. (A,C) or p.o. (B,D)] was administered once a day for three consecutive days starting 24 h after the inflammatory insult (therapeutic protocol). Bars are mean ± SEM of four to five mice for each experimental group. #P < 0.001 versus control; **P < 0.01 and ***P < 0.001 versus DNBS alone.
PEA reduced intestinal permeability
Intracolonic administration of DNBS increased intestinal permeability, as revealed by the abundant concentration of FITC-conjugated dextran in the serum. i.p. (Figure 3C) or p.o. (Figure 3D) PEA (1 mg·kg−1) partially counteracted DNBS-induced increase in intestinal permeability (57 and 47% inhibition, for i.p. and p.o. PEA administration respectively).
PEA stimulated colonic cell regeneration, as shown by immunohistochemistry
The action of PEA was further confirmed by immunohistochemistry. In normal colonic mucosa, the predominant area of cell proliferation was localized to the lower part of the crypts as revealed by Ki-67 distribution (Figure 4A and B). In the colon from DNBS-treated mice, total necrosis with Ki-67 immunoreactivity on inflammatory cells and in a few remaining surface elements was observed (Figure 4C and D). PEA (1 mg·kg−1, i.p. and p.o.) partially counteracted the effect of DNBS on cell proliferation, its mitotic activity being restricted to the two-thirds of the mucosa (i.e. the mature superficial cells were not in a proliferative state) (Figure 4E and F).
Figure 4.
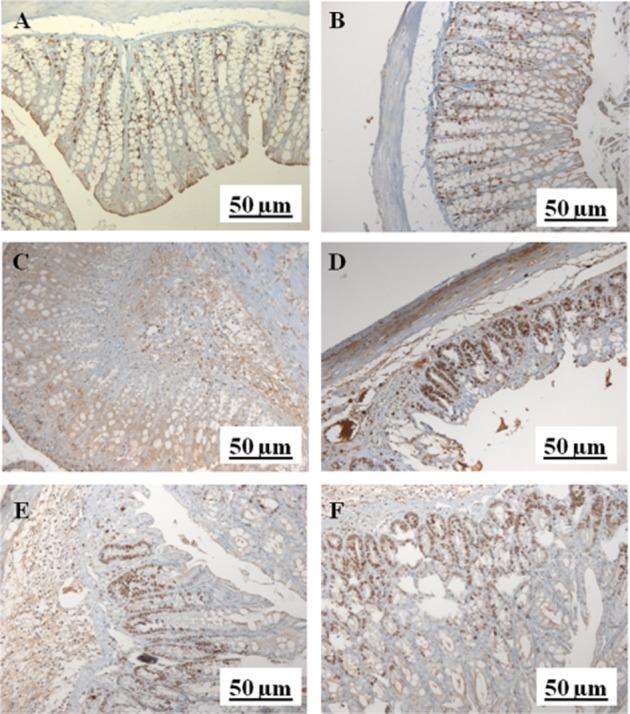
Different patterns of Ki-67 immunoreactivity in the colonic mucosa of control mice [i.e. mice receiving the vehicle used to dissolve PEA i.p. (A) or p.o. (B)], DNBS-treated mice receiving the vehicle used to dissolve PEA [i.p. (C) or p.o. (D)] and mice treated with DNBS plus PEA [i.p. (E) or p.o. (F)]. (A,B) Ki-67-immunopositive cells were localized to the lower part of the crypts. (C,D) Ki-67-immunopositive cells were extended to the superficial part of the crypts. (E,F) Ki-67-immunopositive cells were observed in the two-thirds of the mucosa, only. PEA (1 mg·kg−1) was administered for three consecutive days starting 24 h after the inflammatory insult (therapeutic protocol). The Figure shows results representative of four experiments.
DNBS did not change mRNA expression of enzymes involved in PEA metabolism
In this set of experiments, we verified if changes in PEA levels were accompanied by variations in the mRNA expression of enzymes involved in PEA metabolism. Compared with control mice, DNBS administration did not significantly change the mRNA expression of GDE1, NAPE-PLD (two enzymes involved in PEA biosynthesis), FAAA and NAAA (two enzymes involved in PEA degradation) (Figure 5).
Figure 5.
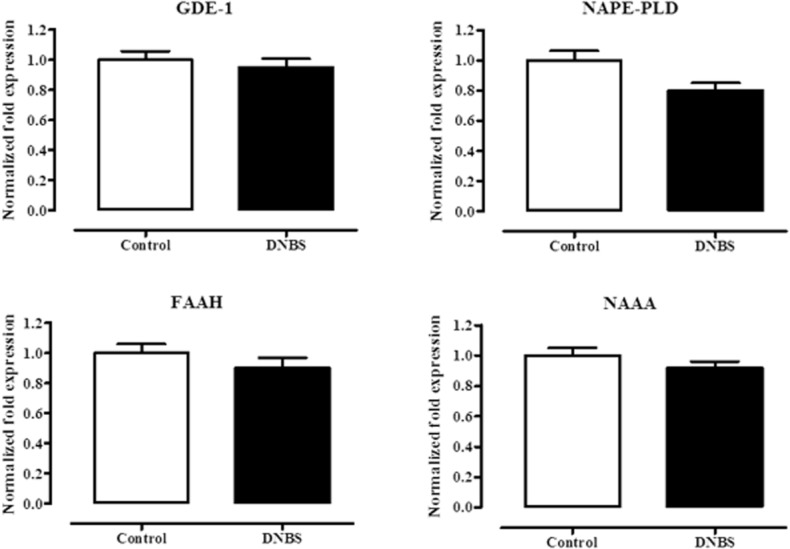
Relative mRNA expression of GDE1, NAPE-PLD, FAAH and NAAA in the colon of control and DNBS-treated mice. Tissues were analysed 3 days after vehicle or DNBS administration. RT-PCR analysis was performed as described in Methods. Results (expressed as fold expression, compared with control, as unity) are mean ± SEM of six experiments. No significant effects of DNBS were found.
DNBS increased colonic PEA levels
Isotope dilution LC-MS analysis of lipid extracts from the colon of control and DNBS-treated mice showed that the endogenous levels of PEA significantly increased (by 2.5-fold) after DNBS administration, compared with control colons (Figure 6A). OEA, another acylethanolamide chemically related to PEA, did not significantly change after DNBS administration (Figure 6B).
Figure 6.
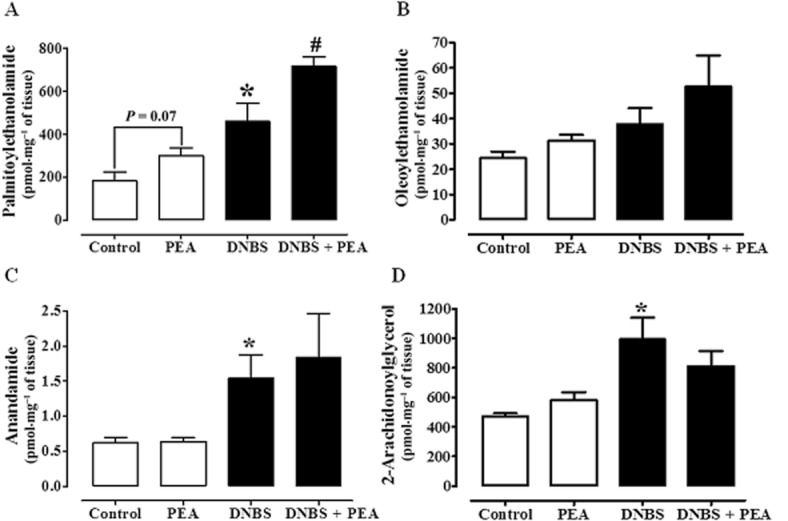
PEA (A), oleoylethanolamide (B), anandamide (C) and 2-arachydonoylglycerol (D) levels in colon of control mice and mice treated intracolonically with the pro-inflammatory agent DNBS: effect of PEA. Tissues were analysed 3 days after vehicle or DNBS administration. PEA (1 mg·kg−1) was administered p.o. once a day for three consecutive days starting 24 h after the inflammatory insult. Data are mean ± SEM of four to five mice. *P < 0.05 versus control; #P < 0.05 versus DNBS alone.
Oral PEA did not change the colonic levels of endocannabinoids
This set of experiments was performed because there is evidence in the literature that PEA can inhibit anandamide inactivation (Bisogno et al., 1997; Di Marzo et al., 2001) and thus indirectly activate endocannabinoid targets (i.e. CB receptors and TRPV1). As previously reported (Borrelli and Izzo, 2009; Borrelli et al., 2009), the levels of both anandamide and 2-AG, compared with control colonic tissues, were significantly increased by treatment with DNBS (2.5-fold increase for anandamide and 2.1-fold increase for 2-AG) (Figure 6C and D). However, PEA (1 mg·kg−1, p.o.) treatment did not affect OEA and endocannabinoid levels, either in control or DNBS-treated mice (Figure 6B–D). As expected, mice receiving p.o. PEA showed higher colonic levels of this acylethanolamide, the effect being significant in DNBS-treated animals (Figure 6A).
Oral PEA affected colonic CB1, GPR55 and TRPV1 (but not CB2 or PPARα) mRNA expression
In these experiments, we measured the mRNA expression of the receptors that are known to be directly or indirectly targeted by PEA. DNBS administration caused colonic down-regulation of GPR55 and TRPV1 mRNA expression, with no changes in CB (CB1 and CB2) receptors and PPARα mRNA expression (Figure 7). PEA administration (1 mg·kg−1, p.o.) counteracted (or partially counteracted) DNBS-induced GPR55 and TRPV1 down-regulation, while it up-regulated, both in control and in DNBS-treated mice, CB1 receptor mRNA expression (Figure 7).
Figure 7.
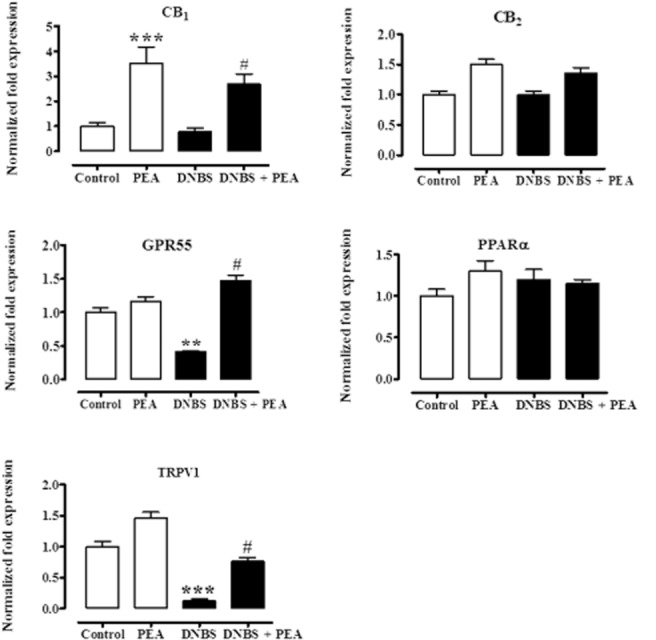
Relative mRNA expression of CB1 receptors, CB2 receptors, GPR55, PPARα, and TRPV1 channels in the DNBS model of colitis: effect of PEA. (PEA, 1 mg·kg−1) was administered p.o. once a day for three consecutive days starting 24 h after the inflammatory insult. Tissues were analysed 3 days after vehicle or DNBS administration. RT-PCR analysis was performed as described in Methods. Results (expressed as fold expression, compared with control equal to one) are mean ± SEM of six experiments. **P < 0.01 and ***P < 0.001 versus control; #P < 0.05 versus DNBS.
The anti-inflammatory action of oral PEA involved CB2 receptors, GPR55 and PPARα
In order to investigate the mode of action of PEA, we evaluated its pharmacological effects in the presence of rimonabant (CB1 receptor antagonist), AM630 (CB2 receptor antagonist), ML-191 (GPR55 antagonist), GW6471 (PPARα antagonist) and capsazepine (TRPV1 channel antagonist). Results showed that AM630, ML-191 and GW6471 abolished or reduced the anti-inflammatory effects of PEA, as evaluated by the colon weight/colon length ratio (Figure 8A–E) and MPO activity (Figure 9A–E). The reversal by the antagonists was significant, except for GW647, which significantly counteracted the effect of PEA on MPO activity (Figure 9D), but not on colon weight/colon length ratio (Figure 8D, P = 0.06). By contrast, capsazepine further increased the anti-inflammatory effect of PEA (Figure 8E and Figure 9E). The antagonists, at the doses used in the present investigation, did not modify, per se, DNBS-induced colitis. However, a higher dose of the GPR55 antagonist ML-191 (i.e. 1 mg·kg−1), per se, slightly, but significantly, ameliorated DNBS-induced colitis, as revealed by the colon weight/colon length ratio [weight:·length ratio (mg·cm−1): control 23.7 ± 3.1, DNBS 75.3 ± 3.2#, DNBS + ML-191 63.4 ± 3.7*; n = 7 for each experimental group. #P < 0.001 vs. control; *P < 0.05 vs. DNBS alone].
Figure 8.
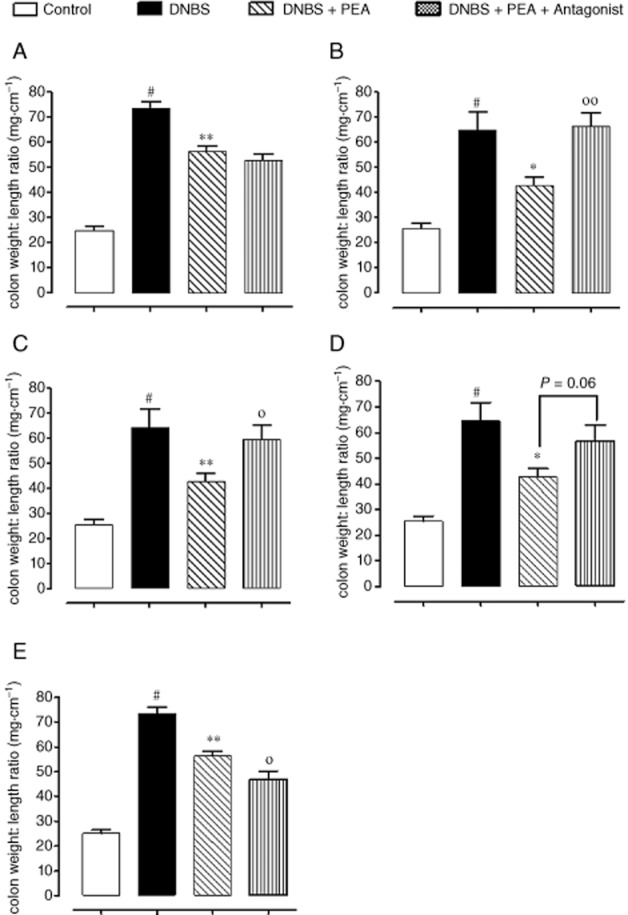
Effect of PEA alone or in the presence of the CB1 receptor antagonist rimonabant (3 mg·kg−1, i.p.) (A), the CB2 receptor antagonist AM630 (10 mg·kg−1, i.p.) (B), the GPR55 receptor antagonist ML-191 (0.5 mg·kg−1, i.p.) (C), the PPARα receptor antagonist GW6471 (1 mg·kg−1, i.p.) (D), or the TRPV1 antagonist capsazepine (10 mg·kg−1, i.p.) (E) on colon weight/colon length ratio in mice with experimental colitis induced by DNBS. PEA (1 mg·kg−1, p.o.) was administered once a day for three consecutive days starting 24 h after the inflammatory insult. The antagonists were given 30 min before PEA administration. Tissues were analysed 3 days after vehicle or DNBS administration. #P < 0.01–0.001 versus control, *P < 0.05 and **P < 0.01 versus DNBS, °P < 0.05 and °°P < 0.01 versus DNBS plus PEA (n = 6–8 mice for each experimental group).
Figure 9.
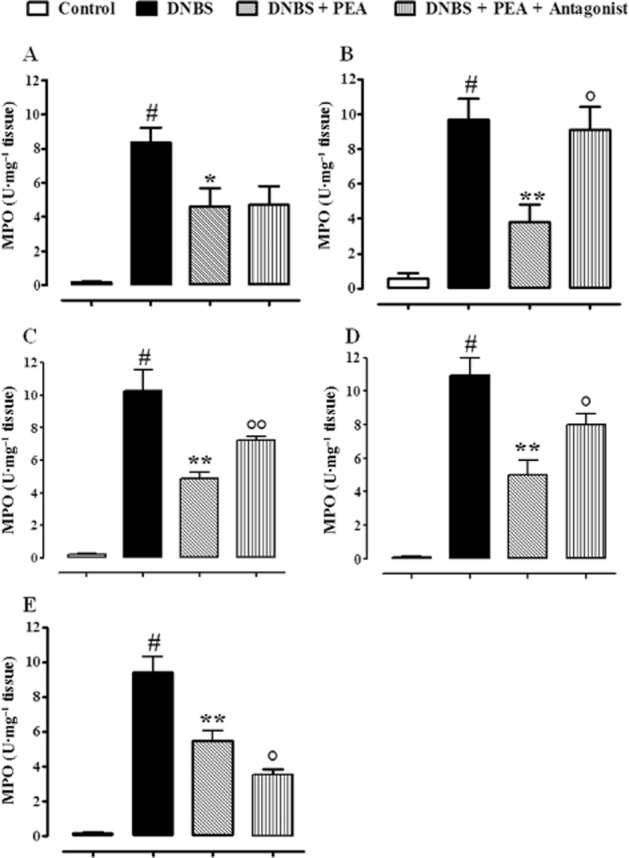
Effect of PEA alone or in the presence of the CB1 receptor antagonist rimonabant (3 mg·kg−1, i.p.) (A), the CB2 receptor antagonist AM630 (10 mg·kg−1, i.p.) (B), the GPR55 receptor antagonist ML-191 (0.5 mg·kg−1, i.p.) (C), the PPARα receptor antagonist GW6471 (1 mg·kg−1, i.p.) (D) or the TRPV1 antagonist capsazepine (10 mg·kg−1, i.p.) (E) on MPO activity in the murine model of colitis induced by DNBS. PEA (1 mg·kg−1, p.o.) was administered once a day for three consecutive days starting 24 h after the inflammatory insult. The antagonists were given 30 min before PEA administration. MPO activity was measured on colonic tissues 3 days after vehicle or DNBS administration (therapeutic protocol). Bars are mean ± SEM of four to five mice for each experimental group. #P < 0.01–0.001 versus control, *P < 0.05 and **P < 0.01 versus DNBS, °P < 0.05 and °°P < 0.01 versus DNBS plus PEA.
Discussion
PEA is a naturally occurring acylethanolamide considered to play protective and homeodynamic roles in the animal and vegetable kingdoms (Esposito and Cuzzocrea, 2013). The anti-inflammatory, analgesic and anti-convulsant properties of this amide are well established and are believed to be of potential therapeutic interest (Skaper and Facci, 2012). Although PEA-containing formulations are promoted for the treatment of intestinal complaints, the actions of this acylethanolamide in the digestive tract have been largely overlooked to date. In the present study, we show that endogenous PEA is produced in the colon in response to an inflammatory insult and that exogenous-administered PEA exerts anti-inflammatory actions in the gut.
We have found that PEA, given i.p. or p.o., reduced the colon weight/colon length ratio of the inflamed colonic tissue, which is considered a reliable and sensitive indicator of the severity and extent of the inflammatory response. PEA was effective when given after the inflammatory insult, suggesting a therapeutic beneficial effect. Furthermore, PEA was significantly more active when given i.p. than when given p.o., a result that can be explained by the presence, in the digestive tract, of NAAA and other amidases able to metabolize PEA (Tsuboi et al., 2007; Borrelli and Izzo, 2009). The effect of PEA was further supported by its ability to (i) reduce the decrease in body weight associated to DNBS administration, thus suggesting a favourable selective effect of this compound on overall mouse health in inflammatory conditions; (ii) decrease the histological signs of colon injury; (iii) lessen MPO activity, a well-established marker of neutrophil infiltration in mouse models of colitis (Krawisz et al., 1984); (iv) partially restore the impaired intestinal permeability; and (v) limit the colonic diffusion of Ki-67, a useful marker for the evaluation of dysplasia in ulcerative colitis (Andersen et al., 1998), as shown by immunohistochemistry, although we cannot exclude the possibility that PEA effect on Ki-67 could be an indirect effect related to its anti-inflammatory action. Previously, it has been shown that PEA (i.p.) significantly reduced intestinal transit in intestinal inflammatory (Capasso et al., 2001) and post-inflammatory conditions (Capasso et al., 2014) as well as the intestinal injury due to ischaemia reperfusion (Di Paola et al., 2012). During the preparation of the present manuscript, others have shown that PEA in vitro exerted anti-inflammatory effects in colonic biopsies from IBD patients and ameliorated dextran sodium sulphate (DSS)-induced colitis when given i.p. to mice (Esposito et al., 2014), an effect abolished by a PPARα, but not a PPARγ, antagonist. Compared with the results obtained by Esposito et al. (2014), we have used a different model of colitis, demonstrated the efficacy of PEA after p.o. administration and, importantly, broadened the investigation of PEA mode of action, by evaluating the potential involvement of endocannabinoids, CB receptors, TRPV1, GPR55 and PPARα (results discussed below).
PEA is biosynthesized and metabolized in animal cells via a number of enzymes which, in many instances, are also involved in the biosynthesis and degradation of the endocannabinoid anandamide and of the anorectic mediator OEA (Ueda et al., 2013). The presence of PEA in the gut is well documented and there is evidence that its intestinal levels may change in response to noxious stimuli (Borrelli and Izzo, 2009; Balvers et al., 2013): for example, PEA has been shown to increase in the duodenum of rats treated with methotrexate (a celiac disease-like model of intestinal atrophy) (D'Argenio et al., 2007) and to decrease in the small intestine of mice treated with the pro-inflammatory agent croton oil (Capasso et al., 2001; Izzo et al., 2012). In the present study, we have observed a nearly threefold increase in colonic PEA levels in mice with experimental colitis, compared with control animals, with no changes in the mRNA expression of GDE1 and NAPE-PLD (two enzymes involved in PEA biosynthesis) as well as in the expression of NAAA and FAAH (two enzymes involved in PEA degradation). Relevant to this point is noteworthy that, although it is well established that FAAH inhibition results in intestinal anti-inflammatory effects (D'Argenio et al., 2006; Storr et al., 2008), there are no data in the literature concerning the effect of NAAA inhibitors in experimental models of colitis. NAAA inhibitors have been recently shown to attenuate tissue reactions to various pro-inflammatory stimuli, both in vitro and in vivo (Solorzano et al., 2009; 2010; Li et al., 2012). The increased levels of PEA in the present study was specific among the acylethanolamides, as the levels of OEA, which is co-released together with PEA, did not change significantly. Importantly, PEA levels are also elevated in patients with ulcerative colitis (Darmani et al., 2005), thus confirming that the model used in the present study can be translated to the human condition, at least as far as the regulation of this mediator is concerned.
In mechanistic terms, there is evidence that PEA activates PPARα (Lo Verme et al., 2005; O'Sullivan and Kendall, 2010) and the orphan receptor GPR55 (Pertwee, 2007) and activates and desensitizes TRPV1 channels in sensory neurons (Ambrosino et al., 2013). Additionally, PEA inhibits the inactivation of the endocannabinoid anandamide (Bisogno et al., 1997; Di Marzo et al., 2001) and potentiates the effect of anandamide at CB receptors or TRPV1 channels (the ‘entourage effect’) (De Petrocellis et al., 2002; Smart et al., 2002; Ho et al., 2008). That said, a further step in our study was to evaluate the mRNA expression of such receptors in the colon of both control and DNBS-treated mice and, more importantly, to evaluate the pharmacological effect of PEA in the presence of the antagonists of the above-mentioned targets. Because the p.o. route of administration is desirable in IBD patients and because PEA is contained in over-the-counter preparations promoted for the treatment of intestinal complaints such as IBD, the experiments, which are discussed below, were performed by administering PEA p.o.
Persuasive evidence suggests that activation of both CB1 and CB2 receptors results in anti-inflammatory effect in the gut (Izzo and Camilleri, 2009; Alhouayek and Muccioli, 2012). In the present study, we have shown that the effect of PEA in the DNBS model of colitis was counteracted by a CB2 receptor antagonist, suggesting an involvement of these receptors in PEA mode of action. Because PEA does not bind efficiently to CB2 receptors (Lambert and Di Marzo, 1999), it is possible that PEA might activate such receptors via the so-called entourage effect, that is, the increase of the endocannabinoid levels and/or modulatory actions at CB receptors (De Petrocellis et al., 2002; Smart et al., 2002). Indeed, we found that DNBS administration caused an increase in endocannabinoid levels, which, however, were not further increased by PEA, probably because they were already maximally increased by the inflammatory insult. Pharmacological effects of PEA counteracted by CB2 receptor antagonists have been previously documented (Calignano et al., 1998; Farquhar-Smith et al., 2002). Conversely, we found that, although PEA up-regulated colonic CB1 mRNA expression, a mechanism which is known to potentially exert anti-inflammatory effects in animal model of colitis (Di Marzo and Izzo, 2006), the CB1 receptor antagonist rimonabant did not change the anti-inflammatory effect of PEA.
The orphan receptor GPR55 has been proposed to mediate some of the pharmacological actions of CBs (Pertwee, 2007; Ryberg et al., 2007; Naderi et al., 2012). Similar to CB1 and CB2 receptors, GPR55 is expressed throughout the rodent gastrointestinal tract (Schicho et al., 2011; Schicho and Storr, 2012). In the present study, we have shown that the recently developed GPR55 antagonist ML-191 (Kotsikorou et al., 2013), at a dose (i.e. 0.5 mg·kg−1) which, per se, did not affect intestinal inflammation, reduced the beneficial effect of PEA in the DNBS model of colitis. The involvement of GPR55 was further supported by the observation that PEA restored GPR55 mRNA down-regulation caused by DNBS. Others have found that GPR55 mRNA expression decreased in the model of pancreatitis induced by cerulein in mice (Li et al., 2013) and increased in the intestine of rats challenged with LPS (Lin et al., 2011).
It is well established that PPARα has a role in controlling mucosal tissue homeostasis and PPARα agonists ameliorate the experimental colitis induced by DNBS and DSS (Cuzzocrea et al., 2004; Azuma et al., 2010). In the present study, we have shown that the effect of PEA on MPO activity in DNBS-treated animals was reduced by GW6471, a PPARα receptor antagonist. PEA-induced changes in colon weight/colon length ratio were also reduced by GW6471, although the effect did not yield a full statistical significance (P = 0.06). Collectively such results suggest an involvement of PPARα in PEA mode of action. Consistent with our results, Esposito et al. (2014) have recently demonstrated that MK866, another PPARα antagonist, almost completely abolished the anti-inflammatory effect of i.p. PEA in the DSS model of murine colitis as well as in human ulcerative colitis biopsies.
There is also good evidence to suggest a pivotal role for TRPV1 in the development and maintenance of IBD (Kaneko and Szallasi, 2014). Increased TRPV1 immunoreactivity was reported in colonic biopsies taken from patients with IBD (Domotor et al., 2005) and TRPV1 antagonists attenuate experimental colitis in mice (Kimball et al., 2004). In the present study, we found that PEA significantly reduced TRPV1 mRNA down-regulation induced by DNBS and, more importantly, it exerted a stronger anti-inflammatory effect in the presence of the TRPV1 antagonist capsazepine, suggesting that this protein may negatively modulate the pharmacological activity of PEA, despite the fact that PEA itself is capable of desensitizing this channel (Ambrosino et al., 2013). Consistent with such result, we have recently demonstrated that 5′-iodoresiniferatoxin, another TRPV1 antagonist, further increased the anti-prokinetic effect of PEA in a post-inflammatory murine model of accelerated gastrointestinal transit (Capasso et al., 2014). Furthermore, de Novellis et al. (2012) have recently reported that PEA decreased the burst and increased the latency of tail flick-evoked onset of ON cell activity in the rostral ventromedial medulla and such effect was enhanced by a TRPV1 antagonist.
In conclusion, our study demonstrates that PEA, an endogenous lipid chemically related to the endocannabinoid anandamide – which is also contained in animal and vegetable foods – is produced in the gut in response to an inflammatory insult and exerts intestinal anti-inflammatory effects when administered i.p. and, more importantly, p.o. The protective effect of p.o. PEA, which might justify the use of PEA as a treatment for IBD, was associated to changes in TRPV1 channels, GPR55 and CB1 receptor mRNA expression. Although we used pharmacological blockade rather than genetic deletion, our results clearly show that the effect of PEA is mediated by multiple targets, including CB2 receptors, GPR55 and PPARα and modulated by a TRPV1 channel antagonist.
Acknowledgments
This work was supported by a grant from Programma Operativo Nazionale ‘Research and development of bioregulators active on epigenetic mechanisms of inflammatory processes in chronic and degenerative diseases (BIAM-EPI)’, nr. 01_02512. BR is grateful to the Enrico and Enrica Sovena Foundation (Rome, Italy).
Glossary
- 2-AG
2-arachidonoylglycerol
- CB
cannabinoid
- DNBS
2,4,6-dinitrobenzenesulfonic acid
- DSS
dextran sodium sulphate
- FAAH
fatty acid amide hydrolase
- GDE1
glycerophosphodiester PDE 1
- IBD
inflammatory bowel disease
- MPO
myeloperoxidase
- NAAA
N-acylethanolamine-hydrolysing acid amidase
- NAPE-PLD
N-arachidonyl-phosphatidylethanolamine PLD
- OEA
oleoylethanolamide
- PEA
palmitoylethanolamide
- TRPV1
transient receptor potential vanilloid type-1
Author contributions
F. B. was responsible for acquisition, analysis and interpretation of data; conception and design; and redaction of the manuscript. B. R. and P. O. conducted acquisition, analysis and interpretation of data, and critical reading of the manuscript. S. P., E. P., R. C., D. C. and G. B. carried out acquisition, analysis and interpretation of data. V. D. M. was responsible for conception and design, analysis and interpretation of data, and critical reading of the manuscript. A. A. I. performed conception and design, analysis and interpretation of data, and redaction of the manuscript.
Conflict of interest
Vincenzo Di Marzo is co-inventor of patents claiming the use of palmitoylethanolamide against inflammatory conditions, and receives research support from Epitech Italia S.r.l., which markets palmitoylethanolamide. Stefania Petrosino is an employee of Epitech Italia S.r.l. The other authors declare that they have no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12907
Table S1 Primer sequences.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear Hormone Receptors. Br J Pharmacol. 2013c;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013d;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhouayek M, Muccioli GG. The endocannabinoid system in inflammatory bowel diseases: from pathophysiology to therapeutic opportunity. Trends Mol Med. 2012;18:615–625. doi: 10.1016/j.molmed.2012.07.009. [DOI] [PubMed] [Google Scholar]
- Alhouayek M, Lambert DM, Delzenne NM, Cani PD, Muccioli GG. Increasing endogenous 2-arachidonoylglycerol levels counteracts colitis and related systemic inflammation. FASEB J. 2011;25:2711–2721. doi: 10.1096/fj.10-176602. [DOI] [PubMed] [Google Scholar]
- Ambrosino P, Soldovieri MV, Russo C, Taglialatela M. Activation and desensitization of TRPV1 channels in sensory neurons by the PPARα agonist palmitoylethanolamide. Br J Pharmacol. 2013;168:1430–1444. doi: 10.1111/bph.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen SN, Rognum TO, Bakka A, Clausen OP. Ki-67: a useful marker for the evaluation of dysplasia in ulcerative colitis. Mol Pathol. 1998;51:327–332. doi: 10.1136/mp.51.6.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviello G, Romano B, Borrelli F, Capasso R, Gallo L, Piscitelli F, et al. Chemopreventive effect of the non-psychotropic phytocannabinoid cannabidiol on experimental colon cancer. J Mol Med (Berl) 2012;90:925–934. doi: 10.1007/s00109-011-0856-x. [DOI] [PubMed] [Google Scholar]
- Azuma YT, Nishiyama K, Matsuo Y, Kuwamura M, Morioka A, Nakajima H, et al. PPARα contributes to colonic protection in mice with DSS-induced colitis. Int Immunopharmacol. 2010;10:1261–1267. doi: 10.1016/j.intimp.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Balvers MG, Verhoeckx KC, Meijerink J, Wortelboer HM, Witkamp RF. Measurement of palmitoylethanolamide and other N-acylethanolamines during physiological and pathological conditions. CNS Neurol Disord Drug Targets. 2013;12:23–33. doi: 10.2174/1871527311312010007. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. J Biol Chem. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Cravatt BF. Chemical probes of endocannabinoid metabolism. Pharmacol Rev. 2013;65:849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrelli F, Izzo AA. Role of acylethanolamides in the gastrointestinal tract with special reference to food intake and energy balance. Best Pract Res Clin Endocrinol Metab. 2009;23:33–49. doi: 10.1016/j.beem.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Borrelli F, Aviello G, Romano B, Orlando P, Capasso R, Maiello F, et al. Cannabidiol, a safe and non-psychotropic ingredient of the marijuana plant Cannabis sativa, is protective in a murine model of colitis. J Mol Med (Berl) 2009;87:1111–1121. doi: 10.1007/s00109-009-0512-x. [DOI] [PubMed] [Google Scholar]
- Borrelli F, Fasolino I, Romano B, Capasso R, Maiello F, Coppola D, et al. Beneficial effect of the non-psychotropic plant cannabinoid cannabigerol on experimental inflammatory bowel disease. Biochem Pharmacol. 2013;85:1306–1316. doi: 10.1016/j.bcp.2013.01.017. [DOI] [PubMed] [Google Scholar]
- Burger D, Travis S. Conventional medical management of inflammatory bowel disease. Gastroenterology. 2011;140:1827–1837. doi: 10.1053/j.gastro.2011.02.045. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Capasso R, Izzo AA, Fezza F, Pinto A, Capasso F, Mascolo N, et al. Inhibitory effect of palmitoylethanolamide on gastrointestinal motility in mice. Br J Pharmacol. 2001;134:945–950. doi: 10.1038/sj.bjp.0704339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso R, Orlando P, Pagano E, Aveta T, Buono L, Borrelli F, et al. Palmitoylethanolamide normalizes intestinal motility in a murine model of post-inflammatory accelerated transit: involvement of CB1 receptors and TRPV1. Br J Pharmacol. 2014;171:4026–4037. doi: 10.1111/bph.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzzocrea S, Di Paola R, Mazzon E, Genovese T, Muià C, Centorrino T, et al. Role of endogenous and exogenous ligands for the peroxisome proliferators activated receptors alpha (PPAR-alpha) in the development of inflammatory bowel disease in mice. Lab Invest. 2004;84:1643–1654. doi: 10.1038/labinvest.3700185. [DOI] [PubMed] [Google Scholar]
- Darmani NA, Izzo AA, Degenhardt B, Valenti M, Scaglione G, Capasso R, et al. Involvement of the cannabimimetic compound, N-palmitoyl-ethanolamine, in inflammatory and neuropathic conditions: review of the available pre-clinical data, and first human studies. Neuropharmacology. 2005;48:1154–1163. doi: 10.1016/j.neuropharm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- D'Argenio G, Valenti M, Scaglione G, Cosenza V, Sorrentini I, Di Marzo V. Up-regulation of anandamide levels as an endogenous mechanism and a pharmacological strategy to limit colon inflammation. FASEB J. 2006;20:568–570. doi: 10.1096/fj.05-4943fje. [DOI] [PubMed] [Google Scholar]
- D'Argenio G, Petrosino S, Gianfrani C, Valenti M, Scaglione G, Grandone I, et al. Overactivity of the intestinal endocannabinoid system in celiac disease and in methotrexate-treated rats. J Mol Med (Berl) 2007;85:523–530. doi: 10.1007/s00109-007-0192-3. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Ligresti A, Bifulco M, Melck D, Di Marzo V. Effect on cancer cell proliferation of palmitoylethanolamide, a fatty acid amide interacting with both the cannabinoid and vanilloid signalling systems. Fundam Clin Pharmacol. 2002;16:297–302. doi: 10.1046/j.1472-8206.2002.00094.x. [DOI] [PubMed] [Google Scholar]
- De Schepper HU, De Man JG, Ruyssers NE, Deiteren A, Van Nassauw L, Timmermans JP, et al. TRPV1 receptor signaling mediates afferent nerve sensitization during colitis-induced motility disorders in rats. Am J Physiol Gastrointest Liver Physiol. 2008;294:G245–G253. doi: 10.1152/ajpgi.00351.2007. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Izzo AA. Endocannabinoid overactivity and intestinal inflammation. Gut. 2006;55:1373–1376. doi: 10.1136/gut.2005.090472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Orlando P, Bisogno T, Zagoory O, Bifulco M, et al. Palmitoylethanolamide inhibits the expression of fatty acid amide hydrolase and enhances the anti-proliferative effect of anandamide in human breast cancer cells. Biochem J. 2001;358:249–255. doi: 10.1042/0264-6021:3580249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Capasso R, Matias I, Aviello G, Petrosino S, Borrelli F, et al. The role of endocannabinoids in the regulation of gastric emptying: alterations in mice fed a high-fat diet. Br J Pharmacol. 2008;153:1272–1280. doi: 10.1038/sj.bjp.0707682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paola R, Impellizzeri D, Torre A, Mazzon E, Cappellani A, Faggio C, et al. Effects of palmitoylethanolamide on intestinal injury and inflammation caused by ischemia-reperfusion in mice. J Leukoc Biol. 2012;91:911–920. doi: 10.1189/jlb.0911485. [DOI] [PubMed] [Google Scholar]
- Diep TA, Madsen AN, Holst B, Kristiansen MM, Wellner N, Hansen SH, et al. Dietary fat decreases intestinal levels of the anorectic lipids through a fat sensor. FASEB J. 2011;25:765–774. doi: 10.1096/fj.10-166595. [DOI] [PubMed] [Google Scholar]
- Domotor A, Peidl Z, Vincze A, Hunyady B, Szolcsányi J, Kereskay L, et al. Immunohistochemical distribution of vanilloid receptor, calcitonin-gene related peptide and substance P in gastrointestinal mucosa of patients with different gastrointestinal disorders. Inflammopharmacology. 2005;13:161–177. doi: 10.1163/156856005774423737. [DOI] [PubMed] [Google Scholar]
- Esposito E, Cuzzocrea S. Palmitoylethanolamide in homeostatic and traumatic central nervous system injuries. CNS Neurol Disord Drug Targets. 2013;12:55–61. doi: 10.2174/1871527311312010010. [DOI] [PubMed] [Google Scholar]
- Esposito G, Capoccia E, Turco F, Palumbo I, Lu J, Steardo A, et al. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-α activation. Gut. 2014;63:1300–1312. doi: 10.1136/gutjnl-2013-305005. [DOI] [PubMed] [Google Scholar]
- Farquhar-Smith WP, Jaggar SI, Rice AS. Attenuation of nerve growth factor-induced visceral hyperalgesia via cannabinoid CB(1) and CB(2)-like receptors. Pain. 2002;97:11–21. doi: 10.1016/s0304-3959(01)00419-5. [DOI] [PubMed] [Google Scholar]
- Fu J, Astarita G, Gaetani S, Kim J, Cravatt BF, Mackie K, et al. Food intake regulates oleoylethanolamide formation and degradation in the proximal small intestine. J Biol Chem. 2007;282:1518–1528. doi: 10.1074/jbc.M607809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi P, Orlando P, Di Siena S, Lolicato F, Petrosino S, Bisogno T, et al. The endocannabinoid system and pivotal role of the CB2 receptor in mouse spermatogenesis. Proc Natl Acad Sci U S A. 2009;106:11131–11136. doi: 10.1073/pnas.0812789106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WS, Barrett DA, Randall MD. ‘Entourage’ effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br J Pharmacol. 2008;155:837–846. doi: 10.1038/bjp.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P. Transient receptor potential (TRP) channels as drug targets for diseases of the digestive system. Pharmacol Ther. 2011;131:142–170. doi: 10.1016/j.pharmthera.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis O, Varnalidis I, Paraskevas G, Botsios D. Nutritional modulation of the inflammatory bowel response. Digestion. 2011;84:89–101. doi: 10.1159/000323456. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Camilleri M. Cannabinoids in intestinal inflammation and cancer. Pharmacol Res. 2009;60:117–125. doi: 10.1016/j.phrs.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Piscitelli F, Capasso R, Marini P, Cristino L, Petrosino S, et al. Basal and fasting/refeeding-regulated tissue levels of endogenous PPAR-alpha ligands in Zucker rats. Obesity (Silver Spring) 2010;18:55–62. doi: 10.1038/oby.2009.186. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Capasso R, Aviello G, Borrelli F, Romano B, Piscitelli F, et al. Inhibitory effect of cannabichromene, a major non-psychotropic cannabinoid extracted from Cannabis sativa, on inflammation-induced hypermotility in mice. Br J Pharmacol. 2012;166:1444–1460. doi: 10.1111/j.1476-5381.2012.01879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y, Szallasi A. Transient receptor potential (TRP) channels: a clinical perspective. Br J Pharmacol. 2014;171:2474–2507. doi: 10.1111/bph.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppel Hesselink JM, de Boer T, Witkamp RF. Palmitoylethanolamide: a natural body-own anti-inflammatory agent, effective and safe against influenza and common cold. Int J Inflam. 2013;2013:151028. doi: 10.1155/2013/151028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball ES, Wallace NH, Schneider CR, D'Andrea MR, Hornby PJ. Vanilloid receptor 1 antagonists attenuate disease severity in dextran sulphate sodium-induced colitis in mice. Neurogastroenterol Motil. 2004;16:811–818. doi: 10.1111/j.1365-2982.2004.00549.x. [DOI] [PubMed] [Google Scholar]
- Koning M, Ailabouni R, Gearry RB, Frampton CM, Barclay ML. Use and predictors of oral complementary and alternative medicine by patients with inflammatory bowel disease: a population-based, case-control study. Inflamm Bowel Dis. 2013;19:767–778. doi: 10.1097/MIB.0b013e31827f27c8. [DOI] [PubMed] [Google Scholar]
- Kotsikorou E, Sharir H, Shore DM, Hurst DP, Lynch DL, Madrigal KE, et al. Identification of the GPR55 antagonist binding site using a novel set of high-potency GPR55 selective ligands. Biochemistry. 2013;52:9456–9469. doi: 10.1021/bi4008885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawisz JE, Sharon P, Stenson WF. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–1350. [PubMed] [Google Scholar]
- Lambert DM, Di Marzo V. The palmitoylethanolamide and oleamide enigmas: are these two fatty acid amides cannabimimetic? Curr Med Chem. 1999;6:757–773. [PubMed] [Google Scholar]
- Li K, Feng JY, Li YY, Yuece B, Lin XH, Yu LY, et al. Anti-inflammatory role of cannabidiol and O-1602 in cerulein-induced acute pancreatitis in mice. Pancreas. 2013;42:123–129. doi: 10.1097/MPA.0b013e318259f6f0. [DOI] [PubMed] [Google Scholar]
- Li Y, Yang L, Chen L, Zhu C, Huang R, Zheng X, et al. Design and synthesis of potent N-acylethanolamine-hydrolyzing acid amidase (NAAA) inhibitor as anti-inflammatory compounds. PLoS ONE. 2012;7:e43023. doi: 10.1371/journal.pone.0043023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin XH, Yuece B, Li YY, Feng YJ, Feng JY, Yu LY, et al. A novel CB receptor GPR55 and its ligands are involved in regulation of gut movement in rodents. Neurogastroenterol Motil. 2011;23:862–e342. doi: 10.1111/j.1365-2982.2011.01742.x. [DOI] [PubMed] [Google Scholar]
- Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- Löwenberg M, D'Haens G. Novel targets for inflammatory bowel disease therapeutics. Curr Gastroenterol Rep. 2013;15:311. doi: 10.1007/s11894-012-0311-3. [DOI] [PubMed] [Google Scholar]
- Massa F, Marsicano G, Hermann H, Cannich A, Monory K, Cravatt BF, et al. The endogenous cannabinoid system protects against colonic inflammation. J Clin Invest. 2004;113:1202–1209. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naderi N, Majidi M, Mousavi Z, Khoramian Tusi S, Mansouri Z, Khodagholi F. The interaction between intrathecal administration of low doses of palmitoylethanolamide and AM251 in formalin-induced pain related behavior and spinal cord IL1-β expression in rats. Neurochem Res. 2012;37:778–785. doi: 10.1007/s11064-011-0672-2. [DOI] [PubMed] [Google Scholar]
- de Novellis V, Luongo L, Guida F, Cristino L, Palazzo E, Russo R, et al. Effects of intra-ventrolateral periaqueductal grey palmitoylethanolamide on thermoceptive threshold and rostral ventromedial medulla cell activity. Eur J Pharmacol. 2012;676:41–50. doi: 10.1016/j.ejphar.2011.11.034. [DOI] [PubMed] [Google Scholar]
- Opheim R, Hoivik ML, Solberg IC, Moum B. Complementary and alternative medicine in patients with inflammatory bowel disease: the results of a population-based inception cohort study (IBSEN) J Crohns Colitis. 2012;6:345–353. doi: 10.1016/j.crohns.2011.09.007. [DOI] [PubMed] [Google Scholar]
- O'Sullivan SE, Kendall DA. Cannabinoid activation of peroxisome proliferator-activated receptors: potential for modulation of inflammatory disease. Immunobiology. 2010;215:611–616. doi: 10.1016/j.imbio.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. GPR55: a new member of the cannabinoid receptor clan? Br J Pharmacol. 2007;152:984–986. doi: 10.1038/sj.bjp.0707464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino S, Iuvone T, Di Marzo V. N-palmitoyl-ethanolamine: biochemistry and new therapeutic opportunities. Biochimie. 2010;92:724–727. doi: 10.1016/j.biochi.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Borrelli F, Pagano E, Di Marzo V, Izzo AA. Effect of Palmitoylethanolamide in a Murine Model of Colitis. Turin, Italy: 36° Congresso Nazionale della Società Italiana di Farmacologia; 2013. [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano B, Borrelli F, Fasolino I, Capasso R, Piscitelli F, Cascio M, et al. The cannabinoid TRPA1 agonist cannabichromene inhibits nitric oxide production in macrophages and ameliorates murine colitis. Br J Pharmacol. 2013;169:213–229. doi: 10.1111/bph.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schicho R, Storr M. A potential role for GPR55 in gastrointestinal functions. Curr Opin Pharmacol. 2012;12:653–658. doi: 10.1016/j.coph.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schicho R, Bashashati M, Bawa M, McHugh D, Saur D, Hu HM, et al. The atypical cannabinoid O-1602 protects against experimental colitis and inhibits neutrophil recruitment. Inflamm Bowel Dis. 2011;17:1651–1664. doi: 10.1002/ibd.21538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, Facci L. Mast cell-glia axis in neuroinflammation and therapeutic potential of the anandamide congener palmitoylethanolamide. Philos Trans R Soc Lond B Biol Sci. 2012;367:3312–3325. doi: 10.1098/rstb.2011.0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart D, Jonsson KO, Vandevoorde S, Lambert DM, Fowler CJ. Entourage' effects of N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and upon anandamide metabolism. Br J Pharmacol. 2002;136:452–458. doi: 10.1038/sj.bjp.0704732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S, et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc Natl Acad Sci U S A. 2009;106:20966–20971. doi: 10.1073/pnas.0907417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solorzano C, Antonietti F, Duranti A, Tontini A, Rivara S, Lodola A, et al. Synthesis and structure-activity relationships of N-(2-oxo-3-oxetanyl)amides as N-acylethanolamine-hydrolyzing acid amidase inhibitors. J Med Chem. 2010;53:5770–5781. doi: 10.1021/jm100582w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storr MA, Keenan CM, Emmerdinger D, Zhang H, Yüce B, Sibaev A, et al. Targeting endocannabinoid degradation protects against experimental colitis in mice: involvement of CB1 and CB2 receptors. J Mol Med. 2008;86:925–936. doi: 10.1007/s00109-008-0359-6. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Takezaki N, Ueda N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA) Chem Biodivers. 2007;4:1914–1925. doi: 10.1002/cbdv.200790159. [DOI] [PubMed] [Google Scholar]
- Ueda N, Tsuboi K, Uyama T. Metabolism of endocannabinoids and related N-acylethanolamines: canonical and alternative pathways. FEBS J. 2013;280:1874–1894. doi: 10.1111/febs.12152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primer sequences.